

Abstract
Locus ceruleus (LC) is the main subcortical site of norepinephrine synthesis. In Alzheimer's disease (AD) patients and rodent models, degeneration of LC neurons and reduced levels of norepinephrine in LC projection areas are significantly correlated with the increase in amyloid plaques, neurofibrillary tangles, and severity of dementia. Activated microglia play a pivotal role in the progression of AD by either clearing amyloid β peptide (Aβ) deposits through uptake of Aβ or releasing cytotoxic substances and proinflammatory cytokines. Here, we investigated the effect of norepinephrine on Aβ uptake and clearance by murine microglia and explored the underlying mechanisms. We found that murine microglia cell line N9 and primary microglia expressed β2 adrenergic receptor (AR) but not β1 and β3AR. Norepinephrine and isoproterenol upregulated the expression of Aβ receptor mFPR2, a mouse homolog of human formyl peptide receptor FPR2, through activation of β2AR in microglia. Norepinephrine also induced mFPR2 expression in mouse brain. Activation of β2AR in microglia promoted Aβ42 uptake through upregulation of mFPR2 and enhanced spontaneous cell migration but had no effect on cell migration in response to mFPR2 agonists. Furthermore, activation of β2AR on microglia induced the expression of insulin-degrading enzyme and increased the degradation of Aβ42. Mechanistic studies showed that isoproterenol induced mFPR2 expression through ERK1/2-NF-κB and p38-NF-κB signaling pathways. These findings suggest that noradrenergic innervation from LC is needed to maintain adequate Aβ uptake and clearance by microglia, and norepinephrine is a link between neuron and microglia to orchestrate the host response to Aβ in AD.
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disease. It is the most frequent cause of dementia in the elderly. AD brain pathology is characterized by amyloid plaques, neurofibrillary tangles, and neuronal loss (Hardy and Higgins, 1992; Selkoe, 1999; Mudher and Lovestone, 2002). Activated microglia and astrocytes were usually found to be associated with the amyloid plaques. Activation of microglia leads to uptake and clearance of amyloid β peptide (Aβ) (Frautschy et al., 1998; Qiu et al., 1998; Simard et al., 2006; Hickman et al., 2008). However, excessive activation of microglia leads to production and release of inflammatory cytokines, nitric oxide, and reactive oxygen species, which contribute to neuronal dysfunction and cell death (Griffin et al., 1998).
The locus ceruleus (LC) is a noradrenergic nucleus located in the pontine tegmentum and serves as the main subcortical site for the synthesis of norepinephrine (NE) and its precursor enzymes (Freedman et al., 1975). Ascending noradrenergic axons project to the hippocampus, the frontal and entorhinal cortices, and to a minor extent to various other brain regions. Degeneration of LC neurons and reduced levels of NE in LC projection areas are well known features of AD. In AD patients the central portion of the LC, which is considered to project predominantly to the hippocampus, frontal and temporal cortex areas that are usually severely affected by senile plaque and neurofibrillary tangle formation, shows the most extensive loss of cells (Marcyniuk et al., 1986). The decreased LC neuron numbers are significantly correlated with the increase of amyloid plaques, neurofibrillary tangles, and severity of dementia (Marcyniuk et al, 1986; Bondareff et al., 1987; Grudzien et al., 2007; Weinshenker, 2008). In rodent AD models, neurotoxin N-(2-chloroethyl)-N-ethyl-bromo-benzylamine (DSP4)-induced LC neuron degeneration and NE deficiency exacerbate Aβ deposition, inflammation, neurodegeneration in brain, and cognitive impairment (Heneka et al., 2006; Kalinin et al., 2007). NE supplement can ameliorate the inflammation induced by LC degeneration in mouse AD models (Heneka et al., 2002). These results suggest that NE has protective effect against AD lesions. In vitro studies showed that NE could promote Aβ uptake by murine microglia (Kalinin et al., 2007; Heneka et al., 2010). However, the underlying mechanisms and the effect of NE on Aβ clearance and the expression of enzymes involved in Aβ degradation by microglia are not clear.
Our previous studies revealed that human G protein-coupled receptor formyl peptide receptor 2 (FPR2) and its mouse homolog mFPR2 are functional receptors for Aβ (Le et al., 2001; Tiffany et al., 2001; Yazawa et al., 2001). FPR2 is expressed at high levels by mononuclear (microglial) cells infiltrating senile plaques in brain tissues from AD patients (Le et al., 2001). FPR2/mFPR2 mediates the uptake of Aβ by microglia and the chemotactic activity of Aβ42 (Le et al., 2001; Yazawa et al., 2001; Iribarren et al., 2005; Chen et al., 2006). In this study, we report that activation of the β2 adrenergic receptor (AR) on microglial cells induces the expression of mFPR2 through ERK1/2-NFκB and p38 NF-κB-dependent signaling pathways (where ERK1/2 is extracellular signal-regulated kinase 1/2 and NF-κB is nuclear factor κB). Microglial cells activated by βAR agonist exhibited a markedly increased capacity to endocytose Aβ42 through mFPR2 and to degrade Aβ42 through upregulation of the Aβ-degrading enzyme, insulin-degrading enzyme (IDE).
Materials and Methods
Reagents.
Aβ42 was purchased from California Peptide Research. Aβ42 was dissolved in DMSO at a concentration of 2 mm as stock solution and kept at −70°C. Norepinephrine, isoproterenol (ISO), and propranolol were purchased from Sigma. W peptide (WKYMVm) and MMK-1 (LESIFRSLLFRVM) were synthesized and purified by the Department of Biochemistry, Colorado State University (Fort Collins, CO). SB203580, PD098059, SP600125, and BAY117082 were obtained from Calbiochem. Antibodies against total or phosphorylated ERK1/2, p38, c-jun N-terminal kinase (JNK), and IκBα were purchased from Cell Signaling Technology. Mouse anti-Aβ antibody and anti-actin monoclonal antibody were from Sigma. The 6E10 antibody against Aβ was from Covance. Antibodies against neprilysin and insulin-degrading enzyme were from Millipore. HRP-conjugated secondary antibodies were from Proteintech. DMEM and Iscove's modified Dulbecco's medium (IMDM) were purchased from Invitrogen. Fetal bovine serum (FBS) was from Hyclone.
Cell culture.
Primary mouse microglia was isolated and purified from whole brain of newborn C57BL/6 mice as described previously (Xu and Drew, 2006). The experiments using animals were in accordance with the National Institutes of Heath Guide for the Care and Use of Laboratory Animals and were approved by the Biological Research Ethics Committee, Institute for Nutritional Sciences, Chinese Academy of Sciences (Shanghai, China). Murine microglial cell line N9 was grown in IMDM supplemented with 5% heat-inactivated FBS, 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 μm 2-mercaptoehtanol (2-ME). mFPR2/293 cells expressing myc-tagged mFPR2 (Ying et al., 2004) were maintained in DMEM containing 10% FCS, 800 μg/ml G418, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Reverse transcription PCR and quantitative real-time PCR.
Total RNA was extracted from cells with TRIzol reagent (Invitrogen) and depleted of contaminating DNA with RNase-free DNase. Reverse transcription (RT) of RNA was performed with Moloney murine leukemia virus reverse transcriptase (Fermentas) and oligo(dT) according to the manufacturer's protocol. The primers of PCR were: β1AR (sense: 5′-TCGCTACCAGAGTTTGCTGACGC, antisense: 5′-AACACCCGCAGGTACACGAAGG); β2AR (sense: 5′-CGACTACAAACCGTCACCAA-3′, antisense:5′-ATAGCGATCCACTGCAATCA-3′); β3AR (sense: 5′-ATGACTAGACCACGAGGTGTAAGG-3′, antisense: 5′-CAGGGTTAGGAAAGCAGTAGGA-3′); mFPR1 (sense: 5′-CAGCCTGTACTTTCGACTTCTCC-3′, antisense: 5′-ATTGGTGCCTGTATCACTGGTCT-3′); mFPR2 (sense: 5′-CTTTATCTGCTGGTTTCCCTTTC-3′, antisense: 5′-CTGGTGCTTGAATCACTGGTTTG-3′); β-actin (sense: 5′-TGTGATGGTGGGAATGGGTCAG-3′, antisense: 5′-TTTGATGTCACGCACGATTTCC-3′). PCR products were visualized by ethidium bromide staining in 1.5% agarose gel and quantified using Gel-Pro Analyzer software (Media Cybernetics). All experiments were replicated at least three times.
Relative quantification of gene expression was conducted with the ABI Prism 7500 sequence detector (Applied Biosystems Inc). PCR results were normalized to the expression of β-actin. Real-time PCR primers for mFPR2 were as follows: 5′-CATGGGCCAGGACTTTCGT-3′ (sense) and 5′-TGGTGCTTGAATCACTGGTTTG-3′ (antisense). Mouse β-actin primers were: 5′-CAACGAGCGGTTCCGAT-3′ (sense); and 5′-GCCACAGGATTCCATACCCA-3′ (antisense).
Intracerebroventricular injection.
Male C57BL/6 mice (8 weeks old) were used in the study. Animals were anesthetized with chloral hydrate (0.18 ml of 6% chloral hydrate per mouse) and immobilized in a stereotaxic apparatus, and the skulls were exposed for trepanation. Five microliters of 1 μm NE (n = 6) or the same volume of PBS (n = 7) was injected into the lateral ventricle. Coordinates were determined according to a mouse stereotactic atlas as follows: −1 mm posterior, 1 mm lateral, 1.7 mm ventral to bregma. After 24 h, mice were killed by cervical dislocation. The brains were removed, washed in PBS, and homogenized in TRIzol reagent. RNA was extracted and examined for mFPR2 mRNA level by real-time PCR.
Calcium mobilization assay.
Ca2+ mobilization was measured with FlexStation II384 system (Molecular Devices) as described previously (Zhu et al., 2009). Briefly, N9 cells were loaded with Fura-3 AM, washed, and resuspended in emission buffer at a density of 4 × 106 cells/L for 15 min. The fluorescence before and after the addition of different concentrations of W peptide was measured with the excitation wavelength at 485 nm and the emission wavelength at 525 nm and presented as relative fluorescence units (RFU).
mFPR2 short hairpin RNA constructs, transfection, and virus transduction.
To construct mFPR2 short hairpin RNA (shRNA) expression plasmid, two complementary mFPR2 DNA oligos (see below) were synthesized, annealed, and inserted between BbsI and XhoI sites of the plasmid pBS-hU6-I. The fragment containing human U6-RNA Pol III promoter and mFPR2 shRNA then were subcloned into FG12 between the XbaI and XhoI sites. FG12 vector also expresses a human UbiC-driven green fluorescent protein (GFP) gene to provide a marker for tracking transduced cells. The resulting plasmids were confirmed by restriction enzyme digestion and DNA sequencing. mFPR2 target sequences are nucleotides 297–315 (ATTAGTTCACATTGTGGTA) (shRNA-a), 537–555 (CTTTGGATCCTGGGCTCAA) (shRNA-b), and 836–854 (TTCTTGACATGTTTGTTAA) (shRNA-c) of mouse FPR2 mRNA (GeneBank accession no. NM_008039). BLAST (Basic Local Alignment Search Tool) search against mouse genome sequences confirmed that only mFPR2 gene was targeted. Recombinant lentiviruses were produced by transient transfection of HEK293T cells with lentiviral mFPR2 shRNA vector, the lentiviral packaging constructs pRSV/REV and pMDLG/pRRE, and the vesicular stomatitis virus glycoprotein (VSVG)-expressing plasmid pHCMVG using the calcium phosphate method, as described previously (Tiscornia et al., 2006). mFPR2/293 cells and N9 cells were transduced with concentrated lentivirus stocks at the same multiplicity of infection in the presence of 4 μg/ml polybrene. After 3∼7 days, GFP-positive cells were sorted by fluorescence-activated cell sorter and kept growing for future use.
Chemotaxis assay.
Chemotaxis assays for N9 cells pretreated with 10 μm ISO or 500 ng/ml lipopolysaccharide (LPS) for 24 h and mFPR2/293 cells transduced with control lentiviruses or mFPR2 shRNA lentiviruses were performed with 48-well chemotaxis chambers (NeuroProbe) as described previously (Cui et al., 2002a). Polycarbonate filters (8 μm pore size, incubated for 90 min at 37°C) were used for measurement of N9 cell migration. For mFPR2/293 cells, 10 μm pore size, collagen type I-coated filters (incubated for 4 h) were used. The results are expressed as the mean ± SD of migrated cells in three high-power fields in triplicate samples or as chemotaxis index, which represents the fold increase in the number of migrated cells in response to chemoattractants over the spontaneous cell migration (to control medium).
Immunofluorescence confocal microscopy.
N9 cells or primary microglia grown on slides in a 24-well plate were treated with 10 μm ISO at 37°C for 24 h, followed by treatment with or without 10 μm Aβ42 for 30 min, and then fixed in 4% paraformaldehyde for 10 min at room temperature. Slides were washed with PBS and incubated with 5% BSA in PBS with 0.05% Tween 20 at room temperature for 1 h to reduce nonspecific binding of antibodies to the cell surface and for cell permeabilization. A mouse anti-Aβ antibody was applied to the slides and incubated overnight at 4°C. The slides were washed and incubated with Alexa Fluor 546-conjugated goat anti-mouse IgG (Invitrogen) for 60 min, washed in PBS, stained with Hoechst 33342, and mounted. Immunofluorescence labeling was observed under a laser scanning confocal microscope (LSM 510 META, Zeiss). Cells in three high-power fields (each includes ∼40 cells) were quantified for amyloid uptake by using ImageJ program (http://rsbweb.nih.gov/ij).
Immunoblot analysis.
Western blotting was performed as described previously (Wang et al., 2008). The following primary antibodies were used: antibodies against total or phosphorylated ERK1/2, p38, JNK, IκBα; antibodies against neprilysin and IDE. Primary antibodies were recognized by HRP-conjugated secondary antibodies. Immunoblot results were quantified by using Gel-Pro Analyzer software (Media Cybernetics).
To examine Aβ42 uptake and degradation by N9 cells and murine primary microglia, Aβ42 was oligomerized as described previously (Tahara et al., 2006). After treatment of the cells with Aβ42 for 12 h, Aβ level in the supernatant and cell lysate was detected by Western blot as described previously (Schägger, 2006). Briefly, 20 μl of supernatant or 50 μg of cell lysate were boiled for 5 min and underwent Tris-Tricine SDS–PAGE and electrotransferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore). The membrane was blocked with 5% nonfat milk and then incubated with anti-Aβ antibody (6E10), or anti-actin monoclonal antibody overnight at 4°C. After incubation with HRP-conjugated secondary antibody, the protein bands were detected with a SuperSignal West Pico chemiluminescent substrate (Pierce) and X-Omat BT film (Eastman Kodak).
Statistics.
The significance of the difference between test and control groups was analyzed with Student's t test, and p values ≤0.05 were considered statistically significant.
Results
Norepinephrine and isoproterenol upregulate mFPR2 expression through activation of β2AR
Human G protein-coupled receptor FPR2 and its mouse homolog mFPR2 have been reported to be the receptor for Aβ42. FPR2/mFPR2 mediates the internalization of Aβ42 by microglia (Yazawa et al., 2001; Iribarren et al., 2005; Chen et al., 2006). We first checked the effect of NE and βAR agonist ISO on mFPR2 mRNA expression in N9 cells by real-time PCR. As shown in Figure 1, NE (Fig. 1A) and ISO (Fig. 1C) both upregulated mFPR2 mRNA level in a dose-dependent manner. The inductive effect of these two compounds on mFPR2 expression was also confirmed in mouse primary microglia (Fig. 1B,D). In C57BL/6 mice injected with 1 μm NE in the lateral ventricle (5 μl/mouse, n = 6), the mFPR2 mRNA level in brain at 24 h after injection was elevated to 1.40 ± 0.29-fold (p = 0.02) of control mice injected with same volume of PBS (n = 7), demonstrating the upregulation of mFPR2 expression by NE in vivo.
Figure 1.

Norepinephrine and isoproterenol induce mFPR2 gene expression in microglia and brain. N9 cells (A, C) or murine primary microglia (B, D) were incubated with different concentrations of NE (A, B) for 6 h or ISO (C, D) for 9 h, and then total RNA was extracted and examined for mFPR2 mRNA level by real-time PCR. Results represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 versus cells cultured with control medium.
To investigate the involvement of βAR in the upregulation of mFPR2 by NE and ISO, we examined the expression of βAR in N9 cells and primary microglia by RT-PCR. The results showed that both N9 cells and primary microglia express β2AR but not β1 and β3 ARs (Fig. 2A). Pretreatment of N9 cells with the β1,2AR antagonist propranolol significantly inhibited the inductive effect of NE and ISO on mFPR2 expression (Fig. 2B and C), suggesting that NE and ISO upregulate mFPR2 through activation of β2AR.
Figure 2.

Norepinephrine and isoproterenol upregulate mFPR2 expression through activation of β2AR. A, The expression of β adrenergic receptors in N9 cells and mouse primary microglia was examined by RT-PCR. B, C, N9 cells pretreated with 10 μm propranolol (Pro) for 1 h were incubated with or without 1 μm NE for 6 h (B) or 10 μm ISO for 9 h (C), and total RNA was extracted and examined for mFPR2 mRNA level by real-time PCR. Results represent the mean ± SD of three independent experiments. *p < 0.05 versus cells cultured with control medium. #p < 0.05, ##p < 0.01, compared with cells treated with NE or ISO alone.
We then checked whether the mFPR2 upregulated by isoproterenol is functional. Calcium mobilization is a signaling event of mFPR2 activation (Tiffany et al., 2001). N9 cells without ISO treatment had no response to W peptide, a ligand for mFPR2 (Fig. 3A). However, after treatment of N9 cells with ISO for 24 h, W peptide induced a robust and transient calcium flux (Fig. 3B). Although W peptide is also reported to be an agonist for mFPR1 (He et al., 2000), ISO had no significant effect on mFPR1 expression in N9 cells (Fig. 3C). These results indicate that mFPR2 induced by ISO could mediate cell response to FPR2 ligand. Together, the above results demonstrated that activation of β2AR on mouse microglia could increase the expression of mFPR2 and its response to agonist. As N9 cells and primary microglia both only express β2AR and respond to NE and ISO in a similar way, we used ISO to treat microglia in most of the following experiments.
Figure 3.

Isoproterenol treatment of microglia increases calcium mobilization but not cell migration in response to mFPR2 agonists. A–C, N9 cells were treated with or without 40 μm ISO for 24 h and then loaded with Fura-3-AM and stimulated with different concentrations of W peptide and measured florescence (RFU) (A, B) or examined for mFPR1 and mFPR2 expression by RT-PCR (C). Data shown in A and B are representative of three independent experiments with similar results. Data in C represent the mean ± SD of three independent experiments. **p < 0.01, compared with cells cultured in medium alone. D, E, N9 cells were cultured in the presence or absence of 500 ng/ml LPS or 10 μm ISO at 37°C for 24 h and then examined for migration in response to W peptide (D), and MMK-1 (E). The results are expressed as the mean ± SD of migrated cells in three high-power fields in triplicate samples, **p < 0.01, cell migration in response to W peptide or MMK-1 compared to medium control; ##p < 0.01, comparison of spontaneous cell migration between N9 cells treated with ISO and LPS. F, Visualization of N9 cell migration in response to W peptide.
As NE and ISO have been reported to enhance microglial cell migration (Heneka et al., 2010), we then examined whether these compounds promote cell migration through upregulation of mFPR2. We found that pretreatment of N9 cells with ISO significantly increased spontaneous cell migration but had no significant effect on cell migration in response to W peptide (Fig. 3D,F) and MMK-1 (Fig. 3E), a potent mFPR2 agonist derived from random peptide library (Hu et al., 2001). As positive control, pretreatment of N9 cells with LPS significantly augmented cell migration in response to W peptide and MMK-1, confirming our previous report that LPS increases microglial cell migration to mFPR2 agonists through upregulation of mFPR2 expression (Cui et al., 2002a). These results suggest that activation of β2AR on microglia enhance cell migration independently of mFPR2.
Isoproterenol enhances Aβ42 uptake by microglia through mFPR2
The effect of ISO on Aβ uptake by microglia was examined by immunofluorescence confocal microscopy. We found that ISO treatment could significantly increase the internalization of Aβ42 in primary murine microglia (Fig. 4A) and N9 cells (data not shown) after incubation with Aβ42 for 30 min. These results were consistent with previous reports (Kalinin et al., 2007; Heneka et al., 2010).
Figure 4.
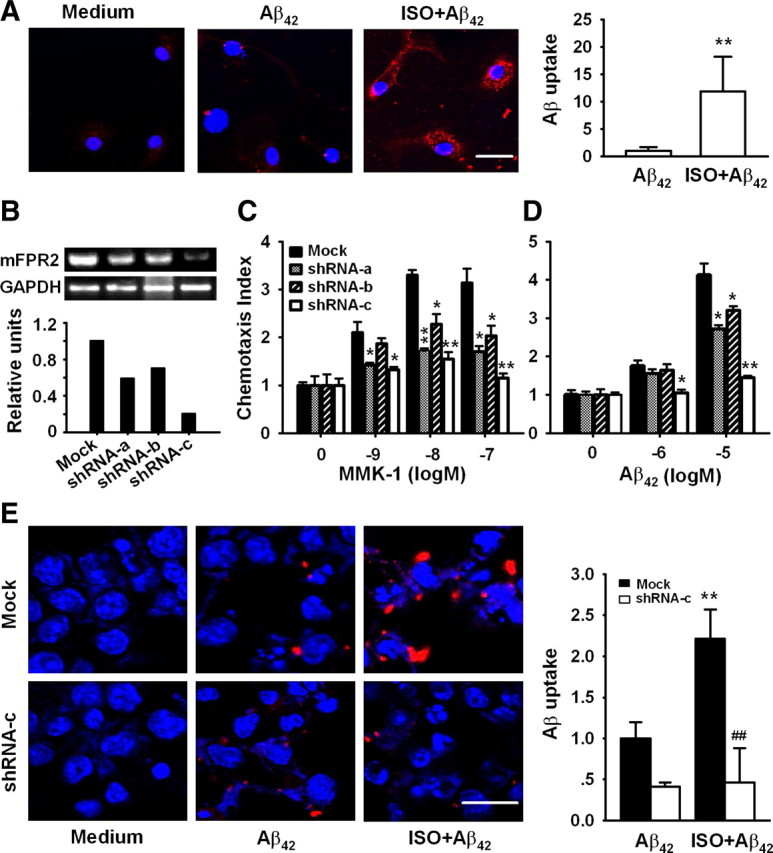
Isoproterenol enhances Aβ42 uptake by microglia. A, E, Primary microglia (A), N9 cells (E) transduced with control lentivirus (Mock), or mFPR2 shRNA-c lentivirus (shRNA-c) were treated with or without 10 μm ISO for 24 h followed by incubation with 10 μm Aβ42 for 30 min and then examined for Aβ uptake with antibody against Aβ (red). Nuclei were stained with Hoechst (blue). Scale bars, 20 μm. Dara are expressed as mean ± SD (n = 2, with duplicate samples in each experiment); **p < 0.01, compared with Aβ uptake by primary microglia or mock transduced N9 cells without ISO treatment; ##p < 0.01, compared with Aβ uptake by mock transduced N9 cells pretreated with ISO. B–D, mFPR2/293 cells were transduced with control lentiviruses (Mock) or mFPR2 shRNA lentiviruses, mFPR2 mRNA level was examined by RT-PCR (B), and cell migration in response to mFPR2 agonists (C: MMK-1; D: Aβ42) was examined by chemotaxis assay. Data are expressed as mean ± SD (n = 3); *p < 0.05, **p < 0.01, chemotaxis index of mFPR2 shRNA lentivirus transduced cells versus control lentivirus transduced cells (Mock).
To examine whether β2AR activation enhances the capability of microglia to uptake Aβ through upregulation of FPR2, we used a RNA interference technique to inhibit mFPR2 expression. Three lentiviral mFPR2 shRNA expression vectors targeting the 297–315 (shRNA-a), 537–555 (shRNA-b), and 836–854 (shRNA-c) regions of mFPR2 coding sequence were constructed. mFPR2/293 cells transduced with various mFPR2 shRNA lentiviruses inhibited mFPR2 expression at mRNA and protein levels with different potency (Fig. 4B and data not shown). The inhibitory effect of mFPR2 shRNA-c is the most potent. To determine whether expression of mFPR2 shRNA could induce functional silencing of mFPR2, we examined the effect of mFPR2 shRNA on chemotactic response to mFPR2 agonists. mFPR2/293 cells expressing mFPR2 shRNA-c showed a markedly reduced chemotactic response to MMK-1 (Fig. 4C) and Aβ42, the major component of amyloid plaque in AD brain (Fig. 4D). mFPR2 shRNA-a and mFPR2 shRNA-b showed certain levels of attenuation of agonist-induced cell migration, albeit with lower efficacy compared with mFPR2 shRNA-c (Fig. 4C,D). Thus, mFPR2 shRNA-c is a selective and highly efficacious mFPR2 shRNA-expressing vector that attenuates both mFPR2 expression and its key function. It was used in the next experiments.
Similar to the results from primary microglia, treatment of control lentivirus-transduced N9 cells with 10 μm ISO markedly increased the uptake of Aβ42 (Fig. 4E, top). However, transduction of N9 cells with mFPR2 shRNA-c lentivirus abrogated the uptake of Aβ42 by ISO-activated microglial cells (Fig. 4E, bottom). These results suggest that ISO enhances Aβ42 uptake by microglia through upregulation of mFPR2.
Activation of β2AR on microglial cells increases the protein level of IDE and enhances Aβ degradation
We then examined the effect of β2AR activation on the expression of enzymes involved in Aβ degradation, including IDE and neprilysin. Western blot analysis showed that pretreatment of N9 cells with 10 μm ISO significantly induced increase of IDE expression at 12 and 24 h and enhanced neprilysin expression at 24 h (Fig. 5A). Treatment with 1 μm NE or 10 μm ISO also significantly induced IDE expression in primary microglia (Fig. 5B), suggesting that β2AR activation enhances IDE expression in microglia. However, neprilysin expression could hardly be detected in primary microglia with or without NE/ISO treatment (data not shown).
Figure 5.

Activation of β2AR on microglia cells increases the protein level of IDE and enhances Aβ degradation. A, N9 cells were treated with 10 μm ISO for 12 and 24 h and cell lysates were subjected to immunoblot analysis with antibodies against IDE, neprilysin (NEP), or β-actin. B, Primary microglia were treated with 10 μm ISO or 1 μm NE for 12 and 24 h and then examined for IDE expression by immunoblot analysis. Data are the mean ± SD (n = 3); *p < 0.05, **p < 0.01, compared with cells cultured in medium alone. C–F, N9 cells (C, D) or primary microglia (E, F) pretreated with control medium, 10 μm ISO, or 10 μm ISO combined with 100 μg/ml insulin for 24 h were incubated with 2 μm Aβ42 for an additional 12 h and then examined for Aβ in cell lysate and supernatant with Western blot. Data are the mean ± SD (n = 3); **p < 0.01, compared with Aβ in cells treated with Aβ42 alone; ##p < 0.01, compared with Aβ in cells treated with ISO followed by Aβ42.
To further check the effect of β2AR activation on Aβ degradation, we incubated microglial cells with Aβ42 for a long time (12 h) and examined the ingested Aβ with Western blot analysis. Intracellular aggregates of Aβ were readily detectable in the cell lysate from N9 cells (Fig. 5C,D) and primary microglia (Fig. 5E,F). Pretreatment of N9 cells and primary microglia with ISO for 24 h significantly decreased Aβ in supernatant and cell lysate (especially the low molecular weight aggregates). These results, together with data in Figure 4, A and E, demonstrated that activation of β2AR on microglia promoted Aβ uptake and degradation. We further evaluate the contribution of IDE and neprilysin to ISO-induced Aβ degradation. Treatment of N9 cells (Fig. 5D) or primary microglia (Fig, 5F) with insulin, a competitive inhibitor of IDE, significantly inhibited ISO-induced Aβ clearance, suggesting ISO enhances Aβ degradation through induction of IDE. However, neprilysin inhibitor phosphoramidon could not suppress ISO-induced Aβ clearance in both N9 cells and primary microglia (data not shown). Together, activation of β2AR on microglia promoted Aβ uptake and degradation by upregulating the expression and function of mFPR2 and IDE, respectively.
Isoproterenol induces FPR2 mRNA expression in microglia through MAP kinases and NF-κB
We further explored the mechanisms underlying FPR2 upregulation by β2AR activation. Because the MAPK (mitogen-activated protein kinase) pathway has been implicated in the induction of mFPR2 in microglial cells via activation of TLR4 by LPS (Cui et al., 2002a) and MAP kinases are downstream signaling molecules of βAR activation (Liu et al., 2006; Gong et al., 2008; Qian et al., 2009), we assessed the role of MAPKs in ISO induction of mFPR2 in microglial cells. As shown in Figure 6A, ISO induced a rapid and potent phosphorylation of p38 MAPK in N9 cells. In cells pretreated with SB203580, a p38 MAPK inhibitor, the level of mFPR2 mRNA induced by ISO was significantly reduced (Fig. 6B). ISO additionally induced increased phosphorylation of ERK1/2, and the MEK (MAPK kinase) inhibitor PD98059 dose-dependently inhibited ERK1/2 phosphorylation and mFPR2 expression induced by ISO (Fig. 6C,D). Although ISO also induced JNK phosphorylation in N9 cells, JNK inhibitor SP600125 had no effect on ISO-induced mFPR2 expression (data not shown). These results indicate that MAPKs p38 and ERK1/2 play an essential role in ISO induction of the mFPR2 gene in mouse microglial cells.
Figure 6.

Isoproterenol induces mFPR2 mRNA expression in microglia through activation of MAP kinases. N9 cells were treated with 10 μm ISO for different periods of time (A) or treated with or without different concentrations of PD98059 (PD) (C) for 1 h followed by 10 μm ISO for 30 min and then examined for p38 or ERK1/2 phosphorylation by Western blot. N9 cells pretreated with different concentrations of SB203580 (SB) (B) or PD (D) for 1 h were stimulated with or without 10 μm ISO for 9 h and then examined for mFPR2 mRNA level by RT-PCR. All data are mean ± SD (n = 3); **p < 0.01, compared with cells without stimulation.
NF-κB has been reported to mediate peptidoglycan-induced mFPR2 expression in microglial cells (Chen et al., 2006). NF-κB is also a molecule of βAR signaling pathway (Takemoto et al., 1999; Chandrasekar et al., 2004). We analyzed the sequence of the promoter region of mFPR2 and found the existence of NF-κB binding sites. Pretreatment of N9 cells with a highly selective IκBα phosphorylation inhibitor, BAY117082, significantly inhibited ISO-induced mFPR2 expression (Fig. 7A). Consistently, stimulation of N9 cells and primary microglia with ISO markedly increased IκBα phosphorylation (Fig. 7B,C) and IκBα degradation (Fig. 7D). These results suggest that NF-κB is an important transcription factor that mediated the upregulation of mFPR2 expression by ISO. Pretreatment of N9 cells with BAY117082 reversed ISO-induced IκBα degradation (Fig. 7D). Pretreatment of N9 cells with PD98059 or SB203580 also reversed IκBα degradation induced by ISO, suggesting that ERK1/2 and p38 are upstream signaling molecules mediating ISO-induced NF-κB activation. Together, these results suggest that ISO induces mFPR2 expression through p38-NF-κB and ERK1/2-NF-κB signaling pathways.
Figure 7.

Isoproterenol induces mFPR2 mRNA expression through ERK/p38-NF-κB pathway. A, N9 cells pretreated with or without different concentrations of BAY117082 (BAY) for 1 h were stimulated with 10 μm ISO for 9 h and then examined for mFPR2 expression by RT-PCR. B, C, N9 cells (B) or primary microglia (C) were stimulated with 10 μm ISO for 30 min and then examined for IκBα phosphorylation by Western blot. D, N9 cells pretreated with or without 50 μm PD98059 (PD), 50 μm SB203580 (SB), or 20 μm BAY for 1 h were stimulated with 10 μm ISO for 30 min and then examined for IκBα level by Western blot. All data are mean ± SD (n = 3); *p < 0.05, **p < 0.01, compared with cells cultured in medium alone; ##p < 0.01, compared with cells treated with ISO alone.
Discussion
In this study, we demonstrated that NE and ISO could induce mFPR2 mRNA expression in microglial cells through β2AR-mediated activation of MAP kinases ERK1/2 and p38, as well as transcription factor NF-κB. Upregulation of mFPR2 expression by β2AR activation in microglia markedly enhanced Aβ42 uptake. We also found that activation of β2AR in microglia increased the expression of Aβ-degrading enzyme IDE and enhanced the degradation of Aβ42.
Microglia are resident brain mononuclear phagocytes and have functions similar to peripheral macrophages. They play a critical role as resident immunocompetent and phagocytic cells in the CNS and serve as scavenger cells in the event of infection, inflammation, trauma, ischemia, and neurodegeneration (Kreutzberg, 1996; González-Scarano and Baltuch, 1999; Aloisi et al., 2001). Although microglial activation has become increasingly regarded as a contributor to AD pathogenesis by producing neurotoxins including reactive oxygen species and proinflammatory cytokines, it also plays a neuroprotective role in AD by secreting proteolytic enzymes that degrade Aβ and express receptors that are involved in the endocytosis and clearance of Aβ. The notion that microglia actively uptake Aβ peptide is supported by experiments using in vitro culture (Frackowiak et al., 1992; Pocock et al., 2002) and animal AD models (Frautschy et al., 1998; Weldon et al., 1998). In mouse AD models, active immunization with Aβ peptide (Schenk et al., 1999; Lombardo et al., 2003) or passive immunization with antibodies to Aβ (Wilcock et al., 2003) resulted in substantial removal of existing Aβ deposits, apparently by microglia. A series of cell membrane receptors has been reported to be able to meditate binding and endocytosis of Aβ, including G protein-coupled receptor formyl peptide receptor 2, scavenger receptors, receptor of advanced glycation end products, Fc receptors, complement receptors, and CD14 (Paresce et al., 1996; Yan et al., 1996; Bard et al., 2000; Webster et al., 2000; Le et al., 2001; Liu et al., 2005). Human FPR2, also called formyl peptide receptor-like 1, is encoded by a gene that shares homology to the human formyl peptide receptor (FPR1) gene. FPR2 was initially known for its low-affinity interaction with formyl-methionyl-leucyl phenylalanine, the high-affinity agonist for FPR1. Subsequent studies have identified many agonists for this receptor, including microbe- and host-derived peptides, lipoxin A4, synthetic peptides, and nonpeptide agonists (Le et al., 2002; Ye, 2009; Ye et al., 2009). Although FPR2 was initially detected in phagocytic leukocytes, other cell types also express this receptor, such as microglia and astrocytes (Le et al., 2002). Aβ42, the key causative factor in AD, has been reported to activate microglia through human FPR2 or its mouse homolog mFPR2 (Le et al., 2001; Tiffany et al., 2001; Yazawa et al., 2001). The amount of FPR2 expressed on the cell surface is regulated through receptor internalization and mobilization from intracellular pools as well as at the transcriptional level (Ye, 2009). For example, the cell surface number of FPR2 is regulated by ligand-induced receptor internalization; stimulation of neutrophils with several agonists that induce degranulation upregulates the FPR2-mediated response to WKYMVm; differentiated HL-60 cells express more FPR2 transcript than undifferentiated HL-60 cells. IL-13 and interferon-γ stimulate the expression of FPR2 in enterocytes. We previously reported that murine microglial cells in the nonstimulated state express low levels of mFPR2 (Cui et al., 2002a) which are upregulated by pathogen-associated molecular patterns and proinflammatory cytokines, such as LPS, TNF-α, PGN, CpG, IFN-γ and CD40 (Cui et al., 2002a,b; Iribarren et al., 2005; Chen et al., 2006; 2007). The upregulation of mFPR2 was associated with a markedly increased microglial chemotaxis to Aβ42 and its endocytosis via mFPR2. These results suggest that mFPR2 in microglia not only mediates the proinflammatory activity of Aβ42 but also may actively participate in the clearance of the amyloid β peptide. Our present study reveals that NE, the endogenous neurotransmitter mainly produced by LC neurons, is able to induce the expression of mFPR2 and promote Aβ uptake by microglia. The inductive effect of NE on mFPR2 expression is mediated by MAPK-NF-κB signaling pathways.
Neurons can influence microglial functions through direct cell-to-cell interactions as well as with the release of soluble mediators. Microglial processes have been reported to be associated with synapses anatomically (Pow et al., 1989), and microglia may also respond to “volume neurotransmission” (Thomas, 1992), in which transmitters may exist and act extrasynaptically. Among signals from neurons that may have an active role in controlling microglial activation are those from two major neurotransmitters: NE and acetylcholine (Carnevale et al., 2007). NE has been reported to be a potent regulator of microglial function. Activation of βAR in microglia has been shown to be capable of inhibiting inflammatory responses induced by various stimuli, including Aβ, IFNγ, and LPS (Loughlin et al., 1993; Mori et al., 2002; Dello Russo et al., 2004; Färber et al., 2005; Madrigal et al., 2005; Heneka et al., 2010). Meanwhile, both in vitro and in vivo studies have demonstrated that βAR activation induces proinflammatory cytokine expression in microglia (Tomozawa et al., 1995; Chandrasekar et al., 2004; Johnson et al., 2005; Blandino et al., 2006). NF-κB has been reported to be involved in both of the anti-inflammatory and proinflammatory effects of βAR agonists (Farmer and Pugin, 2000; Chandrasekar et al., 2004). These results suggest that βAR activation may exert anti-inflammatory and proinflammatory effects under different circumstances. Rat primary microglia express β1 and β2ARs but not β3AR (Tanaka et al., 2002). Our results showed that only β2AR could be detected in both primary mouse microglia and N9 cells. Supporting our results, Hertz et al. (2010) examined the expression of βAR subtypes in acutely isolated murine microglia by microarray and found that microglia mainly express β2AR. Our results demonstrated that NE and ISO upregulated the expression of mFPR2 in murine microglia and enhanced the uptake and clearance of Aβ42 through the activation of β2AR, and ISO enhanced microglial cell migration independent of mFPR2. Thus, NE acts as a link between neuron and microglia to orchestrate the host responses to Aβ in the pathogenesis of AD.
Aβ is produced continuously in brain, and its concentration is determined in part by the activities of several degradative enzymes, including neprilysin, IDE, ECE-1 (endothelin-converting enzyme 1) and ECE-2, and probably plasmin (Leissring et al., 2003; Eckman and Eckman, 2005). IDE mRNA and protein levels in hippocampus were significantly decreased in apolipoprotein E ε4+ AD patients (Cook et al., 2003). In AD mouse models, the expression levels of neprilysin and IDE in microglia were decreased in 14-month-old mice (Hickman et al., 2008). Recent studies demonstrated that IDE expression and activity were complexly regulated by various factors, including aging, Apo E, cytokines, and vitamin E (Eckman and Eckman, 2005; Jiang et al., 2008; Shimizu et al., 2008; Nishida et al., 2009). Therefore, the upregulation of IDE represents a promising strategy for therapy and prevention for AD. Our results showed that NE and ISO could induce IDE expression and promote Aβ degradation through activation of β2AR in primary microglia. To our knowledge, this is the first report demonstrating that the endogenous neurotransmitter NE plays an important role in maintaining expression and activity of IDE in microglia, which contribute to Aβ clearance in brain.
LC neuron degeneration-induced NE deficiency has been linked to more severe brain lesions in both AD patients and animal models (Bondareff et al., 1987; Heneka et al., 2002). In this study we found that by activation of β2AR in microglia, NE enhanced Aβ uptake through upregulation of mFPR2 and increased Aβ42 clearance through induction of IDE. These results suggest that noradrenergic innervation from LC is needed to maintain adequate Aβ uptake and clearance by microglia and that LC neuron degeneration in AD not only impairs neuron-neuron transmission but also may contribute to dysregulation of the normal neuron-glial interaction, abnormal glial reaction, defective Aβ clearance, and, eventually, neurodegeneration. In addition, our findings indicate that the roles of β2AR in regulating microglial functions may be used in developing new therapeutic strategies in the treatment of AD.
Footnotes
This work was supported by grants from National Basic Research Program of China (2010CB529701), National Natural Science Foundation of China (30570557, 30970917), One Hundred Talents Program of Chinese Academy of Sciences and the Chief Scientist Program of Shanghai Institutes for Biological Sciences (SIBS2008006), Chinese Academy of Sciences, and the Science and Technology Commission of Shanghai Municipality (04ZR14151, 07DJ14005, 09ZR1436700). We thank Drs. P. Ricciardi-Castagnoli (Universita Degli Studi di Milano-Bicocca, Milan, Italy) and Ji Ming Wang (National Cancer Institute, Frederick, MD) for the generous gift of N9 cells and mFPR2/293 cells, respectively, and Dr. David Baltimore (California Institute of Technology, Pasadena, CA) for kindly providing plasmids pBS-hU6-I, FG12, pRSV/REV, pMDLG/pRRE, and pHCMVG/VSVG.
References
- Aloisi F, Ambrosini E, Columba-Cabezas S, Magliozzi R, Serafini B. Intracerebral regulation of immune responses. Ann Med. 2001;33:510–515. doi: 10.3109/07853890108995960. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Blandino P, Jr, Barnum CJ, Deak T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol. 2006;173:87–95. doi: 10.1016/j.jneuroim.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Bondareff W, Mountjoy CQ, Roth M, Rossor MN, Iversen LL, Reynolds GP, Hauser DL. Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis Assoc Disord. 1987;1:256–262. doi: 10.1097/00002093-198701040-00005. [DOI] [PubMed] [Google Scholar]
- Carnevale D, De Simone R, Minghetti L. Microglia-neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord Drug Targets. 2007;6:388–397. doi: 10.2174/187152707783399193. [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Marelli-Berg FM, Tone M, Bysani S, Prabhu SD, Murray DR. Beta-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Commun. 2004;319:304–311. doi: 10.1016/j.bbrc.2004.04.185. [DOI] [PubMed] [Google Scholar]
- Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- Chen K, Iribarren P, Huang J, Zhang L, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM. Induction of the formyl peptide receptor 2 in microglia by IFN-gamma and synergy with CD40 ligand. J Immunol. 2007;178:1759–1766. doi: 10.4049/jimmunol.178.3.1759. [DOI] [PubMed] [Google Scholar]
- Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, Schellenberg GD, Jin LW, Kovacina KS, Craft S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003;162:313–319. doi: 10.1016/s0002-9440(10)63822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui YH, Le Y, Gong W, Proost P, Van Damme J, Murphy WJ, Wang JM. Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J Immunol. 2002a;168:434–442. doi: 10.4049/jimmunol.168.1.434. [DOI] [PubMed] [Google Scholar]
- Cui YH, Le Y, Zhang X, Gong W, Abe K, Sun R, Van Damme J, Proost P, Wang JM. Up-regulation of FPR2, a chemotactic receptor for amyloid beta 1-42 (A beta 42), in murine microglial cells by TNF alpha. Neurobiol Dis. 2002b;10:366–377. doi: 10.1006/nbdi.2002.0517. [DOI] [PubMed] [Google Scholar]
- Dello Russo C, Boullerne AI, Gavrilyuk V, Feinstein DL. Inhibition of microglial inflammatory responses by norepinephrine: effects on nitric oxide and interleukin-1beta production. J Neuroinflammation. 2004;1:9. doi: 10.1186/1742-2094-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Eckman CB. Abeta-degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans. 2005;33:1101–1105. doi: 10.1042/BST20051101. [DOI] [PubMed] [Google Scholar]
- Färber K, Pannasch U, Kettenmann H. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci. 2005;29:128–138. doi: 10.1016/j.mcn.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Farmer P, Pugin J. beta-adrenergic agonists exert their “anti-inflammatory” effects in monocytic cells through the IkappaB/NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol. 2000;279:L675–682. doi: 10.1152/ajplung.2000.279.4.L675. [DOI] [PubMed] [Google Scholar]
- Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Foote SL, Bloom FE. Histochemical characterization of a neocortical projection of the nucleus locus coeruleus in the squirrel monkey. J Comp Neurol. 1975;164:209–231. doi: 10.1002/cne.901640205. [DOI] [PubMed] [Google Scholar]
- Gong K, Li Z, Xu M, Du J, Lv Z, Zhang Y. A novel protein kinase A-independent, beta-arrestin-1-dependent signaling pathway for p38 mitogen-activated protein kinase activation by beta2-adrenergic receptors. J Biol Chem. 2008;283:29028–29036. doi: 10.1074/jbc.M801313200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- He R, Tan L, Browning DD, Wang JM, Ye RD. The synthetic peptide Trp-Lys-Tyr-Met-Val-d-Met is a potent chemotactic agonist for mouse formyl peptide receptor. J Immunol. 2000;165:4598–4605. doi: 10.4049/jimmunol.165.8.4598. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL. Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci. 2002;22:2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010;107:6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Lovatt D, Goldman SA, Nedergaard M. Adrenoceptors in brain: Cellular gene expression and effects on astrocytic metabolism and [Ca2+]i. Neurochem Int. 2010 doi: 10.1016/j.neuint.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Le Y, Gong W, Dunlop NM, Gao JL, Murphy PM, Wang JM. Synthetic peptide MMK-1 is a highly specific chemotactic agonist for leukocyte FPRL1. J Leukoc Biol. 2001;70:155–161. [PubMed] [Google Scholar]
- Iribarren P, Chen K, Hu J, Gong W, Cho EH, Lockett S, Uranchimeg B, Wang JM. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1–42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. FASEB J. 2005;19:2032–2034. doi: 10.1096/fj.05-4578fje. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005;135:1295–1307. doi: 10.1016/j.neuroscience.2005.06.090. [DOI] [PubMed] [Google Scholar]
- Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, Feinstein DL. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28:1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Le Y, Gong W, Tiffany HL, Tumanov A, Nedospasov S, Shen W, Dunlop NM, Gao JL, Murphy PM, Oppenheim JJ, Wang JM. Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci. 2001;21:RC123. doi: 10.1523/JNEUROSCI.21-02-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Liu AM, Lo RK, Wong CS, Morris C, Wise H, Wong YH. Activation of STAT3 by G alpha(s) distinctively requires protein kinase A, JNK, and phosphatidylinositol 3-kinase. J Biol Chem. 2006;281:35812–35825. doi: 10.1074/jbc.M605288200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT. Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003;23:10879–10883. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin AJ, Woodroofe MN, Cuzner ML. Modulation of interferon-gamma-induced major histocompatibility complex class II and Fc receptor expression on isolated microglia by transforming growth factor-beta 1, interleukin-4, noradrenaline and glucocorticoids. Immunology. 1993;79:125–130. [PMC free article] [PubMed] [Google Scholar]
- Madrigal JL, Feinstein DL, Dello Russo C. Norepinephrine protects cortical neurons against microglial-induced cell death. J Neurosci Res. 2005;81:390–396. doi: 10.1002/jnr.20481. [DOI] [PubMed] [Google Scholar]
- Marcyniuk B, Mann DM, Yates PO. Loss of nerve cells from locus coeruleus in Alzheimer's disease is topographically arranged. Neurosci Lett. 1986;64:247–252. doi: 10.1016/0304-3940(86)90336-8. [DOI] [PubMed] [Google Scholar]
- Mori K, Ozaki E, Zhang B, Yang L, Yokoyama A, Takeda I, Maeda N, Sakanaka M, Tanaka J. Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors. Neuropharmacology. 2002;43:1026–1034. doi: 10.1016/s0028-3908(02)00211-3. [DOI] [PubMed] [Google Scholar]
- Mudher A, Lovestone S. Alzheimer's disease–do tauists and baptists finally shake hands? Trends Neurosci. 2002;25:22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Ito S, Ohtsuki S, Yamamoto N, Takahashi T, Iwata N, Jishage K, Yamada H, Sasaguri H, Yokota S, Piao W, Tomimitsu H, Saido TC, Yanagisawa K, Terasaki T, Mizusawa H, Yokota T. Depletion of vitamin E increases amyloid beta accumulation by decreasing its clearances from brain and blood in a mouse model of Alzheimer disease. J Biol Chem. 2009;284:33400–33408. doi: 10.1074/jbc.M109.054056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Liddle AC, Hooper C, Taylor DL, Davenport CM, Morgan SC. Activated microglia in Alzheimer's disease and stroke. Ernst Schering Res Found Workshop. 2002;39:105–132. doi: 10.1007/978-3-662-05073-6_7. [DOI] [PubMed] [Google Scholar]
- Pow DV, Perry VH, Morris JF, Gordon S. Microglia in the neurohypophysis associate with and endocytose terminal portions of neurosecretory neurons. Neuroscience. 1989;33:567–578. doi: 10.1016/0306-4522(89)90409-0. [DOI] [PubMed] [Google Scholar]
- Qian L, Hu X, Zhang D, Snyder A, Wu HM, Li Y, Wilson B, Lu RB, Hong JS, Flood PM. beta2 Adrenergic receptor activation induces microglial NADPH oxidase activation and dopaminergic neurotoxicity through an ERK-dependent/protein kinase A-independent pathway. Glia. 2009;57:1600–1609. doi: 10.1002/glia.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Schägger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Kawahara K, Kajizono M, Sawada M, Nakayama H. IL-4-induced selective clearance of oligomeric beta-amyloid peptide(1-42) by rat primary type 2 microglia. J Immunol. 2008;181:6503–6513. doi: 10.4049/jimmunol.181.9.6503. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, Kim S, Yoshikawa J. Increased JNK, AP-1 and NF-kappa B DNA binding activities in isoproterenol-induced cardiac remodeling. J Mol Cell Cardiol. 1999;31:2017–2030. doi: 10.1006/jmcc.1999.1033. [DOI] [PubMed] [Google Scholar]
- Tanaka KF, Kashima H, Suzuki H, Ono K, Sawada M. Existence of functional beta1- and beta2-adrenergic receptors on microglia. J Neurosci Res. 2002;70:232–237. doi: 10.1002/jnr.10399. [DOI] [PubMed] [Google Scholar]
- Thomas WE. Brain macrophages: evaluation of microglia and their functions. Brain Res Brain Res Rev. 1992;17:61–74. doi: 10.1016/0165-0173(92)90007-9. [DOI] [PubMed] [Google Scholar]
- Tiffany HL, Lavigne MC, Cui YH, Wang JM, Leto TL, Gao JL, Murphy PM. Amyloid-beta induces chemotaxis and oxidant stress by acting at formyl peptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J Biol Chem. 2001;276:23645–23652. doi: 10.1074/jbc.M101031200. [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Tomozawa Y, Yabuuchi K, Inoue T, Satoh M. Participation of cAMP and cAMP-dependent protein kinase in beta-adrenoceptor-mediated interleukin-1 beta mRNA induction in cultured microglia. Neurosci Res. 1995;22:399–409. doi: 10.1016/0168-0102(95)00922-g. [DOI] [PubMed] [Google Scholar]
- Wang O, Cai K, Pang S, Wang T, Qi D, Zhu Q, Ni Z, Le Y. Mechanisms of glucose-induced expression of pancreatic-derived factor in pancreatic beta-cells. Endocrinology. 2008;149:672–680. doi: 10.1210/en.2007-0106. [DOI] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. 2000;161:127–138. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr Alzheimer Res. 2008;5:342–345. doi: 10.2174/156720508784533286. [DOI] [PubMed] [Google Scholar]
- Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O'Hare E, Esler WP, Maggio JE, Mantyh PW. Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18:2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Drew PD. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J Neuroimmunol. 2006;171:135–144. doi: 10.1016/j.jneuroim.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yazawa H, Yu ZX, Takeda, Le Y, Gong W, Ferrans VJ, Oppenheim JJ, Li CC, Wang JM. Beta amyloid peptide (Abeta42) is internalized via the G-protein-coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. FASEB J. 2001;15:2454–2462. doi: 10.1096/fj.01-0251com. [DOI] [PubMed] [Google Scholar]
- Ye RD. FPR2/ALX. UCSD-Nature Molecule Pages. 2009 doi: 10.1038/mp.a001648.01. [DOI] [Google Scholar]
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying G, Iribarren P, Zhou Y, Gong W, Zhang N, Yu ZX, Le Y, Cui Y, Wang JM. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J Immunol. 2004;172:7078–7085. doi: 10.4049/jimmunol.172.11.7078. [DOI] [PubMed] [Google Scholar]
- Zhu J, Wang O, Ruan L, Hou X, Cui Y, Wang JM, Le Y. The green tea polyphenol (−)-epigallocatechin-3-gallate inhibits leukocyte activation by bacterial formylpeptide through the receptor FPR. Int Immunopharmacol. 2009;9:1126–1130. doi: 10.1016/j.intimp.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]