

Abstract
Parkinson's disease (PD) is a common neurodegenerative disease that involves the deterioration of dopaminergic neurons in the substantia nigra pars compacta. Although the etiology of PD remains poorly understood, recent genetic, postmortem, and experimental evidence shows that abnormal protein accumulation and subsequent aggregate formation are prominent features of both sporadic and familial PD. While proteasome dysfunction is observed in PD, diverse mutations in the parkin gene are linked to early-onset autosomal-recessive forms of familial PD. We demonstrate that parkin, an E3 ubiquitin ligase, activates the 26S proteasome in an E3 ligase activity-independent manner. Furthermore, an N-terminal ubiquitin-like domain within parkin is critical for the activation of the 26S proteasome through enhancing the interaction between 19S proteasomal subunits, whereas the PD-linked R42P mutant abolishes this action. The current findings point to a novel role for parkin for 26S proteasome assembly and suggest that parkin mutations contribute to the proteasomal dysfunction in PD.
Introduction
Parkinson's disease (PD) is a major neurodegenerative disease characterized by the degeneration of dopaminergic neurons in the substantia nigra pars compacta and the presence of cytoplasmic inclusions known as Lewy bodies (Moore et al., 2005). The etiology of PD remains poorly understood, but several genetic loci such as parkin have been implicated in the pathogenesis of familial forms of PD. Mutations in parkin are responsible for autosomal-recessive juvenile parkinsonism (AR-JP) (Kitada et al., 1998). Parkin contains a C-terminal RING-IBR-RING motif and an N-terminal ubiquitin-like (UBL) domain. Parkin is an E3 ubiquitin ligase promoting the degradation of multiple proteins via the ubiquitin–proteasome system (UPS) (Shimura et al., 2000).
The UPS is a major pathway targeting misfolded and/or damaged proteins for degradation (Chung et al., 2001; Pickart and Cohen, 2004; Ciechanover, 2005). The proteasome is the major proteolytic enzyme in UPS. The 26S proteasome is an ATP-dependent complex made up of the functionally and structurally distinct 20S core particle and 19S regulatory complex (Goldberg, 2003). It contains multiple peptidase activities, including trypsin-like, chymotrypsin-like, and peptidylglutamyl-peptide hydrolyzing-like activities (Hershko and Ciechanover, 1998). The 20S proteasome alone can degrade fully denatured small proteins in an ATP-independent fashion. In contrast, degradation of ubiquitinated proteins is an ATP-dependent process requiring both the 19S regulatory particle and the 20S core (Murata et al., 2009). The 19S regulatory particle is presumed to recognize polyubiquitin-linked proteins, and translocate the substrates into the catalytic chamber of the 20S core (Chung et al., 2001).
The ability of the UPS to recognize and selectively degrade misfolded proteins enables it to protect cells against the toxic effects of protein aggregation (Furukawa et al., 2002; Ciechanover and Brundin, 2003). Aberrations in this system have been implicated in the pathogenesis of several neurodegenerative disorders, including PD (Snyder et al., 2003). Indeed, postmortem analysis of sporadic PD brain samples revealed relatively low proteasomal activities in the substantia nigra (McNaught and Jenner, 2001; McNaught and Olanow, 2003; McNaught et al., 2003). PD-associated proteins have been linked to 26S proteasome dysfunction. For example, in genetic PD mice models such as wild-type and mutant α-synuclein transgenic mice a remarkable downregulation of proteasome activity occurs (Chen et al., 2006). The gene products targeted in familial PD are either degraded via the UPS (α-synuclein, parkin, synphilin-1, mutated DJ-1) or integral components of the degradation pathway (parkin, ubiquitin C-terminal hydrolase L1) (Krüger et al., 2002). Parkin has been reported to interact with proteasomal subunits such as Rpn10 (Sakata et al., 2003), Rpt6 (Tsai et al., 2003), and α 4 (Dächsel et al., 2005). In addition, parkin overexpression was found to enhance the proteasomal activity (Hyun et al., 2002; Dächsel et al., 2005). Although these reports suggest that the proteasome- and PD-associated gene products are functionally linked, the exact relationship between these factors is incompletely understood. Here, we investigated whether a direct mechanistic link exists between parkin and the proteasome. Our results point to a novel role for parkin to modulate the assembly and activity of the 26S proteasome via its UBL domain.
Materials and Methods
Materials.
Peroxidase-conjugated anti-rabbit and anti-mouse antibodies were purchased from Zymed Laboratories. DMEM, fetal bovine serum (FBS), LipofectAMINE PLUS reagents, and anti-V5 antibody were obtained from Invitrogen. Clasto-Lactacystin β-lactone (lactacytin) and MG132 were purchased from A. G. Scientific. Fluorogenic peptides [Suc-LLVY-7-amino-4-methylcoumarin (AMC), Z-LLE-AMC, Bz-VGR-AMC], purified 26S proteasome, antibody to α 1, 2, 3, 5, 6, and 7 subunits of the 20S proteasome, anti-Rpn1, anti-Rpn10, anti-Rpt2, and anti-Rpt5, antibodies were purchased from BIOMOL Research Laboratory. Epoxomicin was purchased from Calbiochem. Anti-hemagglutinin (HA), anti-green fluorescent protein (GFP), anti-α-tubulin, anti-heat shock protein 90 (Hsp90), anti-Myc, and anti-Rpn6 antibodies were purchased from Santa Cruz Biotechnology. Anti-Flag and anti-actin antibodies were from Sigma-Aldrich. Monoclonal anti-parkin antibody was purchased from Cell Signaling Technology. Polyclonal anti-parkin antibody, recognizing the N terminus of parkin, was purchased from Millipore. Rabbit polyclonal anti-T7 antibody was purchased from KOMA Biotech, and mouse monoclonal anti-T7 antibody was from Novagen. The polyclonal antibody to Drosophila parkin was generated in mice by injecting GST-Parkin1-260 and further purified.
Mammalian expression vectors encoding Myc-tagged human wild-type parkin (pcDNA3.1-Myc-parkin) and deletion mutants (parkin77-465, parkin1-415, parkin217-465, and parkin295-465) were kindly provided by K. Tanaka (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). A mammalian expression vector encoding Flag-parkin was kindly provided by G. Lee (Ajou University School of Medicine, Suwon, Korea). Plasmids encoding bacterial recombinant wild-type parkin and parkin deletion mutants (parkin77-465 and parkin1-170) fused with GST were gifts from H. Rhim (The Catholic University School of Medicine, Seoul, Korea). A plasmid encoding UbG76VGFP was a kind gift from N. P. Dantuma (Karolinska Institutet, Stockholm, Sweden). Plasmids encoding various Flag-tagged Rpn1, Rpn10, Rpt3, Rpt5, Rpt6, 20S core subunit α 1, and 20S core subunit β 4; T7-tagged Rpt1, Rpt2, and Rpn6; HA-tagged Rpn1; GFP-fused ornithine decarboxylase (GFP-ODC); and ubiquitinated GFP (GFPu) were prepared as described previously (Park et al., 2005). Bacterial recombinant GST-fused parkin was purified according to the supplier's protocol (GE Healthcare Biosciences).
Cell culture, DNA transfection, and preparation of cell lysates.
HeLa Tet-Off cells (Clontech), human embryonic kidney 293 (HEK293) cells, and dopaminergic neuroblastoma SH-SY5Y cells were maintained in DMEM containing 10% FBS and 100 U/ml penicillin-streptomycin. SH-SY5Y cells stably overexpressing wild-type parkin were generated and maintained as described previously (Um et al., 2009). DNA transfections were performed using LipofectAMINE plus reagent (Invitrogen), and the total amount of DNA in each individual transfection was kept constant with an empty control vector. Cell lysates were prepared by rinsing cells twice with ice-cold PBS and solubilizing cells in lysis buffer containing 50 mm Tris, pH 7.5, 1.0% Nonidet P-40, 150 mm NaCl, 10% glycerol, 1 mm Na3VO4, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 10 mm NaF, and 0.2 mm phenylmethylsulfonyl fluoride.
Immunoprecipitation.
One microgram of appropriate antibody was incubated overnight at 4°C with 1 mg of cell lysate. Thirty microliters of a 1:1 suspension of protein A-Sepharose beads was added, and the mixture was incubated for 2 h at 4°C with gentle rotation. The beads were pelleted and washed five times with cell lysis buffer. The immunocomplexes were then resolved by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked in TBST buffer containing 20 mm Tris, pH 7.6, 137 mm NaCl, 0.05% Tween 20, and 5% nonfat dry milk for 1 h at room temperature. It was then incubated overnight at 4°C in 3% nonfat dry milk containing the appropriate primary antibodies. The membrane was washed several times in TBST buffer and incubated with secondary horseradish peroxidase-coupled antibody. After 1 h, the membrane was washed several times with TBST buffer and visualized with ECL reagents (PerkinElmer).
RNA interference.
The small interfering RNA (siRNA) corresponding to human parkin (product name: 1112370 duplex) and negative control siRNA (AccuTarget Negative control siRNA) were purchased from Bioneer. siRNA transfection into SH-SY5Y cells was performed using Lipofectamine RNAi MAX (Invitrogen).
Measurement of in vitro 26S ubiquitin-dependent proteasomal activity.
Proteasome activity was assayed using ubiquitin-conjugated [14C]-lysozyme as a substrate. Ubiquitin-conjugated [14C]-lysozyme was generated by ubiquitinating [14C]-lysozyme in vitro, using an ubiquitin protein-conjugating kit (Calbiochem). The resulting ubiquitinated proteins were flash frozen in aliquots and thawed just before use. Proteasome activity was assayed using the 26S proteasome degradation kit (Calbiochem) according to the manufacturer's instructions. Briefly, equal amounts of the ubiquitinated [14C]-lysozyme and 26S proteasome (10 nm) were incubated for 60 min at 37°C in buffer containing Mg2+/ATP, in the absence or presence of GST, GST-fused wild-type parkin, GST-fused parkin1-170, or GST-fused parkin77-465. The reaction was quenched by adding buffer containing 10% TCA and 5% BSA. Precipitated proteins were pelleted by centrifugation, and radioactivity incorporated in the soluble fraction was measured using a scintillation counter. The specificity of the assay was ensured by incubating 26S proteasome with 10 μm lactacystin before the addition of ubiquitin-conjugated [14C]-lysozyme to the reaction mixture.
Measurement of 26S proteasome catalytic activity.
Purified GST or GST-fused wild-type parkin were incubated for 1 h at 37°C with a 100 μl reaction mixture containing 10 ng of pure 26S proteasome, 20 mm Tris-HCl, pH 7.4, 2 mm MgCl2, 2 mm ATP, and 80 μm fluorogenic peptide (Suc-LLVY-AMC, Z-LLE-AMC, or Bz-VGR-AMC). The amount of cleaved AMC fragment was quantified using a VictorX3 Multilabel Plate Reader (PerkinElmer) at excitation (EX) = 380/emission (EM) = 460. Reaction mixtures were incubated with 10 μm lactacystin or MG132 before addition of fluorogenic substrates to ensure the specificity of the assays.
Native gel immunoblotting.
Samples were prepared by incubation of 3 μg of purified 26S proteasome with 30 μg of GST, GST-parkin, GST-parkin1-170, or GST-parkin77-465 for 1 h at 37°C. These reaction samples or 15 μg of brain cell lysates from wild-type and parkin knock-out mice were separated on 4–12% NovexR Tris-Glycine Gels (Invitrogen) in 1× NativePAGE running buffer (Invitrogen) for 3 h at 150 V. Proteins were transferred onto a PVDF membrane (GE Healthcare Biosciences) at 25 V for 2 h in 1× NativePAGE transfer buffer (Invitrogen). The PVDF membrane was incubated with methanol for 10 min to fix the proteins, and blocked with 5% milk for 1 h, and incubated overnight with anti-mouse monoclonal antibody to α 1, 2, 3, 5, 6, and 7 subunits of 20S proteasome and anti-Rpn10 (S5a) (19S) subunit antibody in TBST buffer. The blot was then incubated with secondary horseradish peroxidase-coupled antibody for 1 h, washed several times with TBST buffer, and visualized with ECL reagents.
Gel filtration analysis.
SH-SY5Y cells stably overexpressing wild-type parkin were harvested in PBS, washed once with PBS, homogenized with an ultrasonicator in 1 ml of buffer A (25 mm Tris-HCl, pH 7.5; 5 mm DTT; 2 mm MgCl2; 2 mm ATP), and centrifuged at 10,000 × g for 15 min. The resulting supernatant was applied to a gel filtration column (Superdex 200 10/300 GL, GE Healthcare Bioscience) after equilibration with buffer A. One hundred microliter aliquots of fractions were collected and subjected to immunoblotting with anti-20S core, anti-Rpt2, and anti-parkin antibodies.
Generation of parkin knock-out mice, flies, and parkin transgenic flies.
A knock-out mouse harboring an exon 3 deletion in the parkin gene (PaKO) was generated as described previously (Stichel et al., 2007). Experiments were performed using four parkin homozygous (−/−) PaKO mice and their nontransgenic parkin (+/+) littermates. All animal experiments were conducted in accordance with the German guidelines of the Animal Care and Use Committee of the state Nordrhein-Westfalen and were approved by this committee. The parkin loss-of-function mutant flies as well as the four parkin transgenic flies have been generated and provided by J. Chung (Seoul National University, Seoul, Korea). Briefly, flies with a P-element insertion at +988 of the parkin ORF in the parkin locus were obtained from a large-scale P-element-induced mutagenesis (GenExel). The loss-of-function mutants were obtained from P-element excision experiments (Cha et al., 2005). The generation of four parkin transgenic fly lines, parkinWT, the full-length parkin; parkinΔN, N-terminal parkin108-482; parkinN, N-terminal parkin fragment containing UBL domain and Linker; and parkinK71P, a point mutant similar to R42P in AR-JP patients, have also been described previously (Cha et al., 2005). They were all Myc-tagged and subcloned into pUAST vector.
Measurement of 26S proteasome activity of parkin knock-out flies.
Whole-body homogenates from parkin knock-out mutant or control w1118 flies were incubated for 1 h at 37°C with reaction buffer containing 20 mm Tris-HCl, pH 7.4, 2 mm MgCl2, and 80 μm Suc-LLVY-AMC in the absence or presence of 1 mm ATP. The amount of cleaved AMC fragment was quantified using a FL600 fluorescence microplate reader (BioTek Instruments) at EX = 380/EM = 460. A subset of cell lysates was preincubated with 10 μm epoxomicin to ensure the specificity of assay.
Statistical analysis.
Significant differences in in vitro 20S or 26S proteasomal activity or fluorescent intensity were determined with an unpaired Student's t test using Sigma Plot 9.0. Values were expressed as the mean ± SEM.
Results
Parkin modulates proteasome activity in cultured cells
To investigate whether parkin can regulate the activity of 26S proteasome, we used a reporter plasmid encoding an ubiquitin-fused GFP-based substrate (UbG76VGFP). In this substrate, the polyubiquitin chain is covalently attached to GFP to prevent its removal by deubiquitination machinery (Dantuma et al., 2000). Since the sole metabolic destination of UbG76VGFP is degradation by the 26S proteasome, the steady-state level of UbG76VGFP protein is inversely proportional to the intracellular activity of 26S proteasome. Accordingly, the level of UbG76VGFP protein was increased in the presence of the proteasomal inhibitor MG132 (Fig. 1A). Interestingly, the level of UbG76VGFP protein was lower in cells expressing both parkin and UbG76VGFP than in those expressing only UbG76VGFP (Fig. 1A). This indicates that parkin overexpression somehow promotes the degradation of UbG76VGFP by activating the 26S proteasome. Fluorescence microscopy consistently showed a significant reduction in UbG76VGFP levels in the presence of parkin (Fig. 1B). Quantification of UbG76VGFP intensity using a fluorescence reader revealed that parkin reduced UbG76VGFP fluorescence by ∼40% (Fig. 1C). In addition, two other GFP-based constructs such as GFP-ODC and GFPu were used to measure the effect of parkin on 26S proteasome activity. ODC is a well known ubiquitin-independent 26S proteasome substrate. It has a C-terminal 37 residue degron motif, which acts as a signal to allow the binding of ODC to and subsequent degradation by 26S proteasome without polyubiquitination or the action of any other targeting protein (Li et al., 1998; Hoyt et al., 2003). GFPu is constructed by the addition of 16 aa CL1 degron to an enhanced GFP (EGFP) at C terminus to destabilize. The CL1 motif targets GFPu for polyubiquitination and subsequent proteasomal degradation (Bence et al., 2005). Like UbG76VGFP, levels of ODC or GFPu were lower in cells expressing both parkin and ODC/GFPu than in those expressing ODC/GFPu alone (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). As a control for equal transfection efficiency, cotransfection of EGFP vector did not significantly affect GFP expression in the absence or presence of parkin (supplemental Fig. S1C,D, available at www.jneurosci.org as supplemental material). Pretreatment with MG132 also did not affect GFP expression, confirming that UbG76VGFP, GFP-ODC, and GFPu are all specific reporters for the measurement of proteasomal activity (supplemental Fig. S1C,D, available at www.jneurosci.org as supplemental material).
Figure 1.

Parkin promotes the degradation of UbG76VGFP. A, HEK293 cells were cotransfected with each 0.5 μg of UbG76VGFP plus either empty vector or Myc-tagged parkin, and then cultured in the presence or absence of 10 μm MG132. Total cell lysates were immunoblotted with anti-GFP or anti-Myc antibodies. α-Tubulin served as a loading control. The values at the bottom of the top panel indicate the relative intensities of UbG76VGFP bands measured using the ImageJ program (National Institutes of Health). B, C, HEK293 cells were transfected with Myc-parkin and UbG76VGFP and then cultured in the presence or absence of 100 nm epoxomicin. UbG76VGFP degradation was observed by fluorescent microscopy (B) or measured at λEX = 480/λEM = 510 using a fluorescence microplate reader (C). **p < 0.01.
When we measured 26S proteasomal activity using Suc-LLVY-AMC peptide, one of fluorogenic substrates widely used for monitoring 26S proteasomal activity, the 26S proteasome activity was enhanced by ∼25% in the parkin-overexpressing cell, compared with the control vector-overexpressing cell (Fig. 2A). To further examine the specific effect of parkin on 26S proteasome activity, we used siRNA to suppress endogenous parkin expression. The efficiency of siRNA-mediated parkin suppression was monitored by Western blot analysis (Fig. 2B). The 26S proteasomal activity in cells transfected with parkin siRNA was significantly reduced in a dose-dependent manner, but not in cells treated with nonspecific control siRNA (Fig. 2B), indicating that parkin deficiency reduces 26S proteasome activity in cells. Together, these results suggested that parkin promotes 26S proteasome activity in cultured cells.
Figure 2.

The 26S proteasomal activity is altered by parkin overexpression or knockdown. A, HEK293 cells were transfected with empty vector or Myc-tagged wild-type parkin, and then cultured in the presence or absence of 10 μm lactacystin. To confirm the proper expression of parkin, total cell lysates were immunoblotted with anti-Myc antibody. Actin served as a loading control. The 26S proteasomal activity in each sample was determined by assaying Suc-LLVY-AMC cleavage. **p < 0.005 versus vector (Vec). Data are expressed as the mean ± SEM of four experiments. B, After SH-SY5Y cells were mock transfected (-), or transfected with either parkin-siRNA (si-parkin; 100 or 200 nm) or nonspecific control siRNA (nc siRNA; 100 nm), and Western blot analysis was performed with anti-parkin antibodies. Actin served as a loading control. The 26S proteasomal activity in each sample was determined by assaying Suc-LLVY-AMC cleavage. Samples pretreated with 10 μm lactacystin (Lac) served as a positive control. **p < 0.01 versus control sample. Data are expressed as the mean ± SEM of four experiments.
Parkin regulates the 26S proteasome in an E3 ligase activity-independent manner
To identify the domains of the 465 aa parkin protein are critical for 26S proteasome activation, we used the following Myc-tagged deletion mutants: parkin1-100, parkin1-415, parkin77-465, parkin217-465, and parkin295-465 (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Analysis of UbG76VGFP levels in HEK293 cells expressing wild-type parkin and these deletion constructs revealed that only those mutants retaining amino acids 1-100 (i.e., parkin1-100 and parkin1-415) appreciably reduced UbG76VGFP levels (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). GFP fluorescence quantification showed that UbG76VGFP was significantly reduced by parkin1-100, but not by parkin77-465 (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material). These data suggest that the amino acid 1-76 region containing a UBL domain is critical for 26S proteasome activation.
The C-terminal RING domain of parkin, including the RING-IBR-RING structure, is critical for intrinsic ubiquitin E3 ligase activity (Shimura et al., 2000). To test whether 26S proteasome activation is a specific effect of parkin, we investigated the effects of other E3 ubiquitin ligases having either a RING domain (Siah1 or cIAP1), or a homologous to E6-AP C terminus (HECT) domain (Nedd4-2) on UbG76VGFP levels. As a result, none of these E3 ubiquitin ligases significantly affected UbG76VGFP levels (data not shown). Together, these results indicate that parkin selectively activates the 26S proteasome via its N-terminal UBL domain in an E3 ligase-independent manner.
Specific amino acids within the UBL domain of parkin are critical to activate 26S proteasome
Next, we determined whether the 26S proteasome could be activated by other UBL-containing proteins that interact with proteasomes, such as hPLIC-1 or hHR23a (Hiyama et al., 1999; Kleijnen et al., 2000). The amino acid sequences of the UBL domains in hPLIC-1 and hHR23a have 57 and 62% similarities to ubiquitin, respectively. As shown in supplemental Figure 3 (available at www.jneurosci.org as supplemental material), UbG76VGFP levels were slightly reduced by overexpression of wild-type hPLIC-1. However, overexpression of the C-terminal hPLIC-1 fragment containing a UBL domain dramatically reduced UbG76VGFP levels. UbG76VGFP levels were not affected by wild-type hHR23a or its UBL domain-containing mutant (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These results indicate that not all proteins containing a UBL domain can activate the 26S proteasome, even though they may interact with a proteasome.
Parkin does not alter the steady-state levels of proteasomal subunits
To determine whether parkin affects the steady-state protein levels of the 20S or 19S proteasome subunits, we generated SH-SY5Y cell lines stably overexpressing either Myc-tagged wild-type parkin (SH-SY5Y/WT-parkin) or the deletion mutant parkin1-415 (SH-SY5Y/parkin1-415). Immunoblot analysis revealed that levels of the 20S proteasome α subunits and Rpn1 (one of the 19S regulatory subunits) were comparable among SH-SY5Y/WT-parkin cells, SH-SY5Y/parkin1-415 cells, and parental control cells (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). In addition, the steady-state levels of the 20S and 19S proteasomes were not changed by transient overexpression of parkin in HEK293 cells (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). These data suggest that parkin does not enhance 26S proteasomal activity by increasing steady-state levels of the 19S or 20S proteasomal subunits.
Parkin directly activates 26S proteasome in vitro
To test whether parkin may activate 26S proteasome directly or indirectly through the mediation of other factors, we assayed in vitro 26S proteasome activity using polyubiquitinated [14C]-lysozyme as a specific substrate for the 26S proteasome. The assay mixtures included purified 26S proteasome along with GST-wild-type parkin, GST-parkin77-465, or GST-parkin1-170, of which purity was confirmed by Coomassie Blue staining and Western blot analysis with anti-GST or anti-parkin antibodies (data not shown). In this assay, the radioactive signal is directly proportional to the level of proteasomal activity. The addition of the proteasome inhibitor lactacystin significantly reduced radioactivity by >50% (Fig. 3A). 26S proteasomal activity was greater in the presence of GST-wild-type parkin than GST alone by approximately fourfold (Fig. 3A). GST-parkin1-170 increased 26S proteasome activity to a comparable degree as GST-wild-type parkin, whereas GST-parkin77-465 failed to significantly affect 26S proteasome activity (Fig. 3A). These results indicate that parkin can activate the 26S proteasome directly and that the N-terminal UBL-containing domain appears to be critical for this activity.
Figure 3.

Parkin activates ubiquitin-dependent 26S proteasomal activity in vitro. A, In vitro assay of 26S ubiquitin-dependent proteasomal activity in the presence of GST-fused wild-type parkin or its deletion mutants (parkin77-465, parkin1-170). ***p < 0.001 versus control sample incubated with GST. Samples pretreated with 10 μm lactacystin (Lac) served as a positive control. Experiments were repeated a minimum of four times. B–D, Effect of GST-fused wild-type parkin on the chymotryptic activity (B), tryptic activity (C), and PGPH activity (D) of purified 26S proteasome, as measured using fluorogenic substrates. Samples pretreated with 10 μm lactacystin (Lac) or MG132 (MG) served as controls. **p < 0.05, ***p < 0.001 versus control sample incubated with GST. Experiments were repeated a minimum of four times.
Next, we assessed the effect of parkin on three individual ATP-dependent protease activities associated with the 26S proteasome in vitro. For this purpose, chymotryptic activity was measured with Suc-LLVY-AMC peptide, tryptic activity with Bz-VGR-AMC peptide, and peptidylglutamyl hydrolytic (PGPH) activity with Z-LLE-AMC peptide. As shown in Figure 3B–D, the addition of parkin significantly increased all three major catalytic activities by ∼20%, which was not as high as that in Figure 3A, and it might be caused by the differential sensitivities of different assays.
Parkin interacts with 19S regulatory subunits but not 20S core complexes
To investigate how parkin stimulates 26S proteasome activity, we first identified the 26S proteasome subunits with which parkin associates. Gel filtration of lysates from SH-SY5Y cells stably expressing wild-type parkin was performed to resolve and classify the high- and low-molecular-weight protein fractions containing the 26S proteasome and parkin, respectively. Immunoblot analyses of the eluted fractions with anti-20S core, anti-19S Rpt2, and anti-parkin antibodies revealed that the components of the 20S and 19S proteasome coeluted over the same range of high-molecular-weight fractions, while parkin was present in the low-molecular-weight fractions (Fig. 4A). Interestingly, a small portion of parkin coeluted with the high-molecular-weight fractions containing the 19S and 20S proteasome components (Fig. 4A), raising the possibility that parkin interacts with 26S proteasome. Coimmunoprecipitation assay of the equivalent low- and high-molecular-weight fraction revealed that parkin interacts with one of the 19S proteasomal subunit, Rpn10 (Fig. 4B).
Figure 4.

Parkin interacts with the “base” part of 19S proteasome. A, SH-SY5Y cells stably expressing wild-type parkin were homogenized, and soluble proteins were loaded into Superdex 200 column. Fractions were subjected to immunoblot analysis with anti-20S core, anti-19S Rpt2, and anti-parkin antibodies. B, The high-molecular-weight (HMW: fraction number 22–25) or low-molecular-weight (LMW: fraction number 64–67) fractions prepared in A were immunoprecipitated with either preimmune IgG (I) or anti-parkin (P) antibodies, and immunoblotted with anti-Rpn10 or anti-parkin antibodies. Asterisks indicate IgG heavy chains. C, HeLa Tet-Off cells were transfected with 1.5 μg of Myc-parkin alone or together with 1.5 μg of Flag-tagged Rpn1, Rpn10, or Rpt6 for 24 h. Whole lysates (input) and anti-Flag immunoprecipitates were then subjected to immunoblotting with anti-Myc or anti-Flag antibodies. An asterisk indicates IgG heavy chains. D, SH-SY5Y cells were immunoprecipitated with either preimmune I or P antibodies. Whole lysates and anti-parkin immunocomplexes were probed with anti-Rpn1 or anti-parkin antibodies. Asterisks indicate IgG heavy chains. E, SH-SY5Y cells were immunoprecipitated with either preimmune I or P antibodies. Whole lysates and anti-parkin immunocomplexes were probed with anti-Rpn10 or anti-parkin antibodies. Asterisks indicate IgG heavy chains. F, SH-SY5Y cells were immunoprecipitated with either preimmune I or P antibodies. Whole lysates and anti-parkin immunocomplexes were probed with anti-parkin or anti-Rpt5 antibodies. Asterisks indicate IgG heavy chains.
Next, we examined whether parkin can bind directly to the individual 19S or 20S proteasomal subunits, Myc-tagged parkin and a plasmid encoding one of several 19S proteasomal subunits (Rpn1, Rpn6, Rpn10, or Rpt6) or 20S proteasomal subunits (α 1 or β 4) were cotransfected into HeLa cells. Parkin was found to bind to Rpn10 and Rpt6 (Fig. 4C). Parkin also strongly interacted with Rpn1 (Fig. 4C). Moreover, endogenous parkin interacted with endogenous Rpn1 (Fig. 4D), Rpn10 (Fig. 4E), and Rpt5 (Fig. 4F) in SH-SY5Y cells. However, parkin did not bind to Rpn6 (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). Parkin did not bind to the α 1 or β 4 of 20S proteasomal subunits (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material). Moreover, the endogenous α-subunit of the 20S core complex did not bind to parkin in HEK293 cells (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material). Based on the information that Rpn1, Rpn10, Rpt5, and Rpt6 are among the components of the base of 19S complex, while Rpn6 is one of the components of the lid (Murata et al., 2009), these results indicate that parkin is specifically associated with the base of 19S regulatory complex, but not with 20S core particle.
Parkin enhances the formation of 19S regulatory particles
To further understand how parkin stimulates 26S proteasomal activities, we investigated whether parkin affects the in vivo assembly of the 19S proteasome after its binding to 19S regulatory particles. To this end, we determined the relative binding affinity of proteasomal subunits in the absence or presence of parkin. As shown in Figure 5A, parkin overexpression enhanced the binding affinity between Rpt5 and Rpn6. On the contrary, parkin knockdown by siRNA caused the reduced binding between these two subunits (Fig. 5B). Next, we examined whether UBL domain within the parkin affects the binding affinity of these proteins by using R42P, one of the pathogenic mutants of parkin. R42P has also been reported to induce the conformational change of UBL domain (Sakata et al., 2003). Coimmunoprecipitation assay revealed that R42P failed to enhance the binding affinity between Rpt5 and Rpn6 (Fig. 5A). In addition, parkin dramatically increased the interaction between Rpt1 and Rpn1 (Fig. 5C), while not affecting the association between Rpt2 and Rpt3 (Fig. 5D). Since Rpt1, Rpt2, Rpt3, and Rpt5 belong to the 19S proteasomal AAA ATPase family, while Rpn1 and Rpn6 are non-ATPase 19S proteasomal subunits, these data suggest that parkin potentiates the association of ATPases with non-ATPase 19S proteasomal subunits, in which N-terminal UBL domain within the parkin plays a role.
Figure 5.

Parkin accelerates the binding of 19S proteasomal ATPases to non-ATPases. A, HeLa Tet-Off Cells were transfected with plasmids encoding each 1.0 μg of Myc-Parkin-WT, Myc-Parkin-R42P, T7-Rpn6, or/and Flag-Rpt5, as indicated. Whole lysates and anti-Flag immunoprecipitates were probed with anti-Flag, anti-T7, or anti-Myc antibodies. Actin served as a loading control. *IgG heavy chains; #nonspecific bands that were detected by the previous antibodies on the same membrane. B, SH-SY5Y cells were transfected with either parkin-siRNA (200 nm) or nonspecific control siRNA (nc siRNA; 200 nm). Anti-Rpn6 immunoprecipitates were probed with anti-Rpt5 or anti-Rpn6 antibodies. Total cell lysates were probed with anti-Rpn6, anti-Rpt5, and anti-Parkin antibodies. Actin served as a loading control. C, HeLa Tet-Off Cells were transfected with plasmids encoding Myc-parkin, T7-Rpt1, or/and HA-Rpn1. Lysates and anti-HA immunoprecipitates were then probed with anti-HA, anti-T7, or anti-Myc antibodies. Hsp90 served as a loading control. D, HeLa Tet-Off cells were transfected with each 1.0 μg of plasmid encoding Myc-parkin, T7-Rpt2, or/and Flag-Rpt3. Whole lysates (input) or anti-T7 immunoprecipitates from these cells were then subjected to immunoblotting with anti-Flag and anti-T7 antibodies.
Parkin participates in the assembly and maintenance of the 26S proteasome
Since parkin facilitates the formation of the 19S regulatory complex by increasing the binding affinity between 19S proteasomal ATPases and non-ATPases, it may further affect the subsequent formation of the native 26S proteasome. To test this possibility, we analyzed the levels of purified 26S proteasome in the absence and presence of parkin using non-denaturing gel electrophoresis. Native PAGE analysis of the samples using antibodies against the 20S proteasome or the Rpt2 component of the 19S complex showed that the steady-state levels of 26S proteasome (20S proteasome catalytic core plus two 19S regulatory particles) were greater in the presence of GST-wild-type parkin than in the presence of only GST (Fig. 6A,B). In addition, GST-parkin1-170 had a similar effect as GST-wild-type parkin, whereas parkin77-465 had no effect (Fig. 6A,B). These data demonstrate that, in addition to enhancing the assembly of the 19S regulatory complex, parkin stimulates the subsequent association of the 19S and 20S subunits to form the complete 26S proteasome.
Figure 6.
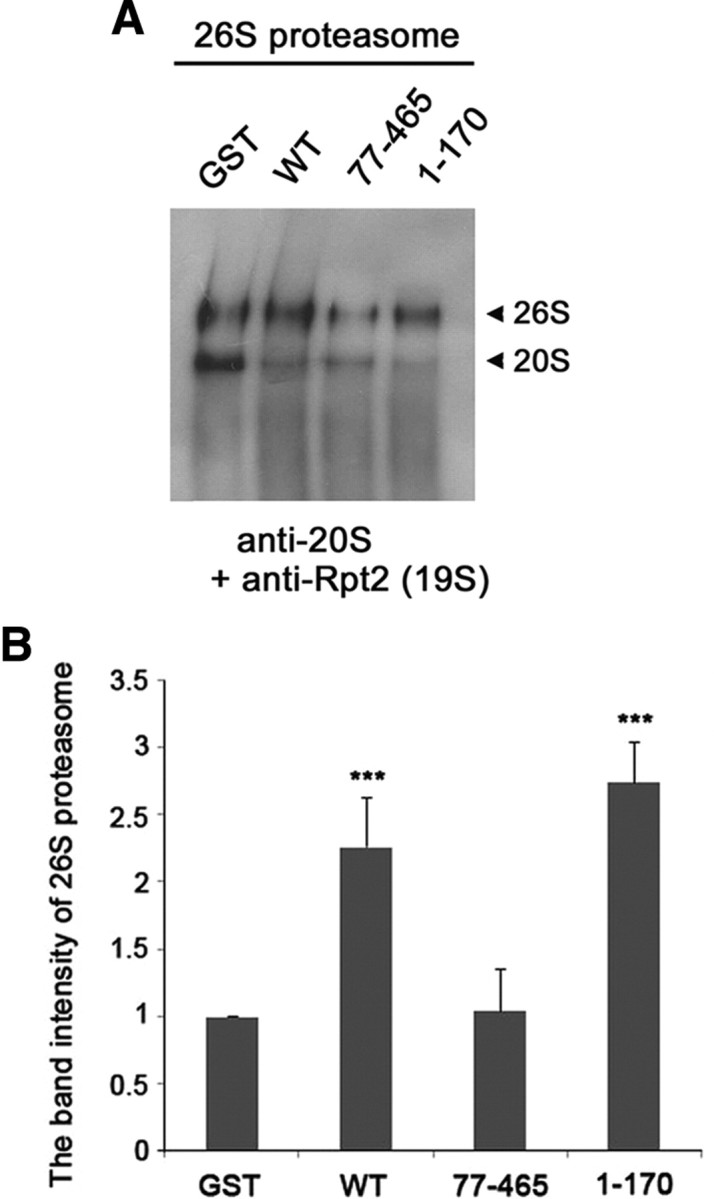
Parkin accelerates the assembly of 26S proteasome. A, Purified 26S proteasome was incubated with GST, GST-fused wild-type parkin, GST-fused parkin77-465, or GST-fused parkin1-170 for 1 h at 37°C. The samples were resolved on a native PAGE gel and subjected to immunoblotting with anti-20S core and anti-Rpt2 antibodies. B, The graph represents the band intensity of 26S proteasome measured by the ImageJ program (National Institutes of Health). Data represent the mean ± SEM of four independent experiments. ***p < 0.001 versus control sample incubated with GST.
Decreased 26S proteasomal activity in parkin KO mice and Drosophila
The physiological significance of our cellular and in vitro findings was tested in mice and Drosophila melanogaster. First, we determined whether 26S proteasomal activity is affected by parkin deficiency using PaKO mice. PaKO mice were generated by targeted deletion of exon 3, which results in a frame shift after amino acid 57 (Stichel et al., 2007). Immunoblot analysis of the 1% NP-40-soluble fraction prepared from brain extracts revealed that parkin was absent in four individual PaKO mice (Fig. 7A). Although the levels of 20S proteasomal α subunits and Rpn10 (one of the 19S regulatory subunits) were comparable between wild-type littermates and PaKO mice (Fig. 7A), 26S proteasome activity was reduced by ∼35% in PaKO mice, relative to control wild-type littermates (Fig. 7B). In control littermate mice treated with the proteasomal inhibitor lactacystin, 26S proteasome activity was reduced by ∼75% (Fig. 7B). Furthermore, native PAGE analysis of control or PaKO mice showed that the steady-state levels of 26S proteasome were significantly reduced in the PaKO mice, compared with the control littermate mice (Fig. 7C,D).
Figure 7.

Downregulation of 26S proteasomal activity in parkin-null mice. A, Immunoblot analysis of brain lysates from parkin-null mice (PaKO) (n = 4) or littermates (Con) (n = 4) using anti-parkin, anti-Rpn10, or anti-20S core antibodies. B, In vitro assay of 26S ubiquitin-dependent proteasomal activity in brain lysates, as measured by degradation of ubiquitin-conjugated [14C]-lysozyme. **p < 0.01 versus control sample. Samples pretreated with 10 μm lactacystin (Lac) served as positive controls. Data are expressed as the mean ± SEM of four experiments. C, The brain lysates from parkin-null mice (PaKO) or littermates (Con) were resolved on a native PAGE gel and subjected to immunoblotting with anti-20S core and anti-Rpn10 antibodies. D, The graph represents the 26S proteasome band intensity from the images of C. The band intensity of 26S proteasome was measured using MultiGauge V3.1. *p < 0.5.
The overall architecture and function of the 26S proteasome are conserved from archaea to eukaryotes (Dahlmann et al., 1991). Drosophila parkin shows a 59% similarity with its human homolog and contains all of the characteristic canonical motifs of human parkin, including the UBL domain and RING-IBR-RING motif. We examined seven parkin-null Drosophila and found that 26S proteasomal activity was significantly lower in these flies than in their control counterparts (Fig. 8A). The treatment of cell lysates from control flies with lactacystin also markedly inhibited (>90%) 26S proteasome activity (Fig. 8A). Immunoblot analysis of the lysates confirmed that Drosophila (dParkin) parkin is absent in parkin-null Drosophila (Fig. 8B).
Figure 8.

Downregulation and upregulation of 26S proteasomal activity in parkin loss-of-function and transgenic flies. A, Whole-body extracts were prepared from parkin knock-out mutant flies (KO) (n = 7) or control w1118 flies (WT) (n = 7), and 26S proteasomal activity was determined by assaying Suc-LLVY-AMC cleavage in the presence or absence of ATP. Samples pretreated with 10 μm lactacystin (Lac) served as a positive control. Differences in the amount of cleaved AMC fragment in the presence and absence of ATP are plotted (**p < 0.01). Data represent the mean ± SEM of a minimum of four experiments. B, Total cell lysates (20 μg) form parkin knock-out mutant flies (KO) or control w1118 flies (WT) were subjected to Western blot analysis using anti-Drosophila Parkin antibody. An asterisk indicates nonspecific bands. Actin served as a loading control. C, Whole-body extracts were prepared from the male flies (n = 7) of hs-GAL4 (hs-GAL4/+) as a control, hs>parkinWT (UAS-parkinWT/Y; hs-GAL4/+), hs>parkinN (UAS-parkinN/Y; hs-GAL4/+), hs>parkinΔN (UAS-parkinΔN/Y; hs-GAL4/+), or hs>parkinK71P (UAS-parkinK71P/Y; hs-GAL4/+), and 26S proteasomal activity was determined by assaying Suc-LLVY-AMC cleavage in the presence of ATP (***p < 0.001). Before performing the assays, heat shock was applied at 37°C for 2 h to express the proteins. Samples pretreated with 10 μm MG132 served as a positive control. D, Whole-body extracts (20 μg) from the male flies of hs-GAL4 (hs-GAL4/+), hs>parkinWT (UAS-parkinWT/Y; hs-GAL4/+), hs>parkinN (UAS-parkinN/Y; hs-GAL4/+), hs>parkinΔN (UAS-parkinΔN/Y; hs-GAL4/+), or hs>parkinK71P (UAS-parkinK71P/Y; hs-GAL4/+) were subjected to Western blot analysis using anti-Myc antibody to confirm the proper expression.
Increased 26S proteasomal activity in parkin transgenic Drosophila
Next, we measured the activity of 26S proteasome in several parkin transgenic flies, which express Myc-tagged deleted parkin mutant (parkinΔN, N-terminal deleted parkin108-482; parkinN, N-terminal parkin fragment containing UBL domain and Linker), one-point mutant (parkinK71P), or wild-type parkin (parkinWT). The expression of these proteins was confirmed by Western blot analysis with anti-Myc antibody (Fig. 8D). Compared with the control flies, the 26S proteasome activity was increased by ∼25% in parkinWT and parkinN flies, whereas parkinΔN, which has no UBL domain, did not have the same effect on 26S proteasome activity as wild-type or parkinN (Fig. 8C). In addition, the proteasome activity in parkinK71P fly was similar to that of control flies (Fig. 8C). Based on the fact that the K71P mutation within UBL domain of Drosophila parkin is correspondent to the R42P mutation in AR-JP patients (Cha et al., 2005), these data support our hypothesis that the UBL domain of parkin is critical to upregulation of 26S proteasome activity in vivo.
Familial PD-linked mutant of parkin with a mutation in the UBL domain fails to activate 26S proteasome
The effect of PD-linked parkin mutants on 26S proteasome activity was investigated by transfecting HEK293 cells with UbG76VGFP alone or together with one of the familial PD-linked parkin mutants (i.e., K161N, T240R, or R42P). Whereas R42 is located in the UBL domain, K161 and T240 are located in the RING1 domain. UbG76VGFP levels were significantly reduced in cells expressing wild-type parkin as well as in those expressing K161N and T240R, which have intact UBL domains (Fig. 9). However, they remained unaffected by expression of R42P mutant, which has a point mutation within the UBL domain (Fig. 9). Thus, Arg42 residue may play a key role in regulating parkin activity for 26S proteasome activation.
Figure 9.

The PD-linked R42P parkin mutant does not alter proteasome activation. HEK293 cells were cotransfected with each 0.5 μg of UbG76VGFP plus either empty vector, or with Myc-tagged wild-type parkin or its PD-linked mutants (R42P, K161N, or T240R) for 24 h. Total cell lysates were subjected to immunoblotting with anti-GFP or anti-parkin antibodies. Actin served as a loading control. The values at the bottom of the top panel indicate the relative quantities of UbG76VGFP bands, as measured with the ImageJ program (National Institutes of Health).
Discussion
In the present study, we demonstrate that familial PD-associated protein, parkin, activates the 26S proteasome by increasing the binding affinity between the 19S proteasomal subunits via its UBL domain. Familial PD-associated proteins were first linked to proteasome by finding that α-synuclein interacts with the S6 proteasomal protein (Snyder et al., 2003). In addition, several groups demonstrated that wild-type and mutant α-synuclein induce proteasome dysfunction in neuronal systems (Tanaka et al., 2001; Petrucelli et al., 2002; Snyder et al., 2003; Smith et al., 2005; Zhang et al., 2008). Another familial PD-associated protein, synphilin-1, has also recently been reported to bind to the S6 proteasomal protein (Marx et al., 2007). Here, we demonstrate that parkin interacts with 19S regulatory units (i.e., Rpn1, Rpn10, Rpt5, and Rpt6), consistent with reports that parkin interacts with Rpn10 and Rpt6 subunits of the 19S regulatory complexes (Sakata et al., 2003; Tsai et al., 2003). Moreover, our finding that parkin1-76 (which contains the UBL domain) is necessary for activation of 26S proteasome is consistent with the report that parkin interacts with Rpn10 via a UBL domain (Sakata et al., 2003). Importantly, our data show that parkin binds to the base of 19S regulatory subunits, but not to the lid of 19S regulatory subunits and 20S core complexes. This differs from a previous report that parkin interacts with the α 4 of the 20S proteasome (Dächsel et al., 2005). This interaction was discovered by transfecting cells with mammalian expression vectors encoding the α 4 subunit and then performing immunoanalysis with anti-α 4 antibody. In contrast, in the present study, cells were transfected with parkin, and parkin immunoprecipitates were probed with antibody recognizing the endogenous whole α subunits of the 20S proteasome. Therefore, the discrepancy in these findings could be explained by the ability of parkin to bind only the free form of the α 4 subunit. In this way, the affinity of parkin for the α 4 subunit may be reduced when α 4 is incorporated into the 26S proteasome, resulting in an apparent decrease of parkin for the α 4 component of the intact 20S proteasome complex. We found that the binding interaction between parkin and Rpn1 was stronger than that between parkin and Rpn10 or Rpt6, suggesting that parkin associates with the 19S proteasome primarily via the Rpn1 subunit. This result is supported by a recent report that proteins containing a UBL domain interact reversibly with the proteasome principally through Rpn1 as well as through Rpn10 (Husnjak et al., 2008).
The base of the 19S complexes is composed of six homologous AAA ATPases (Rpt1-6), and three non-ATPases (Rpn1, Rpn2, and Rpn13). The ATPase subunits are required for substrate unfolding and α-ring channel opening, prerequisites for threading substrates into the 20S proteasome. Rpt5, Rpn1, Rpn10, and Rpn13 capture ubiquitinated proteins, and Rpn10 is thought to sit at the interface of the lid and base. The lid is composed of nine non-ATPase subunits (Rpn3, Rpn5-9, Rpn11-12, and Rpn15) (Ciechanover, 1998, 2005). In our experiment, parkin promoted the binding between Rpt1, one of the proteasomal ATPases, and Rpn1, one of the non-ATPases. However, parkin did not affect the binding affinity between proteasomal ATPases. These data indicate that parkin elevates proteasome activity by enhancing the association between the ATPases and non-ATPases of the 19S regulatory subunits. Although our data indicate that parkin actively promotes the assembly of the 26S proteasome, it is possible that the disassembly of the 26S proteasome into the 20S catalytic core and 19S regulatory units may also be inhibited by parkin.
Many studies have shown that ubiquitin E3 ligases are associated to some extent with the 26S proteasome, leading to recruitment of the ubiquitination machinery (Xie and Varshavsky, 2000; You and Pickart, 2001). Therefore, the direct binding of parkin to the proteasome, more specifically to the 19S regulatory subunits, may have a similar role for the subsequent substrate presentation to the proteasome. However, in vitro assays of 26S proteasomal activity showed that parkin directly activates 26S proteasome without the participation of additional factors. Furthermore, our data revealed that the N-terminal UBL domain retains the ability to activate the 26S proteasome, consistent with a previous report that parkin binds to Rpn10 via a UBL domain (Husnjak et al., 2008). Thus, although the RING2 domain (including a RING-IBR-RING structure) is necessary for the intrinsic ubiquitin E3 ligase activity of parkin (Shimura et al., 2000), this C-terminal region appears not to be required for activation of the 26S proteasome. Together, these findings suggest that parkin promotes the interaction between the 19S regulatory subunits in an E3 ligase activity-independent manner.
Several proteins containing ubiquitin-like domain, including hPLICs and HR23a, have been found to bind proteasome (Schauber et al., 1998; Hiyama et al., 1999; Kleijnen et al., 2000; Elsasser et al., 2002). However, the exact role of these interactions remains to be clarified. Some of these proteins inhibited the degradation of proteasome-targeting substrates (Kleijnen et al., 2003), whereas hPLIC1 has been reported to enhance the ubiquitination and degradation of proteasome-targeting substrates (Gao et al., 2003). In our experiment, hPLIC and parkin had similar stimulatory effects on proteasomal activity, while hHR23a was inactive. Interestingly, an analysis of BLAST sequence searches revealed that there is ∼56% similarity in the UBL domains of parkin and hPLIC-1. However, no significant similarity was found between parkin and hHR23a. Multiple sequence alignments of UBL domains within parkin (human parkin, NP_004553; rat parkin, NP_064478; mouse parkin, AAI13205; Drosophila parkin, NP_730601), PLIC-1 from several species (human PLIC-1, AAG02473; rat PLIC-1, NP_446199; mouse PLIC-1, AAF01365), and human ubiquitin protein (CAA44911) revealed that ∼10 specific amino acids are well conserved among those proteins. This suggests that the amino acid sequences shared between UBL domains of parkin and hPLIC-1 may be critical for proteasome activation. However, the finding that the R42P mutation abolished stimulatory activity indicates that additional residues may be required for full 26S proteasome stimulatory activity. This idea should be tested and confirmed through further experiments.
Although several parkin mutants have been reported to induce proteasome dysfunction (Kyratzi et al., 2007) and parkin downregulation renders cells more sensitive to proteasome inhibition-induced toxicity (Yang et al., 2007), the detailed biochemical interaction between parkin and the proteasome as well as the functional role of parkin remains to be elucidated. The findings that 26S proteasomal activity is reduced in parkin-null mice and fruit flies, and enhanced in parkin transgenic flies, not only confirm the physiological relevance of our in vitro and cellular studies, but also points to a novel and conserved role for parkin in the activation of 26S proteasome. As parkin has been reported to act as an E3 ubiquitin ligase to date, parkin mutation is widely assumed to cause the nondegradation of substrates and consequent accumulation in cells, which may be a cause of PD. The present study demonstrated that parkin has an additional function as a positive regulator of 26S proteasome. We also showed that PD-linked mutations of parkin, especially those affecting critical amino acids within the UBL domain, are unable to enhance the basal 26S proteasome activity. These findings are important for PD as well as neurodegeneration in general. In cases of familial PD related to parkin mutations, such as early-onset AR-JP, mutations may result in a “loss of function” of the regulatory action of parkin on intracellular proteasome. This, in turn, may cause a significant defect in normal protein turnover, leading to abnormal protein accumulation and the formation of toxic aggregates, which could eventually contribute to neuronal cell death and PD pathogenesis. The current findings suggest that there can be multiple differential processes underlying the development of PD induced by diverse parkin mutations depending on the location of functional domain.
Footnotes
This study was supported by the Brain Research Center of the 21st Century Frontier Research Program Technology (2009K-001251 to K.C.C.) funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea. This work was also supported by a National Research Foundation of Korea (NRF) grant funded by MEST (2010-0018916 to K.C.C.) and by the Basic Science Research Program through NRF (2010-0001668 to K.C.C.). This work was also partially supported by a grant of the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A092004 to K.C.C.). We thank K. Tanaka, G. Lee, H. Rhim, and N.P. Dantuma for providing plasmids; J. Chung for providing parkin mutant flies; and Y. Park for technical assistance.
References
- Bence NF, Bennett EJ, Kopito RR. Application and analysis of the GFPu family of ubiquitin-proteasome system reporters. Methods Enzymol. 2005;399:481–490. doi: 10.1016/S0076-6879(05)99033-2. [DOI] [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Thiruchelvam MJ, Madura K, Richfield EK. Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol Dis. 2006;23:120–126. doi: 10.1016/j.nbd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Chung KK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson's disease and other neurodegenerative disorders. Trends Neurosci. 2001;24(11 Suppl):S7–S14. doi: 10.1016/s0166-2236(00)01998-6. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Dächsel JC, Lücking CB, Deeg S, Schultz E, Lalowski M, Casademunt E, Corti O, Hampe C, Patenge N, Vaupel K, Yamamoto A, Dichgans M, Brice A, Wanker EE, Kahle PJ, Gasser T. Parkin interacts with the proteasome subunit alpha4. FEBS Lett. 2005;579:3913–3919. doi: 10.1016/j.febslet.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Dahlmann B, Kopp F, Kuehn L, Hegerl R, Pfeifer G, Baumeister W. The multicatalytic proteinase (prosome, proteasome): comparison of the eukaryotic and archaebacterial enzyme. Biomed Biochim Acta. 1991;50:465–469. [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- Elsasser S, Gali RR, Schwickart M, Larsen CN, Leggett DS, Müller B, Feng MT, Tübing F, Dittmar GA, Finley D. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol. 2002;4:725–730. doi: 10.1038/ncb845. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Vigouroux S, Wong H, Guttman M, Rajput AH, Ang L, Briand M, Kish SJ, Briand Y. Brain proteasomal function in sporadic Parkinson's disease and related disorders. Ann Neurol. 2002;51:779–782. doi: 10.1002/ana.10207. [DOI] [PubMed] [Google Scholar]
- Gao L, Tu H, Shi ST, Lee KJ, Asanaka M, Hwang SB, Lai MM. Interaction with a ubiquitin-like protein enhances the ubiquitination and degradation of hepatitis C virus RNA-dependent RNA polymerase. J Virol. 2003;77:4149–4159. doi: 10.1128/JVI.77.7.4149-4159.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hiyama H, Yokoi M, Masutani C, Sugasawa K, Maekawa T, Tanaka K, Hoeijmakers JH, Hanaoka F. Interaction of hHR23 with S5a. The ubiquitin-like domain of hHR23 mediates interaction with S5a subunit of 26 S proteasome. J Biol Chem. 1999;274:28019–28025. doi: 10.1074/jbc.274.39.28019. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Zhang M, Coffino P. Ubiquitin-independent mechanisms of mouse ornithine decarboxylase degradation are conserved between mammalian and fungal cells. J Biol Chem. 2003;278:12135–12143. doi: 10.1074/jbc.M211802200. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH, Lee M, Hattori N, Kubo S, Mizuno Y, Halliwell B, Jenner P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem. 2002;277:28572–28577. doi: 10.1074/jbc.M200666200. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell. 2000;6:409–419. doi: 10.1016/s1097-2765(00)00040-x. [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Alarcon RM, Howley PM. The Ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol Biol Cell. 2003;14:3868–3875. doi: 10.1091/mbc.E02-11-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Eberhardt O, Riess O, Schulz JB. Parkinson's disease: one biochemical pathway to fit all genes? Trends Mol Med. 2002;8:236–240. doi: 10.1016/s1471-4914(02)02333-x. [DOI] [PubMed] [Google Scholar]
- Kyratzi E, Pavlaki M, Kontostavlaki D, Rideout HJ, Stefanis L. Differential effects of Parkin and its mutants on protein aggregation, the ubiquitin-proteasome system, and neuronal cell death in human neuroblastoma cells. J Neurochem. 2007;102:1292–1303. doi: 10.1111/j.1471-4159.2007.04620.x. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–34975. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- Marx FP, Soehn AS, Berg D, Melle C, Schiesling C, Lang M, Kautzmann S, Strauss KM, Franck T, Engelender S, Pahnke J, Dawson S, von Eggeling F, Schulz JB, Riess O, Krüger R. The proteasomal subunit S6 ATPase is a novel synphilin-1 interacting protein—implications for Parkinson's disease. FASEB J. 2007;21:1759–1767. doi: 10.1096/fj.06-6734com. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001;297:191–194. doi: 10.1016/s0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Olanow CW. Proteolytic stress: a unifying concept for the etiopathogenesis of Parkinson's disease. Ann Neurol. 2003;53:S73–S84. doi: 10.1002/ana.10512. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson's disease. Exp Neurol. 2003;179:38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. 2009;10:104–115. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- Park Y, Hwang YP, Lee JS, Seo SH, Yoon SK, Yoon JB. Proteasomal ATPase-associated factor 1 negatively regulates proteasome activity by interacting with proteasomal ATPases. Mol Cell Biol. 2005;25:3842–3853. doi: 10.1128/MCB.25.9.3842-3853.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- Sakata E, Yamaguchi Y, Kurimoto E, Kikuchi J, Yokoyama S, Yamada S, Kawahara H, Yokosawa H, Hattori N, Mizuno Y, Tanaka K, Kato K. Parkin binds the Rpn10 subunit of 26S proteasomes through its ubiquitin-like domain. EMBO Rep. 2003;4:301–306. doi: 10.1038/sj.embor.embor764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature. 1998;391:715–718. doi: 10.1038/35661. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lübbert H. Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:2377–2393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003;278:22044–22055. doi: 10.1074/jbc.M212235200. [DOI] [PubMed] [Google Scholar]
- Um JW, Stichel-Gunkel C, Lübbert H, Lee G, Chung KC. Molecular interaction between parkin and PINK1 in mammalian neuronal cells. Mol Cell Neurosci. 2009;40:421–432. doi: 10.1016/j.mcn.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Xie Y, Varshavsky A. Physical association of ubiquitin ligases and the 26S proteasome. Proc Natl Acad Sci U S A. 2000;97:2497–2502. doi: 10.1073/pnas.060025497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhou HY, Li B, Niu GZ, Chen SD. Downregulation of parkin damages antioxidant defenses and enhances proteasome inhibition-induced toxicity in PC12 cells. J Neuroimmune Pharmacol. 2007;2:276–283. doi: 10.1007/s11481-007-9082-2. [DOI] [PubMed] [Google Scholar]
- You J, Pickart CM. A HECT domain E3 enzyme assembles novel polyubiquitin chains. J Biol Chem. 2001;276:19871–19878. doi: 10.1074/jbc.M100034200. [DOI] [PubMed] [Google Scholar]
- Zhang NY, Tang Z, Liu CW. alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem. 2008;283:20288–20298. doi: 10.1074/jbc.M710560200. [DOI] [PubMed] [Google Scholar]