

Abstract
Adrenomedullin (AM) belongs to calcitonin gene-related peptide (CGRP) family and is a pronociceptive mediator. This study investigated whether AM plays a role in the development of tolerance to morphine-induced analgesia. Repetitive intrathecal injection of morphine increased the expression of AM-like immunoreactivity (AM-IR) in the spinal dorsal horn and dorsal root ganglion (DRG) neurons. Ganglion explant culture study showed that this upregulation of AM-IR was μ-opioid receptor dependent through the use of another agonist, fentanyl, and a selective antagonist, CTAP (d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2). The coadministration of the selective AM receptor antagonist AM22-52 markedly attenuated the development of morphine tolerance, associated thermal hyperalgesia, and increase in AM-IR. A likely autocrine mechanism is supported by the finding that AM-IR is colocalized with AM receptor components in DRG neurons. Furthermore, opiate-induced increase in AM content was blocked by protein kinase C (PKC) inhibitors, whereas a PKC activator increased AM synthesis and release. A treatment with AM22-52 also inhibited increases in the expression of CGRP-IR in the spinal cord and DRGs as well as in culture ganglion explants, whereas exposure to CGRP failed to alter AM content. Together, these results reveal that a sustained opiate treatment induces an upregulation of AM through the activation of μ-opioid receptors and the PKC signaling pathway. This phenomenon contributes to the development of tolerance to the antinociceptive effects of opiates at least partially via the upregulation of CGRP. Targeting AM and its receptors should be considered as a novel approach to preserve the analgesic potency of opiates during their chronic use.
Introduction
Opiates are the most effective analgesics for the treatment of moderate to severe pain. However, chronic administration of opiates results in the development of tolerance to their analgesic effects, thus limiting their use (Carroll et al., 2004). The precise mechanisms underlying the development of opiate tolerance are not fully understood. It has been established that chronic administration of opiates induces the upregulation of pronociceptive mediators. Opiate tolerance is actually accompanied with pain hypersensitivity called opioid-associated hyperalgesia (Chang et al., 2007), which displays the similar alterations in neurochemical markers and neurobiological responses as inflammatory hyperalgesia (Ossipov et al., 2005). The changes in neuronal plasticity after sustained exposure to morphine are attributable to an increase in the levels and/or activities of glutamate (Zeng et al., 2006), calcitonin gene-related peptide (CGRP) (Menard et al., 1996), substance P (Powell et al., 2000), nitric oxide (Wong et al., 2000), and TRPV1 (Chen et al., 2008) in dorsal root ganglia (DRGs) or spinal dorsal horn. The inhibition of their activities by intrathecal administration of corresponding antagonist or inhibitor can attenuate or reverse tolerance. It is believed that an increase in these pronociceptive mediators at the spinal level results in the prolonged excitation of dorsal horn neurons or central sensitization, which could produce hyperalgesia (Baranauskas and Nistri, 1998) and counteract the antinociceptive effects of opiates (King et al., 2005).
Adrenomedullin (AM) may be associated with the development of tolerance to opiate-induced analgesia. AM is a 52 aa peptide and is broadly distributed in the CNS (Serrano et al., 2000; Shan and Krukoff, 2001). This peptide belongs to a family that includes AM2 (intermedin) (Roh et al., 2004), CGRP, amylin (Poyner et al., 2002), and calcitonin receptor-activating peptides (Katafuchi et al., 2003a,b). AM shares some of the cardiovascular properties of CGRP and has vasodilator as well as hypotensive effects (Brain and Grant, 2004). We have previously shown that AM is expressed in superficial laminae of the spinal cord and both peptidergic and nonpeptidergic (IB4) neurons in DRGs. Moreover, the intrathecal administration of the AM receptor agonist, AM1–50, produces long-lasting hyperalgesia (Ma et al., 2006). The expression of AM has been shown to be enhanced in the spinal cord and/or DRGs in acute (Ma et al., 2006) and chronic (Hong et al., 2009) inflammation, whereas blockade of AM receptors abolishes inflammatory hyperalgesia. These data suggest that AM is a potent nociceptive mediator.
The present study aimed to investigate the putative role of AM in the development of morphine-induced tolerance and associated hyperalgesia as well as the possible mechanisms involved in these effects. As AM induces its biological effects by acting on calcitonin receptor-like receptor (CLR)/receptor activity-modifying protein 2 or 3 (RAMP2 or RAMP3) receptor complex (Poyner et al., 2002), their colocalization in DRG neurons was also examined. Some of these results have been presented previously in abstract form (Chabot et al., 2008; Hong et al., 2008).
Materials and Methods
Animals.
Adult male Sprague Dawley rats (250–320 g; Charles River Breeding Laboratories) were housed individually at 22°C with 50% humidity under a 12 h light/dark cycle and given ad libitum access to food and water. The experimental procedures described in the present study were approved by animal care committees of McGill University, the Canadian Council on Animal Care and Fujian Normal University, and were in accordance with the guidelines of the treatment of animals of the International Association for the Study of Pain (Zimmermann, 1983). All efforts were made to minimize animal suffering and the number of animals used in our experiments.
Intrathecal catheter implantation.
Animals were implanted with chronic indwelling catheters using a modification of the previously described technique (Pogatzki et al., 2000). Briefly, intrathecal implantation of catheter was performed under anesthesia of a mixture of ketamine (100 mg/kg), acepromazine (2 mg/kg), and xylazine (10 mg/kg). The lumbar skin was longitudinally incised for 2–3 cm in the midline at the level of the iliac crests. The space between the L5 and L6 was punctured with a 21 gauge hypodermic needle. Tail flick indicated the penetration of the needle into an intrathecal space. After withdrawal of the needle, PE-10 polyethylene tubing (Stoelting) was cranially advanced 3 cm through the hole. Back skin was tunneled to allow the distal end of the catheter to reach the higher back. A small loop was formed in the end of tubing and sutured to the neck muscle. Rats were allowed to recover for 7 d before being used for behavioral testing. Only the animals with no evidence of neurological deficits after catheter placement were used for behavioral testing.
The chronic morphine protocol consisted of intrathecal administration of morphine sulfate (20 μg) to each rat per day for 6 consecutive days. This protocol has been proven to generate tolerance to morphine antinociception (Johnston et al., 2004; Q. Cai et al., 2007). Drug or vehicle was administered under conscious condition.
Assessment of nociceptive behavior.
Tail flick latency (TFL) was determined by the radiant heat tail flick assay using a Tail Flick Meter (IITC Life Science). A radiant heat was focused on the underside of the tail 3 cm from its distal end and tail flick latency automated by equipment. Radiant heat intensity was adjusted to produce on average a baseline of 2–3 s (high intensity) or 7.0–9.0 s (low intensity) in naive rats. The cutoff latency was established at 10 and 20 s, respectively, to avoid tissue damage. TFL at any test time point was measured three times at 1.5 min intervals, and the mean value of these measurements was taken.
Animals were given a bolus intrathecal injection of vehicle or drugs once daily for 6 d. Nociceptive testing was performed before intrathecal injection to establish the baseline response level, and 20 min after injection to determine the drug effect. On day 7, cumulative morphine dose–response curves were generated to determine acute opiate potency. Each animal was given ascending doses of morphine at 30 min intervals and tested 20 min after each injection. This protocol was continued until a maximal antinociceptive response was reached. The morphine dose–response curves were constructed, and the ED50 values of the agonist were determined from these curves. TFL was determined 20 min after morphine administration because the peak effect of morphine is achieved at 20 min (Jiang et al., 2006; M. Cai et al., 2007).
Ganglion explant cultures.
Animals were killed by decapitation. Trigeminal ganglia and DRGs (C2–L6) were dissected under sterile technique and collected in Hanks solution. After a wash in Hanks solution, ganglia were transferred to DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. The ganglia were grown at 37°C in 5% CO2. Media (including ligands when indicated) were replaced every other day. The ganglia explant cultures were exposed to various drugs, such as morphine, fentanyl, d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP), 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo (2,3-a)pyrrolo(3,4-c)-carbazole (Gö-6976), chelerythrine chloride, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H-89), phorbol ester, AM, and CGRP for 1 or 6 d. The ganglion explants were then harvested, frozen on dry ice, and stored at −80°C until additional processing.
Immunohistochemistry.
Rats were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.). The animals were perfused intracardially with cold 0.01 m PBS and subsequently with 4% paraformaldehyde in 0.1 m phosphate buffer (PB). The L4–L5 segments of the spinal cord and DRGs were removed and postfixed in the same fixative overnight. Tissues were then transferred into 30% sucrose in PB for cryoprotection. Spinal cord segments at a 40 μm thickness and DRGs at 10 μm were cut on a cryostat. Immunohistochemistry was performed at room temperature on free-floating sections (spinal cord) or slides (DRGs) using an avidin–biotin complex technique as described previously (M. Cai et al., 2007). To permit comparisons across treatment groups, sections from different groups were processed simultaneously. After pretreatment with 0.3% H2O2 and 10% normal goat serum, sections were incubated with polyclonal antisera in rabbit anti-AM (1:1000; Bachem) or CGRP (1:4000; Peninsula Laboratories) for 24–36 h at 4°C. The tissues were then transferred to biotinylated secondary IgG complex (1:200 in 10% goat serum in PBS) for 2 h followed by exposure to avidin–biotin horseradish peroxidase complex (1:100; Vector Laboratories) for 1 h. The chromogen was developed with 0.01% H2O2 and 0.05% diaminobenzidine. After being thoroughly rinsed with PBS, spinal sections were mounted on gelatin-coated slides, air dried, dehydrated in a series of graded alcohol, cleared in xylene, and coverslipped. Using different sections (n = 2), the primary antibody against either AM or CGRP was omitted in the immunohistochemical process for control. This procedure resulted in the absence of staining.
For double immunostaining of CLR with RAMP2 or RAMP3, or AM with RAMP2, RAMP3, or CLR, DRG sections were first incubated in 10% normal goat serum and next in a mixture of purified rabbit polyclonal antibody against CLR (1:100; Santa Cruz) with goat antisera of RAMP2 or RAMP3 (1:50–100) or rabbit polyclonal antisera of AM (1:150–200; Bachem) with goat antisera of CLR or RAMP2 or RAMP3 (1:50; Santa Cruz) for 24–36 h at 4°C. Sections were then incubated in goat anti-rabbit IgG conjugated with Alexa Fluor 488 and streptavidin conjugated with Alexa Fluor 568 (1:200; Invitrogen) for 1.5–2 h at room temperature. CLR immunoreactivity (CLR-IR) and AM-IR were labeled as green, whereas RAMP2-IR or RAMP3-IR or/and CLR-IR were labeled as red.
For control, omission of the primary antibody resulted in negative staining in all the sections tested. Incubation of purified AM or CLR antiserum was also preabsorbed with rAM1-50, CLR, RAMP2, and RAMP3 proteins (1 μm). These procedures resulted in the complete absence of staining.
Enzyme immunoassay (ELISA).
The dorsal half of the spinal cord segments or DRGs were minced and homogenized using a sonicator in ice-cold PBS containing Tris-HCl (40 mm), pH 7.5, SDS (2%), aprotinin (2 mg/ml), leupeptin (2 mg/ml), pepstatin A (2 mg/ml), phenylmethylsulfonyl fluoride (1 mm), dithiothreitol (1 mm), and EDTA (1 mm). Homogenates were shaken at 4°C followed by centrifugation at 15,000 × g for 30 min. The supernatant was then aliquoted and stored at −80°C.
Quantitative determination of AM (Phoenix Pharmaceuticals) and CGRP (Peninsula Laboratories) was performed using a commercially available ELISA kit. All ELISA procedures were performed according to manufacturer's instructions. Microplates were read using a microplate reader (Molecular Devices). Average value (picograms/milligram sample tissue) from three or four rats was taken to express the level of peptide in each group. The amount of protein in tissue samples was determined in the crude supernatants using the BCA Protein Assay kit (Bio-Rad). The level of AM and CGRP in the tissues is expressed as picograms of peptide per milligram of tissue protein. To compare data among different treatments, actual values were divided by the value of control group and expressed as percentage of control (control group being 100%).
Drugs and chemicals.
Ketamine, acepromazine, xylazine, and isoflurane were obtained from Animal Care Resources (McGill University). Morphine sulfate, CTAP, Gö-6976, chelerythrine chloride (CLT), H-89, phorbol-12,13-dibutyrate (phorbol ester), hydrogen peroxide, diaminobenzidine, aprotinin, leupeptin, pepstatin A, phenylmethylsulfonyl fluoride, dithiothreitol, and EDTA were purchased from Sigma-Aldrich. Fentanyl citrate was obtained from Yichang Humanwell Pharmaceutical. Hanks solution and DMEM were from Invitrogen. Human CGRP, AM1-50, and AM22-52 were obtained as a gift from Dr. Alain Fournier (Institut National de la Recherche en Santé, Quebec, Montreal, Canada) or purchased from Bachem. Antisera against AM and CGRP were obtained from Bachem and other antibodies (CLR, RAMP1, and RAMP2) from Santa Cruz Biotechnology. All antibodies were diluted in 0.9% sterile physiological saline. ABC-Elite kit was obtained from Vector Laboratories. The AM ELISA kit was from Phoenix Pharmaceuticals, and the CGRP ELISA kit was purchased from Peninsula Laboratories.
Statistical analysis.
Inhibition of the tail flick response (i.e., analgesia) was expressed as the percentage of the maximum possible effect (% MPE) using the following formula: % MPE = ([postdrug latency − baseline latency]/[cutoff time − baseline latency]) × 100%. The ED50 values are defined as the dose that produces 50% of the maximal effect (Emax) and were determined using a nonlinear regression analysis.
To determine quantitative measurement of AM-IR profile in the spinal dorsal horn, six spinal cord sections were randomly selected from each rat. Images were captured from laminae I–II of the dorsal horn at 250× magnification using a SPOT digital camera. The number of pixels occupied by AM-IR profiles was counted automatically using an image analysis software (imaging software Image-Pro Plus). Values are expressed as mean ± SEM.
Quantification of the AM- or CGRP-IR-positive and -negative DRG neurons was performed using image analysis software. Eight sections (870 × 640 μm each) per animal were randomly selected. The optical density of IR product was quantified using a 255 level grayscale. To determine the percentage of positive AM- or CGRP-IR neurons in each DRG, a threshold of average cytoplasmic density level of AM-IR product was set using an image of a normal DRG. The optical density threshold was then applied to all other DRG sections. All neurons sectioned through their nucleus for which mean optical density exceeded the threshold were counted as positive. Otherwise, cells were considered as negative. Approximately 150 cells were measured from each one of eight DRG sections of each rat (1200 cells in total per rat). The number of positive cells was expressed as a percentage of total DRG neurons in the corresponding subpopulation. For neuron size measurement, the neuronal soma area was calculated using imaging software Image-Pro Plus. Small neurons were <600 μm2, medium neurons were 600–1200 μm2, and large neurons were >1200 μm2 (Gendron et al., 2006). Data are expressed as mean ± SEM. Statistical significance between groups was examined using one-way ANOVA followed by Tukey's test for multiple comparisons. To detect changes over time between two groups (treatment group by time), data were analyzed using a two-way ANOVA. A value of p < 0.05 was considered statistically significant.
Results
Blockade of AM receptors by AM22-52 prevents the development of morphine-induced tolerance and heat hyperalgesia
Bolus saline (10 μl) or morphine (20 μg) was given intrathecally daily for 6 d, and the nociceptive threshold was assessed 20 min later by TFL. The cumulative dose–response of morphine was generated on day 7. After a 6 d treatment with saline, morphine ED50 value calculated from the dose–response curve was 7.3 ± 0.6 μg. In a separate group, morphine produced a maximal antinociceptive effect on day 1. Thereafter, morphine-induced response progressively declined over the following days, being completely abolished on day 6 (Fig. 1A). The cumulative dose–response curve showed a major shift to the right and an 11-fold increase in morphine ED50 value (81.5 ± 3.4 μg) compared with the saline-treated group (Fig. 1B, Table 1).
Figure 1.

Effects of AM receptor antagonist AM22-52 on the development of morphine tolerance and heat hyperalgesia in the tail flick test. Morphine (20 μg) or saline was administered intrathecally once daily with or without AM22-52. A, Tolerance to morphine-induced analgesia was prevented by the coadministration of AM22-52 in a dose-dependent manner (*p < 0.05 vs saline group). B, The antinociceptive effect of cumulative doses of morphine was generated on day 7. C, Six day injection of morphine induced heat hyperalgesia on day 7 (***p < 0.001 vs vehicle group), which was suppressed by a cotreatment with AM22-52 (###p < 0.001 vs morphine alone). Injection of AM22-52 for 6 d had no effect by itself. N = 6–9. Error bars indicate SEM.
Table 1.
Comparison of potency of morphine antinociception (ED50)
| Groups | Baseline latency (s) | ED50 (μg) (mean ± SEM) | 95% CI |
|---|---|---|---|
| Saline | 2.46 ± 0.2 | 7.3 ± 0.6 | 6.1–8.8 |
| Morphine (20 μg) | 2.75 ± 0.37 | 81.5 ± 3.4# | 69.7–94.3 |
| Morphine (20 μg) + AM22-52 (2 nmol) | 2.41 ± 0.11 | 68.7 ± 2.7* | 58.1–88.4 |
| Morphine (20 μg) + AM22-52 (5 nmol) | 2.30 ± 0.07 | 31.4 ± 1.0* | 24.9–39.3 |
| Morphine (20 μg) + AM22-52 (10 nmol) | 2.30 ± 0.12 | 8.5 ± 0.3* | 7.1–10.0 |
CI, Confidence interval.
#p < 0.05 versus saline group;
*p < 0.05 versus morphine group.
To determine the possible role of AM in the development of morphine tolerance, morphine was coadministered with the AM receptor antagonist AM22-52 for 6 d. As illustrated in Figure 1, AM22-52 prevented the development of morphine tolerance and shifted the dose–response curve of morphine to the left in a dose-dependent manner (2, 5, and 10 nmol) (Fig. 1A,B) (F(4,30) = 364.6; p < 0.001). The cotreatment with AM22-52 at a dose of 10 nmol maintained analgesic effect of morphine with an ED50 value being almost equal (8.5 ± 0.3 μg) (Fig. 1B, Table 1) to the saline group on day 7. By itself, intrathecally administered AM22-52 at a dose of 10 nmol did not alter TFL (n = 7) (data not shown).
Saline, morphine (20 μg), morphine (20 μg) plus AM22-52 (10 nmol), or AM22-52 (10 nmol) were administered intrathecally once daily for 6 d. TFL was assessed each day before intrathecal administration and on day 7 as well. No significant changes in TFLs were detected throughout the whole observation period in saline-injected rats. Chronic morphine injection significantly decreased TFLs compared with saline group (Fig. 1C) (F(3,24) = 32.26; p < 0.001), indicating thermal hyperalgesia. However, the concurrent administration of AM22-52 inhibited the hyperalgesic effect of morphine as reflected by a significant difference with morphine alone group (Fig. 1C) (p < 0.001). AM22-52 by itself had no effect on thermal pain sensitivity compared with the saline treatment (p > 0.05).
Chronic administration of morphine increases the level of AM-IR in DRGs and spinal dorsal horn
After a 6 d treatment with saline or morphine, AM-IR-positive neurons in L4–L5 DRGs were examined. AM-IR was mainly expressed in small- to medium-sized neurons (Fig. 2A,D). In morphine (20 μg)-treated rats, the percentages of AM-IR neurons in both small- and medium-sized subpopulations were significantly increased (Fig. 2B,D) (p < 0.001 or 0.05). A coadministration of AM22-52 (10 nmol) with morphine significantly suppressed morphine-induced increase in AM-IR in small- and medium-sized neurons (Fig. 2C,D) (F(2,11) = 7.1; p < 0.05). ELISA data revealed that the chronic treatment with morphine induced a twofold increase in AM content in DRGs compared with saline treatment (Fig. 2E) (F(2,11) = 37.5; p < 0.05). The coadministration of AM22-52 abolished morphine-induced increase in AM content (Fig. 2E) (p < 0.05).
Figure 2.

Effect of chronic morphine on AM levels in lumbar DRGs. AM-IR was expressed in small (small arrows), medium (large arrows), and large (arrowheads) subtypes of DRG cells. Compared with saline injection (A, D), morphine (20 μg) significantly increased the percentage of small- and medium-sized AM-IR DRG neurons (B, D) (*p < 0.05). The coadministration of AM22-52 (10 nmol) significantly suppressed morphine-induced increase in AM-IR-positive neurons (C, D) (#p < 0.05). Similarly, chronic morphine considerably increased AM content in lumbar DRG homogenates assayed by ELISA (E) (*p < 0.05). Morphine-induced increase in AM content was suppressed by the coadministration of AM22-52 (E) (#p < 0.05). Scale bar, 100 μm. N = 4–5. Error bars indicate SEM.
AM-IR was localized in the superficial laminae of the spinal cord in saline-administered rats (Fig. 3A). A significant increase in AM-IR was seen in the superficial dorsal horn after a 6 d treatment with morphine compared with saline (Fig. 3B,D) (F(2,12) = 48.6; p < 0.05). This increase was abolished by the coadministration of AM22-52 (Fig. 3C,D) (p < 0.05).
Figure 3.

Effects of chronic morphine on the expression of AM-IR in lumbar spinal dorsal horn. Compared with saline injection (A, D), morphine (20 μg) significantly increased AM-IR in spinal dorsal horn (B, D) (*p < 0.05). The coadministration of AM22-52 (10 nmol) significantly suppressed morphine-induced increase in AM-IR in the dorsal horn of the lumbar spinal cord (C, D) (#p < 0.05). Scale bar, 500 μm. N = 4–5. Error bars indicate SEM.
Morphine increases AM content in cultured ganglion explants through the activation of μ-opiate receptors and the protein kinase C (PKC) signaling pathway
To confirm morphine-induced upregulation of AM, cultured ganglion explants were exposed to vehicle or morphine for 6 d and AM content was assayed. Figure 4 shows that the morphine treatment significantly increased AM content in ganglion explants in a concentration-dependent manner (Fig. 4A) (p < 0.05). To further establish that morphine-induced increase in AM by the activation of μ-opiate receptors, ganglion explants were cultured with morphine (3.3 μm) in the presence or absence of the selective μ-opiate receptor antagonist CTAP for 6 d. CTAP attenuated morphine-induced increase in AM content at concentrations of 10 and 100 nm (Fig. 4B) (F(4,13) = 25.4; p < 0.001). Chronic treatment with fentanyl, another μ-opiate agonist, at concentrations of 10, 100, and 1000 nm for 6 d also significantly increased AM content in ganglia explants compared with vehicle treatment (Fig. 4C) (F(3,12) = 27.6; p < 0.001).
Figure 4.

Effect of chronic treatment with a μ-opiate receptor agonist or antagonist or PKC activator or inhibitor on AM content in ganglion explant cultures. Morphine significantly increased AM content (A), an effect blocked by a cotreatment with CTAP (B) and PKC inhibitors Gö-6976 and CLT (D). A selective μ-opiate agonist fentanyl mimicked the effect of morphine on AM content (C). The PKC activator phobor ester increased AM content in cultured explants (E) and medium (F). *p < 0.05, **p < 0.01, and ***p < 0.001 compared to vehicle group; ##p < 0.01 and ###p < 0.001 compared to morphine alone group. N = 4–5. Error bars indicate SEM.
To determine the signal transduction pathway underlying the morphine-induced upregulation of AM, cultured ganglion explants were cotreated with morphine (3.3 μm) and a PKC inhibitor, Gö-6976, for 6 d. Gö-6976 (100 nm) completely inhibited morphine-induced increase in AM content (Fig. 4D) (F(2,8) = 112.0; p < 0.05). The inhibitor by itself had no effect on AM content compared with vehicle. A cotreatment with another PKC inhibitor, CLT (500 nm), similarly abolished morphine-induced increase in AM content (Fig. 4D) (F(2,6) = 61.4; p < 0.05), whereas CLT by itself did not alter AM content in ganglion explants. However, a cotreatment with a protein kinase A (PKA) inhibitor, H-89, failed to alter morphine-induced increase in AM content at concentrations of 250 nm and 5 μm (n = 3) (data not shown).
To further establish the role of PKC in morphine-induced increase in AM content, ganglion explants were treated with a PKC activator phorbol ester for 6 d. Figure 4 shows that phorbol ester significantly increased AM content in cultured ganglion explants (Fig. 4E) (F(3,8) = 6.38; p < 0.05) and medium (Fig. 4F) (F(3,8) = 62.5; p < 0.001) in a concentration-dependent manner.
AM may act via CGRP to modulate the development of tolerance to morphine-induced analgesia
As CGRP has been documented to play a pivotal role in the development of morphine tolerance (Menard et al., 1996), the present study investigated whether increase in AM level produced by chronic exposure to morphine affects the expression of CGRP. Intrathecal injection of morphine (20 μg) for 6 consecutive days significantly increased the percentage of CGRP-IR in small (F(2,10) = 56.7; p < 0.05)- and medium (F(2,10) = 40.6; p < 0.05)-sized neurons in L4–L5 DRGs (Fig. 5B,D) compared with saline treatment (Fig. 5A,D). The coadministration of AM22-52 significantly reduced the number of CGRP-IR-positive neurons (Fig. 5C,D) (p < 0.05) compared with morphine alone. ELISA assay confirmed that chronic morphine significantly increased CGRP content (Fig. 5E) (F(2,7) = 7.0; p < 0.05), and this increase was abolished by a cotreatment with AM22-52 (Fig. 5E) (p < 0.05).
Figure 5.

Effect of the antagonism of AM receptors on the expression of CGRP-IR in lumbar DRGs. CGRP-IR was expressed in small (small arrows), medium (large arrows), and large (arrowheads) subtypes of DRG cells. Compared with saline injection (A, D, E), chronic morphine (20 μg) administration significantly increased the percentages of CGRP-IR-positive neurons in DRGs (B, D) (*p < 0.05–0.001) and CGRP content in DRG homogenates (E) (*p < 0.05). A cotreatment with AM22-52 (10 nmol) suppressed morphine-induced increase in the expression of CGRP-IR or CGRP content (C–E) (#p < 0.05–0.001). Scale bar, 100 μm. N = 4–5. Error bars indicate SEM.
Repeated exposure to morphine for 6 d also significantly increased CGRP-IR in superficial layers of the spinal cord (Fig. 6B,D) (F(2,12) = 9.7; p < 0.05) compared with saline treatment (Fig. 6A,D). This effect was significantly suppressed by the coadministration of AM22-52 (Fig. 6C,D) (p < 0.05). ELISA data confirmed that chronic morphine treatment significantly increased CGRP content in the dorsal half of the spinal cord compared with saline (Fig. 6E) (p < 0.05) and AM22-52 inhibited this increase (Fig. 6E) (F(2,6) = 20.2; p < 0.05).
Figure 6.

Effect of the antagonism of AM receptors on CGRP-IR level in the spinal cord. Compared with saline injection (A, D, E), chronic morphine (20 μg) administration significantly increased the optical densities of CGRP-IR in superficial laminae of the spinal cord (B, D) (*p < 0.05) and CGRP content in spinal dorsal horn homogenates (E) (*p < 0.05). The coadministration of AM22-52 (10 nmol) suppressed morphine-induced increase in CGRP levels (C–E) (#p < 0.05). A chronic treatment with the AM receptor agonist AM1-50 concentration-dependently increased CGRP content in cultured ganglion explants (F) (*p < 0.05, ***p < 0.001). Scale bar, 100 μm. N = 4–5. Error bars indicate SEM.
Since in vivo data showed that the blockade of AM receptors reduced morphine-induced upregulation of CGRP, the stimulation of AM receptors on CGRP expression was investigated further in ganglion culture. Figure 6F illustrates that a treatment with AM for 24 h increased CGRP content in ganglion explants in a concentration-dependent manner (0.1–100 nm; F(4,10) = 31.7; p < 0.05). However, a treatment with CGRP (1–1000 nm) for 24 h failed to alter AM content in ganglion explants (n = 3) (data not shown).
AM colocalizes with CLR, RAMP2, and RAMP3 in DRG neurons
The results showing that chronic treatment with morphine failed to increase AM and CGRP contents in DRGs after blockade of AM receptors suggest that AM modulates itself as well as CGRP expression through an autocrine or paracrine mechanism. As functional AM receptors are generated by CLR associated with RAMP2 (AM1 receptors) or RAMP3 (AM2 receptors) (Husmann et al., 2003), the possible colocalization of AM with CLR, RAMP2, and RAMP3 was examined. AM-IR (Fig. 7A,D,G), CLR-IR (Fig. 7B), RAMP2-IR (Fig. 7E), or RAMP3-IR (Fig. 7H) was detected in the soma of small- and medium-sized DRG neurons. Staining was diffusely distributed in small vesicles throughout the cytoplasm but was excluded from the nucleus. Moreover, AM-IR was frequently found to be colocalized with CLR-IR (Fig. 7C), RAMP2-IR (Fig. 7F), and RAMP3-IR (Fig. 7I) in the soma of neurons, suggesting that AM released by DRG neurons acts primarily on AM receptors present in the DRG neurons.
Figure 7.
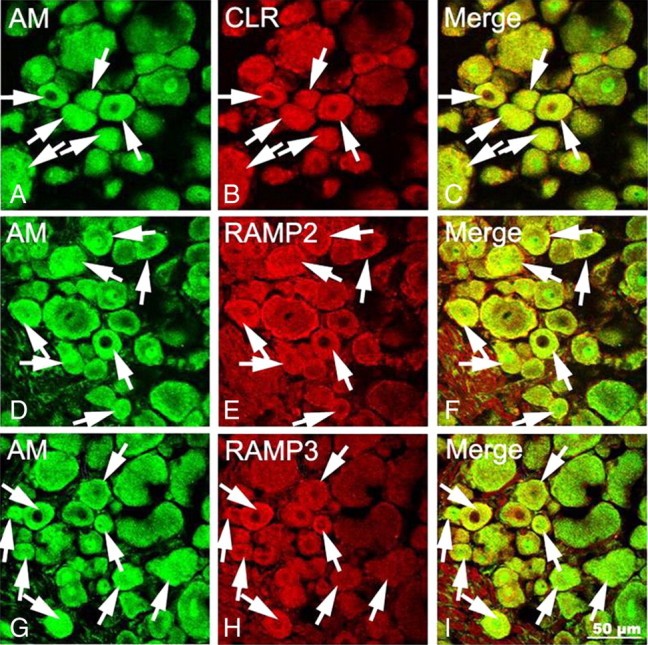
Confocal images showing localization of AM-IR and AM receptor components in DRG neurons. AM-IR neurons are identified by Alexa Fluor 488 (A, D, G, arrows; green) fluorescence, whereas CLR-IR (B)-, RAMP2-IR (E)-, and RAMP3-IR (H)-positive neurons are labeled by Alexa Fluor 568 (arrows; red) fluorescence. AM-IR is extensively colocalized with CLR-IR, RAMP2-IR, or RAMP3-IR in DRG neurons (C, F, I, arrows; yellow). Scale bar, 50 μm.
Discussion
The present study was designed to investigate the role of AM receptor signaling in tolerance to morphine-induced analgesia. We have demonstrated that the chronic morphine treatment induced an increase in AM-IR expression in the spinal cord and/or DRGs in vivo and in vitro. The morphine-induced increase in AM content was blocked by CTAP in ganglion explant culture, whereas fentanyl mimicked the effect of morphine. The increase in AM content in the cultured ganglia was abolished by the PKC but not PKA inhibitors and the PKC activator upregulated AM content. Furthermore, the blockade of AM receptors by AM22-52 prevented the development of morphine tolerance and the increases in the expression of AM-IR. The coadministration of AM22-52 also inhibited opiate-associated thermal hyperalgesia and morphine-induced increase in CGRP-IR in the spinal cord and DRGs. These results for the first time suggest that the upregulation of AM, which is induced by chronic morphine administration via PKC signaling pathway, contributes to morphine tolerance and associated hyperalgesia.
This study shows that the chronically administered morphine to the rat increased AM-IR levels in superficial layers of the spinal cord as well as in small- and medium-sized DRG neurons, the key structures involved in nociceptive processing (Millan, 1999). The observations that morphine increased the number of AM-IR-positive neurons by 22–36% but AM content by 208% in DRGs suggest that AM content in individual AM neuron was also increased. Similarly, an in vitro study showed that sustained exposure to morphine concentration-dependently increased AM level in cultured ganglion explants, an effect suppressed by the selective μ-opioid receptor antagonist CTAP (Pelton et al., 1985), whereas another selective μ-opioid receptor agonist, fentanyl, also upregulated AM synthesis. The finding that the morphine-induced increase in AM level is mediated through the activation of μ-opioid receptors is consistent with the purported role of this receptor subtype in the development of tolerance (Taylor and Fleming, 2001; Martini and Whistler, 2007). Together, these results suggest that chronic morphine treatment upregulates AM in the spinal cord and DRGs via μ-opioid receptor mechanism.
Next, we investigated the role of PKC and PKA signaling transduction pathways in morphine-induced upregulation of AM. The pivotal role of PKC in the development of morphine tolerance has been well documented. A chronic morphine treatment was shown to increase the level of PKC in the spinal dorsal horn (Mao et al., 1995; Granados-Soto et al., 2000), whereas the inhibition of its synthesis by gene knock-out (Zeitz et al., 2001) or antisense treatment (Hua et al., 2002) prevents the development of morphine tolerance. However, it remains to be clear whether PKC activation is related to an upregulation of pronociceptive mediators involved in plastic changes seen in tolerant animals. Our in vitro study showed that morphine-induced upregulation of AM was blocked by two PKC inhibitors, Gö-6976 and CLT. Furthermore, the PKC activator phorbol ester increased AM expression and release, similar to that observed in cultured murine monocyte/macrophage cell line (Kubo et al., 1998). These results suggest that the activation of the PKC signaling pathway is an upstream regulator of AM synthesis after long exposure to opiates. Our data are consistent with a previous report showing that opiate-induced activation of PKC signaling is involved in the increased synthesis of CGRP seen in cultured DRGs (Belanger et al., 2002). However, it seems unlikely that PKA is a major player since a treatment with the PKA inhibitor H-89 failed to regulate morphine-induced upregulation of AM in cultured ganglia.
Most interestingly, a cotreatment with the AM receptor antagonist AM22-52 maintained the potency of morphine-induced analgesia as evidenced by shifting the dose–response curve to the left. AM22-52 is currently the only available antagonist specific for AM receptors with a preferential affinity for AM1 over AM2 receptors (Hay et al., 2003). This antagonist demonstrates only low (Husmann et al., 2000; Takhshid et al., 2006) or no (Hay et al., 2003) affinity for CGRP receptors. The AM1 (CLR with RAMP2) and AM2 (CLR with RAMP3) receptor components are enriched in the spinal dorsal horn and DRGs (Owji et al., 1996; Cottrell et al., 2005; Ma et al., 2006). Therefore, antagonism of AM, most likely AM1, receptors in the spinal cord and DRGs likely explains the effects observed with AM22-52. Our results showed that the blockade of AM receptors suppressed morphine-induced increase in AM levels, implying that this peptide can facilitate its own synthesis by acting on AM receptors in DRG neurons. This notion is supported by the anatomical colocalization of AM-IR with CLR-IR, RAMP2-IR, or RAMP3-IR in DRG neurons. Acting via an autocrine or paracrine pathway, AM could induce a positive feedback to stimulate its own production, thus keeping AM concentration at a high level. This could lead to AM playing a role in the maintenance of the hypersensitivity of spinal nociceptive neurons. Accordingly, the blockade of AM receptors could inhibit this positive cycle maintaining the synthesis of AM at a minimal level. The results showing that the antagonism of AM receptors preserved the analgesic potency of morphine and inhibited morphine-induced increase in AM-IR in DRGs and spinal dorsal horn demonstrate that the augmented production of AM is involved in the loss of antinociceptive potency of morphine. Therefore, the present study suggests that AM/AM receptor signaling plays a key role in the development of morphine tolerance.
AM has been demonstrated to be a potent pronociceptive mediator. The upregulation of AM in the spinal dorsal horn and DRGs could contribute to the induction of hyperalgesia observed during acute (Ma et al., 2006) and chronic (Hong et al., 2009) inflammation. In accordance with these observations, the present study showed that the blockade of AM receptors inhibited opiate-associated hyperalgesia, a phenomenon that has been well documented to occur after chronic morphine administration (Chang et al., 2007). We further tested the hypothesis that the effects of AM are also mediated via the production of CGRP on the basis of the purported role of this peptide in opiate tolerance (Trang et al., 2005). It is well established that repeated administration of morphine increases the expression of CGRP in the spinal dorsal horn and DRG neurons (Menard et al., 1996; Powell et al., 2000) and its release in the spinal cord (Gardell et al., 2002). More importantly, the antagonism of CGRP receptors not only prevents and reverses morphine-induced tolerance but also suppresses morphine-induced upregulation of CGRP in the spinal cord (Menard et al., 1996; Powell et al., 2000) and DRGs (Powell et al., 2003). In accordance with these previous reports, a chronic morphine treatment increased CGRP-IR in DRG neurons and spinal dorsal horn. We further observed that the level of CGRP in individual CGRP-IR neurons in DRGs was also increased as revealed by the comparison between the increase in the number of CGRP-IR neurons (10–20%) and the content of CGRP (167%). Similarly, chronic morphine increased the expression of CGRP-IR in nerve fibers by 25% but its content by 89% in the spinal dorsal horn. All these increases were inhibited after the blockade of AM receptors, suggesting that the stimulation of AM receptor signaling is mediated by the upregulation of CGRP, and that AM is able to stimulate CGRP synthesis in DRG neurons and the release of CGRP from nerve terminals. Our in vitro study showed that AM increased CGRP content in cultured ganglion explants in a concentration-dependent manner. As CGRP failed to alter AM content in cultured ganglion explants, AM must have a direct effect on CGRP synthesis and is likely an upstream intermediary in chronic morphine-induced CGRP release. These observations are in agreement with a report showing that exogenous AM increased the mRNA and peptide levels of CGRP in human adipocyte tissue, whereas CGRP failed to modulate AM mRNA (Linscheid et al., 2005). Since AM and CGRP were found to be coexpressed in DRG neurons (Ma et al., 2006) and AM is extensively colocalized with AM receptor components CLR, RAMP2, or RAMP3, it is likely that AM can stimulate CGRP synthesis through an autocrine or paracrine mechanism. Together, these data suggest that morphine-induced hyperalgesia seen after a chronic exposure occurs as a result of increased synthesis and release of the pronociceptive mediators CGRP and AM. This is consistent with the notion that enhanced excitatory neurotransmission at the spinal level counteracts the antinociceptive potency of morphine leading to the development of morphine tolerance (King et al., 2005).
As illustrated in Figure 8, our results suggest that chronically administered morphine activates PKC signaling transduction pathway in DRG neurons via μ-opioid receptors, resulting in the upregulation of AM synthesis. The increased AM production in turn binds to its receptors expressed in DRG neurons to further stimulate its own production and that of CGRP via an autocrine or paracrine mechanism. During a chronic exposure to morphine, the synthesis and release of both AM and CGRP are facilitated, leading to the development of hyperalgesia and antinociceptive tolerance. Thus, targeting AM/AM receptor signaling could be a useful therapy to prevent the development of opiate tolerance during sustained administration of these drugs.
Figure 8.

A proposed mechanism highlighting the role of AM in the development of morphine tolerance and morphine-associated hyperalgesia. Morphine binds to opiate μ-opioid receptors (μOR) in DRG neurons and activates the PKC, but not PKA, signaling transduction pathway, leading to the upregulation of the synthesis of AM. In turn, AM binds to AM receptors (AMR) expressed in DRG neurons to stimulate the production of AM and CGRP through autocrine and/or paracrine mechanism(s). Accordingly, a chronic morphine treatment can induce the synthesis of both AM and CGRP, leading to the development of hyperalgesia and tolerance to morphine-induced analgesia.
Footnotes
This study was supported by grants from the Canadian Institutes of Health Research (R.Q.) and the Natural Science Foundation of China (Y.H.). We thank Mira Thakur for proofreading the English text.
The authors declare no competing financial interests.
References
- Baranauskas G, Nistri A. Sensitization of pain pathways in the spinal cord: cellular mechanisms. Prog Neurobiol. 1998;54:349–365. doi: 10.1016/s0301-0082(97)00067-1. [DOI] [PubMed] [Google Scholar]
- Belanger S, Ma W, Chabot JG, Quirion R. Expression of calcitonin gene-related peptide, substance P and protein kinase C in cultured dorsal root ganglion neurons following chronic exposure to mu, delta and kappa opiates. Neuroscience. 2002;115:441–453. doi: 10.1016/s0306-4522(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84:903–934. doi: 10.1152/physrev.00037.2003. [DOI] [PubMed] [Google Scholar]
- Cai M, Chen T, Quirion R, Hong Y. The involvement of spinal bovine adrenal medulla 22-like peptide, the proenkephalin derivative, in modulation of nociceptive processing. Eur J Neurosci. 2007;26:1128–1138. doi: 10.1111/j.1460-9568.2007.05755.x. [DOI] [PubMed] [Google Scholar]
- Cai Q, Jiang J, Chen T, Hong Y. Sensory neuron-specific receptor agonist BAM8–22 inhibits the development and expression of tolerance to morphine in rats. Behav Brain Res. 2007;178:154–159. doi: 10.1016/j.bbr.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Carroll IR, Angst MS, Clark JD. Management of perioperative pain in patients chronically consuming opioids. Reg Anesth Pain Med. 2004;29:576–591. doi: 10.1016/j.rapm.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Chabot JG, Hong Y, Fournier A, Quirion R. Mechanisms underlying increase in levels and effects of adrenomedullin in morphine tolerance. Soc Neurosci Abstr. 2008;34:133. [Google Scholar]
- Chang G, Chen L, Mao J. Opioid tolerance and hyperalgesia. Med Clin North Am. 2007;91:199–211. doi: 10.1016/j.mcna.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Chen Y, Geis C, Sommer C. Activation of TRPV1 contributes to morphine tolerance: involvement of the mitogen-activated protein kinase signaling pathway. J Neurosci. 2008;28:5836–5845. doi: 10.1523/JNEUROSCI.4170-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GS, Roosterman D, Marvizon JC, Song B, Wick E, Pikios S, Wong H, Berthelier C, Tang Y, Sternini C, Bunnett NW, Grady EF. Localization of calcitonin receptor-like receptor and receptor activity modifying protein 1 in enteric neurons, dorsal root ganglia, and the spinal cord of the rat. J Comp Neurol. 2005;490:239–255. doi: 10.1002/cne.20669. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Lucido AL, Mennicken F, O'Donnell D, Vincent JP, Stroh T, Beaudet A. Morphine and pain-related stimuli enhance cell surface availability of somatic δ-opioid receptors in rat dorsal root ganglia. J Neurosci. 2006;26:953–962. doi: 10.1523/JNEUROSCI.3598-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granados-Soto V, Kalcheva I, Hua X, Newton A, Yaksh TL. Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. doi: 10.1016/S0304-3959(99)00281-X. [DOI] [PubMed] [Google Scholar]
- Hay DL, Howitt SG, Conner AC, Schindler M, Smith DM, Poyner DR. CL/RAMP2 and CL/RAMP3 produce pharmacologically distinct adrenomedullin receptors: a comparison of effects of adrenomedullin22-52, CGRP8–37 and BIBN4096BS. Br J Pharmacol. 2003;140:477–486. doi: 10.1038/sj.bjp.0705472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Liu Y, Chabot JG, Fournier A, Quirion R. Upregulation of adrenomedullin in the spinal cord and dorsal root ganglia in the early phase of CFA-induced inflammation in rats. Pain. 2009;146:105–113. doi: 10.1016/j.pain.2009.07.015. [DOI] [PubMed] [Google Scholar]
- Hua XY, Moore A, Malkmus S, Murray SF, Dean N, Yaksh TL, Butler M. Inhibition of spinal protein kinase Calpha expression by an antisense oligonucleotide attenuates morphine infusion-induced tolerance. Neuroscience. 2002;113:99–107. doi: 10.1016/s0306-4522(02)00157-4. [DOI] [PubMed] [Google Scholar]
- Husmann K, Sexton PM, Fischer JA, Born W. Mouse receptor-activity-modifying proteins 1, -2 and -3: amino acid sequence, expression and function. Mol Cell Endocrinol. 2000;162:35–43. doi: 10.1016/s0303-7207(00)00212-4. [DOI] [PubMed] [Google Scholar]
- Husmann K, Born W, Fischer JA, Muff R. Three receptor-activity-modifying proteins define calcitonin gene-related peptide or adrenomedullin selectivity of the mouse calcitonin-like receptor in COS-7 cells. Biochem Pharmacol. 2003;66:2107–2115. doi: 10.1016/j.bcp.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Jiang J, Huang J, Hong Y. Bovine adrenal medulla 22 reverses antinociceptive morphine tolerance in the rat. Behav Brain Res. 2006;168:167–171. doi: 10.1016/j.bbr.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katafuchi T, Kikumoto K, Hamano K, Kangawa K, Matsuo H, Minamino N. Calcitonin receptor-stimulating peptide, a new member of the calcitonin gene-related peptide family. Its isolation from porcine brain, structure, tissue distribution, and biological activity. J Biol Chem. 2003a;278:12046–12054. doi: 10.1074/jbc.M207970200. [DOI] [PubMed] [Google Scholar]
- Katafuchi T, Hamano K, Kikumoto K, Minamino N. Identification of second and third calcitonin receptor-stimulating peptides in porcine brain. Biochem Biophys Res Commun. 2003b;308:445–451. doi: 10.1016/s0006-291x(03)01413-x. [DOI] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- Kubo A, Minamino N, Isumi Y, Katafuchi T, Kangawa K, Dohi K, Matsuo H. Production of adrenomedullin in macrophage cell line and peritoneal macrophage. J Biol Chem. 1998;273:16730–16738. doi: 10.1074/jbc.273.27.16730. [DOI] [PubMed] [Google Scholar]
- Linscheid P, Seboek D, Zulewski H, Keller U, Müller B. Autocrine/paracrine role of inflammation-mediated calcitonin gene-related peptide and adrenomedullin expression in human adipose tissue. Endocrinology. 2005;146:2699–2708. doi: 10.1210/en.2004-1424. [DOI] [PubMed] [Google Scholar]
- Ma W, Chabot JG, Quirion R. A role for adrenomedullin as a pain-related peptide in the rat. Proc Natl Acad Sci U S A. 2006;103:16027–16032. doi: 10.1073/pnas.0602488103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Phillips LL, Lu J, Mayer DJ. Increases in protein kinase C gamma immunoreactivity in the spinal cord of rats associated with tolerance to the analgesic effects of morphine. Brain Res. 1995;677:257–267. doi: 10.1016/0006-8993(95)00161-i. [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Menard DP, van Rossum D, Kar S, St. Pierre S, Sutak M, Jhamandas K, Quirion R. A calcitonin gene-related peptide receptor antagonist prevents the development of tolerance to spinal morphine analgesia. J Neurosci. 1996;16:2342–2351. doi: 10.1523/JNEUROSCI.16-07-02342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Owji AA, Gardiner JV, Upton PD, Mahmoodi M, Ghatei MA, Bloom SR, Smith DM. Characterisation and molecular identification of adrenomedullin binding sites in the rat spinal cord: a comparison with calcitonin gene-related peptide receptors. J Neurochem. 1996;67:2172–2179. doi: 10.1046/j.1471-4159.1996.67052172.x. [DOI] [PubMed] [Google Scholar]
- Pelton JT, Gulya K, Hruby VJ, Duckles SP, Yamamura HI. Conformationally restricted analogs of somatostatin with high μ-opiate receptor specificity. Proc Natl Acad Sci U S A. 1985;82:236–239. doi: 10.1073/pnas.82.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogatzki EM, Zahn PK, Brennan TJ. Effect of pretreatment with intrathecal excitatory amino acid receptor antagonists on the development of pain behavior caused by plantar incision. Anesthesiology. 2000;93:489–496. doi: 10.1097/00000542-200008000-00029. [DOI] [PubMed] [Google Scholar]
- Powell KJ, Ma W, Sutak M, Doods H, Quirion R, Jhamandas K. Blockade and reversal of spinal morphine tolerance by peptide and non-peptide calcitonin gene-related peptide receptor antagonists. Br J Pharmacol. 2000;131:875–884. doi: 10.1038/sj.bjp.0703655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci. 2003;18:1572–1583. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, Muff R, Fischer JA, Foord SM. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004;279:7264–7274. doi: 10.1074/jbc.M305332200. [DOI] [PubMed] [Google Scholar]
- Serrano J, Uttenthal LO, Martínez A, Fernández AP, Martínez de Velasco J, Alonso D, Bentura ML, Santacana M, Gallardo JR, Martínez-Murillo R, Cuttitta F, Rodrigo J. Distribution of adrenomedullin-like immunoreactivity in the rat central nervous system by light and electron microscopy. Brain Res. 2000;853:245–268. doi: 10.1016/s0006-8993(99)02273-8. [DOI] [PubMed] [Google Scholar]
- Shan J, Krukoff TL. Distribution of preproadrenomedullin mRNA in the rat central nervous system and its modulation by physiological stressors. J Comp Neurol. 2001;432:88–100. doi: 10.1002/cne.1090. [DOI] [PubMed] [Google Scholar]
- Takhshid MA, Poyner DR, Chabot JG, Fournier A, Ma W, Zheng WH, Owji AA, Quirion R. Characterization and effects on cAMP accumulation of adrenomedullin and calcitonin gene-related peptide (CGRP) receptors in dissociated rat spinal cord cell culture. Br J Pharmacol. 2006;148:459–468. doi: 10.1038/sj.bjp.0706750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DA, Fleming WW. Unifying perspectives of the mechanisms underlying the development of tolerance and physical dependence to opioids. J Pharmacol Exp Ther. 2001;297:11–18. [PubMed] [Google Scholar]
- Trang T, Quirion R, Jhamandas K. The spinal basis of opioid tolerance and physical dependence: involvement of calcitonin gene-related peptide, substance P, and arachidonic acid-derived metabolites. Peptides. 2005;26:1346–1355. doi: 10.1016/j.peptides.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Wong CS, Hsu MM, Chou YY, Tao PL, Tung CS. Morphine tolerance increases [3H]MK-801 binding affinity and constitutive neuronal nitric oxide synthase expression in rat spinal cord. Br J Anaesth. 2000;85:587–591. doi: 10.1093/bja/85.4.587. [DOI] [PubMed] [Google Scholar]
- Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI. Reduced development of tolerance to the analgesic effects of morphine and clonidine in PKC gamma mutant mice. Pain. 2001;94:245–253. doi: 10.1016/S0304-3959(01)00353-0. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental painin conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]