

Abstract
We had described a muscarinic-mediated long-term synaptic enhancement at Schaffer collateral synapses caused by the insertion of AMPARs in spines of rat hippocampal CA1 pyramidal neurons that requires Ca2+ release from IP3-sensitive stores (Fernández de Sevilla et al., 2008). We now show that this AMPA-mediated LTPIP3 is precisely matched by an amplification of NMDAR-mediated transmission. The enhanced AMPAR transmission involves SNARE protein activity and CaMKII activation. The amplification of NMDA transmission requires combined CaMKII, PKC, and SRC kinase activity without detectable surface incorporation of NMDARs, suggesting that changes in receptor properties mediate this process. The enhanced AMPAR- and NMDAR-mediated transmission markedly reduce the induction threshold of “Hebbian” LTP. We conclude that both modes of glutamatergic synaptic potentiation may play a critical functional role in the regulation of the learning machinery of the brain by adding flexibility to the demands of the hippocampal network.
Introduction
Although considerable effort has been made to elucidate the causal and expression mechanisms of long-term potentiation (LTP), they remain controversial and more varied than previously considered (Johnston et al., 1992; Cohen et al., 1998; Chevaleyre et al., 2006; Bayazitov et al., 2007; Campanac and Debanne, 2007; Fernández de Sevilla et al., 2008; Fidzinski et al., 2008). The classical “Hebbian” LTP (LTPH) (Hebb, 1949) requires activation of NMDA receptors (NMDARs) and is caused by an enhancement of AMPA receptor (AMPAR)-mediated transmission (Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Collingridge et al., 2004; Malenka and Bear, 2004). Other varieties of LTP are typified by a long-term enhancement of NMDAR-mediated transmission (O'Connor et al., 1994; Aniksztejn and Ben-Ari, 1995; Clark and Collingridge, 1995; Foy et al., 1999; Skeberdis et al., 2001; Grosshans et al., 2002; MacDonald et al., 2006; Kwon and Castillo, 2008). A new form of LTP is mediated by Ca2+ release from IP3-sensitive endoplasmic reticulum (ER) stores triggered by activation of muscarinic cholinergic receptors (mAChRs) (Fernández de Sevilla et al., 2008).
The CA1 region of the rodent hippocampus receives a robust cholinergic projection from the medial septal nuclei (Lewis and Shute, 1967; Frotscher and Léránth, 1985). In CA1 pyramidal cells, mAChR activation enhances NMDA-mediated responses (Harvey et al., 1993; Auerbach and Segal, 1996; Marino et al., 1998) and induces Ca2+ release from ER stores (Fernández de Sevilla et al., 2008). Therefore, mAChR activation might induce a parallel long-term enhancement of both AMPAR- and NMDAR-mediated transmission. Because of its Ca2+ permeability and slow kinetics, increments in NMDAR-mediated transmission contribute strongly to postsynaptic depolarization, especially during repetitive activity that results in an increased Ca2+ influx with important functional consequences in Ca2+-dependent cellular processes as the induction threshold and magnitude of LTP (Abraham and Bear, 1996; Salter, 1998).
In this study, we analyzed in vitro the changes in NMDAR-mediated transmission at SC synapses induced by a brief local application of ACh on the main branch of the apical dendrites of CA1 pyramidal cells. ACh caused a Ca2+ wave that rapidly propagated along the main apical dendrite toward the soma (Zucker, 1999; Rose and Konnerth, 2001) followed by a long-term enhancement of both AMPAR- (Fernández de Sevilla et al., 2008) and NMDAR-mediated transmission (present results). These effects were dependent on activation of M1 mAChRs and Ca2+ release from IP3-sensitive ER stores. The amplified AMPAR-mediated transmission relied on a SNARE protein-mediated increase in the surface expression of AMPARs (Fernández de Sevilla et al., 2008) and required protein CaM kinase II (CaMKII) activity. The increased NMDA response required the combined activation of CaMKII, protein kinase C (PKC), and SRC family protein kinase (SRC). The enhanced NMDAR-mediated transmission did not include surface incorporation of NMDARs and it was mediated most likely by changes in receptor properties. The amplified glutamatergic transmission markedly reduced the LTPH induction threshold, revealing interactions between two mechanistically different forms of LTP that add flexibility to the regulation of the cellular learning machinery of the brain.
Materials and Methods
Procedures of animal care, surgery, and slice preparation were in accordance with the guidelines laid down by the European Communities Council (Directive 86/609/EEC) on the ethical use of animals with every effort being made to minimize the suffering and number of animals used. The procedures will be described briefly because they have been extensively detailed previously (Borde et al., 1995; Fernández de Sevilla and Buño, 2003).
Slice preparation.
Young Wistar rats (14–16 d old) of either sex were decapitated, and the brain was removed and submerged in cold (∼4°C) artificial CSF (ACSF, in mm: 124.00 NaCl, 2.69 KCl, 1.25 KH2PO4, 2.00 Mg2SO4, 26.00 NaHCO3, 2.00 CaCl2, 10.00 glucose). The pH of the ACSF was stabilized at 7.4 by bubbling with carbogen (95% O2, 5% CO2). Picrotoxin (PTX; 50 μm) was added to the ACSF to block GABAA-mediated inhibition. In these conditions, epileptiform activity was never observed in our sample. In Mg2+-free solution, MgSO4 was omitted and the amplitude of the AMPA component of the EPSC (EPSCAMPA) was measured at a delay of 20 ms, while the NMDA component (EPSCNMDA) was measured at 100 ms when EPSCNMDA can be recorded in isolation. Transverse hippocampal slices (300–350 μm thick) were cut with a vibratome (Pelco 3000) and incubated in the ACSF (>1 h, at room temperature, 20–22°C). Slices were transferred to a 2 ml chamber fixed to an upright microscope stage (Olympus BX51WI) equipped with infrared differential interference contrast (DIC) video microscopy and a 40× water-immersion objective. Slices were superfused with carbogen-bubbled ACSF (∼2 ml/min) and maintained at room temperature. In some cases higher temperatures in the physiological range of 32 ± 2°C were used. d-(−)-2-Amino-5-phosphonopentanoic acid (d-AP5; 50 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm), (5S,10R)- (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-iminemaleate [(+)-MK-801 maleate 80 μm], pirenzepine (75 nm), methoctramine (1 μm), methyllycaconitine (MLA, 125 μm), thapsigargin (1 μm), PP2 [5 μm; also called 3-(4-chlorophenyl)1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine], tetrodotoxin (TTX; 100 nm), 12-O-tetradecanoylphorbol-13-acetate (TPA; 100 nm), and okadaic acid (okadaic; 100 nm) were added to the ACSF as needed. Thapsigargin (1 μm), which depletes Ca2+ stores by inhibiting sarco-endoplasmic reticulum Ca2+ ATPase, was dissolved in DMSO (0.01%). DMSO at the final concentrations used had no effects on synaptic responses or postsynaptic conductance (n = 3).
Recordings and analysis.
Patch-clamp recordings from the soma of CA1 pyramidal neurons were performed in both the whole-cell voltage- and current-clamp configurations with 3–7 MΩ fire-polished pipettes connected to a Cornerstone PC-ONE amplifier (Dagan). Fast and slow capacitances were neutralized and series resistance was compensated (∼80%). Recordings were rejected when the access resistance (10–20 MΩ) increased >10% during the experiment. Patch pipettes were either filled with an internal solution that contained in mm: 135 K-MeSO4, 10 HEPES, 0.2 EGTA, 4 Na2-ATP, 0.3 Na3-GTP buffered to pH 7.2–7.3 with KOH or a solution that had the following: 107.5 Cs-gluconate, 8 NaCl, 0.2 EGTA, 20 HEPES, 10 TEA-Cl, 4 Na2-ATP, and 0.3 Na3-GTP at pH 7.3. In experiments with the cesium-based internal solution the amplitude of EPSCAMPA was measured at a delay of 20 ms and at −60 mV, whereas EPSCNMDA was measured at 100 ms and at +60 mV (see in Figs. 1A and 3C black and gray arrows, respectively).
Figure 1.

ACh induces a long-lasting synaptic enhancement of EPSCAMPA and EPSCNMDA. A, Top, Representative averaged EPSCs (n = 20; as in all other cases) recorded at −60 mV (inward currents) and at +60 mV (outward currents) before (1) and during (2) synaptic inhibition, and following the enhancement (3) induced by ACh. The black and gray arrows indicate where EPSCAMPA and EPSCNMDA were measured, respectively. A, Bottom, Time course of the EPSC amplitude changes during cholinergic LTP (EPSCAMPA filled circles; EPSCNMDA empty circles); the arrow indicates the ACh “puff” (n = 20). B, Top left, Representative superimposed paired EPSCNMDA (R1 and R2) recorded at +60 mV under CNQX (20 μm) before (black trace) and 30 min after the ACh “puff” (gray trace). B, Top right, Same as top left but scaled to the peak of the R1 EPSC. B, Bottom, Summary data showing the effect of ACh on R1 (empty bars) and the paired-pulse ratio (R2/R1, filled bars) at room temperature (RT, n = 8), and at 32°C (n = 8). C, Time course of EPSCAMPA (filled circles) and EPSCNMDA (empty circles) amplitudes, before and during d-AP5, following the ACh “puff” (arrow), and after a washout of the d-AP5 (n = 8). D, Time course of the EPSCNMDA amplitude before and during TTX, following the ACh “puff” (arrow), and after a washout of the TTX (n = 10).
Figure 3.

EPSCNMDA potentiation is MK-801 sensitive and Botox resistant. A, Left, Superimposed EPSCs recorded in Mg2+-free solution before (ACSF 1, black trace) and during (2, MK-801; dark gray trace) MK-801 (80 μm) superfusion and 20 min after starting a washout of MK-801 and of the ACh “puff” (3, Post-ACh; light gray trace). A, Right, As in A (left), but scaled to the peak showing the lack of recovery of the NMDAR-mediated transmission (arrow) 20 min after a washout of MK-801 and the ACh “puff.” Black and gray arrows indicate where EPSCAMPA and EPSCNMDA amplitudes were measured, respectively. B, Plot showing the time course of the decay of EPSCNMDA amplitude induced by superfusion with MK-801 (1), during a 20 min interval with no synaptic stimulation that started with a washout of MK-801 (2), and that continued after resuming stimulation (3). Two conditions are shown, with and without the ACh “puff” (empty and filled circles, respectively; n = 6 in both cases). C, Left, Representative control averaged EPSCs (1) from a Botox (0.5 μm) loaded cell recorded at −60 mV (black traces), showing EPSPAMPA, and at +60 mV (gray traces), where the NMDA component amplitude was measured. C, Right, Same as C (left), but 30 min after the ACh “puff” (2, Post-ACh). Black and gray arrows indicate where EPSCAMPA and EPSCNMDA were measured, respectively. D, Time course of the peak amplitude of EPSCAMPA (filled circles) and EPSCNMDA (empty circles) before (1, Control) and after (2, Post ACh; arrow) ACh application in Botox-loaded cells (n = 7).
In some experiments the intracellular solutions either contained heparin (5 mg/ml), which inhibits IP3 receptors (IP3Rs) in the ER, ruthenium red (400 μm), which inhibits ryanodine receptors in the ER, 1,2-bis (o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA; 50 mm), which is a fast Ca2+ chelator, the nonpermeable CaMKII peptide inhibitor 281–309 (10 μm) (Colbran et al., 1988), the PKC inhibitor chelerythrine (1 μm), or the light chain of the B type botulinum toxin (i.e., Botox 0.5 μm), which inhibits the SNARE protein-mediated membrane fusion of endosome complexes. Pipettes were placed with a mechanical micromanipulator (Narishige). The holding potential (Vh) was adjusted to −60 mV. Data were low-pass filtered at 3.0 kHz and sampled at 10.0 kHz, through a Digidata 1322A (Molecular Devices). The pClamp programs (Molecular Devices) were used to generate stimulus timing signals and transmembrane current pulses, and to record and analyze data. EPSCs were evoked by stimulation of SCs with a nichrome bipolar electrode (60 μm diameter, tip separation ∼100 μm) placed in the stratum radiatum ∼100–200 μm from the soma of the recorded neuron. In some experiments a bipolar stimulation electrode pulled from theta borosilicate glass capillary (diameter of the tip, 5–8 μm), filled with ACSF, and connected to a digital stimulator through chloride-silver electrodes was used. No significant differences in the results were observed with the two stimulation methods. A Grass S88 and SIU generated the stimulation pulse protocols. Stimulation intensity was adjusted to evoke EPSC amplitudes between 30 and 100 pA. In some experiments, a second bipolar stimulation electrode was placed in the alveus ∼200–300 μm from the soma of the recorded neuron to stimulate cholinergic inputs from the medial septum (Fernández de Sevilla and Buño, 2003). Ten theta burst stimulation (TBS) protocols were applied through the alveus electrode at 0.2 Hz. Each TBS protocol consisted in four 100-ms-duration pulse barrages delivered at 5 Hz (5 pulses, 0.3 ms duration pulses at 50 Hz). A brief, localized, ACh “puff” (300 ms duration) was applied through a pipette (tip diameter, ∼5 μm) loaded with ACh (1 mm) and connected to a Picospritzer II (General Valve). The pipette tip was placed during the “puff” with a hydraulic micromanipulator (Narishige) close (∼20–50 μm) to the shaft of the apical dendrite ∼20 μm from the soma of the recorded CA1 pyramidal neuron. The pipette was withdrawn from the tissue immediately after the “puff.” Chemicals were purchased from Sigma-Aldrich Química, Tocris Bioscience (Biogen Científica), and Alomone Labs.
Extracellular recordings.
In some experiments, extracellular recordings were performed to avoid the dialysis that occurs with the whole-cell configuration. Patch pipettes filled with the extracellular solution were placed at the stratum pyramidal–radiatum border for extracellular recording. The fiber volley and the field EPSP (fEPSP) evoked by SC stimulation were recorded and analyzed before, during, and 30 min after the ACh “puff” applied in the stratum radiatum close to the extracellular electrode (∼100 μm).
Ca2+ imaging and photolysis of caged IP3.
Simultaneous electrophysiology recordings and intracellular Ca2+ imaging were obtained in selected experiments. The latter was performed by fluorescence microscopy with the Ca2+ indicator fluo-3 (Invitrogen). Patch pipettes were filled with the internal solution containing 50–100 μm fluo-3. Imaging experiments were performed after a 10 min stabilization period that allowed the equilibration of the dye in the soma and apical dendritic shaft. Cells were illuminated during 40 ms every 200 ms at 490 nm with a monochromator (Polychrome IV; TILL Photonics). Successive images were obtained at 5 s−1 with a cooled monochrome CCD camera (Luca, Andor Technologies) attached to the Olympus microscope that was equipped with a filter cube (Chroma Technology) optimized for fluo-3. Camera control, synchronization with electrophysiological measurements and quantitative epifluorescence measurements were made with the ImagingWorkbench software (INDEC-BioSystems). Changes in fluorescence signals were expressed as the proportion (%) of relative change in fluorescence (ΔF/F0), where F0 is the prestimulus fluorescence level when the cell is at rest and ΔF is the change in fluorescence during activity. Plots of Ca2+ signal variations versus time were obtained “off-line” at specified regions of interest from stored image stacks and expressed as ΔF/F0. Corrections were made for indicator bleaching during trials by subtracting the signal measured under the same conditions when the cell was not stimulated. Flash photolysis of caged IP3 was performed after filling pyramidal neurons with a pipette solution that contained fluo-3 and caged IP3 (Calbiochem) at 100 μm. The cell was illuminated with a flash unit (UV Flash II; TILL Photonics) attached to the microscope through a dual port condenser, and simultaneous electrophysiological recordings and Ca2+ imaging were carried out.
An average baseline of control EPSCs was obtained by repeated stimulation at 0.3 Hz and the magnitude of the average changes in peak EPSC amplitude induced by the various procedures tested were expressed as a proportion (%) of the control amplitude and plotted as a function of time. Statistical estimations were performed both with the maximum average peak EPSC values (n = 20) and with values measured at delays of 100 ms from the peak when EPSCNMDA can be recorded in isolation. The presynaptic or postsynaptic origin of the changes in EPSC amplitude was estimated both at room temperature and at 32°C by measuring the variations in the paired-pulse ratio (PPR, defined as R2/R1, where R1 and R2 were the average peak amplitudes of the first and second EPSCs, respectively) (Fernández de Sevilla et al., 2002). Statistical analysis was performed using the Origin 7.0 program (OriginLab).
Results
ACh induces a parallel long-term enhancement of AMPAR- and NMDAR-mediated transmission
We first determined whether the previously described enhancement of the AMPAR-mediated transmission (Fernández de Sevilla et al., 2008) was accompanied by an increase in the NMDA-mediated component. We recorded under voltage clamp several control EPSCs evoked by single SC stimulation at 0.3 Hz both at −60 and +60 mV (averages of 20) to check for response stability. We then applied an ACh “puff” <20 min after “break-in.” ACh induced a brief initial inhibition of the EPSCs followed by an inward current and a rapid Ca2+ wave that started at or close to the first bifurcation point of the apical dendritic shaft and ended at the soma (Fernández de Sevilla et al., 2008). These responses were followed by a gradually developing and sustained enhancement of both EPSCAMPA and EPSCNMDA. Both enhancements followed similar time courses and reached a steady state in 30–40 min (208.7 ± 20.8% and 239.7 ± 20.9% of control, respectively, p < 0.001; n = 20) without changes in the AMPA/NMDA ratio (from 5.1 ± 0.4 in control to 4.8 ± 0.6 after ACh; p > 0.05; n = 20) (Fig. 1A). This long-term enhancement of NMDAR-mediated transmission could last up to 4 h (extracellular recordings; n = 3, data not shown) and was observed at more physiological temperatures of 32°C (236.2 ± 22.5% of control; p < 0.001; n = 8) (Fig. 1B). Finally, we tested whether the enhancement of NMDAR-mediated transmission was due to a presynaptic increase in release probability by comparing the PPR in the control and during potentiation. There were not significant changes in the PPR of the EPSCNMDA isolated with CNQX, neither at room temperature (from 1.1 ± 0.2 in control to 1.2 ± 0.3 after ACh; p > 0.05; n = 8) or at more physiological temperatures of 32°C (from 1.1 ± 0.4 in control to 1.3 ± 0.6 after ACh; p > 0.05; n = 8). These results may indicate the absence of a contribution of presynaptic mechanisms to the enhancement of NMDAR-mediated transmission (Fig. 1B).
We had shown that the ACh-mediated long-term enhancement of EPSCAMPA was insensitive to inhibition of NMDARs with d-AP5 and resistant to the block of Na+ channels with tetrodotoxin (TTX) during ACh application (Fernández de Sevilla et al., 2008). To test whether the enhancement of EPSCNMDA was also insensitive to blockade of NMDARs, we superfused d-AP5 (50 μm), which eliminated the NMDAR-mediated transmission, and subsequently applied ACh followed by a washout in control solution (Fig. 1C). During the washout, EPSCNMDA recovered gradually from the inhibition by d-AP5, exceeded control amplitudes in ∼18 min, and increased to a steady state in ∼30–40 min (212.2 ± 21.2% of control; p < 0.001; n = 8) (Fig. 1C). Moreover, the potentiation of the EPSCNMDA isolated with CNQX in Mg2+-free solution persisted when Na+-mediated spike activity was blocked under TTX (0.5 μm) during ACh application (Fig. 1D). Under blockade of Na+ channels, ACh induced a potentiation of EPSCNMDA (from 222.2 ± 20.2 in control to 198.8 ± 21; p < 0.001; n = 10) as measured following a washout of TTX.
The above results together indicate that the enhancement EPSCNMDA does not require activation of NMDARs nor presynaptic or postsynaptic Na+-dependent action potential activity, thus departing from the classical LTPH.
M1 mAChR and IP3R mediate the LTP of AMPA- and NMDAR-mediated transmission
We had shown that the LTPIP3 of EPSCAMPA needed the activation of the M1 subgroup of mAChRs, synthesis of IP3, and Ca2+ release from IP3-sensitive ER stores (Fernández de Sevilla et al., 2008). We tested whether the LTP of EPSCNMDA relied on similar mechanisms. Blockade of the M1 subgroup of mAChRs with the specific antagonist pirenzepine (75 nm) prevented the LTP of EPSCNMDA, which measured before and 30 min after the ACh challenge was essentially identical under pirenzepine (p > 0.05; n = 8 in both cases). Pirenzepine also suppressed the Ca2+ signal without modifying control synaptic responses (Fig. 2A,B). Superfusion with the M2 mAChR antagonist methoctramine (1 μm) or high doses of the α-7 nicotinic receptor antagonist MLA (125 μm) did not modify the Ca2+ wave induced by ACh (139.2 ± 10.8% of control in methoctramine and a 135.6 ± 8.8% of control in MLA; p < 0.001; n = 8), nor the enhancement of the NMDAR-mediated transmission (210.8 ± 10.5% of control in methoctramine and 201.0 ± 15.0% of control in MLA; p < 0.001; n = 8) (Fig. 2B).
Figure 2.

Activation of M1 mAChR and Ca2+ release from ER are required for the ACh-induced EPSCNMDA potentiation. A, Averaged somatic Ca2+ signal (n = 6) induced by the ACh “puff” and superimposed averaged EPSCs before (control, black trace) and after (Post-ACh, gray trace) ACh in control solution and during superfusion with pirenzepine (75 nm). B, Summary data showing the effects of ACh on the Ca2+ signal (empty bars) and post-ACh EPSC amplitude (filled bars) under superfusion with MLA (n = 8; p < 0.01), pirenzepine (n = 8; p > 0.05), methoctramine (n = 8; p < 0.01), intracellular ruthenium red (400 μm; RuRed n = 8; p < 0.01), and intracellular heparin (5 mg/ml; n = 6; p < 0.01). C, Representative somatic Ca2+ signal (left) induced by IP3 uncaging (100 μm) and superimposed EPSC (right) in control conditions (black trace) and 30 min after IP3 uncaging (Post-Uncaging, gray trace). D, Time course of the EPSCNMDA peak amplitude changes following IP3 uncaging (UV flash); note that although higher than controls, post-uncaging values are not significantly different (n = 20, p > 0.05).
We checked whether the enhancement of the NMDAR-mediated transmission required Ca2+ release from IP3-sensitive stores (Fig. 2B). We loaded pipettes with the fast Ca2+ chelator BAPTA (50 mm) that prevented the Ca2+ wave and the sustained enhancement of EPSCNMDA (p < 0.001; n = 8, data not shown). Similar results were obtained by depleting ER stores with bath applied thapsigargin (1 μm, p < 0.001; n = 6, data not shown). Loading the cells with the IP3R antagonist heparin (5 mg/ml) had comparable effects (p < 0.001; n = 6). Contrastingly the LTP of EPSCNMDA was insensitive to inhibition of ryanodine receptors with ruthenium red (400 μm) (p > 0.05; n = 8). The above results together imply that the ACh-induced long-term enhancement of glutamatergic transmission required Ca2+ release from IP3-sensitive ER stores, whereas the contribution of Ca2+-induced Ca2+ release through ryanodine receptors was insignificant. The results also indicate that M1 mAChRs activation is obligatory for the long-term enhancement of EPSCNMDA. Contrastingly, an involvement of M2 mAChRs and α-7 nicotinic receptors was not detected.
We tested the effects of photolysis of caged IP3 that directly activates IP3Rs and bypasses the intracellular synthesis of IP3. Uncaging of IP3 directly releases Ca2+ from IP3-sensitive ER stores and induces EPSCAMPA potentiation (Fernández de Sevilla et al., 2008). However, although IP3 uncaging induced a Ca2+ rise, the EPSCNMDA amplitude did not change in the ∼30 min after uncaging analyzed (110.1 ± 10.9%; p > 0.05; n = 20) (Fig. 2C,D). Therefore, although activation of IP3-sensitive ER stores and the subsequent release of Ca2+ are obligatory for the induction of the LTPIP3 of EPSCAMPA (Fig. 2A,B, heparin), IP3 uncaging did not induce the potentiation of the EPSCNMDA. This result indicates that the Ca2+ rise is necessary but not sufficient to trigger the LTPIP3 of EPSCNMDA (see Discussion).
Different intracellular mechanisms mediate the LTPIP3 of EPSCAMPA and EPSCNMDA
We had shown that the long-term enhancement of EPSCAMPA with LTPIP3 was mediated by an increased functional surface expression of AMPARs in spines of activated SC synapses (Fernández de Sevilla et al., 2008). To test whether similar mechanisms regulate the enhancement of EPSCNMDA, we superfused MK-801 (80 μm) in Mg2+-free solution and stimulated SCs at 1 Hz while holding the cell at −60 mV. MK-801 caused a gradual inhibition of the NMDA component that was complete in ∼15 min [Fig. 3A (scaled gray arrow),B]. A few minutes later, we retuned to stimulation at 0.3 Hz and applied the ACh “puff” that had no effect on NMDAR-mediated transmission, as indicated by the absence of potentiation of EPSCNMDA or recovery, following a prolonged washout (∼40 min; p > 0.05; n = 6) in MK-801 and Mg2+-free solution (Fig. 3A,B, filled circles). In contrast in the same experiments the ACh “puff” induced a long-term enhancement of EPSCAMPA that reached a steady state in 30–40 min (205.4 ± 25.4% of control; p < 0.001; n = 6) (Fig. 3A, unscaled black arrow). In the absence of the ACh “puff,” there also was no recovery of the EPSCNMDA from the MK-801 blockade (Fig. 3B, empty circles), suggesting no detectable insertion of NMDARs in the spines in our control conditions. Since the activity-dependent inhibition of NMDARs by MK-801 is irreversible, the above results imply that following the complete blockade of NMDARs by MK-801, ACh did not induce a detectable insertion of NMDARs in spines of activated SC synapses. To check whether with this methodology we are able to detect the insertion of new NMDARs, we repeated the MK-801 experiments and applied phorbol esters that are known to increase the surface expression of NMDARs (Lan et al., 2001). In these conditions, the EPSCNMDA recovered from the MK-801 blockade indicating the de novo insertion of NMDARs (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Moreover, we also show that ACh does not interfere with the surface expression of new NMDARs induced by phorbol esters. A possible interpretation of these results is that the ACh-mediated enhancement of EPSCNMDA is caused by modifications of the biophysical properties without surface insertion of NMDARs (Wang et al., 1996; Salter, 1998; Lu et al., 1999; Qiu et al., 2006). In contrast, inhibition of EPSCNMDA with MK-801 did not prevent the LTPIP3 of EPSCAMPA, confirming that the long-term enhancement of AMPAR-mediated transmission did not rely on NMDAR activation (Fernández de Sevilla et al., 2008).
The increased surface expression of AMPARs in spines of potentiated synapses induced by ACh (Fernández de Sevilla et al., 2008) mainly relies on SNARE protein-mediated membrane fusion of AMPAR-containing endosomes, which is blocked by inhibition of SNARE protein activity with botulinum neurotoxins (Lüscher et al., 1999; Yao et al., 2008). Therefore, to further test for possible differences in the mechanisms mediating the LTPIP3 of EPSCAMPA and EPSCNMDA, we loaded the cells with the light chain of the B type botulinum toxin (i.e., Botox 0.5 μm). Botox loading did not modify baseline transmission of control AMPA and NMDA responses (Fig. 3D) (p > 0.05; n = 7) but prevented the ACh-mediated long-term enhancement of EPSCAMPA, measured at the EPSC peak at −60 mV, where this component may be recorded in isolation (101.5 ± 12.5%, p > 0.05, n = 7). However, intracellular Botox did not modify the LTPIP3 of the NMDAR-mediated transmission, measured at delays of 100 ms from the EPSC peak at +60 mV, where this component may be recorded in isolation (201.5 ± 14.0%, p < 0.001, n = 7) (Fig. 3C,D, empty and filled circles, respectively).
The above results together allow us to conclude that the long-term enhancement of EPSPAMPA required SNARE protein activity. In contrast, the increase of EPSCNMDA did not rely on SNARE protein-mediated NMDAR insertion. Thus, a possible interpretation of these results is that the enhanced EPSCNMDA was caused by modifications of the biophysical properties of receptors.
Different protein kinases regulate AMPAR- and NMDAR-mediated transmission
NMDARs function as “coincidence detectors” in activity-dependent synaptic plasticity. The strong spine depolarization triggered by LTP induction protocols causes a supralinear Ca2+ influx mediated by the activation of NMDARs and voltage-gated Ca2+ channels (Yuste et al., 1999; Sjöström and Nelson, 2002; Malenka and Bear, 2004; Kampa et al., 2006; Nevian and Sakmann, 2006). The increased spine Ca2+ concentration activates intracellular kinases, which trigger both enhanced surface availability of functional AMPARs and changes in the biophysical properties of NMDARs (Abbott and Nelson, 2000; Malinow and Malenka, 2002; Fuenzalida et al., 2007).
We tested whether different kinases mediated the long-term amplification of AMPAR- and NMDAR-mediated transmission. We first investigated the effects of inhibiting the autophosphorylation of CaMKII, which is known to be obligatory for the expression of LTPH (Otmakhov et al., 1997; Nicoll and Malenka, 1999; Fukunaga and Miyamoto, 2000; Lisman et al., 2002). Blockade of CaMKII autophosphorylation, with the peptide inhibitor 281–309 (10 μm) (Colbran et al., 1988) included in the intracellular solutions, suppressed the LTPIP3 of both EPSCAMPA and EPSCNMDA (control and post-ACh EPSCs were essentially identical; p > 0.05; n = 8) (Fig. 4A). Therefore, the autophosphorylation of CaMKII represents the common intracellular trigger for the LTPIP3 of both EPSCAMPA and EPSCNMDA.
Figure 4.
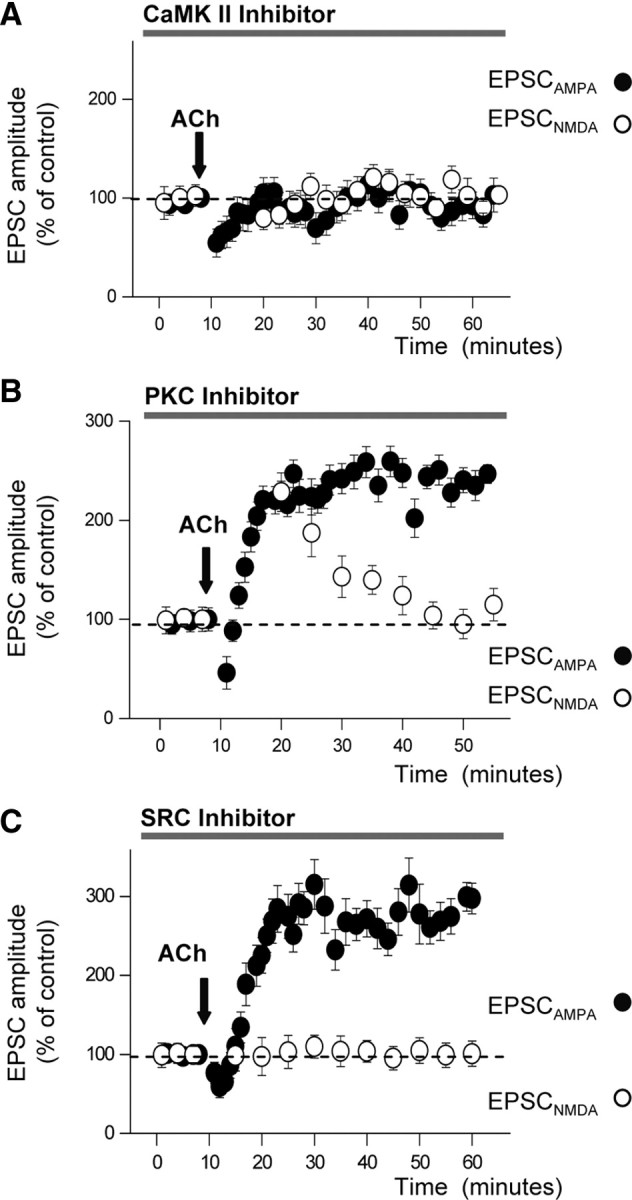
CaMKII activation is obligatory for the potentiation of EPSCAMPA and EPSCNMDA; the potentiation of EPSCNMDA also requires PKC and SRC activation. A, Peak amplitudes of EPSCAMPA (filled circles) and EPSCNMDA (empty circles) as a function of time following ACh application (arrow) under intracellular blockade of CaMKII with intracellular p281–309 (10 μm; n = 8). B, Same as A, under blockade of PKC with intracellular chelerythrine (1 μm; n = 7). C, Same as A, under blockade of SRC kinase with superfusion of PP2 (5 μm; n = 9).
Evidence has been reported that PKC plays a key role in the regulation of recombinant NMDARs and in the induction of LTPH by enhancing the expression of functional NMDARs (Köhr and Seeburg, 1996; Lan et al., 2001). Therefore, PKC might also play a role in the induction of the ACh-mediated enhancement of the NMDAR-mediated transmission. Chelerythrine (1 μm), a PKC inhibitor included in the intracellular solutions, blocked the LTPIP3 of the NMDAR-mediated transmission after a transient potentiation that started immediately after the ACh “puff” and decayed, reaching control values in ∼25 min. Control and post-ACh EPSCNMDA amplitudes were similar (p > 0.05; n = 7) (Fig. 4B, empty circles) 30 min after the ACh “puff.” In contrast, chelerythrine did not modify the ACh-mediated enhancement of AMPAR-mediated transmission (249.5 ± 20.5% of control, p < 0.001, n = 7) (Fig. 4B, filled circles).
It has also been shown that NMDA responses are enhanced by SRC kinase activity (Köhr and Seeburg, 1996; Wang et al., 1996; Salter, 1998; Lu et al., 1999; Grosshans et al., 2002; MacDonald et al., 2006; Qiu et al., 2006). Therefore, we tested whether SRC kinase inhibition could prevent the long-term ACh-mediated enhancement of NMDAR-mediated transmission. Superfusion with the SRC kinase antagonist PP2 (5 μm) prevented the LTPIP3 of EPSPNMDA (control and post-ACh EPSCs were similar; p > 0.05; n = 9) (Fig. 4C, empty circles) without modifying the enhancement of the AMPAR-mediated transmission (274.6 ± 22.8% of control, p < 0.001, n = 9) (Fig. 4C, filled circles). Finally, we tested the time course of the washout of PP2 by checking the changes in the slope of extracellular recorded field EPSPNMDA isolated with CNQX and Mg2+-free solution. The ACh “puff” potentiated the EPSPNMDA (156.5 ± 14.3% of control, p < 0.01, n = 6) only after 60 min of a washout of PP2 in control solution (data not shown).
Therefore, two different intracellular routes involving the activity of different kinases arising from a common trigger—i.e., the Ca2+-dependent activation of CaMKII—induce the enhancement of both AMPAR- and NMDAR-mediated transmission. In addition, both intracellular routes act through two different final mechanisms, the former via a SNARE protein-dependent insertion of functional AMPARs in spines, and the latter by functional modifications of NMDARs, most likely related to changes in biophysical properties of the receptor channel. In all the above pharmacological experiments, the EPSCAMPA and EPSCNMDA amplitudes were measured as indicated for Figure 3C.
LTPIP3 reduces the induction threshold of the LTPH of EPSCAMPA
We have previously shown that the EPSP magnitude and waveform regulate the threshold and extent of the LTPH of EPSCAMPA induced by STDP (Fuenzalida et al., 2007). Therefore, it is conceivable that the ACh-mediated long-term amplification of EPSPAMPA and EPSPNMDA might reduce the threshold of the LTPH of EPSCAMPA. We tested the consequence of the LTPIP3 on the induction threshold of the LTPH of EPSCAMPA induced by STDP. We first checked, with whole-cell recordings, that in control conditions STDP protocols consisting in a subthreshold EPSP followed by a backpropagating action potential (BAP) at delays of 10 ms repeated 30 and 20 times at 1 s−1 were unable to induce an LTPH of EPSCAMPA (p > 0.05; n = 10) (Fig. 5A, gray squares and triangles, respectively). However, 40 presentation of the same protocol induced a robust LTPH of EPSCAMPA (253.3 ± 18.4% of control, p < 0.001; n = 10) (Fig. 5A, black squares).
Figure 5.

ACh lowers the number of EPSP-BAP pairings required to induce LTPH. A, Top, Representative record showing the EPSP-BAP pairing at 10 ms delay applied at 1 Hz to test LTP induction with STDP. A, Bottom, Time course of the EPSC amplitude changes induced by STDP protocol (empty arrow) in control conditions with 20 pairings (empty triangles n = 10), 30 pairings (gray squares n = 10), and 40 pairings (black squares n = 10). Note that LTP was only induced with 40 pairings. B, Top, Schematic diagram showing the timing of the experimental protocol; ACh was “puffed” (filled arrow) 60 min before patching a cell and applying the STDP protocol (empty arrow). B, Bottom, Time course of the EPSC amplitude changes induced by STDP (empty arrow) applied 60 min after the ACh “puff” with 20 pairings (empty triangles n = 10) and 10 pairings (filled triangles n = 10). Note that ACh reduced the number of pairings required for LTPH.
Using extracellular recordings, to avoid dialysis associated with prolonged whole-cell recordings, we applied an ACh “puff” in the stratum radiatum close to the extracellular electrode that was placed in the radiatum–somatic border (see Materials and Methods). ACh generated an initial presynaptic inhibition followed by a sustained enhancement of the fEPSP slope indicating the induction of LTPIP3, without changes of the fiber volley (Fernández de Sevilla et al., 2008). When LTPIP3 had reached a steady amplitude in ∼30 min (166.5 ± 18.3% of control, p > 0.01, n = 10, data not shown), a pyramidal neuron close to the extracellular recording electrode (<100 μm) was “patched” (<50 min) and several control EPSC were recorded under voltage clamp. The recording was then switched to current clamp and the STDP protocol applied with a EPSP-BAP delay of 10 ms presented 20 times at 1 s−1. The STDP protocol invariantly induced a strong LTPH of EPSCAMPA, as judged by the enhanced EPSC amplitude recorded after returning to the voltage-clamp configuration (222.3 ± 10.8% of control, p < 0.001, n = 10) (Fig. 5B, empty triangles), suggesting that these pyramidal neurons had been “primed” by the LTPIP3. In contrast, 10 presentations of the same protocol were unable to induce an LTPH (p > 0.05; n = 10) (Fig. 5B, filled triangles). These primed neurons showed a reduction of the threshold of the LTPH of EPSCAMPA most likely caused by the cholinergically induced long-term enhancement of both AMPAR- and NMDAR-mediated transmission. The threshold reduction of the LTPH of EPSCAMPA was absent when ACh was applied in the presence of pirenzepine, thus favoring the above idea. Under pirenzepine, 40 stimulations at 1 s−1 were necessary to induce LTPH of EPSCAMPA (n = 5, p < 0.001), while 20 times were unable to induce LTPH of EPSCAMPA (n = 5, p > 0.05). Finally, we also checked that the same STDP protocols were unable to induce an LTPH of EPSCNMDA in control conditions and after ACh potentiation in Mg2+-free solution (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
The enhanced NMDAR- and AMPAR-mediated transmission regulate the LTPH of EPSCAMPA
Both AMPAR- and NMDAR-mediated transmission may contribute to the changes in the induction threshold of the LTPH of EPSCAMPA, the former by regulating spine depolarization and the latter by generating the necessary Ca2+ influx and increasing the amplitude and duration of the EPSP. Therefore, a cooperative interaction between the two EPSP components regulates Ca2+ influx by depolarizing the pyramidal cell and relieving the Mg2+-mediated blockade of the modified NMDARs in spines. We investigated the role of this interaction and of the activation of the AMPA and NMDA components of SC EPSPs in the regulation of the threshold of LTPH of EPSCAMPA before and after inducing LTPIP3.
The control average amplitude of the AMPA and NMDA components was measured under voltage clamp and LTPIP3 was induced with an ACh “puff” (as above, Fig. 6A, left plot, filled and empty circles, respectively). When the peak amplitude of the EPSC had increased and stabilized in ∼20 min (191.9 ± 17.4% of control, p < 0.001, n = 6), gradually increasing concentrations of CNQX were added to the solution until the amplitude of the EPSCAMPA was reduced back to control values (Fig. 6A, left plot, filled circles). Then a neighbor pyramidal neuron (<100 μm) was patched and control EPSCs were recorded. Finally we switched to the current-clamp configuration and STDP protocols with EPSP-BAP delays of 10 ms repeated 20 times at 1 s−1 were applied, resulting in a strong LTPH of EPSCAMPA (183.6 ± 20.4% of control, p < 0.001, n = 6) (Fig. 6A, right plot, empty triangles). However the same STDP protocol repeated 10 times at 1 s−1 was ineffective (p > 0.05; n = 6) (Fig. 6A, right plot, filled triangles). Therefore the LTPIP3-mediated functional enhancement of NMDARs was able to overcome the partial inhibition of AMPARs by CNQX, and reduced the number of coincidences necessary to induce LTPH of EPSCAMPA. One possible interpretation of these results is that the cholinergic-induced enhanced EPSPNMDA, via increasing Ca2+ influx, lowers the threshold of the LTPH of EPSCAMPA.
Figure 6.

The LTPH threshold is lowered by the potentiation of EPSCAMPA and EPSCNMDA. A, Left, Time course of the peak amplitudes of EPSCAMPA (filled circles) and EPSCNMDA (empty circles) before and during ACh application (arrow), and ∼20 min after superfusing gradually increasing concentrations of CNQX (gray bar) until the EPSCAMPA amplitude attained control values. A, Right, Time course of the EPSC peak amplitude before and after applying the STDP protocols (empty arrow) in a neighbor cell that was patched 90 min later (i.e., when EPSCNMDA had been potentiated by the ACh “puff” and CNQX had reduced EPSCAMPA to control values). Note that the potentiation is induced by 20 STDP pairings (empty triangles; n = 6), but not by 10 pairings (filled triangles; n = 6). B, Left, Schematic diagram showing the experiment protocol during superfusion with PP2 (SRC inhibitor; gray bar) and ACh (filled arrow). A neighboring cell (<100 μm) was patched 50 min after a washout of PP2 and the STDP protocols applied 60 min after ACh “puff” and the PP2 washout (empty arrow). B, Right, Time course of the EPSC potentiation induced when 20 STDP pairings were applied following the 60 min ACh puff and the PP2 washout (empty triangles n = 6) and absence of effects with 10 pairings (filled triangles n = 6).
Using similar procedures, we tested the effects of the enhanced AMPAR-mediated transmission on the threshold of the LTPH of EPSCAMPA. We first blocked the cholinergic-mediated enhancement of EPSCNMDA with PP2 and induced the LTPIP3 of the AMPA component (data not shown but similar to Fig. 4C). Then, we patched a neighbor neuron after 60 min of PP2 washout, when LTPIP3 and LTPH of EPSCAMPA could be induced (data not shown). We recorded control EPSCs under voltage clamp and switched to current clamp. The usual STDP protocol (EPSP-BAP delays of 10 ms repeated 20 times at 1 s−1) induced a robust LTPH of EPSCAMPA (199.9 ± 15.2% of control; p < 0.001, n = 6), suggesting an important decrease in the induction threshold of LTPH of EPSCAMPA (Fig. 6B, empty triangles). However, the same STDP protocol repeated 10 times at 1 s−1 was ineffective (p < 0.05; n = 6) (Fig. 6B, filled triangles). Therefore, a possible interpretation of these results is that the cholinergic-induced enhanced EPSPAMPA, via increasing the spine depolarization, reduces the induction threshold of the LTPH of EPSCAMPA.
A similar cholinergic LTP can be induced by synaptic stimulation of alveus
We tested the changes of SC EPSCs evoked by stimulation of medial septum cholinergic inputs in the alveus with TBS protocols (see Materials and Methods). We recorded a stable baseline of the EPSCAMPA and EPSCNMDA (measured as in Figs. 1A and 3C). We then applied 10 TBSs at 0.2 Hz in the alveus, each protocol followed by SC stimulation (Fig. 7A). Alveus TBS induced a brief initial inhibition of both EPSCAMPA and EPSCNMDA (75.5 ± 19.5% and 77.7 ± 25.4% of control, respectively, p < 0.001; n = 6) followed by a gradually developing and sustained enhancement of both components of synaptic transmission (Fig. 7B,C). EPSCAMPA and EPSCNMDA potentiation followed similar time courses and reached a steady state in 30–40 min (205.5 ± 19.5% and 289.7 ± 25.4% of control, respectively, p < 0.001; n = 6).
Figure 7.

An atropine-sensitive LTP of EPSCAMPA and EPSCNMDA is induced by stimulation of medial septum output fibers in the alveus. A, Representative response of a voltage-clamped CA1 pyramidal neuron evoked stimulation of the alveus with the TBS protocol followed by SC stimulation. B, Representative superimposed averaged EPSCs recorded at −60 mV (inward currents) and at +60 mV (outward currents, n = 20) before [black trace, Control (1)] and 40 min after alveus TBS [gray trace, Post Alveus TBS (2)]. C, Time course of EPSCAMPA (filled circles) and EPSCNMDA (empty circles) amplitudes during the induction of LTP by alveus TBS (gray arrow). D, Summary data showing the effect of alveus TBS on EPSCAMPA (filled bars) and EPSCNMDA (empty bars) in control ACSF (n = 6, p < 0.01), d-AP5 (n = 6; p < 0.01), and atropine (n = 6; p > 0.05).
To determine whether the LTP of EPSCAMPA and EPSCNMDA induced by the alveus TBS was mediated via activation of mAChRs, we repeated the experiments under atropine. In these conditions, alveus TBS was unable to induce LTP of SC EPSCs (p > 0.05, n = 6) (Fig. 7D), suggesting that the induction of this LTP was mediated by activation of mAChRs. Moreover, alveus TBS was unable to induce LTP of SC EPSCs (p > 0.05, n = 5) in the presence of pirenzepine, suggesting that the induction of this LTP was mediated by activation of M1 type mAChRs. Finally, we checked whether this LTP was independent of NMDARs activation by applying the alveus TBS under superfusing with d-AP5 (50 μm). In the presence of d-AP5, alveus TBS was able to induce LTP of SC EPSCs (p < 0.001, n = 6) (Fig. 7D), suggesting that the induction of this LTP was NMDAR independent.
Together these results suggest that an NMDAR-independent and mAChR-dependent LTP of EPSCAMPA and EPSCNMDA with similar time course and comparable increases in synaptic efficacy can be induced by synaptic stimulation of cholinergic inputs to CA1 pyramidal neurons.
Discussion
We provide original evidence indicating that both NMDA- and AMPA-mediated components of SC synaptic transmission are enhanced for a prolonged period of time by a brief “puff” of ACh applied close to the apical dendrite of CA1 pyramidal neurons. We also show that the ACh-mediated long-term enhancement of the NMDA- and AMPA-mediated components is initiated in SC synapses by two different mechanisms, both triggered by activation of the M1 subgroup of mAChRs, Ca2+ release from IP3-sensitive ER stores and CaMKII activation. However, while the amplification of AMPAR-mediated transmission requires a SNARE protein-mediated raise in the surface expression of AMPARs, the enhancement of NMDAR-mediated transmission involves the combined activation of PKC and SRC kinase activity without detectable surface incorporation of NMDARs. The final functional outcome of the combined amplification of the NMDAR- and AMPAR-mediated transmission is both a cholinergic mediated increase in the synaptic efficacy and a marked reduction of the induction threshold of LTPH evoked by STDP protocols.
Several groups have reported a long-term enhancement of NMDAR-mediated synaptic transmission that presents analogies with the one reported here. First, an M2 mAChR-mediated enhancement of NMDAR-mediated transmission induced by carbachol has been described at SCs by Auerbach and Segal (1994, 1996). However, the effect described by those authors was paralleled only by a minor enhancement of the AMPA component. Given that ACh but not carbachol is rapidly inactivated in the slice, the discrepancy between our results and those of Auerbach and Segal (1994, 1996) could reside in the prolonged presence of carbachol, which may have activated different mechanisms than those contributing to the LTPIP3 (Fernández de Sevilla et al., 2008). Another possible cause is that muscarinic innervation of CA1 pyramidal cells changes markedly during development (Amenta et al., 1995) and rats used by Auerbach and Segal were much older (>7 weeks and up to 36 months) than our sample (14–16 d). A specific short-lived potentiation of the NMDA component induced by ACh that involves IP3 production has also been reported (Markram and Segal, 1990). This potentiation is also different from the LTPIP3 reported here because it is transient and requires coactivation of M2 mAChRs and NMDARs. Finally, a long-term enhancement of NMDAR-mediated transmission has been reported in mossy fiber–CA3 synapses (Kwon and Castillo, 2008). This potentiation also requires a postsynaptic Ca2+ rise, but differs from the enhanced NMDAR-mediated transmission reported here because it is specific for NMDARs, requires coactivation of NMDARs and mGlu5Rs, and is caused by a PKC-dependent recruitment of NMDARs by a SNARE-dependent process.
Role of Ca2+ release from IP3-sensitive stores
Activation of IP3Rs triggers the major mechanism of Ca2+ release from ER stores in CA1 pyramidal neurons (Nakamura et al., 1999). We show that the M1 mAChR-mediated enhancement of both AMPAR- and NMDAR-mediated transmission is linked to the production of IP3, since both potentiations are abolished by blocking IP3 receptors with heparin. In addition, we demonstrate with IP3 uncaging that activation of IP3Rs is necessary but not sufficient to induce the sustained enhancement of NMDAR-mediated transmission. A possible interpretation of the failure to induce the LTPIP3 of EPSCNMDA with IP3 uncaging is that Ca2+ release from IP3-sensitive ER stores is not sufficient to activate PKC, which also requires diacylglycerol, which is only produced by the activation of M1 mAChRs by ACh. Therefore, these results could indicate that adequate levels of diacylglycerol must be attained to activate PKC and SRC to induce the enhancement of NMDAR-mediated transmission.
Several neurotransmitters can trigger IP3 production and Ca2+ release from IP3-sensitive stores and induce Ca2+ waves and long-term facilitation of AMPAR-mediated transmission. In hippocampal pyramidal cells, these actions can be induced by activation of metabotropic glutamate receptors (Bashir et al., 1993; Nakamura et al., 1999, 2000; Fitzpatrick et al., 2009) and purinergic and adrenergic receptors (Nishizaki and Mori, 1998). Therefore, different membrane receptors may trigger the same intracellular signaling cascades as those activated by mAChRs. However, what functional demands require signaling through the different membrane receptors to trigger the IP3-mediated enduring modifications in synaptic efficacy remains to be determined.
IP3R activation triggers Ca2+ waves
The enhancement of the NMDAR-mediated transmission is closely linked to the genesis of Ca2+ waves in the apical dendrites of CA1 pyramidal cells. The ER extends throughout the dendritic arbor and spines in CA1 pyramidal cells (Spacek and Harris, 1997) and colocalizes with IP3Rs (Sharp et al., 1993). The colocalization could support a rapid rise in spine Ca2+ concentration during ACh application. BAPTA loading, intracellular heparin, and thapsigargin inhibit Ca2+ waves and suppress the ACh-induced NMDAR potentiation. In contrast, inhibition of Ca2+ release from ryanodine-sensitive ER stores and of Ca2+ influx through NMDARs does not interfere with the Ca2+ signals induced by ACh or with the long-term enhancement of NMDAR-mediated transmission. Similarly to the mAChRs-mediated AMPA potentiation, the potentiation of NMDAR-mediated transmission does not require presynaptic and postsynaptic spike activity or Ca2+ influx through voltage-gated Ca2+ channels. Therefore, the intracellular Ca2+ elevation that induces this form of LTP is exclusively mediated via release from IP3-sensitive ER stores (Nakamura et al., 1999; Fernández de Sevilla et al., 2008).
We had demonstrated that ACh enhances the surface expression of both GluR1 and GluR2 AMPARs subunits at the spines of CA1 pyramidal neurons (Fernández de Sevilla et al., 2008). AMPAR trafficking is also modified by synaptic activity (Lissin et al., 1999; Ehlers, 2000; Lin et al., 2000) linked to LTPH (Malinow and Malenka, 2002). In contrast, we now show that the increased NMDAR-mediated transmission does not rely on an enhanced number of available receptors at the synapse but on biophysical modifications of preexisting surface expressed receptors. Our results suggest that the enhanced NMDAR-mediated transmission is probably mediated by phosphorylation of preexisting receptors triggered by the combined activation of CaMKII, PKC, and SRC kinase. Contrastingly, the amplification of AMPAR-mediated transmission only requires CaMKII activation and a SNARE protein-mediated insertion of AMPARs. Interestingly the initial common trigger for both enhancements of AMPAR- and NMDAR-mediated transmission is CaMKII activation.
Functional role of the enhanced NMDAR-mediated transmission
We show that the enhancement of both EPSCAMPA and EPSCNMDA lowers the threshold of the LTPH of EPSCAMPA induced by STDP protocols. Interestingly, several processes triggered by ACh converge to cooperate in this action. First, the enhanced AMPAR-mediated transmission increases spine depolarization and may contribute to augment Ca2+ influx by increasing the relief of the voltage-dependent Mg2+ blockade of NMDARs. Second, the increased NMDAR component amplifies the depolarization and duration of the EPSP, thus also contributing to the increased intracellular Ca2+ concentration by further expanding Ca2+ influx.
Because of the Ca2+ permeability of NMDARs, ACh-induced long-term increases in NMDAR-mediated synaptic transmission may have important functional consequences in Ca2+-dependent processes. Therefore, a cooperative multiplicative interaction between the two enhanced glutamatergic EPSP components would cause the supralinear enhancement of spine Ca2+ concentration possibly required to reduce the LTPH threshold. These results agree with reports that show that upregulation of the NMDA receptor function by activation of SRC mediates the induction of LTP in the CA1 region (Yu et al., 1997; Lu et al., 1998; Salter, 1998; MacDonald et al., 2006).
In conclusion, this long-lasting enhancement of NMDAR-mediated transmission is expressed postsynaptically and requires Ca2+ release from IP3-sensitive stores and PKC and SRC activation. In contrast, it does not require depolarization, presynaptic or postsynaptic spike activity, or activation of voltage-gated Ca2+ conductance or of NMDARs. This LTPIP3 reveals yet another cascade of postsynaptic events that might be present during behavior and can drive AMPARs into synapses, boost the function of NMDARs, and enhance synaptic transmission through Ca2+ release from ER stores and the activation of PKC and SRC. A similar cholinergic long-lasting enhancement of SC EPSPs may be induced by stimulation of septohippocampal cholinergic fibers (Fernández de Sevilla et al., 2008), suggesting that LTPIP3 may be functional in the natural condition. Moreover, we now demonstrate that alveus theta burst stimulation induces an NMDAR-independent and mAChR-dependent potentiation of both EPSCAMPA and EPSCNMDA (present results). This cholinergic potentiation of both the AMPAR- and NMDAR-mediated synaptic transmission could be the mechanism underlying the evidence showing that activation of AChRs lowers the induction threshold of Hebbian LTP. Therefore, novel dynamic interactions between two mechanistically different forms of long-term enhancement of glutamatergic transmission might play key roles in the regulation of the threshold and operational power of the cellular learning machinery of the brain.
Footnotes
Financial support was from “Dirección General de Investigación Científica y Tecnológica, Ministerio de Ciencia and Tecnología” (BFU2005-07486) and “Comunidad Autónoma de Madrid” (GR/SAL/0877/2004) grants (Spain) to W.B. and “Ministerio de Ciencia e Innovación” grants to D.F.d.S. (BFU2008-03488). D.F.d.S. was a postdoctoral fellow funded by a Comunidad Autónoma de Madrid grant (GR/SAL/0877/2004), a “Ministerio de Ciencia and Tecnología” grant (BFU2005-07486) at the Instituto Cajal, Consejo Superior de Investigaciones Científicas, and is currently supported by a Ramón y Cajal Contract at the Departamento de Anatomía, Histología y Neurociencia, Facultad de Medicina, Universidad Autónoma de Madrid.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Amenta F, Liu A, Giannella M, Pigini M, Tayebati SK, Zaccheo D. Age-related changes in the density of muscarinic cholinergic M1 and M2 receptor subtypes in pyramidal neurons of the rat hippocampus. Eur J Histochem. 1995;39:107–116. [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y. Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptors activation during the tetanus. J Neurophysiol. 1995;74:2349–2357. doi: 10.1152/jn.1995.74.6.2349. [DOI] [PubMed] [Google Scholar]
- Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus. J Neurophysiol. 1994;72:2034–2040. doi: 10.1152/jn.1994.72.4.2034. [DOI] [PubMed] [Google Scholar]
- Auerbach JM, Segal M. Muscarinic receptors mediating depression and long-term potentiation in rat hippocampus. J Physiol. 1996;492:479–493. doi: 10.1113/jphysiol.1996.sp021323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci. 2007;27:11510–11521. doi: 10.1523/JNEUROSCI.3077-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde M, Cazalets JR, Buño W. Activity-dependent response depression in rat hippocampal CA1 pyramidal neurons in vitro. J Neurophysiol. 1995;74:1714–1729. doi: 10.1152/jn.1995.74.4.1714. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Campanac E, Debanne D. Plasticity of neuronal excitability: Hebbian rules beyond the synapse. Arch Ital Biol. 2007;145:277–287. [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Clark KA, Collingridge GL. Synaptic potentiation of dual-component excitatory postsynaptic currents in the rat hippocampus. J Physiol. 1995;482:39–52. doi: 10.1113/jphysiol.1995.sp020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Raymond CR, Abraham WC. Priming of long-term potentiation induced by activation of metabotropic glutamate receptors coupled to phospholipase C. Hippocampus. 1998;8:160–170. doi: 10.1002/(SICI)1098-1063(1998)8:2<160::AID-HIPO8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Colbran RJ, Fong YL, Schworer CM, Soderling TR. Regulatory interactions of the calmodulin-binding, inhibitory, and autophosphorylation domains of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1988;263:18145–18151. [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Buño W. Presynaptic inhibition of Schaffer collateral synapses by stimulation of hippocampal cholinergic afferent fibres. Eur J Neurosci. 2003;17:555–558. doi: 10.1046/j.1460-9568.2003.02490.x. [DOI] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Cabezas C, de Prada AN, Sánchez-Jiménez A, Buño W. Selective muscarinic regulation of functional glutamatergic Schaffer collateral synapses in rat CA1 pyramidal neurons. J Physiol. 2002;545:51–63. doi: 10.1113/jphysiol.2002.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Núñez A, Borde M, Malinow R, Buño W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. J Neurosci. 2008;28:1469–1478. doi: 10.1523/JNEUROSCI.2723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidzinski P, Shor O, Behr J. Target-cell-specific bidirectional synaptic plasticity at hippocampal output synapses. Eur J Neurosci. 2008;27:1111–1118. doi: 10.1111/j.1460-9568.2008.06089.x. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JS, Hagenston AM, Hertle DN, Gipson KE, Bertetto-D'Angelo L, Yeckel MF. Inositol-1,4,5-trisphosphate receptor-mediated Ca2+ waves in pyramidal neuron dendrites propagate through hot spots and cold spots. J Physiol. 2009;587:1439–1459. doi: 10.1113/jphysiol.2009.168930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Léránth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J Comp Neurol. 1985;239:237–246. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Fuenzalida M, Fernandez de Sevilla D, Buño W. Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. J Neurosci. 2007;27:11940–11948. doi: 10.1523/JNEUROSCI.0900-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Miyamoto E. A working model of CaM kinase II activity in hippocampal long-term potentiation and memory. Neurosci Res. 2000;38:3–17. doi: 10.1016/s0168-0102(00)00139-5. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- Harvey J, Balasubramaniam R, Collingridge GL. Carbachol can potentiate N-methyl-D-aspartate responses in the rat hippocampus by a staurosporine and thapsigargin-insensitive mechanism. Neurosci Lett. 1993;162:165–168. doi: 10.1016/0304-3940(93)90586-a. [DOI] [PubMed] [Google Scholar]
- Hebb DO. The organization of behavior. New York: Wiley; 1949. [Google Scholar]
- Johnston D, Williams S, Jaffe D, Gray R. NMDA-receptor-independent long-term potentiation. Annu Rev Physiol. 1992;54:489–505. doi: 10.1146/annurev.ph.54.030192.002421. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574:283–290. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol. 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Role of glutamate autoreceptors at hippocampal mossy fiber synapses. Neuron. 2008;60:1082–1094. doi: 10.1016/j.neuron.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lewis PR, Shute CC. The cholinergic limbic system: projections to hippocampal formation, medial cortex, nuclei of the ascending cholinergic reticular system, and the subfornical organ and supra-optic crest. Brain. 1967;90:521–540. doi: 10.1093/brain/90.3.521. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lissin DV, Carroll RC, Nicoll RA, Malenka RC, von Zastrow M. Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci. 1999;19:1263–1272. doi: 10.1523/JNEUROSCI.19-04-01263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol. 2006;18:71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Segal M. Acetylcholine potentiates responses to N-methyl-D-aspartate in the rat hippocampus. Neurosci Lett. 1990;113:62–65. doi: 10.1016/0304-3940(90)90495-u. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Mori M. Diverse signal transduction pathways mediated by endogenous P2 receptors in cultured rat cerebral cortical neurons. J Neurophysiol. 1998;79:2513–2521. doi: 10.1152/jn.1998.79.5.2513. [DOI] [PubMed] [Google Scholar]
- O'Connor JJ, Rowan MJ, Anwyl R. Long-lasting enhancement of NMDA receptor-mediated synaptic transmission by metabotropic glutamate receptor activation. Nature. 1994;367:557–559. doi: 10.1038/367557a0. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zhao LF, Korwek KM, Weeber EJ. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J Neurosci. 2006;26:12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Konnerth A. Stores not just for storage. intracellular calcium release and synaptic plasticity. Neuron. 2001;31:519–522. doi: 10.1016/s0896-6273(01)00402-0. [DOI] [PubMed] [Google Scholar]
- Salter MW. Src, N-methyl-D-aspartate (NMDA) receptors, and synaptic plasticity. Biochem Pharmacol. 1998;56:789–798. doi: 10.1016/s0006-2952(98)00124-5. [DOI] [PubMed] [Google Scholar]
- Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993;13:3051–3063. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Nelson SB. Spike timing, calcium signals and synaptic plasticity. Curr Opin Neurobiol. 2002;12:305–314. doi: 10.1016/s0959-4388(02)00325-2. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Opitz T, Zheng X, Bennett MV, Zukin RS. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology. 2001;40:856–865. doi: 10.1016/s0028-3908(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Yu XM, Salter MW. Ca(2+)-independent reduction of N-methyl-D-aspartate channel activity by protein tyrosine phosphatase. Proc Natl Acad Sci U S A. 1996;93:1721–1725. doi: 10.1073/pnas.93.4.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]