

Abstract
We discovered a nonpeptidic compound, TAK-070, that inhibited BACE1, a rate-limiting protease for the generation of Aβ peptides that are considered causative for Alzheimer's disease (AD), in a noncompetitive manner. TAK-070 bound to full-length BACE1, but not to truncated BACE1 lacking the transmembrane domain. Short-term oral administration of TAK-070 decreased the brain levels of soluble Aβ, increased that of neurotrophic sAPPα by ∼20%, and normalized the behavioral impairments in cognitive tests in Tg2576 mice, an APP transgenic mouse model of AD. Six-month chronic treatment decreased cerebral Aβ deposition by ∼60%, preserving the pharmacological efficacy on soluble Aβ and sAPPα levels. These results support the feasibility of BACE1 inhibition with a noncompetitive inhibitor as disease-modifying as well as symptomatic therapy for AD.
Introduction
The accumulation of amyloid-β peptides (Aβ) in the brain is strongly implicated in the pathogenesis of Alzheimer's disease (AD), and considered as a prime target for the disease-modifying therapy of AD (Selkoe and Schenk, 2003). Aβ is proteolytically produced through sequential cleavages by β- and γ-secretases from amyloid precursor protein (APP). The β-secretase cleavage of APP is executed by a membrane-bound aspartic protease, β-site APP-cleaving enzyme 1 (BACE1), which is considered to be the rate-limiting step in the production of Aβ (Cole and Vassar, 2008), whereas a majority of APP is cleaved by α-secretase at the midportion of Aβ sequence in a way to preclude Aβ production, by competing with BACE1.
γ-Secretase generates the C termini of Aβ with different length, e.g., Aβ40 or Aβ42, the latter being considered as the pathogenic species (Iwatsubo et al., 1994). Inhibition of γ-secretase may potentially cause side effects, because genetic knock-out (KO) of presenilin 1 and 2, the catalytic subunits of γ-secretase, leads to embryonic lethality due to failure in activation of Notch, which is essential for development and differentiation (Shen et al., 1997; Wong et al., 1997; Donoviel et al., 1999). Furthermore, cognitive deficits associated with synaptic degeneration have been documented in PS1/PS2 conditional KO mice with or without APP transgenic background (Saura et al., 2004, 2005; Chen et al., 2008). In contrast, BACE1 KO mice do not show such fatal phenotypes despite its complete ablation, except for partial hypomyelination at the developmental stage (Hu et al., 2006; Sankaranarayanan et al., 2008) or schizophrenia-like behavior in homozygous BACE1 KO mice (Savonenko et al., 2008), whereas cognitive deficits are ameliorated on APP transgenic background (Ohno et al., 2004, 2006, 2007). Furthermore, it has been well documented that the protein levels or activities of BACE1 are upregulated in the brains of patients with sporadic AD (Stockley and O'Neill, 2007). Therefore, BACE1 is considered as a promising target for the mechanism-based therapy for AD. So far, several BACE1 inhibitors have been reported (Hussain et al., 2007; Sankaranarayanan et al., 2009; Silvestri, 2009), although no compound that is orally active and highly penetrable to brain tissues with functional ameliorations has been documented.
We conducted a cell-based assay in the IMR32 human neuroblastoma cell line for small chemical compounds that reduce the secretion of Aβ and increase that of sAPPα, the latter being recognized as neurotrophic with ameliorative effects on cognitive behaviors (Isacson et al., 2002; Postina, 2008). Finally we discovered a nonpeptidic compound, (R)-6-[(1,1′-biphenyl)-4-ylmethoxy]-1,2,3,4-tetrahydro-N,N-dimethyl-2-naphthalene-ethan-amine hydrochloride monohydrate (TAK-070) (Fig. 1), as a novel noncompetitive BACE1 inhibitor. TAK-070 ameliorated Aβ pathology and behavioral deficits in Tg2576, an APP transgenic model mice of AD, although the reduction in Aβ levels was modest, unlike those observed by complete ablation of BACE1. We propose that the partial reduction in Aβ as well as increase in sAPPα by a noncompetitive BACE1 inhibition may be sufficient to modify amyloid pathology and ameliorate cognitive deficits, without causing potential adverse events by complete BACE1 ablation.
Figure 1.

Chemical structure of TAK-070.
Materials and Methods
Compound
The chemical TAK-070 was made by Takeda Pharmaceutical Company Limited (Takeda), and the chemical structure is shown in Figure 1. The chemical synthesis and related information are described in the patent of JP-A 11-80098 (WO98/38156). γ-Secretase inhibitor IX (DAPT) was purchased from Calbiochem.
Cell cultures and sample preparation
IMR32 human neuroblastoma cell line was obtained from American Type Culture Collection (ATCC), and mouse Neuro-2a neuroblastoma cells stably expressing human Swedish mutant APP (N2aAPPsw cells) were generated as described previously (Tomita et al., 2002). For ELISA analysis, cells were cultured on 48-well multi-plates at 5 × 104 cells/cm2 to reach near total confluence in DMEM (Nikken Biomedical Laboratory) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Wako) in a humid atmosphere containing 10% CO2. The culture medium was replaced with DMEM/0.2% bovine serum albumin (BSA) (Wako) containing various concentrations of TAK-070, and the cells were cultured for 24 h. The conditioned media were subjected to ELISA quantitation.
Quantitation of sAPPα and Aβ by ELISA
To quantitate human sAPPα, we used LN27 that recognizes the N-terminal portion of APP (Zymed) as a capture antibody. ELISA plates (high binding, clear plate, Greiner) were filled with LN27 (0.5 μg/ml, 75 μl/well) in carbonated buffer (100 mmol/L, pH 9.6) and incubated at 4°C overnight. After washing the plates with PBS (Invitrogen) three times, each well was blocked with 100 μl of BlockAce solution (Dai-Nippon) diluted fourfold (v/v) for >2 h. After washing the plates with PBS twice, 50 μl samples or standards prepared from conditioned media containing sAPPα were mixed with 50 μl of buffer A (20 mmol/L phosphate buffer, pH 7.2, 10% BlockAce, 0.2% protease-free BSA, 0.05% thimerosal, 0.4 mol/L NaCl, 0.076% CHAPS, 2 mmol/L EDTA-2Na, 0.2% SDS, and 4 mmol/L DTT) in each well. Buffer A contains DTT to break the S-S bond of sAPPα to enhance the recognition by the LN27 antibody. The mixture was incubated in the plate overnight at 4°C. After washing the plates with PBS four times, BAN50-HRP (75 μl/well) [which recognizes the C-terminal portion of human sAPPα (Asami-Odaka et al., 1995)] diluted in the detection buffer (20 mmol/L phosphate buffer, pH 7.2, 1% protease-free BSA, 2 mmol/L EDTA-2Na, 0.05% thimerosal, and 0.4 mol/L NaCl) was added to each well. The plates were incubated at room temperature for 3–4 h. After washing the plates with PBS six times, substrates were added and the reaction mixtures were developed. To measure sAPPα in brain lysates, the homogenate buffer free of detergents was used to preclude contamination of membrane-associated APP.
Aβ40 or Aβ42 was quantitated by two-site sandwich ELISA using a capture antibody BNT77, which recognizes the midportion of Aβ without detecting Aβ17-40/42 (i.e., the cleaved products by α- and γ-secretases) (Fukumoto et al., 1999), and the detector antibodies of BA27-HRP or BC05-HRP that specifically detect the C termini of Aβ40 or Aβ42, respectively, as described previously (Asami-Odaka et al., 1995). TMB substrate (Pierce) was used as a chromogenic substrate. After stopping the reaction with phosphoric acid solution (1 mol/L, 75 μl/well), the enzymatic products were measured using a multi-label counter at OD450 (WALLAC Arvo Sx; PerkinElmer Life Sciences).
Immunoblot analysis
Quantification of the levels of sAPPβ, sAPPα, APP, APP C-terminal fragment (CTF) (e.g., C83 and C99), BACE, or ADAM10 was performed on conditioned media or cell lysates of N2aAPPsw cells treated with vehicle DMSO (0.1% v/v), 3 μmol/L TAK-070, or 3 μmol/L DAPT for 24 h. SeeBlue Plus2 (Invitrogen) was used as a molecular weight standard. Protein samples separated by SDS-PAGE were electrophoretically transferred to an Immobilon PVDF membrane (Millipore). The membranes were blocked with 5% (w/v) skim milk solution (Wako) in TBS-T (20 mmol/L Tris-buffer, pH 7.0, containing 50 mmol/L NaCl and 0.1% Tween 20) and reacted overnight with a detector antibody. The following monoclonal or polyclonal antibodies were used: monoclonal antibodies that specifically react with the C terminus of Swedish mutant sAPPβ (sAPPβsw) [clone 6A1, IBL, 1:100 dilution (Lakshmana et al., 2009)], the C terminus of human sAPPα [BAN50, Takeda, 0.5 μg/ml (Asami-Odaka et al., 1995)], α-tubulin (clone AA4.3, Developmental Studies Hybridoma Bank, cultured medium from hybridoma), respectively, and polyclonal antibodies to the C terminus of APP [APP(C), No. 18961; IBL, 1:1000 dilution] that detect total APP and APP-CTFs, anti-mouse/rat APP [APP(597), No. 28055; IBL, 1:1000 dilution] raised against the C-terminal 16 aa of rodent sAPPα that specifically recognizes rodent, but not human, sAPPα, anti-sAPPβ [No. 18957; IBL, 1:100 dilution (Lakshmana et al., 2009)] specific for sAPPβ derived from wild-type APP (sAPPβwt), ADAM10 (735–749) (No. 422751; Calbiochem, 0.5 μg/ml), and the C terminus of BACE1 (No. 28051; IBL, 0.1 μg/ml, 1:200). Specificity of anti-human/mouse APP antibodies is shown in supplemental Figure S1 (available at www.jneurosci.org as supplemental material). The hybridoma clone AA4.3 was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology (Iowa City, IA). After washing with TBS-T, the membranes were further incubated with TBS (20 mmol/L Tris-buffer, pH 7.0, containing 50 mmol/L NaCl) buffer containing an anti-mouse IgG antibody-HRP (1/5000) for a monoclonal antibody or an anti-rabbit IgG antibody-HRP (1/5000) (GE Healthcare) for polyclonal antibodies. The membranes were washed with TBS-T, and then immunoreactive bands were visualized using Immunostar, Immunostar LD (Wako), or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) according to the manufacturer's instructions. The intensity of bands on the membrane was captured and quantitated using LAS-1000plus (FUJIFILM).
Cell-based assay for α-secretase activity
The assay (Doedens et al., 2003) was performed with a slight modification. N2aAPPsw cells were cultured in DMEM supplemented with 10% FCS until grown to confluence. The cells were collected by PBS (−) (Ca2+, Mg2+ free) buffer and centrifuged for 5 min at 300 × g. After washing with PBS (−), the cells were suspended in PBS (−) at a final concentration of 4 × 107 cells/ml. The enzymatic reaction was initiated by combining an equal volume (100 μl) of cell suspension and reaction mixture at a final cell concentration of 2 × 107 cells/ml, 10 μmol/L each of leupeptin (Peptide Institute), aprotinin (Roche Diagnostics), and α-secretase fluorogenic substrate [MCA-HQKLVFFA (K-DNP), BioSource], with vehicle of DMSO, TAK-070 (final concentration: 3 μmol/L), or (−)-epigallocatechin-3-gallate (catechin, Wako) (final concentration: 20 μmol/L). After each incubation time point, the cells were centrifuged, the cell-free supernatants of each 100 μl were added to a 96-well black plate (Greiner), and fluorescence intensity after cleavage by α-secretase was measured (excitation 320 nm, emission 400 nm) (WALLAC Arvo Sx; PerkinElmer Life Sciences).
Expression and purification of FLAG-tagged full-length BACE1 or truncated BACE1 (1-454)
The plasmid containing cDNA encoding the entire coding frame of human BACE1 (clone No. FG04087) was obtained from KAZUSA DNA Research Institute. The full-length BACE1 (1-501) and C-terminally truncated BACE1 (1-454, 460, 465, 471 and 474) lacking the transmembrane domain were cloned into pcDNA3.1 (−) (Invitrogen) vector with a C-terminal FLAG tag [pcDNA3.1(−)BACE1-flag and pcDNA3.1(−)BACE1(1-454, 460, 465, 471, or 474)-flag, respectively]. COS-7 cells were cultured in DMEM supplemented with 10% (v/v) heat-inactivated FBS at 37°C in a humid atmosphere of 5% CO2. Cells were grown in an F225 cell culture flask (225 cm2) and transfected with 22.5 μg of pcDNA3.1(−)BACE1-flag or pcDNA3.1(−)BACE1(1-454, 460, 465, 471, or 474)-flag, using Fugene6 (Roche Diagnostics). Forty-eight hours after transfection, cells were scraped in PBS and centrifuged for 10 min at 1870 × g. The supernatant was used as a source for further purification of the truncated BACE1 (1-454, 460, or 465). To purify full-length BACE1 or truncated BACE1 (1-471 or 474), the pellet was resuspended in 50 mmol/L Tris-HCl buffer, pH 7.4, containing 0.15 mol/L NaCl, 1 mmol/L EDTA, and 0.1 mmol/L PMSF. The cells were disrupted by sonication and centrifuged at 1870 × g for 10 min. The supernatant was centrifuged at 100,000 × g for 45 min to yield crude membrane pellets. The membrane was solubilized in 50 mmol/L Tris-HCl buffer, pH 7.4, containing 50 mmol/L octyl-β-glucoside, 0.15 mol/L NaCl, 1 mmol/L EDTA, and 0.1 mmol/L PMSF at 4°C for 2 h, centrifuged at 100,000 × g for 45 min. The fractions containing full-length of BACE1, C-terminally truncated BACE1 (1-454, 460, 465, 471, or 474) fused with FLAG tag were then loaded on an Anti-FLAG M2 affinity gel (Sigma) column. The column was washed with 50 mmol/L Tris-HCl buffer, pH 7.4, containing 0.15 mol/L NaCl, and purified FLAG-tagged recombinant BACE1 proteins were obtained by elution with 100 μg/ml FLAG peptides.
Cell-free assay for BACE1 activity
A statine substrate analog inhibitor PI (TEEISEVNXVAEF; X = statine) (Sinha et al., 1999) and the fluorogenic substrate for BACE1 [Nma-SEVKMDAEK(Dnp)RR-NH2] were purchased from the Peptide Institute. The substrate was dissolved in 125 mmol/L acetic acid. TAK-070 and PI were dissolved in dimethylformamide (DMF). Assays were performed in black 96-well microplates (Greiner) in a final volume of 50 μl. Each well contained 25 μl of acetate buffer (pH 5.5, 50 mmol/L), 10 μl of recombinant BACE1, 10 μl of substrate (250 μmol/L), and 5 μl of various concentrations of compounds at a final DMF concentration of 0.5%. The assay mixtures were incubated at 37°C for 20 h. After incubation, the fluorescence of the enzymatic product was measured at 460 nm (excitation at 325 nm) using Fluoroskan Ascent (Labsystems). The percentage of inhibition was calculated by an equation of 100 × [1 − (test − blank)/(control − blank)], where test, control, and blank are fluorescence intensities in the presence of a compound, absence of a compound, and absence of both the BACE1 enzyme and a compound, respectively. IC35 values were calculated by linear regression analysis using a BSAS program. To clarify the inhibition profile, double-reciprocal (Lineweaver–Burk) plot analysis was performed using 10 μl substrate of 100, 150, 250, 500, or 1000 μmol/L (a final concentration of 20, 30, 50, 100, or 200 μmol/L, respectively) and 5 μl of TAK-070 of 100 or 300 μmol/L (a final concentration of 10 or 30 μmol/L) in total assay solution of 50 μl. The reciprocal of change in the fluorescence value in the presence of TAK-070 at each concentration was plotted on the vertical axis, and the reciprocal of the substrate concentration was plotted on the longitudinal axis.
Surface plasmon resonance binding assay
We used a Biacore3000/BiacoreA100 instrument to generate sensorgrams for binding of TAK-070 onto full-length BACE1, C-terminally truncated BACE1 (1-454), (1-460), (1-465), (1-471), (1-474), APP688 [Leu18-Leu688 with a C-terminal 6-His tag, also referred to as protease nexin II containing Kunitz-type Protease Inhibitor (KPI) domain, #3466-PI, R&D Systems], and sAPPβ containing KPI domain (BACE1-cleaved N-terminal product of APP, #SIG-39938, Sigma). Each protein was immobilized on a Sensor Chip CM5 (carboxymethylated dextran matrix chip) using amine-coupling kit (Biacore). The sensorgrams were recorded at a flow rate of 30 μl/60 s in a solution of PBS containing 10% DMSO and 0.005% Surfactant P20 (Biacore) at room temperature. TAK-070 was initially dissolved in DMSO and diluted in PBS containing 0.005% Surfactant P20 at a final concentration of 0.5–8, 5, or 10 μmol/L. Specific binding to each protein was calculated as signal to each protein subtracted by signal to vehicle (DMSO).
Animals
All animals were housed in rooms maintained at 24°C with a 12 h light/dark cycle. Food (chow containing TAK-070; Oriental Yeast) and tap water were provided ad libitum. In each experiment, mice were randomly grouped, avoiding differences in body weight among groups. All experiments using animals were reviewed and approved by the Internal Animal Care and Use Committee of Takeda Pharmaceutical Research Laboratories.
Short-term treatment of Tg2576 by TAK-070
Female Tg2576 mice at 2 months of age were used for short-term treatment with TAK-070. Tg2576 were fed either chow containing TAK-070 (5.6 ppm or 56 ppm, corresponding to ∼0.87 or 8.2 mg/kg, p.o., respectively; n = 15) or chow without TAK-070 (n = 15) for 7 weeks. Then, each mouse was decapitated and the cerebral cortex was dissected out on ice. Each sample was immediately frozen on dry ice and stored at −80°C until assay. Halves of the cerebral cortices were homogenized in ice-cold Tris-extraction buffer (50 mmol/L Tris, pH 7.2, 200 mmol/L sodium chloride, 2% protease-free bovine serum albumin, and 0.01% thimerosal) containing protease inhibitor cocktails (1 mmol/L PMSF, 40 KIU aprotinin, 10 μmol/L pepstatin A, 1 mmol/L phosphoramidon, 10 mmol/L 1,10-phenanthroline, 2 mmol/L EDTA) without detergents. After centrifugation at 21,000 × g for 5 min, the supernatants were further diluted and subjected to sandwich ELISAs for Aβ40, Aβ42, or sAPPα.
Long-term treatment of Tg2576 by TAK-070
Male and female Tg2576 mice at 7 months of age (n = 16–17 for each group, n = 8–9, male; n = 8, female) were used for long-term treatment with TAK-070. Tg2576 mice were fed chow containing TAK-070 (56 ppm, corresponding to ∼7 mg/kg/d, p.o., when evaluated at 6 months of treatment) for 6 months and a week from 7 months of age, or chow without TAK-070 (vehicle control). Male Tg2576 mice at 8 months of age (n = 9) were used as a young control. After decapitation, the brains were removed and the left cerebral hemisphere was immediately frozen on dry ice and stored at −80°C until biochemical assays; the right hemisphere was fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and subjected to immunohistochemical analysis. Biochemical quantitation of Aβ and sAPPα was performed as follows: the cerebral cortex was initially homogenized with ice-cold Tris-extraction buffer and centrifuged as in the short-term treatment study to obtain the supernatants for quantitation of soluble Aβ and sAPPα. The pellet was then homogenized in a 19-fold volume of ice-cold 70% formic acid, and centrifuged at 44,000 × g for 5 min. The supernatant was further diluted, neutralized with 1 mol/L Tris-based solution, and the levels of insoluble Aβ40 and Aβ42 were quantitated by ELISA.
Immunohistochemistry
Immunohistopathological analysis was performed on two distinct coronal sections from the right hemisphere at the level of the hippocampus and thalamus of Tg2576 mice. Sample preparation and quantitation of Aβ plaques were conducted under blinded conditions for the examiner. Four-micrometer-thick sections were deparaffinized and pretreated with 99% formic acid for 5 min. The section was blocked with 10% fetal calf serum for 30 min and then reacted with BAN50 (0.5 μg/ml) at 4°C overnight. BAN50-positive plaques were visualized with Dako REAL EnVision Detection Kit (Dako) using diaminobenzidine as a chromogen. The amyloid burden with a diameter more than ∼30 μm (percentage of immunopositive areas that comprised the total area) and the number of plaques throughout the right cerebral neocortices were quantitated using Vanox (AH-2, Olympus) connected to a digital video camera (Prog Res 3012, Carl Zeiss) and image analysis software (Win ROOF, Mitani).
Y-maze and Morris water maze tests
Male Tg2576 mice of 18 weeks of age were divided into three groups, i.e., vehicle-treated (n = 14), TAK-070 1 mg/kg treated (n = 14), and TAK-070 3 mg/kg treated (n = 14). Wild-type littermates (n = 15) were used as a nontransgenic control group. Tg2576 mice were treated with TAK-070 (1 or 3 mg/kg, p.o.) or vehicle (0.5% methylcellulose; MC) once a day for 9 d before the behavioral test. Each mouse was treated with drugs after all trials were completed every day during the test period. Each mouse was sequentially subjected to Y-maze test on day 10, and then in Morris water maze test from day 11 to day 13. On day 14, the mice were decapitated. The brains were dissected out on ice immediately and stored at −80°C.
Y-maze test.
To measure spontaneous alternation behavior and exploratory activity, a black Y-maze with arms of 40 cm length, 3 cm width, with 12.5 cm walls was used. Each animal underwent one trial, during which the animal was placed into one of the three alleys and allowed free exploration of the maze for 5 min, and alternations and total numbers of arm choices were recorded. Spontaneous alternation, expressed as a percentage, refers to ratio of arm choices differing from the previous two choices to the total number of arm entries.
Morris water maze test.
The water maze pool comprised a circular plastic water tank, 120 cm in diameter and 20 cm in depth. The pool was filled with water at room temperature to a height of 15 cm. A transparent acrylic platform (10 × 10 cm), its top surface being 0.5 cm below the surface of water, was located in a constant position in the middle of one quadrant from the center and edge of the pool, and was invisible for mice inside the pool. Each mouse was given four trials daily for 3 consecutive days with an interval of ∼20 min. The sequence of the starting points was randomly selected. The escape latency and the swimming distance for mice to find the hidden platform were automatically recorded by the computer analyzing system (Target/2, Neuroscience). The value for each session was defined as the mean of four trials. The probe test was not conducted because the deficits were too modest to evaluate the effects of compounds.
Novel object recognition test
Male Tg2576 mice of 5 months of age were divided into two groups, vehicle treated (n = 14) and TAK-070 3 mg/kg treated (n = 15). As a nontransgenic control group, wild-type littermates (n = 15) were used. Tg2576 mice were treated with TAK-070 (3 mg/kg, p.o.) or vehicle (0.5% MC) once a day for 15 d before the test. During the test, each mouse was treated with TAK-070 or vehicle after all trials were completed.
Each mouse was subjected to the novel object recognition test from day 16 to day 17. In the acquisition session on day 16, the same two objects were placed in the back corner of the test box (30 × 30 × 30 cm). The mouse was then placed in another corner of the box and the time exploring each object was recorded for 5 min. After 24 h later on day 17, animals were placed back into the same box, except that one of the familiar objects used during the acquisition was replaced with a novel object. The animals were then allowed to explore freely for 5 min. A preference ratio of the time exploring the novel object to the time exploring both objects was calculated as an index of cognitive function.
Statistical analysis
Statistical analysis was performed by the one-tailed Williams' test for analysis of multiple groups in dose–response study, by Tukey's test for analysis of multiple groups in no dose–response study or Student's t test for analysis of two groups under the BSAS program.
Results
TAK-070 reduced Aβ secretion and increased that of sAPPα in cell cultures
We treated human IMR-32 neuroblastoma cells with TAK-070 for 24 h, and measured the levels of Aβ and sAPPα in the conditioned media by ELISA. We observed a concentration-dependent suppression of the secretion of Aβ, with minimum effective concentrations (MECs) for Aβ40 and Aβ42 of ∼100 and ∼1000 nmol/L, respectively (Fig. 2A). TAK-070 also stimulated sAPPα production in a concentration-dependent manner with MEC of ∼100 nmol/L. The percentage reduction in the levels of Aβ40 and Aβ42, and percentage increase in that of sAPPα by treatment with 3 μmol/L TAK-070 were ∼50, ∼70, and ∼30%, respectively. Similarly significant effects at submicromolar to micromolar ranges of TAK-070 on APP processing (∼25% reduction in Aβ secretion and ∼90% increase in sAPPα at 3 μmol/L TAK-070) were observed in mouse Neuro-2a neuroblastoma cells stably overexpressing human APP carrying Swedish-type familial Alzheimer mutation (APPsw; N2aAPPsw cells) (Fig. 2B).
Figure 2.

Effects of TAK-070 on secretion of Aβ and sAPPα in cultured cells. The levels of Aβ40, Aβ42, and sAPPα secreted in conditioned media were quantitated by ELISAs. A, Human IMR32 neuroblastoma cells were treated with TAK-070 for 24 h. Vehicle control levels for Aβ40 and Aβ42 were 17.3 and 5.8 fmol/ml on average, respectively. Levels of sAPPα were determined as arbitrary unit values. Values are mean percentages relative to levels in the control (±SEM) in four independent experiments. B, Mouse Neuro2a neuroblastoma cells stably expressing human APPsw (N2aAPPsw cells) were treated with TAK-070 for 24 h. Vehicle control levels of Aβ40 and Aβ42 were 447.6 and 114.6 fmol/ml, respectively. Levels of sAPPα were determined as arbitrary unit values. Values are mean percentages of the control (±SEM) in six independent experiments. *p < 0.025, compared with the vehicle control (one-tailed Williams' test).
TAK-070 inhibited BACE1 activity in cultured cells
We next examined the effects of TAK-070 in N2aAPPsw cells by immunoblot analysis. Treatment with TAK-070 (3 μmol/L) significantly decreased the secreted level of both human Swedish sAPPβ and mouse endogenous sAPPβ, N-terminal counterparts of APP generated by BACE1 cleavage, by ∼16 and ∼19%, respectively. Simultaneously, the levels of human and mouse endogenous sAPPα were increased by ∼70% and ∼30%, respectively (Fig. 3A). We then examined the effects of TAK-070 on the levels of membrane-bound APP and its C-terminal stubs (e.g., C83 and C99), BACE1, and ADAM10 [a neuronal α-secretase candidate (Jorissen et al., 2010)] in lysates of N2aAPPsw cells. TAK-070 decreased the level of C99 by ∼15%, in contrast to the prominent increase in the levels of C83 and C99 (by ∼2.1- and ∼7.1-fold, respectively) by inhibition of γ-secretase by DAPT (Fig. 3B). TAK-070 treatment did not significantly affect the protein levels of APP, C83, BACE1, or ADAM10 (Fig. 3B). The levels of mouse sAPPβ in the conditioned media of TAK-070-treated naive N2a cells also was decreased (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). We further examined the effects of TAK-070 on α-secretase activity using a cell-based, peptide cleavage assay (Doedens et al., 2003). Although (−)-epigallocatechin-3-gallate induced the enzymatic activity, in line with the reported increase in the active form of ADAM10 (Obregon et al., 2006), TAK-070 did not show any incremental effects on the α-secretase-cleaved product (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), suggesting that TAK-070 is not an α-secretase activator.
Figure 3.

Immunoblot analysis of the protein levels of APP derivatives and those of secretases in N2aAPPsw cells. A, Immunoblots of sAPPβ (sAPPβsw derived from transfected human APPsw and sAPPβwt from endogenous mouse APP) and sAPPα (human sAPPα derived from transfected human APPsw and mouse sAPPα from endogenous APP) in media after treatment with TAK-070 (3 μmol/L) or vehicle from three independent experiments are shown. Values in the graph (right) show the mean percentages of band intensities analyzed by densitometry relative to those in vehicle control (±SEM) in the three independent experiments. *p < 0.05, **p < 0.01, versus vehicle control (Student's t test). B, Immunoblots of APP, C-terminal fragments of APP (C99 and C83), BACE1 and high- and low-molecular-weight forms of ADAM10 (pro- and matured forms, respectively) from lysates of N2aAPPsw cells treated with vehicle, DAPT (3 μmol/L), or TAK-070 (3 μmol/L) are shown. Levels of α-tubulin are shown as an internal control. Note that all the immunoblot data are obtained from a single membrane replica with identical exposure. Values for C99 and C83 (right) are mean percentages of band intensities analyzed by densitometry relative to those in vehicle control (±SEM) in the three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, versus vehicle control (Student's t test).
Noncompetitive BACE1 inhibition by TAK-070 in a cell-free assay
To confirm that TAK-070 has a direct inhibitory effect on BACE1, we developed a cell-free assay, using recombinant full-length human BACE1 and a quenching type fluorogenic BACE1 substrate based on ∼10 aa residues flanking the β-cleavage site of wild-type human APP. TAK-070 inhibited the BACE1 activity in a concentration-dependent manner, with IC35 of ∼3.15 μmol/L and MEC of ∼100 nmol/L (Fig. 4A), the latter being a similar effective concentration to that in cell culture studies (Fig. 2A,B). Under the same experimental conditions, a peptidic BACE1 inhibitor (TEEISEVNXVAEF; X = statine) inhibited BACE1 activity with IC35 value of 38.8 nmol/L, which was consistent with the previously published data (Sinha et al., 1999). To further examine the inhibitory profile of TAK-070, we conducted a Lineweaver–Burk plot analysis by incubating the fluorogenic BACE1 substrate with recombinant full-length human BACE1 in the presence of 10 or 30 μmol/L TAK-070. All fitted lines converged at an identical point on the x-axis with an estimated Km value of 156 μmol/L (Fig. 4B), indicating that TAK-070 inhibits BACE1 in a noncompetitive manner. The Ki value estimated from the y-axis values with an intercept of (1 + [I]/Ki)/Vmax was 19 μmol/L.
Figure 4.

Noncompetitive inhibition of BACE1 activity by TAK-070 in cell-free assay. A, Concentration-dependent inhibition of BACE1 activity by TAK-070. Human recombinant full-length BACE1 purified from COS-7 cells (rhBACE1) was incubated with a fluorogenic BACE1 substrate based on the amino acid sequence of wild-type human APP flanking the BACE1 cleavage site (Nma-SEVKMDAEK(Dnp)RR-NH2) in the presence of various concentrations of TAK-070 (indicated in abscissa, in nanomoles per liter). Values are mean percentage inhibition (±SEM) in three independent experiments. B, Lineweaver–Burk plot analysis of the mode of inhibition by TAK-070. rhBACE1 was incubated with 20–200 μmol/L BACE1 substrate in the presence (10 or 30 μmol/L) or absence of TAK-070. The plots of 1/V versus 1/[S] were fitted by the Lineweaver–Burk straight line. Result of a representative experiment is shown.
TAK-070 did not inhibit other aspartic proteases (e.g., cathepsin D and E, renin, and γ-secretase (Takahashi et al., 2003)), nor activated enzymatic activity of human TACE in cell-free assays even at the concentration of 100 μmol/L (data not shown), in agreement with the cell culture data described above.
Binding of TAK-070 to full-length BACE1, but not to its extracellular domain
To gain further insight into the mechanism of the noncompetitive BACE1 inhibition by TAK-070, we examined the binding of TAK-070 to BACE1 using a surface plasmon resonance assay. Since TAK-070 inhibited the proteolytic activity of full-length BACE1 [BACE1 (1-501)] in a noncompetitive manner, but not that of the truncated BACE1 (1-454), lacking the transmembrane domain (data not shown), we first compared the binding of TAK-070 to BACE1 (1-501) or truncated BACE1 (1-454). Surface Plasmon resonance assay clearly showed that TAK-070 was specifically bound to BACE1 (1-501) in a concentration-dependent manner (0.5–8 μmol/L), but not to BACE1 (1-454) within the same concentration range (Fig. 5A). To further narrow down the binding site of TAK-070 within the C-terminal region of BACE1, we examined the binding of TAK-070 to a series of C-terminally truncated BACE1, i.e., BACE1 (1-460), (1-465), (1-471), and (1-474). The binding of TAK-070 to BACE1 (1-460) and (1-465) was completely lost, whereas BACE1 (1-474) retained a comparable affinity to TAK-070 as BACE1 (1-501), and the binding of BACE1 (1-471) was partially impaired (Fig. 5B). These data suggest that the critical region within the C terminus of BACE1 for binding to TAK-070 resides around residues 465-474, a subdomain of the membrane spanning region. We also examined the binding of TAK-070 to recombinant proteins of APP (18-688) containing Kunitz-type protease inhibitor domain and the BACE1-cleavage site or sAPPβ, and found that neither APP (18-688) nor sAPPβ showed significant binding to TAK-070 (5 μmol/L) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 5.
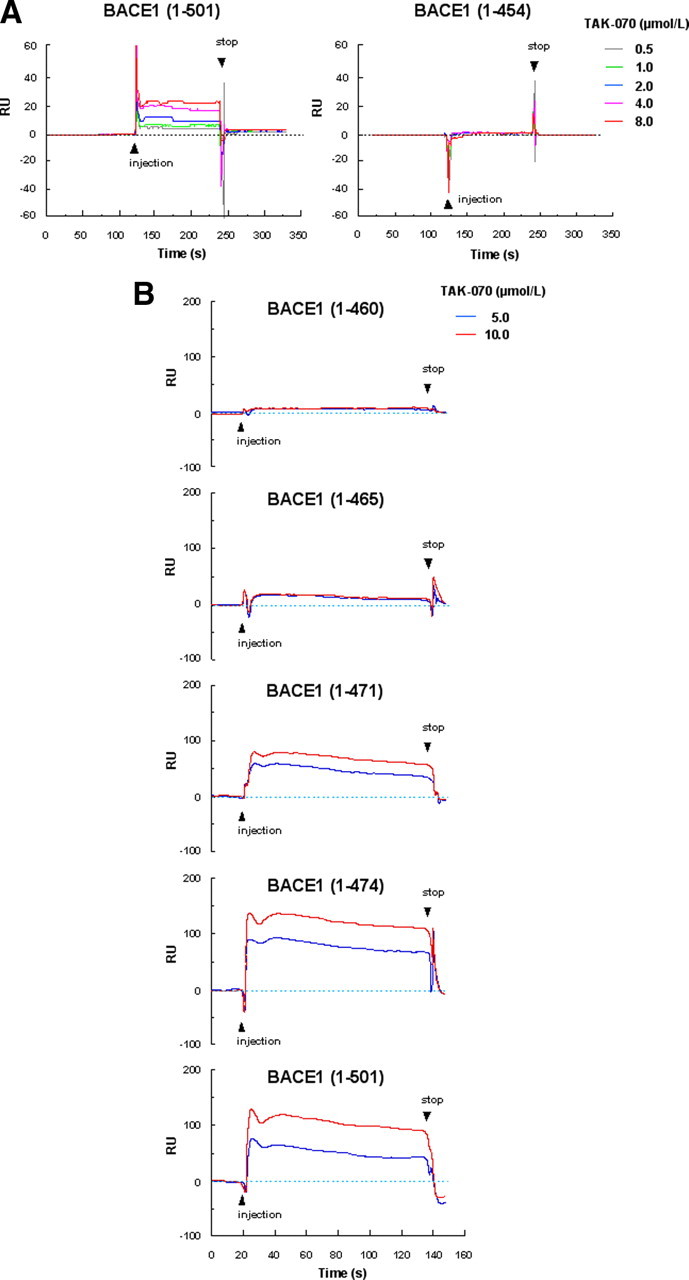
Surface plasmon resonance assay of the binding of TAK-070 to BACE1. A, Sensorgram showing a binding of TAK-070 to full-length BACE1 (1-501) (left panel), but not to C-terminally truncated BACE1 (1-454) lacking the membrane spanning region (right panel). TAK-070 bound to full-length BACE1 in a concentration-dependent manner (within the range of 0.5–8 μmol/L). B, Binding of TAK-070 (5 and 10 μmol/L) to full-length BACE1 (1-501) or C-terminally truncated BACE1 (1-474), (1-471), (1-465), and (1-460). One relative unit (RU) corresponds to 1 pg/mm2.
These data from cell-based and cell-free studies collectively indicate that TAK-070 is a direct, noncompetitive inhibitor for BACE1 that acts by binding to the noncatalytic site of BACE1, presumably to the transmembrane domain.
TAK-070 reduced Aβ and increased sAPPα in the brains of Tg2576 mice
We then examined whether TAK-070 is effective on Aβ and sAPPα in the brains of Tg2576 mice, a transgenic mouse model of AD that overexpresses APPsw. We first performed a short-term treatment, feeding young female Tg2576 mice with chow containing TAK-070 (5.6 and 56 ppm, corresponding to 0.87 and 8.2 mg/kg/d, p.o., respectively) starting at 2 months of age for 7 weeks. All mice survived without any differences in body weight and food consumption among cohorts. Oral administration of TAK-070 significantly reduced the levels of soluble Aβ40 and Aβ42 in Tris buffer-soluble fractions of the cerebral cortex (average ± SEM: 7707 ± 334 and 1825 ± 100 fmol/g wet weight, respectively, in vehicle group) by ∼16–23%, and increased that of sAPPα by ∼15–21% at both doses (Fig. 6A).
Figure 6.

Effects of TAK-070 on Aβ and sAPPα levels in the brains of Tg2576 mice. A, Levels of Tris-soluble Aβ40, Aβ42, and sAPPα in the cerebral cortices of young female Tg2576 mice after short-term administration. Values are mean percentages (±SEM) relative to levels in vehicle control (n = 15 for both cohorts). *p < 0.025, versus vehicle control (one-tailed Williams test). B, Levels of Tris-soluble Aβ40, Aβ42, and sAPPα in cerebral cortices of 13-month-old Tg2576 mice after long-term treatment. The number of 13-month-old Tg2576 mice with vehicle or TAK-070 (56 ppm, corresponding to ∼7 mg/kg/d, p.o.) were 13 (male 6, female 7) and 16 (male 10, female 6), respectively after 6 months treatment. Values are mean percentages (±SEM) relative to levels in young controls (8-month-old nontreated Tg2576, n = 9). C, Levels of Tris-insoluble, formic acid-extractable Aβ40 and Aβ42 in cerebral cortices examined in B. Values are the fold increase (±SEM) relative to levels in young controls (8-month-old nontreated Tg2576). D, Aβ immunohistochemistry of coronal sections from brains of TAK-070 (bottom panel) or vehicle (top panel) treated-Tg2576 mice (13 months old). E, Amyloid burden (% of area covered by Aβ immunoreactivity; left panel) or density of plaque (number per mm2 area; right panel) in the cerebral neocortices of Tg2576 mice. Mean values (±SEM) are shown. ##p < 0.01, versus those in 8-month-old mice; *p < 0.05, **p < 0.01, versus those in vehicle control (Student's t test) in B, C, and E.
We next conducted a long-term treatment of Tg2576 mice with TAK-070. We started treatment at the age of ∼7 months, just before Tg2576 mice develop the Aβ deposition as amyloid plaques (at ∼8 months). Tg2576 mice were fed with chow containing 56 ppm TAK-070 until 13 months of age for ∼6 months. Tg2576 mice tolerated chronic treatment with TAK-070, and the mean survival rates were at similar levels after ∼6 months treatment by vehicle or TAK-070 (81% or 94%, respectively), without any differences in body weights and food consumption between cohorts.
We first quantitated the levels of Tris-soluble Aβ in the brains of untreated 13-month-old Tg2576 mice, which were dramatically increased by 68% and 129%, respectively for Aβ40 and Aβ42, compared with those at 8 months (Fig. 6B). Notably, the level of sAPPα was decreased by 32% at 13 months. Consistent with the results in young Tg2576 mice (Fig. 6A), TAK-070 reduced the levels of Tris-soluble Aβ40 and Aβ42 by ∼15 and ∼25%, respectively, and increased that of sAPPα by ∼22% even after the 6 months of treatment (Fig. 6B).
We next quantitated the levels of insoluble Aβ that was extracted from the Tris-insoluble pellets by formic acid denaturation. The levels of insoluble Aβ40 and Aβ42 in untreated Tg2576 mice were markedly increased at 13 months by ∼35-fold and ∼23-fold, respectively, compared with those of young control mice (6367 ± 720 and 3513 ± 317 pmol/g wet weight, in 8 months of Tg2576). No gender differences were noted in the extent of age-related Aβ increase in our cohort (data not shown). Chronic TAK-070 treatment significantly reduced the levels of insoluble Aβ40 and Aβ42 by ∼30% (Fig. 6C).
We then analyzed the effects of TAK-070 on the formation of Aβ plaques using immunohistochemistry and unbiased morphometric analysis. The numbers of Aβ plaques in the cerebral neocortex and hippocampus in TAK-070-treated cohort were markedly reduced compared to those in the vehicle-treated mice (Fig. 6D). Quantitative analysis demonstrated that the Aβ burden (i.e., percentage area covered by Aβ immunoreactivity), as well as the number of plaques per area, were reduced by ∼60% upon treatment with TAK-070 (Fig. 6E), in agreement with the biochemical data.
TAK-070 ameliorated behavioral deficits in Tg2576 mouse model of AD
We finally assessed the effects of TAK-070 on the behavioral deficits in Tg2576 mice. For this purpose, we conducted three different types of behavioral tests, i.e., Y-maze test, Morris water maze test and a novel object recognition test in relatively young (∼5 months old) Tg2576 mice, in which behavioral impairments, along with synaptic deficits, have been documented at this stage, preceding Aβ deposition (Westerman et al., 2002; Ohno et al., 2004; Jacobsen et al., 2006).
We initially conducted Y-maze test, which has been considered as a test for spatial memory. The total arm entries of vehicle-treated Tg2576 mice (n = 14) were not significantly different from those of the wild-type control mice (n = 15). Treatment with TAK-070 for 9 d did not affect the total arm entries in Tg2576 mice (data not shown), suggesting that repeated treatment with TAK-070 did not have any effects on the basal level of exploring activity. However, the spontaneous alternation in vehicle-treated Tg2576 was significantly reduced to ∼50%. This reduction was recovered by treatment with TAK-070 in a dose-dependent manner, and the ameliorating effect was significant at both dosages of 1 (n = 14) or 3 mg/kg (n = 14) (Fig. 7A).
Figure 7.

Effects of TAK-070 on impaired behavior of Tg2576 mice in Y-maze test and Morris water maze test. A, Spontaneous alternations (as a percentage) in Y-maze test. B, C, Escape latency (in seconds) (B) and swimming distance (in centimeters) (C) of mice in the invisible Morris water maze test. Male Tg2576 mice (18 weeks old) were treated with TAK-070 (1 or 3 mg/kg, p.o.) or vehicle for 9 d and then sequentially tested in Y-maze on day 10 and Morris water maze tests on days 11–13. Mean values (±SEM) in 14 animals in each Tg2576 mice group and in 15 wild-type mice (Wild) are shown. *p < 0.05, **p < 0.01, versus those in Wild (Student's t test); +p < 0.025, versus those in the vehicle-treated Tg2576 mice (Williams' test).
We then assessed the effects of TAK-070 on impairments in spatial memory by sequentially subjecting the same cohorts to the Morris water maze test. The ability of Tg2576 mice to find an invisible platform was impaired compared to that in wild-type mice. On training day 2, significant differences in both escape latency and swimming distance remained between Tg2576 and wild-type mice, whereas they diminished on day 3. Treatment with TAK-070 reduced the latency (Fig. 7B), as well as the distance (Fig. 7C), in a dose-dependent manner. On training day 2, the reduction in the swimming distance in TAK-070-treated Tg2576 mice (3 mg/kg) was statistically significant (p < 0.025, Williams' test). No significant effects were observed on the swimming speed between the vehicle- and TAK-070-treated mice (data not shown). On the next day of Morris water maze test, we obtained brains from all Tg2576 mice and measured the brain levels of Tris buffer-soluble Aβ peptides, which were decreased by ∼9–16% for Aβ40, and ∼8–12% for Aβ42, by administration of 1 and 3 mg/kg TAK-070, respectively, compared with those in vehicle-treated mice. These values were at similar levels to those observed in short-term treatment (see Fig. 6A).
We further assessed the effects of TAK-070 on recognition memory by a novel object recognition test using new cohorts. After a 15 d successive treatment with vehicle (n = 15; wild type mice, n = 14; Tg2576) or TAK-070 (3 mg/kg, p.o., n = 15; Tg2576), all mice were subjected to an acquisition trial on day 1, in which mice were allowed to get access to the two identical objects in the test box. As expected, all mice equally interacted with both objects in the exploration (data not shown). On the following day, one of the two objects was replaced with a novel one and retention test was conducted. Whereas wild-type mice more frequently interacted with a novel object than a familiar object, with the novel object preference ratio of 78% (Fig. 8A,B), vehicle-treated Tg2576 mice showed a markedly decreased preference ratio of 44% (Fig. 8B), indicating an apparent impairment in recognition memory in Tg2576. By contrast, TAK-070 treatment significantly recovered the preference ratio to a normal range of 71% (Fig. 8B).
Figure 8.

Effects of TAK-070 on impaired behavior of Tg2576 mice in a novel object recognition test. Mean (±SEM) time spent interacting with familiar or novel objects (A) and the novel object preference ratio (±SEM) (B) in the retention test conducted 24 h after the acquisition trial are shown. A, **p < 0.01, versus the familiar control object. B, **p < 0.01, versus the wild control, ##p < 0.01, versus the vehicle-treated control (Student's t test).
Discussion
We show that TAK-070 is an orally active BACE1 inhibitor that effectively lowers the levels of soluble Aβ and increases that of sAPPα, inhibits cerebral deposition of insoluble Aβ, and rescues behavioral deficits in vivo in a transgenic mouse model of AD. Notably, the partial inhibition in the levels of soluble Aβ eventually resulted in a significant reduction in Aβ deposition after a 6 month chronic treatment, preserving the pharmacological efficacy at a similar level to that in a short-term treatment. We also suggest that TAK-070 exerts a unique noncompetitive inhibitory activity by interacting presumably with the transmembrane region of BACE1 outside the catalytic domain.
Multiple lines of genetic, clinical, and cell biological evidence support the causative role of Aβ in the pathogenesis of AD (for review, see Selkoe and Schenk, 2003). In contrast, sAPPα has been reported to have neurotrophic effects, e.g., promotion of synapse formation or amelioration of cognitive deficits [for review, see Isacson et al. (2002) and Postina (2008)]. In our present study, untreated, aged Tg2576 mice had lower brain levels of sAPPα and higher soluble Aβ with aging, in agreement with previous observations that BACE1 activity is upregulated with aging in the brains of animals as well as humans (Fukumoto et al., 2004; Zohar et al., 2005). Hence, manipulation of APP processing by BACE1 inhibition in a way to reduce Aβ and increase sAPPα would be a rational strategy for the treatment and prevention of AD.
The chemical structure of TAK-070 differs markedly from that of peptide-based BACE1 inhibitors (for review, see Silvestri, 2009). However our cellular and cell-free assay data clearly indicated that TAK-070 is a bona fide BACE1 inhibitor. Cell-free study showed that TAK-070 directly and specifically inhibited full-length BACE1 without affecting other aspartic proteases. TAK-070 reduced levels of secreted Aβ and sAPPβ, together with an increase in sAPPα in cultured cells (Fig. 3), which are in agreement with the previous results of antisense oligonucleotide study for BACE1 (Vassar et al., 1999). The Lineweaver–Burk plot analysis revealed that TAK-070 is a noncompetitive inhibitor (Fig. 4), which was supported by the surface plasmon resonance assay. TAK-070 did bind to the full-length BACE1 (1-501) and truncated BACE1 (1-471 and 474), but not to the truncated BACE1 (1-454, 460, and 465) (Fig. 5). This suggests that TAK-070 inhibits BACE1 activity in a unique mode of interaction by binding to the ∼10 aa residues in the C-terminal region (residues 465–474) within the transmembrane domain, but not to the catalytic center (located in residues 93–96 and 289–293). Surface plasmon resonance assay also showed that TAK-070 does not interact with APP(18-688) or sAPPβ (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). This suggests that TAK-070 does not affect APP processing by binding to subdomain of APP containing the BACE1-cleavage sites. We were not able to completely rule out the possibility that TAK-070 interacts with the transmembrane domain of APP, like benzofuran-containing compounds that bind C99 (Espeseth et al., 2005). However, TAK-070 failed to inhibit Aβ secretion from HEK293 cells overexpressing C99 (data not shown), supporting the notion that TAK-070 does not target C99 in APP. In addition, the possibility that TAK-070 is an α-secretase activator was excluded by (1) the lack of increase in the protein levels of α-secretase candidate, i.e., ADAM10 (Fig. 3), (2) lack of inhibition of TACE activity using a peptidic substrate in a cell-free assay (data not shown), and (3) the lack of increase in α-secretase activity in a cell-based assay (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
The potency of TAK-070 to reduce the Aβ secretion in cell cultures was modest (i.e., ∼25% reduction was achieved at 3 μmol/L with a MEC of ∼0.1–0.3 μmol/L in N2aAPPsw cells) (Fig. 2). These results were in agreement with the relatively modest BACE1-inhibitory effect in the cell-free assay with IC35 of ∼3.15 μmol/L and MEC at ∼0.1 μmol/L (Fig. 4). Interestingly, however, we observed similar levels of reduction in soluble Aβ by ∼20% in the brains of Tg2576 mice (Fig. 6A,B). Although small chemicals generally have less potency in brains, hampered by the blood–brain-barrier and cell-penetration issues, this relatively high potency of TAK-070 is likely to be attributable to the highly lipophilic structure bearing N-alkyl-amine moiety. In fact, a single administration of TAK-070 in rat (3 mg/kg, p.o.) yielded effective concentration of ∼2 μmol/L in brain with the Tmax of ∼24 h using 14C-TAK-070, and the brain exposure levels in short-term- and long-term-treated Tg2576 mice were ∼8 μmol/L and ∼6–11 μmol/L, respectively (56 ppm of TAK-070, corresponding to ∼7–8 mg/kg) (Fig. 6) (our unpublished observations). Furthermore, it has been reported that full-length BACE1, forming a high-molecular-weight complex associated with lipid, exhibits higher enzymatic activity than that of C-terminally truncated BACE1 (1-454) (Marlow et al., 2003; Westmeyer et al., 2004). This may support the view that lipophilic TAK-070 effectively reaches the membrane-associated BACE1 complex.
TAK-070 exhibits ceiling effects on reduction in Aβ and increase in sAPPα (Figs. 2, 6A), which may partly be explained by the noncompetitive inhibitory profile for BACE1. We have also observed similar plateau effects in normal rats with a minimum effective dose of 0.1 mg/kg after 4 week administration (our unpublished observation). However, long-term treatment with TAK-070 led to more pronounced Aβ-lowering effects on insoluble Aβ (Fig. 6C–E) than on soluble Aβ (Fig. 6A,B). This finding dovetails with the observation in BACE1 heterozygous KO crossed with PDAPP transgenic mice, in which soluble Aβ levels were lowered only by 12% at a young age, whereas Aβ-accumulation was eventually reduced by ∼50–90% with synaptic amelioration in elderly animals (McConlogue et al., 2007). Together, these results strongly suggest that partial inhibition of BACE1, causing partial reduction in Aβ and increase in sAPPα, has sufficient pharmacological efficacy on normalization of APP processing and cognitive functions.
Behavioral deficits in Tg2576 mice have been reported to occur before the deposition of Aβ plaques (Westerman et al., 2002; Ohno et al., 2004; Jacobsen et al., 2006), which may be due to the accumulation of toxic forms of Aβ, e.g., oligomers, that leads to the deterioration of synaptic functions and behaviors (Walsh et al., 2002; Cleary et al., 2005; Venkitaramani et al., 2007). In the present study, relatively young (∼5 months) Tg2576 mice showed impairment in behaviors both in Y-maze and novel object recognition tests, whereas the deficits in Morris water maze test were modest, with no differences in the acquisition trial on day 3 between Tg2576 and wild-type cohorts. TAK-070 ameliorated all these behavioral deficits by a short-term treatment at biochemically effective doses (1–3 mg/kg, p.o.) (Figs. 7, 8). TAK-070 had ameliorative effects in the Y-maze and Morris water maze tests that reflect the hippocampal-dependent learning, in line with observations in BACE1 homozygous KO/APP transgenic bigenic mice (Ohno et al., 2004, 2006, 2007). However, there were pivotal differences: TAK-070 treatment affected neither the total number of arm entry in Y-maze test (Ohno et al., 2004) nor the swimming speed in Morris water maze test (Ohno et al., 2006), which were documented to be abnormal in BACE1-homozygous KO regardless of APP-transgenic background. Furthermore, BACE1-homozygous KO in nontransgenic background have been reported to show cognitively deteriorative (Ohno et al., 2004, 2006, 2007), schizophrenia-like (Savonenko et al., 2008), or hypomyelination (Hu et al., 2006; Sankaranarayanan et al., 2008) phenotypes, underscoring the necessity of BACE1 activity for physiological functions, probably due to multiplicity of substrates for BACE1 (for review, see Marks and Berg, 2008). Also in nontransgenic aged rats, TAK-070 ameliorated behavioral deficits in the water maze test (our unpublished observation). Hence, TAK-070 appears to be pharmacologically effective and safe by partial BACE1 inhibition, avoiding adverse events due to complete inhibition of BACE1.
It is noteworthy that the pharmacological effects of orally administered TAK-070 for ∼6 months on the brain levels of soluble Aβ and sAPPα were similar to those in short-term treatment (Fig. 6A,B). Under the chronic treatment, mice were tolerable to TAK-070 and survived comparable to vehicle control after ∼6 months. These profiles should be a merit of this compound, considering the long period of AD medication. The sustained efficacy of TAK-070 markedly differs from those documented in other BACE1 inhibitors (Sankaranarayanan et al., 2008) or on the higher efficacy of a compound in the presence of inhibitors of P-glycoprotein (Hussain et al., 2007), that determines exposure levels of compounds in brains.
In sum, the successful treatment by a noncompetitive BACE1 inhibitor, TAK-070, provides strong support for the validity of partial BACE1 inhibition as a disease-modifying as well as symptomatic therapy for AD. TAK-070 will also provide a clue for the elucidation of the mechanism of noncompetitive regulation of the activity of BACE1.
Footnotes
We thank the Mayo Clinic for supplying the Tg2576 mice, Kozo Shimakawa for breeding the Tg2576 mice, Satoko Osawa for technical assistance, Dr. Gopal Thinakaran for valuable advice, and Drs. Zen-ichi Terashita, Yasuhiro Sumino, and Shigenori Ohkawa for continuous support to the study.
All authors except for T. Tomita and T. Iwatsubo are employees of Takeda Pharmaceutical Company, which was engaged in the research of BACE1 inhibitors for potential use as AD therapeutics.
References
- Asami-Odaka A, Ishibashi Y, Kikuchi T, Kitada C, Suzuki N. Long amyloid β-protein secreted from wild-type human neuroblastoma IMR-32 cells. Biochemistry. 1995;34:10272–10278. doi: 10.1021/bi00032a022. [DOI] [PubMed] [Google Scholar]
- Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP. Loss of presenilin function causes Alzheimer's disease-like neurodegeneration in the mouse. J Neurosci Res. 2008;86:1615–1625. doi: 10.1002/jnr.21601. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cole SL, Vassar R. BACE1 structure and function in health and Alzheimer's disease. Curr Alzheimer Res. 2008;5:100–120. doi: 10.2174/156720508783954758. [DOI] [PubMed] [Google Scholar]
- Doedens JR, Mahimkar RM, Black RA. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun. 2003;308:331–338. doi: 10.1016/s0006-291x(03)01381-0. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeseth AS, Xu M, Huang Q, Coburn CA, Jones KLG, Ferrer M, Zuck PD, Strulovici B, Price EA, Wu G, Wolfe AL, Lineberger JE, Sardana M, Tugusheva K, Pietrak BL, Crouthamel M-C, Lai M-T, Dodson EC, Bazzo R, Shi X-P, et al. Compounds that bind APP and inhibit Aβ processing in vitro suggest a novel approach to Alzheimer disease therapeutics. J Biol Chem. 2005;280:17792–17797. doi: 10.1074/jbc.M414331200. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Tomita T, Matsunaga H, Ishibashi Y, Saido TC, Iwatsubo T. Primary cultures of neuronal and non-neuronal rat brain cells secrete similar proportions of amyloid β peptides ending at Aβ40 and Aβ42. Neuroreport. 1999;10:2965–2969. doi: 10.1097/00001756-199909290-00017. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. β-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol. 2004;164:719–725. doi: 10.1016/s0002-9440(10)63159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hussain I, Hawkins J, Harrison D, Hille C, Wayne G, Cutler L, Buck T, Walter D, Demont E, Howes C, Naylor A, Jeffrey P, Gonzalez MI, Dingwall C, Michel A, Redshaw S, Davis JB. Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases β-cleavage of amyloid precursor protein and amyloid-β production in vivo. J Neurochem. 2007;100:802–809. doi: 10.1111/j.1471-4159.2006.04260.x. [DOI] [PubMed] [Google Scholar]
- Isacson O, Seo H, Lin L, Albeck D, Granholm AC. Alzheimer's disease and Down's syndrome: roles of APP, trophic factors and ACh. Trends Neurosci. 2002;25:79–84. doi: 10.1016/s0166-2236(02)02037-4. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30:4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmana MK, Yoon IS, Chen E, Bianchi E, Koo EH, Kang DE. Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid β peptide generation. J Biol Chem. 2009;284:11863–11872. doi: 10.1074/jbc.M807345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks N, Berg MJ. Neurosecretases provide strategies to treat sporadic and familial Alzheimer disorders. Neurochem Int. 2008;52:184–215. doi: 10.1016/j.neuint.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Marlow L, Cain M, Pappolla MA, Sambamurti K. β-Secretase processing of the Alzheimer's amyloid protein precursor (APP) J Mol Neurosci. 2003;20:233–239. doi: 10.1385/JMN:20:3:233. [DOI] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- Obregon DF, Rezai-Zadeh K, Bai Y, Sun N, Hou H, Ehrhart J, Zeng J, Mori T, Arendash GW, Shytle D, Town T, Tan J. ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced (alpha)-secretase cleavage of amyloid precursor protein. J Biol Chem. 2006;281:16419–16427. doi: 10.1074/jbc.M600617200. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postina R. A closer look at α-secretase. Curr Alzheimer Res. 2008;5:179–186. doi: 10.2174/156720508783954668. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Price EA, Wu G, Crouthamel MC, Shi XP, Tugusheva K, Tyler KX, Kahana J, Ellis J, Jin L, Steele T, Stachel S, Coburn C, Simon AJ. In vivo beta-secretase 1 inhibition leads to brain Aβ lowering and increased α-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324:957–969. doi: 10.1124/jpet.107.130039. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Holahan MA, Colussi D, Crouthamel MC, Devanarayan V, Ellis J, Espeseth A, Gates AT, Graham SL, Gregro AR, Hazuda D, Hochman JH, Holloway K, Jin L, Kahana J, Lai MT, Lineberger J, McGaughey G, Moore KP, Nantermet P, et al. First demonstration of cerebrospinal fluid and plasma Aβ lowering with oral administration of a β-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates. J Pharmacol Exp Ther. 2009;328:131–140. doi: 10.1124/jpet.108.143628. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Silvestri R. Boom in the development of non-peptidic β-secretase (BACE1) inhibitors for the treatment of Alzheimer's disease. Med Res Rev. 2009;29:295–338. doi: 10.1002/med.20132. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Stockley JH, O'Neill C. The proteins BACE1 and BACE2 and β-secretase activity in normal and Alzheimer's disease brain. Biochem Soc Trans. 2007;35:574–576. doi: 10.1042/BST0350574. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T. Sulindac sulfide is a noncompetitive γ-secretase inhibitor that preferentially reduces Aβ42 generation. J Biol Chem. 2003;278:18664–18670. doi: 10.1074/jbc.M301619200. [DOI] [PubMed] [Google Scholar]
- Tomita T, Katayama R, Takikawa R, Iwatsubo T. Complex N-glycosylated form of nicastrin is stabilized and selectively bound to presenilin fragments. FEBS Lett. 2002;520:117–121. doi: 10.1016/s0014-5793(02)02802-8. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. β-Amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmeyer GG, Willem M, Lichtenthaler SF, Lurman G, Multhaup G, Assfalg-Machleidt I, Reiss K, Saftig P, Haass C. Dimerization of β-site β-amyloid precursor protein-cleaving enzyme. J Biol Chem. 2004;279:53205–53212. doi: 10.1074/jbc.M410378200. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Zohar O, Pick CG, Cavallaro S, Chapman J, Katzav A, Milman A, Alkon DL. Age-dependent differential expression of BACE splice variants in brain regions of tg2576 mice. Neurobiol Aging. 2005;26:1167–1175. doi: 10.1016/j.neurobiolaging.2004.10.005. [DOI] [PubMed] [Google Scholar]