

Abstract
Attention deficit/hyperactivity disorder (ADHD) is characterized by inattention, impulsivity, and motor hyperactivity. Several lines of research support a crucial role for the dopamine transporter (DAT) gene in this psychiatric disease. Consistently, the most commonly prescribed medications in ADHD treatment are stimulant drugs, known to preferentially act on DAT. Recently, a knock-in mouse [DAT-cocaine insensitive (DAT-CI)] has been generated carrying a cocaine-insensitive DAT that is functional but with reduced dopamine uptake function. DAT-CI mutants display enhanced striatal extracellular dopamine levels and basal motor hyperactivity. Herein, we showed that DAT-CI animals present higher striatal dopamine turnover, altered basal phosphorylation state of dopamine and cAMP-regulated phosphoprotein 32 kDa (DARPP32) at Thr75 residue, but preserved D2 receptor (D2R) function. However, although we demonstrated that striatal D1 receptor (D1R) is physiologically responsive under basal conditions, its stimulus-induced activation strikingly resulted in paradoxical electrophysiological, behavioral, and biochemical responses. Indeed, in DAT-CI animals, (1) striatal LTP was completely disrupted, (2) R-(+)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide (SKF 81297) treatment induced paradoxical motor calming effects, and (3) SKF 81297 administration failed to increase cAMP/protein kinase A (PKA)/DARPP32 signaling. Such biochemical alteration selectively affected dopamine D1Rs since haloperidol, by blocking the tonic inhibition of D2R, unmasked a normal activation of striatal adenosine A2A receptor-mediated cAMP/PKA/DARPP32 cascade in mutants. Most importantly, our studies highlighted that amphetamine, nomifensine, and bupropion, through increased striatal dopaminergic transmission, are able to revert motor hyperactivity of DAT-CI animals. Overall, our results suggest that the paradoxical motor calming effect induced by these drugs in DAT-CI mutants depends on selective aberrant phasic activation of D1R/cAMP/PKA/DARPP32 signaling in response to increased striatal extracellular dopamine levels.
Introduction
Attention deficit/hyperactivity disorder (ADHD) is the most commonly diagnosed psychiatric illness in childhood, affecting 5–10% of school-aged children that often maintain symptoms into adolescence and adulthood (Biederman, 2005). ADHD is characterized by the coexistence of attention problems, impulsivity, and motor hyperactivity, or by the prevalence of one of these domains over the others (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision, 2000). So far, the treatment for calming hyperactivity and reducing attentional-cognitive deficits of ADHD patients mainly involves the administration of psychostimulants (Greenhill, 1992). The paradoxical calming response to stimulant medications in ADHD subjects has always been considered enigmatic, since these compounds are able to strongly evoke motor stimulation in healthy subjects. Despite the still mostly unclear molecular bases of ADHD, convergent results from neuroimaging, neuropsychological, genetic, and neurochemical studies suggest that dysfunctions in connected catecholamine-enriched frontostriatal regions, such as prefrontal cortex and caudate–putamen, are primarily involved in this mental disorder (Sowell et al., 2003; Bush et al., 2005). Alteration of catecholaminergic neurotransmission has also been pointed out by genetic studies that supported the association between ADHD and polymorphisms in genes encoding dopamine (DA) D4 and D5 receptors, DA β-hydroxylase, and noradrenaline (NA) and DA transporters (NET and DAT, respectively) (Faraone, 2004; Yang et al., 2004; Thapar et al., 2005). Among these genes, DAT1 is probably one of the most intriguing candidates since it is the principal target for amphetamine and methylphenidate, the most widely used anti-ADHD medications (Swanson et al., 2007). To further support the involvement of an altered DAT function in this psychiatric syndrome, clinical studies indicate that DAT density is often abnormal in ADHD brains (Ernst et al., 1999; Volkow et al., 2007).
In the attempt to investigate possible gene defects or environmental conditions accounting for this psychiatric illness, different animal models with ADHD-like phenotypes have been proposed (Viggiano et al., 2003; Russell, 2007). Certainly, one of the most valuable ADHD models is represented by mice with a null mutation in DAT gene (DAT-KO) (Giros et al., 1996; Gainetdinov et al., 1999a). Although DAT-KO mice show several features resembling those found in ADHD subjects, including cognitive deficits (Gainetdinov et al., 1999a; Morice et al., 2007), the genetic DAT deletion underlying the generation of this mutant animal does not match in patients, where only DAT polymorphisms have been described (Thapar et al., 2005; Gizer et al., 2009). For this reason, ADHD animal models with subtle genetic modifications in DAT gene would be more suitable to better mimic clinical observations, thus providing additional information on the neurobiological substrates strictly required to develop the ADHD-like syndrome.
Through a multidisciplinary approach, herein we further examined the possibility to consider DAT-cocaine-insensitive (DAT-CI) mice, previously shown to be hyperactive and calmed by methylphenidate (Chen et al., 2006; Tilley and Gu, 2008a,b), as a putative animal model of ADHD motor symptoms. Remarkably, we found that the paradoxical motor calming effect induced by amphetamine, nomifensine, and bupropion in DAT-CI mutants is mainly associated with enhanced striatal dopaminergic neurotransmission and, notably, caused by a selective aberrant phasic D1 receptor-dependent cAMP/protein kinase A (PKA)/dopamine and cAMP-regulated phosphoprotein 32 kDa (DARPP32) signaling.
Materials and Methods
Animals
Adult DAT-CI male mice were generated by homologous recombination in 129/SvJ embryonic stem cells as previously described (Chen et al., 2006). DAT-CI mice bear a triple point mutation (L104V/F105C/A109V) within DAT protein that is ∼90-fold more insensitive to cocaine inhibition than wild type (WT) (Chen et al., 2006). The mutant mice have been backcrossed to C57BL/6J mice for 10 or more generations; thus, they are generally considered to be in the C57BL/6J background (Tilley and Gu, 2008b). Animals were housed in a maximum of five per cage, at a constant temperature (22 ± 1°C) and maintained on a 12 h light/dark cycle, with food and water ad libitum. Experiments were performed during the light phase, in accordance to protocols approved by the veterinary department of the Italian Ministry of Health and in line with the ethical and safety rules and guidelines for the use of animals in biomedical research provided by the relevant Italian laws and European Union directives (number 86/609/EC). All efforts were made to minimize the animals' suffering.
Drugs
Cocaine hydrochloride, amphetamine sulfate, nomifensine maleate, nisoxetine hydrochloride, fluoxetine hydrochloride, haloperidol, R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride [R(+)-SCH 23390 hydrochloride], R-(+)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide [R(+)-SKF 81297 hydrobromide], and 2-p-(2-carboxyethyl) phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride hydrate (CGS 21680 hydrochloride hydrate) were purchased from Sigma-Aldrich. (−)-Quinpirole hydrochloride and 2-(2-furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine (SCH 58261) were purchased from Tocris Bioscience. Wellbutrin (GlaxoSmithKline) was used to prepare bupropion solution. Haloperidol was dissolved in a solution of 10% acetic acid in saline and the pH was brought to 6.0 with 1 m NaOH. SCH 58261 was dissolved in DMSO and subsequently diluted 1:10 with saline (0.9% NaCl). All other compounds were dissolved in saline. All drugs were administered intraperitoneally in a volume of 10 ml/kg, except fluoxetine, injected subcutaneously at the same volume.
Behavior
Novelty-induced exploration.
Spontaneous motor activity was measured by novelty-induced exploratory task (Usiello et al., 2000), in WT and DAT-CI naive mice (n = 16 per genotype). Animals were individually placed into the experimental cage (35 × 25 × 30 cm) and videotaped for 120 min, under dim lighting (∼100 lux on the cage floor). Subsequently, movies were visually analyzed by an experimenter blinded to genotype. The total number of sector crossings (the floor of the experimental cage was subdivided on the monitor in six identical squares), as index of horizontal motor activity, and total counts of seated, wall and free rearing, as index of vertical activity, were scored every 10 min for a total of 12 intervals. Novelty-induced horizontal and vertical motor activity was analyzed by two-way ANOVA (time × genotype) with repeated measures. Statistical analyses were performed with StatView software (version 5.0.1.0; SAS Institute).
Object recognition test.
To evaluate attention, memory, and discriminative abilities, WT (n = 9) and DAT-CI (n = 10) naive mice were submitted to the object recognition (Bevins and Besheer, 2006). Mice were gently handled 5 min per day for a week before the start of the experiment. To provide mice the familiarity to the testing environment, a 3 d habituation phase was conducted by exposing each animal to the experimental cage (40 × 40 cm Plexiglas chamber with white walls and black floor) for at least 20 min per day. On the training session (day 4), the box was enriched with two identical green, squared Lego objects (3 × 6 × 6 cm), positioned in the back left and right corners of the apparatus, 6 cm far from the walls. Each animal was placed in the middle point of the wall opposite to the sample objects and left to freely explore and familiarize with the objects for 15 min. Twenty-four hours later (testing session), one of the two objects (familiar) was substituted by a new one (novel), different in color and shape (a gray, metal pyramid, 5 × 5 × 6 cm). Similarly to the training procedure, each mouse was placed in the apparatus and left free to explore it for 5 min. Both training and testing phases were video recorded and the time spent exploring each of the two objects was visually measured by a blinded experimenter. Objects exploration was scored when the mouse nose was in contact with the object or directed at the object within a distance ≤2 cm. The following two measures were considered: (1) the total exploration time (in seconds) that animals spent interacting with the two familiar objects during the training phase; (2) the exploration time (in percentage) that animals spent interacting with the novel object over the total exploration time (e.g., [novel/(familiar + novel)] × 100) during the testing phase. Mice that had <8 s of exploration during the training phase were excluded from the analysis. Statistical analysis of total exploration time during training phase was performed by Student's t test. Total exploration time during testing phase was analyzed by two-way ANOVA (object × genotype) with repeated measures, followed by one-way ANOVA (genotype). Statistical analyses were performed with StatView software (version 5.0.1.0; SAS Institute).
Accelerating rotating rod test.
To evaluate balance, motor coordination, and motor learning, DAT-CI (n = 14) and WT (n = 15) naive mice were tested on the accelerating rotarod (Ugo Basile; Biological Research Apparatus), as previously described (Errico et al., 2008). The test was performed by placing mice on a rotating drum (3 cm of diameter) and measuring the time that each mouse was able to achieve walking on the top of the rod. The time at which each animal fell from the drum was recorded automatically when it contacted the plate at the base of the rod, which stopped the session trial. The speed of the rotarod accelerated from 4 to 40 rpm over a 5 min period. Mice were given four consecutive trials with a maximum time of 300 s; a minimum of 30 min intertrial rest interval was used to avoid fatigue and exhaustion. The fall latency (expressed in seconds) obtained from each of four trials was used as dependent variable and analyzed by two-way ANOVA (trial × genotype) with repeated measures. Statistical analyses were performed with StatView software (version 5.0.1.0; SAS Institute).
Motor responses induced by drugs.
To measure motor effects induced by stimulant drugs, a set of mice was randomly assigned to treatment group as follows: cocaine (40 mg/kg: WT, n = 8; DAT-CI, n = 7), amphetamine (5 mg/kg: n = 8 per genotype), and nomifensine (15 mg/kg: n = 8 per genotype). Mice treated with cocaine, amphetamine, or nomifensine shared the same vehicle-treated group (n = 16 per genotype). All other treatments were compared with a specific vehicle-treated group. Different groups of animals were randomly injected with bupropion (30 mg/kg: n = 8 per genotype; vehicle: WT, n = 4; DAT-CI, n = 8), fluoxetine (20 mg/kg: n = 7 per genotype; vehicle: n = 4 per genotype), nisoxetine (10 mg/kg: WT, n = 7; DAT-CI, n = 11; vehicle: WT, n = 7; DAT-CI, n = 8), SKF 81297 (2.5 mg/kg: WT, n = 6; DAT-CI, n = 7; 5 mg/kg: WT, n = 6; DAT-CI, n = 9; vehicle: n = 8 per genotype), and SCH 58261 (5 mg/kg: WT, n = 7; DAT-CI, n = 8; vehicle: WT, n = 7; DAT-CI, n = 8). Mice were habituated to the experimental cage (35 × 25 × 30 cm) for 60 min, and then injected with one of the testing drugs and replaced in the same cage, to videotape their activity for 60 min. Conversely, to evaluate the motor response associated with the pharmacological manipulation of dopamine D1, D2, and adenosine A2A receptors (D1R, D2R, and A2AR, respectively), WT and DAT-CI mice were randomly assigned to treatment group as follows: quinpirole (0.5 mg/kg: n = 5 per genotype; vehicle: n = 8 per genotype), haloperidol (0.1 mg/kg: n = 5 per genotype), SCH 23390 (0.05 mg/kg: n = 8 per genotype), and CGS 21680 (0.25 mg/kg: WT, n = 8; DAT-CI, n = 7; vehicle: n = 7 per genotype). Mice treated with haloperidol or SCH 23390 shared the same vehicle-treated group (n = 8 per genotype). In this set of experiments, mice were analyzed in novel test cage. Thus, animals were drug injected with one of the testing compounds and immediately exposed to the experimental cage (35 × 25 × 30 cm) to videotape their activity for 30 min (quinpirole and CGS 21680) or 60 min (haloperidol and SCH 23390). Behavioral procedures were performed according to the study by Usiello et al. (2000). As for novelty-induced exploration, also motor responses induced by drugs were tested under dim lighting (∼100 lux on the cage floor). At the end of the behavioral tests, all movies were visually analyzed by an experimenter blinded to treatment and genotype. The number of sector crossings (the floor of the experimental cage was subdivided on the monitor in six identical squares), as index of horizontal motor activity, and the total counts of seated, wall and free rearing, as index of vertical activity, were scored every 10 min, over the total test time. Number of sector crossing was analyzed within genotypes by two-way ANOVA (time × treatment) with repeated measures, followed by Fisher's post hoc analysis when required. Total sector crossing was analyzed by two-way ANOVA (genotype × treatment), followed by one-way ANOVA (treatment). Statistical analyses were performed with StatView software (version 5.0.1.0; SAS Institute).
HPLC analysis
Animals were killed by decapitation, and striata were rapidly dissected out, frozen on dry ice, and stored at −80°C until analysis with HPLC. At such time, tissue samples were homogenized in 0.1 m perchloric acid and centrifuged at 10,000 rpm for 10 min before filtering through minispin filters for additional 3 min at 10,000 rpm. Samples were analyzed as previously described (Carta et al., 2006) with minor modifications. Briefly, 25 μl of each sample were injected by a cooled autosampler (Midas) into an ESA Coulochem III coupled with an electrochemical detector for detection of DA, dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), NA, and serotonin (5HT). The mobile phase (5 g/L sodium acetate, 30 mg/L Na2-EDTA, 100 mg/L octane-sulfonic acid, 10% methanol, pH 4.2) was delivered at a flow rate of 500 μl/min to a reverse phase C18 column (4.6 mm inner diameter, 150 mm length; Chrompack). Peak identification and quantification were conducted using the Clarity chromatographic software (DataApex). Data were analyzed by Student's t test.
Western blotting
WT and DAT-CI mice were injected with 20 mg/kg fluoxetine, 0.1 or 0.5 mg/kg haloperidol, 5 mg/kg SKF 81297, 10 mg/kg amphetamine or vehicle, and killed by decapitation 30 min later. The heads were immediately immersed in liquid nitrogen for 5–6 s, the brains were removed, and the striata dissected out within 20 s on an ice-cold surface, sonicated in 1% SDS and boiled for 10 min, as previously described (Errico et al., 2009). This extraction procedure prevents protein phosphorylation and dephosphorylation, hence ensuring that the level of phosphoproteins measured ex vivo reflects the in vivo situation (Svenningsson et al., 2000; Santini et al., 2009). Aliquots (2 μl) of the homogenate were used for the protein determination by Bio-Rad Protein Assay kit (Bio-Rad). Equal amounts of total proteins for each sample were loaded onto 10% polyacrylamide gels. Proteins were separated by SDS-PAGE and transferred overnight to membranes [PVDF (polyvinylidene difluoride)] (GE Healthcare). Membranes were immunoblotted overnight using selective antibodies against P-Thr202/Tyr204-ERK42/44 (1:1000; Cell Signaling Technology), P-Ser40-TH (1:1000; Millipore), P-Thr34-DARPP32 (1:1000; PhosphoSolution), P-Thr75-DARPP32 (1:1000; PhosphoSolution), or P-Ser845-GluR1 (1:1000; PhosphoSolution). Blots were then incubated in horseradish peroxidase-conjugated secondary antibodies and target proteins visualized by ECL detection (Pierce), followed by quantification by Quantity One software (Bio-Rad). Antibodies against ERK42/44 (1:1000; Cell Signaling Technology), TH (1:1000; Millipore), DARPP32 (1:1000; Cell Signaling Technology), and GluR1 (1:1000; Millipore Biotechnology) that are not phosphorylation state-specific were used to estimate the total amount of proteins. All optical density values were normalized to DARPP32 for variation in loading and transfer. Normalized values were then averaged and used as dependent variable. The same procedure of killing, dissection, and Western blotting was used to detect basal levels of P-Thr202/Tyr204-ERK42/44, Golf (1:1000; kindly provided by Dr. Hervé, Université Pierre et Marie Curie, Paris, France), P-Thr34-DARPP32, P-Thr75-DARPP32, and P-Ser845-GluR1 in naive WT and DAT-CI mice. Data from basal protein levels, fluoxetine-induced P-Thr202/Tyr204-ERK42/44, SKF 81297- and amphetamine-induced P-Thr34-DARPP32 and P-Thr75-DARPP32 were analyzed by Student's t test; all other results were analyzed by two-way ANOVA (genotype × treatment), followed by one-way ANOVA (treatment). Statistical analyses were performed with StatView software (version 5.0.1.0; SAS Institute).
Voltammetry
WT and DAT-CI mice were anesthetized and killed by decapitation. Brains were rapidly removed and coronal corticostriatal slices (300–400 μm thick) were prepared using a vibratome (Leica VT1000S; Leica Microsystems). Individual slices were transferred to a recording chamber and submerged in a continuously flowing Krebs' solution (35°C, 2–3 ml/min) gassed with 95% O2/5% CO2, containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, 25 NaHCO3. The tips of the carbon fiber electrodes (WPI Carbon Fiber CF 30–100) were gently positioned to a depth of 50–150 μm in the striatal slices of mice. Constant potential amperometry (CPA) was obtained with a WPI Micro C holding the electrodes at an oxidation potential of 0.55–0.60 V versus reference electrode Ag/AgCl. To limit a possible underestimation of DA concentrations caused by an ascorbic acid-dependent signal (Kawagoe and Wightman, 1994; Schmitz et al., 2001; Venton et al., 2002), calibration was obtained within the slices by superfusing DA (1–30 μm), therefore leaving unaffected the endogenous ascorbic acid concentration. Under these conditions, it is commonly assumed that CPA principally measures striatal extracellular content of DA rather that of NA. Indeed, electrodes are more sensitive to DA than to NA, and the striatal DA innervation and DA tissue content are much greater than NA ones (DA:NA content is 100:1 in the caudate of mammals) (Kuhr et al., 1986; Garris et al., 1993). Striatal slices of WT and DAT-CI mice (n = 6 per genotype and treatment) were superfused with 100 μm amphetamine, 30 μm nomifensine, or 100 μm bupropion (diluted in the artificial CSF). A single electrical shock (0.01–0.006 ms, 10–50 V, controlled by a stimulus isolation unit) (Grass S88 Stimulator; Grass Instruments) was delivered every 3 min. Such stimulation protocol caused a rapid rise in signal that generally decayed back to baseline in 2–4 s (data not shown). Variation in the amplitude of the baseline (unstimulated) DA signal was detected. The stable DA signal was stored and digitized with a Digidata 1322A (Molecular Devices). Data were analyzed by Student's t test.
Electrophysiology
Hippocampal LTP.
WT and DAT-CI mice (n = 6 per genotype) were anesthetized and killed by cervical dislocation. Brains were rapidly removed and parasagittal hippocampal slices (400 μm thick) were cut by using a vibratome. Slices were kept submerged at 29–30°C and superfused (2–3 ml/min) with oxygenated (95% O2, 5% CO2) artificial CSF containing the following (in mm): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 10 glucose. A bipolar tungsten stimulating electrode was placed in the CA1 stratum radiatum to stimulate the Schaffer collateral fibers, and extracellular field EPSPs (fEPSPs) were recorded with a glass microelectrode (2–3 MΩ, filled with 2 m NaCl) positioned in the stratum radiatum. Long-term potentiation (LTP) was electrically induced by repetitive high-frequency stimulation (HFS) protocol (one train, 100 Hz), and the effect of conditioning train was expressed as the mean ± SEM percentage of baseline EPSP slopes measured 60 min after stimulation protocol. A two-tailed unpaired t test was used for statistical comparisons of mean fEPSP slopes.
Striatal LTP.
Slices were prepared and maintained as described previously (Calabresi et al., 1998; Martella et al., 2009). WT (n = 16) and DAT-CI (n = 12) mice were anesthetized and killed by cervical dislocation. Brains were rapidly removed, and coronal or parasagittal corticostriatal slices were cut by using a vibratome in an ice-cold (0°C) Krebs' solution whose composition was the following (in mm): 126 NaCl, 2.5 KCl, 1.3 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 18 NaHCO3. Current-clamp recordings were performed using sharp microelectrodes filled with 2 m KCl (40–60 MΩ). Signal acquisition and off-line analysis were performed using an Axoclamp 2B amplifier and pClamp9.2 software (Molecular Devices). To optimize LTP induction, magnesium was omitted from the external medium (Martella et al., 2009). EPSPs were suppressed by a combination of AMPA and NMDA receptor antagonists, CNQX (10 μm) and MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate] (50 μm), and no significant differences were found in the two groups of mice (data not shown) (p > 0.05). HFS was performed by three trains, 3 s duration, 100 Hz frequency, at 20 s intervals; stimulus intensity was raised to suprathreshold levels. The amplitude of EPSPs was averaged and plotted as percentage of the control amplitude for 15 min before protocol induction. Student's t test was used to compare values before and after HFS protocol. Drugs were from Tocris Bioscience and were applied by switching the control perfusion to drug-containing solution.
Sharp-electrode recordings of DA neurons.
WT and DAT-CI mice (n = 9 per genotype) were used. An individual slice of the ventral midbrain (300–400 μm thick) was transferred to a recording chamber and submerged in a continuously flowing Krebs' solution (35°C, 2–3 ml/min) (in mm: 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, 25 NaHCO3) gassed with 95% O2/5% CO2. Intracellular recordings were performed on dopaminergic neurons from substantia nigra pars compacta using standard procedures previously described (Mercuri et al., 1995). The recording electrodes, filled with a 2 m KCl solution, had a tip resistance of 40–80 MΩ. Membrane voltage and current signals were recorded using an Axoclamp-900A amplifier (Molecular Devices). The electrophysiological and pharmacological characteristics of these cells have been described previously (Lacey et al., 1987; Grace and Onn, 1989; Mercuri et al., 1995).
Results
Spontaneous motor activity and discriminative abilities of DAT-CI mutants
It has been previously shown that saline-injected DAT-CI mice, when tested in an open-field arena over a 15 min test session and recorded through a video tracking system, display motor hyperactivity (Chen et al., 2006). To further investigate the spontaneous motor repertoire of DAT-CI mice, here we analyzed over a 2 h test session naive mutant and control animals through a nonautomated behavioral analysis, performed in a nonstressful environment such as a novel home cage (Fig. 1a,b). Overall, two-way ANOVA with repeated measures indicated that DAT-CI mice displayed significantly higher horizontal activity, compared with WT (genotype effect, F(1,330) = 34.506, p < 0.0001) (Fig. 1a). Moreover, statistical analysis of the vertical activity counts, including seated, wall and free rearing, showed similar responses, with DAT-CI mice displaying a robust hyperactive vertical motor phenotype compared with controls (genotype effect, F(1,330) = 22.723, p < 0.0001) (Fig. 1b). Interestingly, we also found that the motor hyperactive phenotype of mutants was accompanied by normal coordination and motor learning, as assessed by accelerating rotating rod task (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Spontaneous motor activity and discriminative abilities of DAT-CI mutants. a, b, Horizontal (number of sector crossings) (a) and vertical (counts of seated, wall and free rearing) (b) activity in a novel cage performed by WT and DAT-CI naive mice (n = 16 per genotype). Data are presented as time course with 10 min interval over a 120 min test. c, d, Memory and discriminative abilities of DAT-CI were evaluated in an object recognition test. c, Time of exploration (expressed in seconds) spent by WT (n = 9) and DAT-CI (n = 10) mice contacting the objects to be familiarized, over a 15 min training phase. d, Exploratory preference for the novel object (percentage of total exploration time) spent by WT and DAT-CI mice, over a 5 min test. The dashed line indicates the chance level (50%) of object exploration. All values are expressed as mean ± SEM. **p < 0.01 versus WT mice (one-way ANOVA). Genotypes are as indicated.
Then, we analyzed sensorial-discriminative and memory abilities of DAT-CI animals in an object recognition paradigm (Dere et al., 2007). Overall, mutants displayed a slight but not significant reduction in the object exploration time, compared with control group (Student's t test, p = 0.1530) (Fig. 1c). Moreover, on the retention test performed 24 h after the first exposure to the familiar object, both WT and mutant mice showed a significant preference for the novel object (ANOVA: objects effect, F(1,17) = 61.669, p < 0.0001) (Fig. 1d). However, we also found a significant genotype × object interaction, indicating different responsiveness between genotypes in novel object-induced exploration (F(1,17) = 9.565, p = 0.0066). Indeed, one-way ANOVA evidenced that DAT-CI mice spent less time than WT animals in approaching the unfamiliar object (F(1,17) = 9.464, p = 0.0068).
Paradoxical motor effect of cocaine, amphetamine, nomifensine, and bupropion in DAT-CI mutants
Psychostimulants are known to strongly enhance motor activity in normal individuals and WT animals by increasing dopaminergic, noradrenergic, and serotonergic neurotransmission (Amara and Sonders, 1998; Han and Gu, 2006). Nevertheless, these compounds exert a paradoxical calming effect in ADHD patients and in animals that mimic such psychiatric disorder (Solanto, 1998; Gainetdinov, 2008). Here, to further validate DAT-CI mutants as a potential mouse model for ADHD motor symptoms, we evaluated the motor responses to amphetamine, nomifensine, and bupropion, involved in the medication of this mental disorder (Shekim et al., 1989; Wilens et al., 2005; Wilens, 2006). In this regard, we performed behavioral experiments on WT and DAT-CI animals in habituated experimental cage, where the effects of cocaine, amphetamine, nomifensine, and bupropion were analyzed by visual observation. As shown in Figure 2, all compounds administered, cocaine (40 mg/kg), amphetamine (5 mg/kg), nomifensine (15 mg/kg), and bupropion (30 mg/kg), strongly enhanced horizontal motor activity of WT mice, compared with their respective vehicle-treated group (two-way ANOVA with repeated measures: cocaine treatment effect, F(1,110) = 49.588, p < 0.0001; amphetamine treatment effect, F(1,110) = 42.595, p < 0.0001; nomifensine treatment effect, F(1,110) = 49.989, p < 0.0001; bupropion treatment effect, F(1,50) = 55.605, p < 0.0001). In contrast, all these drugs were able to induce a severe paradoxical motor sedation in DAT-CI animals, referred to their respective vehicle-treated group (Fig. 2a,c,e,g). Interestingly, the effect of cocaine was less relevant than other drugs (Fig. 2a). Indeed, statistical analysis evidenced that, in DAT-CI mutants, cocaine lost its calming effect in the last 20 min of observation (Fisher's post hoc analysis: 10, 20, and 30 min, p < 0.01; 40 min, p < 0.05; 50 and 60 min, p > 0.05). However, reduction of basal hyperactivity in DAT-CI mice was robust along the entire session of analysis using amphetamine (treatment effect, F(1,110) = 45.232, p < 0.0001), nomifensine (treatment effect, F(1,110) = 15.039, p = 0.0008), and bupropion (treatment effect, F(1,70) = 26.264, p = 0.0002) (Fig. 2c,e,g). Analysis of total sector crossing over 1 h test session recapitulates the clear opposite effect of cocaine, amphetamine, nomifensine, and bupropion on motor behavior of WT and DAT-CI mice (Fig. 2b,d,f,h).
Figure 2.
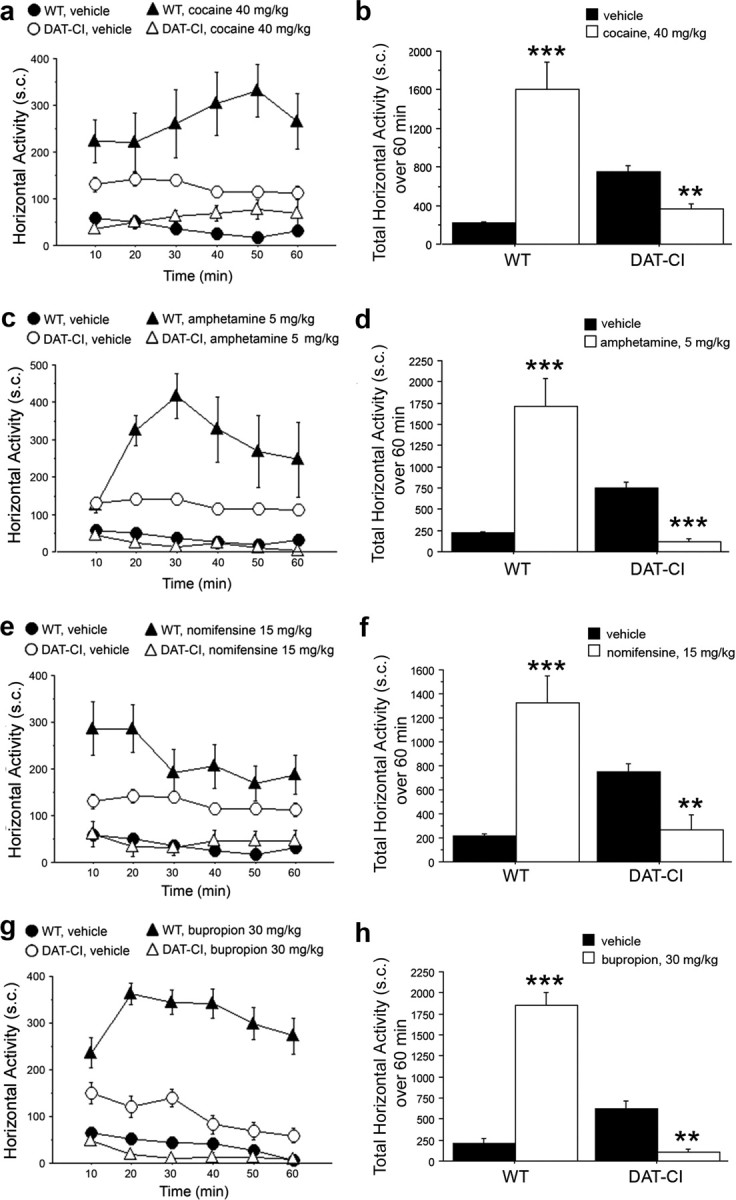
Paradoxical effect of cocaine, amphetamine, nomifensine, and bupropion on motor activity of DAT-CI mutants. Horizontal motor activity induced in WT and DAT-CI mice by intraperitoneal administration of 40 mg/kg cocaine (WT, n = 8; DAT-CI, n = 7) (a, b), 5 mg/kg amphetamine (n = 8 per genotype) (c, d), 15 mg/kg nomifensine (n = 8 per genotype) (e, f), or 30 mg/kg bupropion (n = 8 per genotype) (g, h), after 1 h of cage habituation. Cocaine, amphetamine, and nomifensine shared the same vehicle group (n = 16 per genotype), whereas bupropion-treated mice were compared with a different vehicle group (WT, n = 4; DAT-CI, n = 8). Locomotion is expressed as number of sector crossings, measured every 10 min over a 1 h test, and presented as time course (a, c, e, g) or total activity (b, d, f, h). All values are expressed as mean ± SEM. **p < 0.01, ***p < 0.0001 versus vehicle group within genotype (one-way ANOVA). Genotypes and treatments are as indicated.
In parallel to sector crossing, we also visually scored vertical activity of WT and mutant mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Similar to the horizontal activity, statistical analyses revealed that amphetamine, nomifensine, and bupropion enhanced vertical activity in treated WT, but depressed it in DAT-CI treated mice, compared with their respective vehicle-treated groups (supplemental Fig. 2c–h, available at www.jneurosci.org as supplemental material). Once again, this genotype-dependent effect, although still present, was less evident after cocaine administration (supplemental Fig. 2a,b, available at www.jneurosci.org as supplemental material).
Effect of selective noradrenaline and serotonin transporter inhibitors on DAT-CI motor hyperactivity
Cocaine, amphetamine, nomifensine, and bupropion are known to exert their influence by acting on DAT as well as on NET and serotonin transporter (SERT), even though with peculiar specificity and different mechanisms of action (Amara and Sonders, 1998; Han and Gu, 2006). In the light of the motor calming effect induced by these drugs in DAT-CI mice, we firstly evaluated the potential contribution of noradrenergic and/or serotonergic transmission in this paradoxical response, treating animals with selective NET or SERT inhibitors. Overall, statistical analysis indicated that administration of the selective NET blocker nisoxetine (10 mg/kg) was able to significantly reduce horizontal (treatment effect, F(1,85) = 4.751, p = 0.0436) (Fig. 3a,b) but not vertical motor hyperactivity of DAT-CI mice (supplemental Fig. 3a,b, available at www.jneurosci.org as supplemental material). In contrast to nisoxetine, administration of the selective SERT inhibitor fluoxetine, at the same dose (20 mg/kg) and route of administration (subcutaneous) previously used in DAT-KO mice (Gainetdinov et al., 1999a; Beaulieu et al., 2006), was ineffective to attenuate hyperlocomotion in both WT and DAT-CI animals (treatment effect, WT: F(1,45) = 1.885, p = 0.230; DAT-CI: F(1,45) = 1.873, p = 0.2043) (Fig. 3c,d). Similarly, fluoxetine treatment did not evoke any detectable calming response on vertical activity of mutant mice (supplemental Fig. 3c,d, available at www.jneurosci.org as supplemental material).
Figure 3.

Effect of selective NET and SERT inhibitors on DAT-CI motor activity. Horizontal motor activity induced in WT and DAT-CI mice by intraperitoneal injection of 10 mg/kg nisoxetine (WT, n = 7; DAT-CI, n = 11) or vehicle (WT, n = 7; DAT-CI, n = 8) (a, b) or subcutaneous administration of 20 mg/kg fluoxetine (n = 7 per genotype) or vehicle (n = 4 per genotype) (c, d), after 1 h of cage habituation. Locomotion is expressed as number of sector crossings, measured every 10 min over a 1 h test, and presented as time course (a, c) or total activity (b, d). P-Thr202/Tyr204-ERK42/44 protein levels were determined by Western blotting in the striatum of WT (n = 8) and DAT-CI (n = 7) naive mice (e), 20 mg/kg fluoxetine- or vehicle-injected WT (n = 6 per treatment) (f) and DAT-CI (g) mice (20 mg/kg fluoxetine: n = 5; vehicle: n = 6). The top panels show representative blots comparing the different genotypes or treatments, for each protein detected. All data are expressed as mean ± SEM. *p < 0.05 versus vehicle group within genotype (one-way ANOVA). Genotypes and treatments are as indicated.
Therefore, the lack of calming effect exerted by fluoxetine in DAT-CI animals strikingly contrasts with the results obtained in DAT-KO mice, whose hyperactivity was dramatically blocked by enhancing serotonergic neurotransmission (Gainetdinov et al., 1999a). The calming action of fluoxetine on hyperactivity of DAT-KO mice has been linked to aberrant regulation of striatal ERK42 signaling (Beaulieu et al., 2006). Thus, we evaluated the consequences of increased serotonergic transmission on the phosphorylation state of ERK42/44 in DAT-CI striata. We analyzed phosphorylated levels of ERK42 and ERK44 (P-Thr202/Tyr204-ERK42/44) under naive conditions (Fig. 3e) and after vehicle or 20 mg/kg fluoxetine treatment (Fig. 3f,g). Overall, the results indicated no significant alteration on physiological ERK signaling between genotypes (Student's t test, p > 0.05, per each phosphoprotein) (Fig. 3e). Moreover, in line with the absence of motor influence induced by fluoxetine, our biochemical results showed that striatal content of both P-Thr202/Tyr204-ERK42 and P-Thr202/Tyr204-ERK44 was comparable between treated and untreated striata in both genotypes (p > 0.05, per each phosphoprotein within genotypes) (Fig. 3f,g).
Altered dopaminergic homeostasis in DAT-CI striatum
Previous data demonstrated that the lack of dopamine transporter in DAT-KO mice resulted in ∼95% reduction of striatal DA, accompanied by a concomitant increase of HVA levels and by reduced 5HT content (Jones et al., 1998). Based on this evidence, here we analyzed the striatal levels of DA, HVA, DOPAC, NA, and 5HT by HPLC (Fig. 4a). Similar to DAT-KO mice, our results in DAT-CI mutants indicated a significant reduction of DA (WT, 44.8 ± 10.1 pmol/mg tissue; DAT-KO, 15.4 ± 1.2 pmol/mg tissue; p < 0.05, Student's t test), coupled to increased HVA levels (p < 0.0001) (Fig. 4a). Moreover, in line with previous observation on DAT-KO mice (Jones et al., 1998), the ratio analysis between DOPAC and DA revealed enhanced catabolism of this neurotransmitter in DAT-CI striata (p < 0.0001) (Fig. 4b). Interestingly, the DOPAC/DA ratio, measured in cortical homogenates, did not reveal any difference between genotypes (data not shown). However, total striatal NA and 5HT levels resulted to be, respectively, unaffected or slightly decreased in mutants compared with controls (NA, p > 0.05; 5HT, p < 0.05) (Fig. 4c).
Figure 4.

Altered dopaminergic homeostasis in DAT-CI striatum. a, Basal levels of striatal DA, DOPAC, and HVA content, expressed as picomoles per milligram of tissue. b, Dopaminergic catabolism measured as DOPAC/DA ratio. c, Basal levels of striatal NA and 5HT, expressed as picomoles per milligram of tissue. Neurochemical measurements were performed by HPLC in homogenates from WT (n = 8) and DAT-CI (n = 7) naive mice. d, Striatal extracellular DA levels (in nanomolar concentration) after the administration of 100 μm amphetamine (left), 30 μm nomifensine (middle), or 100 μm bupropion (right) in WT and DAT-CI mice (n = 6 per genotype and treatment), measured by voltammetry. *p < 0.05, **p < 0.01, ***p < 0.0001 versus WT (Student's t test). Values are expressed as mean ± SEM. Genotypes and treatments are as indicated.
Previous evidence indicated that DAT mutant protein is insensitive to cocaine inhibition in DAT-CI mice, since it does not provoke changes in their striatal extracellular DA levels (Chen et al., 2006). Therefore, our behavioral data obtained with nisoxetine, while explaining the calming effect induced by cocaine administration, in turn suggested only a partial contribution of enhanced noradrenergic neurotransmission on the more pronounced motor calming effect induced by amphetamine, nomifensine, and bupropion in DAT-CI mutants. Therefore, to explain this difference, we challenged the hypothesis that, in addition to NA, enhanced dopaminergic transmission in mutants may be substantially involved in the aberrant motor effect triggered by amphetamine, nomifensine, and bupropion. Thus, to ascertain the involvement of changes in the dopaminergic signaling associated with abnormal motor responses found in DAT-CI animals, we measured the levels of striatal DA release in response to these drugs. In support to our idea, voltammetry detection indicated that these stimulants were all able to consistently enhance striatal dopaminergic transmission in WT and DAT-CI mice (Fig. 4d). However, we also found that all the tested compounds elicited a smaller extracellular DA release in mutant slices compared with controls (30 μm nomifensine, p < 0.01; 100 μm amphetamine and 100 μm bupropion, p < 0.0001) (Fig. 4d).
Dopamine D2R-mediated functions in DAT-CI mutants
We reported that enhanced noradrenergic and serotonergic transmissions induced by NET and SERT selective inhibitors seem to have, respectively, mild or no influence on motor hyperactivity of DAT-CI animals. Conversely, we showed that amphetamine, nomifensine, and bupropion exerted a severe calming effect in mutants. Together, such results pointed out a potential relevance of an abnormal dopaminergic transmission in the paradoxical sedative effect exerted by these drugs in DAT-CI mice. Therefore, in the attempt to identify selective alterations in the mutant dopaminergic system involved in such paradoxical response, here we studied the downstream in vitro and in vivo events related to activation/inhibition of the most relevant DA receptor types, such as D1R and D2R.
As first step, we analyzed the D2R-mediated presynaptic function. It is well known that DA D2R, when located on dopaminergic neurons, regulates DA homeostasis by controlling firing, as well as DA synthesis and synaptic release (Jackson and Westlind-Danielsson, 1994; De Mei et al., 2009). Accordingly, we monitored in DAT-CI animals, the firing of DA neurons by intracellular electrophysiological recordings in response to bath application of the D2R-like agonist quinpirole (Fig. 5a). Interestingly, despite the complete loss of D2-autoreceptor function in DAT-KO mice (Jones et al., 1999), our results showed that quinpirole was able to inhibit the firing of DAT-CI dopaminergic neurons of substantia nigra pars compacta by a membrane hyperpolarization/outward current (Lacey et al., 1987). Then, we measured in single-electrode voltage-clamped neurons, held at −60 mV, the amplitude of the outward current caused by quinpirole (1 μm). Overall, our data indicated that, although D2-autoreceptor-mediated function is mainly preserved in mutant DA neurons, the magnitude of quinpirole-induced outward current was smaller in DAT-CI than in WT mice (WT, 103.0 ± 3.9 pA; DAT-CI, 68.6 ± 5.7 pA; p < 0.0001, Student's t test).
Figure 5.

Dopamine D2R-mediated functions in DAT-CI mutants. a, Firing of DA neurons (outward current; in picoamperes) measured by sharp microelectrode recordings in response to bath application of 1 μm quinpirole in WT and DAT-CI mice (n = 9 per genotype). The top panel shows representative trace recordings. b, Horizontal motor activity in WT and DAT-CI mice in response to intraperitoneal administration of 0.5 mg/kg quinpirole (n = 5 per genotype) or vehicle (n = 8 per genotype), in a novel cage. Locomotion is expressed as total number of sector crossings over a 30 min test. c, P-Ser40-TH protein levels determined by Western blotting in the striatum of WT and DAT-CI mice, after intraperitoneal injection of 0.1 mg/kg haloperidol or vehicle (n = 6 per genotype and treatment). The top panels show representative blots comparing the different treatments. d, Horizontal motor activity in WT and DAT-CI mice in response to intraperitoneal administration of 0.1 mg/kg haloperidol (n = 5 per genotype) or vehicle (n = 8 per genotype), over a 60 min test, in a novel cage. All values are expressed as mean ± SEM. a, ***p < 0.0001 versus WT (Student's t test); b–d, *p < 0.05, **p < 0.01, ***p < 0.0001 versus vehicle group within genotype (one-way ANOVA). Genotypes and treatments are as indicated.
Furthermore, we examined the motor suppression effect of 0.5 mg/kg quinpirole induced through D2-autoreceptor activation (Usiello et al., 2000). In line with electrophysiological recordings, ANOVA analysis indicated that quinpirole elicited in both genotypes a consistent reduction of locomotion (WT, F(1,11) = 80.970, p < 0.0001; DAT-CI, F(1,11) = 8.814, p = 0.0128) (Fig. 5b). Such in vivo result further highlights a preserved D2R presynaptic-mediated function and, most importantly, indicates that the hyperactive phenotype of DAT-CI mutants is tightly regulated by their higher extracellular DA content (Chen et al., 2006). D2 autoreceptors, located on dopaminergic terminals, are also involved in the regulation of phosphorylation state of tyrosine hydroxylase (TH), the rate-limiting enzyme in DA synthesis. Blockade of D2Rs with the antagonist haloperidol increases TH phosphorylation (Håkansson et al., 2004), an effect requiring a normal D2-autoreceptor function (Lindgren et al., 2003). Thus, we analyzed the phosphorylation state of TH at Ser40 residue (P-Ser40-TH) in animals treated with vehicle or 0.5 mg/kg haloperidol (Fig. 5c). Overall, Western blotting analysis revealed significant treatment effect (two-way ANOVA: treatment effect, F(1,20) = 29.881, p < 0.0001). Moreover, one-way ANOVA indicated that haloperidol treatment was effective in enhancing P-Ser40-TH levels in both genotypes (WT, F(1,10) = 7.520, p = 0.0208; DAT-CI, F(1,10) = 31.828, p = 0.0002). Although we found unchanged levels of total TH between genotypes, Fisher's post hoc test displayed a reduction of P-Ser40-TH content in vehicle-treated DAT-CI mice, compared with vehicle-treated controls (p < 0.01), indicating that basal levels of this phosphoprotein are influenced by genotype. Finally, we studied D2R-dependent postsynaptic function by analyzing the motor inhibition induced by 0.1 mg/kg haloperidol treatment (Fig. 5d). Our behavioral data showed a robust effect of haloperidol in both genotypes (ANOVA analysis: WT, F(1,11) = 103.833, p < 0.0001; DAT-CI, F(1,11) = 17.001, p = 0.0017), thus indicating a functional D2R postsynaptic responsiveness in mutants.
Abnormal phasic dopamine D1R-mediated functions in DAT-CI mutants
Herein, we investigated the functional state of DA D1R in DAT-CI animals. Basal activation of D1R was evaluated by administering to animals the selective D1R antagonist SCH 23390 (0.05 mg/kg) (Fig. 6a). In line with a normal D1R-mediated transmission, our data revealed that its blockade by SCH 23390 treatment was effective in strongly decreasing locomotion in DAT-CI animals (ANOVA: WT, F(1,14) = 113.702, p < 0.0001; DAT-CI, F(1,14) = 33.488, p < 0.0001), thus suggesting that a functional tonic activation of striatal D1R sustains hyperactivity found in mutant mice.
Figure 6.

Dopamine D1R-mediated functions in DAT-CI mutants. Horizontal activity analyzed in WT and DAT-CI mice after intraperitoneal challenge with 0.05 mg/kg SCH 23390 or vehicle (n = 8 per genotype and treatment) (a) and 2.5 mg/kg (WT, n = 6; DAT-CI, n = 7), 5 mg/kg (WT, n = 6; DAT-CI, n = 9) SKF 81297 or vehicle (n = 8 per genotype) (b). Locomotion is expressed as total number of sector crossings, over a 60 min test, performed in a novel home cage for SCH 23390 and after 1 h of cage habituation for SKF 81297. c, Time course (expressed in seconds) of EPSP amplitude, recorded from coronal corticostriatal slices of WT (n = 16) and DAT-CI (n = 12) mice. The arrow shows the time at which HFS protocol (3 trains, 100 Hz, 3 s duration, 20 s interval) was applied. The bottom traces are representative of EPSP amplitudes recorded before and after HFS induction, in WT (left) and DAT-CI mice (right). Calibration: 5 mV, 15 ms. d, Time course (expressed in minutes) of fEPSP slope recorded from parasagittal hippocampal slices of WT and DAT-CI mice (n = 6 per genotype). The arrow shows the time at which HFS protocol (1 train, 100 Hz, 1 s) was applied. Data are expressed as percentage of baseline EPSP slopes measured 60 min after stimulation protocol. The bottom traces are representative of the average fEPSP, recorded 1 min before (pre) and 60 min after (post) the tetanus stimulation, in WT (left) and DAT-CI (right) mice. Calibration: 0.5 mV, 10 ms. All values are expressed as mean ± SEM. a, ***p < 0.0001 versus vehicle group within genotype (one-way ANOVA). b, *p < 0.05, **p < 0.01, ***p < 0.0001 versus vehicle group within genotype (Fisher's post hoc comparison). Genotypes and treatments are as indicated.
Next, we analyzed phasic D1R-dependent responses through administration of the selective D1R agonist SKF 81297 (2.5 and 5 mg/kg) (Fig. 6b). Strikingly, behavioral analysis indicated a robust motor calming effect induced in mutants by D1R stimulation, as revealed by a significant genotype × treatment interaction (F(2,38) = 42.968; p < 0.0001). Thus, subsequent one-way ANOVA displayed that SKF 81297 was able to enhance horizontal motor activity in WT (F(2,17) = 19.672; p < 0.0001), whereas it significantly suppressed hyperlocomotion in DAT-CI mice (F(2,21) = 28.008; p < 0.0001). Moreover, Fisher's post hoc comparison revealed a dose-dependent effect of SKF 81297 in increasing locomotion of WT (2.5 mg/kg, p < 0.05; 5 mg/kg, p < 0.0001, compared with vehicle-treated group). In contrast, both doses of this D1R agonist similarly reduced sector crossing in mutant animals (2.5 and 5 mg/kg, p < 0.0001, compared with vehicle-treated group). We also explored the effect of SKF 81297 on vertical motor activity of mice. In line with locomotion, WT control mice displayed a strong dose-dependent stimulation of vertical activity induced by SKF 81297, whereas hyperactive behavior of DAT-CI mice was reduced by D1R stimulation (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
In addition to motor function, it is well established that DA D1R plays a crucial role on the control of striatal LTP via cAMP/PKA/DARPP32 pathway activation (Calabresi et al., 2000; Centonze et al., 2003). Thus, to further investigate phasic D1R-dependent responses, we examined LTP in the striatum of mutant mice (Fig. 6c). Interestingly, electrophysiological data indicated that this form of striatal synaptic plasticity was completely abolished in DAT-CI mice (WT, 155.8 ± 3.9%; DAT-CI, 97.0 ± 7.8%; p < 0.001, ANOVA, followed by Tukey's post hoc test) (Fig. 6c). Furthermore, to ascertain that disruption of striatal LTP in DAT-CI mice was a selective D1R/cAMP/PKA/DARPP32-dependent event, we also measured the NMDAR-dependent form of LTP at CA1 synapses in hippocampal slices (Fig. 6d) (Lynch et al., 1990). Here, we found that HFS at CA1 synapses induced a comparable long-lasting potentiation in both genotypes (WT, 144.4 ± 5.4%; DAT-CI, 150.4 ± 1.2%; p > 0.05) (Fig. 6d). Overall, our data indicated that striatal D1R-dependent transmission in DAT-CI mice is preserved under basal conditions, but it displays an aberrant functioning under a phasic activation state.
Aberrant dopamine D1R-dependent PKA signaling in the striatum of DAT-CI mutants
Our in vivo and in vitro observations suggested that a phasic deregulated D1R-mediated transmission occurs in the striatum of mice with point mutations in the DAT gene. Herein, to study the possible molecular features underlying the aberrant D1R-dependent responses, we analyzed the striatal PKA signaling downstream the activation of this DA receptor. Stimulation of DA D1Rs and adenosine A2ARs, respectively, in the direct and indirect pathway of basal ganglia, overall results in Golf-mediated triggering of adenylyl cyclase, increased cAMP synthesis and activation of PKA (Zhuang et al., 2000; Hervé et al., 2001). Based on this, we first measured total Golf levels in the striatum of naive WT and DAT-CI mice. Despite what was observed in DAT-KO animals (Hervé et al., 2001), results indicated comparable levels of this G-stimulatory protein between genotypes (Student's t test, p > 0.05) (Fig. 7a). As a measure of basal striatal PKA activity, deriving from both direct and indirect circuits of basal ganglia (Gerfen, 1992), we detected in naive mice the phosphorylation state of the AMPAR subunit GluR1 at Ser845 (P-Ser845-GluR1), one of the main known targets of PKA (Roche et al., 1996; Snyder et al., 2000). Our data pointed out unchanged striatal content of P-Ser845-GluR1 in DAT-CI, compared with WT animals (p > 0.05) (Fig. 7b).
Figure 7.
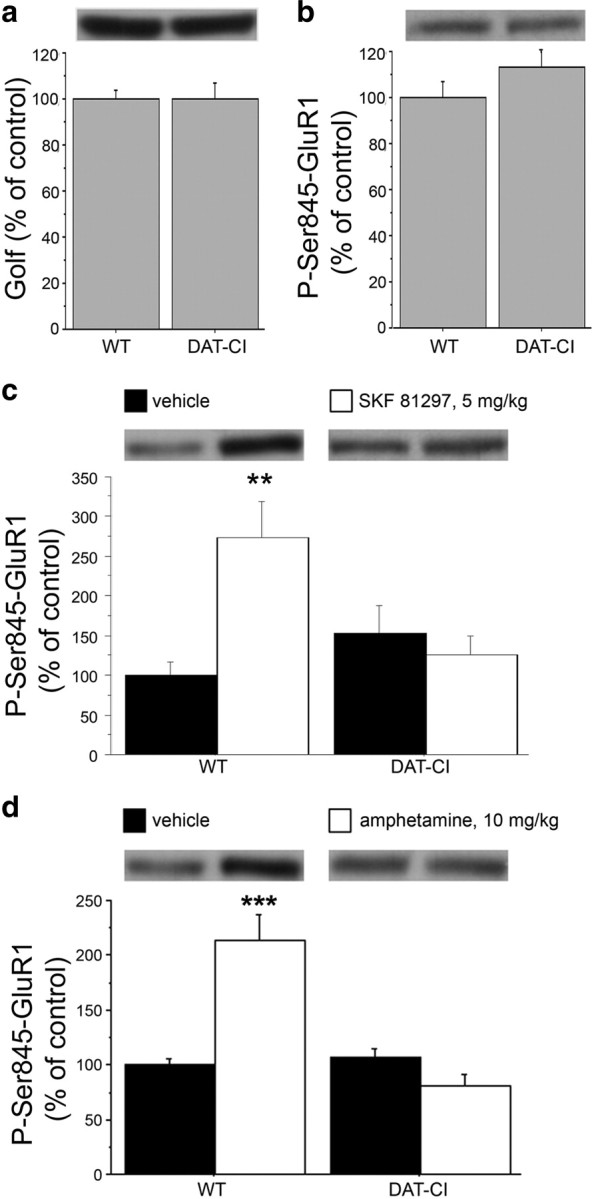
Aberrant D1R-dependent signaling in the striatum of DAT-CI mutants. Western blotting analysis performed on striatal homogenates in WT and DAT-CI mice, showing basal levels of Golf (n = 32 per genotype) (a) and P-Ser845-GluR1 (WT, n = 8; DAT-CI, n = 7) (b). Western blotting analysis of striatal P-Ser845-GluR1 content in WT and DAT-CI mice, after intraperitoneal challenge with 5 mg/kg SKF 81297 (n = 8 per genotype) or vehicle (n = 8 per genotype) (c); 10 mg/kg amphetamine (n = 15 per genotype) or vehicle (WT, n = 16; DAT-CI, n = 14) (d). The top panels show representative blots comparing the different genotypes (a, b) or treatments (c, d) for each protein detected. All data are expressed as mean ± SEM. **p < 0.01, ***p < 0.0001 versus vehicle group within genotype (one-way ANOVA). Genotypes and treatments are as indicated.
In addition to basal biochemical analysis, which involved both A2AR and D1R effect on P-Ser845-GluR1 striatal levels, we then investigated the selective functionality of D1R-mediated phasic activation of cAMP/PKA signaling. In this respect, we measured striatal P-Ser845-GluR1 levels in response to treatment with the D1R agonist SKF 81297 (5 mg/kg) (Fig. 7c). Statistical analysis revealed a significant genotype × treatment interaction (F(1,28) = 9.986; p = 0.0038), indicating a different PKA activation in response to SKF 81297 treatment between genotypes (Fig. 7c). Indeed, the following one-way ANOVA performed within genotypes, unmasked a significant increase of P-Ser845-GluR1 content only in SKF 81297-treated WT (F(1,14) = 13.262; p = 0.0027) but revealed no changes between treated and untreated mutants (F(1,14) = 0.416; p = 0.5292).
We have previously shown that amphetamine was able to produce an evident motor calming effect in DAT-CI mice. In the light of this evidence, we investigated the biochemical effect of this anti-ADHD drug on the stimulation of PKA activity (Fig. 7d). Overall, statistical analysis indicated significant genotype × treatment interaction (F(1,56) = 25.436; p < 0.0001). Moreover, subsequent one-way ANOVA evidenced that amphetamine increased P-Ser845-GluR1 levels selectively in WT mice (F(1,29) = 22.758; p < 0.0001). Conversely, this effect was absent in treated DAT-CI mutants, in which a paradoxical decrease of P-Ser845-GluR1 content was observed (F(1,27) = 3.986; p = 0.0561). We also evaluated the locomotion of DAT-CI mice at the same dose of amphetamine used for biochemical studies (10 mg/kg). The results evidenced that, also at a higher dose, amphetamine was still able to produce in mutants a paradoxical motor calming effect (data not shown).
Abnormal regulation of DARPP32 phosphorylation in DAT-CI striata
In addition to P-Ser845-GluR1, another major substrate of PKA in the striatum is DARPP32, which plays a complex role in controlling and integrating dopaminergic signaling (Svenningsson et al., 2004). The specific mode of DARPP32 function has been demonstrated to be strictly regulated by two main phosphorylation sites, Thr34 and Thr75 (Svenningsson et al., 2004). Indeed, PKA-dependent phosphorylation at Thr34 site converts DARPP32 into a potent inhibitor of PP-1 (protein phosphatase-1) (Hemmings et al., 1984); conversely, phosphorylation at Thr75 residue, by cyclin-dependent kinase 5 (Cdk5), switches DARPP32 into an inhibitor of PKA (Bibb et al., 1999).
Here, we investigated the consequences of persistent higher DA extracellular levels on the phosphorylation state of DARPP32 in DAT-CI striata. According to previous literature (Bibb et al., 1999; Svenningsson et al., 2003), in WT mice, activation of D1R with SKF 81297 or amphetamine induced a remarkable enhancement in DARPP32 phosphorylation at the PKA target site Thr34 (p < 0.01, per each drug) (Fig. 8b), but not at the Cdk5 site Thr75 (p > 0.05, per each genotype and drug) (Fig. 8e,f). However, in DAT-CI mice, we found no changes in the levels of P-Thr34-DARPP32 between vehicle and SKF 81297 or amphetamine treatment (p > 0.05, per each drug) (Fig. 8c). Western blotting experiments also indicated no difference between genotypes in the basal striatal P-Thr34-DARPP32 (Student's t test, p > 0.05) (Fig. 8a). Interestingly, and in contrast to what observed on Thr34 phosphorylation, we found increased basal levels of phosphorylated Thr75 residue in the striata of mutant mice (p < 0.01) (Fig. 8d).
Figure 8.

Altered DARPP32 phosphorylation in DAT-CI striata. Western blotting analysis performed on striatal homogenates in WT and DAT-CI mice. Basal levels of P-Thr34-DARPP32 (n = 12 per genotype) (a) and P-Thr75-DARPP32 (n = 12 per genotype) (d) are shown. Western blotting analysis of striatal P-Thr34-DARPP32 (b, c) and P-Thr75-DARPP32 (e, f) levels after intraperitoneal challenge with 5 mg/kg SKF 81297 (n = 6 per treatment) or vehicle (n = 6 per treatment), and amphetamine 10 mg/kg (n = 6 per treatment) or vehicle (n = 6 per treatment) in WT (b, e) and DAT-CI (c, f) mice. The top panels indicate representative blots comparing different genotypes (a, d) or treatments (b, c, e, f). All data are expressed as mean ± SEM. b, **p < 0.01 versus vehicle-treated group (Student's t test). d, **p < 0.01 versus WT (Student's t test).
Unaltered biochemical and behavioral A2AR-dependent responses in DAT-CI mice
In addition to DA D1Rs, also adenosine A2ARs are positively coupled to adenylyl cyclase and activate cAMP/PKA/DARPP32 signaling in the striatum (Svenningsson et al., 1999), where these receptors are expressed in the same population of medium spiny neurons bearing D2Rs (Schiffmann et al., 1991; Fink et al., 1992). To unmask the involvement of basal A2AR-mediated transmission in PKA-dependent GluR1 phosphorylation in DAT-CI striata, we treated animals with 0.5 mg/kg haloperidol to block the inhibitory tone exerted by D2Rs on cAMP/PKA signaling (Håkansson et al., 2006). The results confirmed a normal D2R postsynaptic function in mutants (Fig. 9a) and, in turn, excluded defects on A2AR transmission since biochemical analysis revealed nonsignificant treatment × genotype interaction (F(1,26) = 0.244; p = 0.6257). Accordingly, one-way ANOVA revealed that haloperidol increased P-Ser845-GluR1 levels in both genotypes (WT, F(1,13) = 4.896, p = 0.0454; DAT-CI, F(1,13) = 6.149, p = 0.0276).
Figure 9.

Physiological A2AR-dependent response in DAT-CI mice. a, Western blotting analysis of striatal P-Ser845-GluR1 content in WT and DAT-CI mice, after intraperitoneal injection of 0.5 mg/kg haloperidol (WT, n = 8; DAT-CI, n = 7) or vehicle (WT, n = 7; DAT-CI, n = 8). The top panels show representative blots comparing the different treatments. Horizontal activity in WT and DAT-CI mice in response to intraperitoneal injection of 5 mg/kg SCH 58261 (WT, n = 7; DAT-CI, n = 8) or vehicle (WT, n = 7; DAT-CI, n = 8) (b), and 0.25 mg/kg CGS 21680 (WT, n = 8; DAT-CI, n = 7) or vehicle (n = 7 per genotype) (c). Data are presented as total number of sector crossing over 60 min test, performed after 1 h of cage habituation (b), or over 30 min test, performed in a novel cage (c). All data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.0001 versus vehicle group within genotype (one-way ANOVA). Genotypes and treatments are as indicated.
Then, to evaluate the in vivo functioning of A2ARs in mutants, we also treated mice with selective A2AR antagonist or agonist, known to elicit, respectively, hyperactivity or motor depression in WT animals. Thus, in accordance to unaltered responsiveness of A2AR-dependent transmission in DAT-CI mutants, behavioral results evidenced that 5 mg/kg of the selective A2AR antagonist SCH 58261 significantly increased horizontal activity in DAT-CI mice (one-way ANOVA: F(1,14) = 6.512, p = 0.0230) (Fig. 9b). Moreover, we also administered to mice the selective A2AR agonist CGS 21680 (Lindskog et al., 2002). In line with a normal A2AR-mediated response, the results indicated that CGS 21680, at the dose of 0.25 mg/kg, was able to significantly reduce horizontal activity of DAT-CI animals (DAT-CI, F(1,12) = 26.948, p = 0.0002), thus confirming a proper A2AR-dependent response in mutants (Fig. 9c).
Discussion
Genetic studies indicate that ADHD is a highly inheritable, polygenic psychiatric disease that results from complex gene–gene and gene–environment interactions (Faraone, 2004; Thapar et al., 2005). Today, several lines of research point out an involvement of DAT1 gene in ADHD. Consistently, DAT-KO mice have been considered a reliable animal model for this psychiatric disorder (Giros et al., 1996; Gainetdinov et al., 1999a; Gainetdinov, 2008). Recent studies have revealed that DAT-CI mutants with three point mutations in the cocaine binding site of the DAT gene show increased basal motor activity, absence of cocaine-induced extracellular DA elevation, and loss of cocaine- and methylphenidate-induced conditioned place preference (Chen et al., 2006; Tilley and Gu, 2008b). In this work, we further confirm that DAT-CI mutants show basal horizontal and vertical hyperactivity, accompanied by a mild reduction of cognitive ability, whereas no alterations were observed in motor coordination. Most importantly, we showed that the motor hyperactive phenotype of DAT-CI mice was dramatically reverted by amphetamine, nomifensine, and bupropion treatments. Together, previous and present data suggest that a triple point mutation in DAT gene, encoding for a mutant DAT with reduced functionality (Chen et al., 2006), is a sufficient genetic factor to mimic some cardinal features of ADHD-like symptoms.
We also suggest that some of the severe modifications found in DAT-KO brains, including the loss of D2-autoreceptor functions (Jones et al., 1999), may not represent an essential neuronal milieu for determining ADHD-like phenotype. Indeed, our electrophysiological and biochemical data showed an overall preserved D2-autoreceptor activity in mutants, since DAT-CI dopaminergic neurons are still able to respond to quinpirole-dependent inhibition of firing and to haloperidol-dependent increase of TH phosphorylation. However, similarly to DAT-KO (Jones et al., 1998), our HPLC experiments evidenced that DAT-CI mutants display a significant decrease of striatal DA content, accompanied by enhanced DA turnover. In this respect, higher levels of the DA metabolite HVA in the CSF of ADHD individuals support the idea that an altered reuptake may lead to increased extracellular DA content, which, in turn, causes inattention and hyperactivity (Castellanos et al., 1996). Thus, according to Castellanos et al. (1996), our and previous results on DAT-CI (Chen et al., 2006) and DAT-KO mice (Gainetdinov et al., 1999b) suggest that basal DA excess at striatal synapses represents the common leading force that drives hyperactivity, in humans and animals. In this line, we found that basal motor hyperactivity can be severely attenuated in DAT-CI mutants through the reduction of the extracellular DA levels, evoked by quinpirole administration. Indeed, quinpirole is known to activate the presynaptic D2R and to exert, in turn, a negative feedback on DA accumulation at the synaptic cleft (Mercuri et al., 1997; Usiello et al., 2000; Rouge-Pont et al., 2002).
In DAT-KO mice, the calming effect of psychostimulants was fully explained through increased serotonergic neurotransmission (Gainetdinov et al., 1999a). Our results, conversely, indicated the absence of sedative effects induced by increased 5HT transmitter in DAT-CI mice. Indeed, the treatment with a selective inhibitor of SERT, fluoxetine, did not affect basal hyperactivity of these animals. We also observed that pharmacological enhancement of 5HT transmission did not alter P-Thr202/Tyr204-ERK42/44 levels in DAT-CI striata. Consistent with previous data (Tilley and Gu, 2008b), we also found that a selective NET blocker, nisoxetine, through enhancement of NA transmission, attenuated the motor hyperactivity of DAT-CI mice. However, this effect results to be moderate, if compared with the strong motor sedation induced by amphetamine, nomifensine, and bupropion. In this regard, in addition to NA, we questioned whether DA itself could have a concomitant role in mediating the profound calming effect of these drugs in DAT-CI mice. In agreement with our idea, voltammetry data indicated that amphetamine, nomifensine, and bupropion are all able to trigger a significant increase of extracellular DA in DAT-CI striata, although to a lesser extent than WT. In the light of previous mutagenesis and modeling studies (Sen et al., 2005; Beuming et al., 2008), reduced release of DA in response to amphetamine, nomifensine, and bupropion found in mutants, leads to the hypothesis that point mutations in the transmembrane domain 2 of DAT protein (Chen et al., 2005) may result in conformational changes with indirect and partial effects on the binding of these drugs.
To understand the biological nature of the sedative effect evoked by amphetamine, nomifensine, and bupropion through the increase of extracellular DA levels, we examined the functioning of D1R and D2R in mutant brains. In addition to a preserved D2-autoreceptor function, we found that also responsiveness of postsynaptic D2R is normal in DAT-CI striata, since haloperidol was able to consistently suppress motor hyperactivity of mutant mice. Similarly, basal functioning of D1R was not altered in DAT-CI animals, as demonstrated by the strong decrease of their hyperlocomotion induced by the selective D1R antagonist SCH 23390. In contrast, we found a strikingly abnormal response of D1R under phasic stimulation, since the treatment with the selective D1R agonist SKF 81297 induced a strong paradoxical calming effect in mutants. Furthermore, in support of an altered response selectively involving a stimulated D1R transmission, we observed that HFS-induced LTP was absent in the striatum but normally induced in the hippocampus of DAT-CI mice. On the basis of previous literature (Calabresi et al., 2000; Centonze et al., 2003), this result supports the idea of an altered PKA-dependent signaling downstream striatal D1R stimulation in mutant brains. Accordingly, we found that phosphorylation of GluR1 at Ser845, one of the main targets of PKA action, is not induced or even decreased in DAT-CI striata, in response to SKF 81297 and amphetamine, respectively. In line with a dramatic alteration in the activity of D1R/cAMP/PKA cascade in DAT-CI striata, we found that both SKF 81297 and amphetamine were also unable to increase phosphorylation at PKA-selective site of DARPP32. However, our biochemical data showed enhanced basal levels of P-Thr75-DARPP32 in DAT-CI striata. We suggest that this change might be part of an adaptive negative-feedback mechanism to counteract the persistent higher DA extracellular levels coupled to abnormal overstimulation of D1R. The basal upregulation of phospho-Thr75 could be attributable either to reduced PP-2A (protein phosphatase 2A) activity (Nishi et al., 1999) or to enhanced Cdk5 signaling (Nishi et al., 2000). Regardless of the precise mechanism underlying abnormal basal DARPP32 phosphorylation at Thr75, the present results suggest the existence of a constitutive enhanced inhibitory tone on PKA activity in striatal D1R-bearing DAT-CI neurons. In line with our data, a previous report by Bibb et al. (1999) indicated a significant upregulation of basal phosphorylation at Thr75 site in rats chronically treated with cocaine.
Interestingly, our results rule out a potential contribution of A2AR on abnormal cAMP/PKA cascade in DAT-CI striata, since A2AR-dependent phosphorylation of Ser845-GluR1, unmasked by haloperidol challenge, was undistinguishable between genotypes.
In this regard, it will be important to ascertain whether the aberrant phasic D1R-dependent responses found in DAT-CI mice may also develop, as a common pathophysiological feature, in other animal models of ADHD.
An accredited hypothesis to explain the calming action of anti-ADHD drugs implies that psychostimulant-dependent elevation of extracellular DA activates presynaptic D2R. In turn, D2-autoreceptor function would determine a robust reduction of dopaminergic tone, thus leading to an overall attenuation of postsynaptic D1R- and D2R-mediated transmission associated with a motor sedative effect (Solanto, 1998). In addition, we suggest that the calming effect of psychostimulants may directly result also from aberrant striatal phasic D1R/cAMP/PKA/DARPP32-dependent response that, in turn, inhibits events depending on its activation, such as motor stimulation. In the light of our results, we suggest that DAT-CI mice may represent a useful experimental in vivo tool, complementary to DAT-KO animals, for modeling behavioral and molecular defects in ADHD, closely associated with DAT functioning. In particular, both functional abnormality and lack of DAT protein account for similar paradoxical responses to psychostimulants that, nevertheless, seem to depend on different neurotransmitter systems, such as dopaminergic/noradrenergic in DAT-CI and serotonergic in DAT-KO mice.
Although reducing ADHD symptoms, the use of stimulant medications has always generated controversy, since ADHD patients can experience severe side effects and manifest the highest risk of abusing or diverting their stimulant prescriptions in adulthood. These detrimental effects demand that new therapeutic approaches should be unveiled for ADHD. In this line, the discovery of D1R as a potential selective dopaminergic target directly controlling ADHD-like hyperactive state may serve to avoid undesirable effects caused by stimulation of different DA receptors and neurotransmitter systems.
Footnotes
We thank Dr. G. Fisone, Dr. A. Bertolino, Dr. B. Al Mehdawy, and Dr. D. Viggiano for helpful discussions on this manuscript. We also thank Dr. C. Mazzola, D. Vitucci, and M. A. Ricci for their excellent technical support.
A.U. represents the Mariano Scippacercola Foundation.
References
- Amara SG, Sonders MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 1998;51:87–96. doi: 10.1016/s0376-8716(98)00068-4. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Paradoxical striatal cellular signaling responses to psychostimulants in hyperactive mice. J Biol Chem. 2006;281:32072–32080. doi: 10.1074/jbc.M606062200. [DOI] [PubMed] [Google Scholar]
- Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.”. Nat Protoc. 2006;1:1306–1311. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- Biederman J. Attention-deficit/hyperactivity disorder: a selective overview. Biol Psychiatry. 2005;57:1215–1220. doi: 10.1016/j.biopsych.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Bush G, Valera EM, Seidman LJ. Functional neuroimaging of attention-deficit/hyperactivity disorder: a review and suggested future directions. Biol Psychiatry. 2005;57:1273–1284. doi: 10.1016/j.biopsych.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Blockade of M2-like muscarinic receptors enhances long-term potentiation at corticostriatal synapses. Eur J Neurosci. 1998;10:3020–3023. doi: 10.1111/j.1460-9568.1998.00348.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, Chergui K, Svenningsson P, Fienberg AA, Greengard P. Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J Neurosci. 2000;20:8443–8451. doi: 10.1523/JNEUROSCI.20-22-08443.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Stancampiano R, Tronci E, Collu M, Usiello A, Morelli M, Fadda F. Vitamin A deficiency induces motor impairments and striatal cholinergic dysfunction in rats. Neuroscience. 2006;139:1163–1172. doi: 10.1016/j.neuroscience.2006.01.027. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Elia J, Kruesi MJ, Marsh WL, Gulotta CS, Potter WZ, Ritchie GF, Hamburger SD, Rapoport JL. Cerebrospinal fluid homovanillic acid predicts behavioral response to stimulants in 45 boys with attention deficit/hyperactivity disorder. Neuropsychopharmacology. 1996;14:125–137. doi: 10.1016/0893-133X(95)00077-Q. [DOI] [PubMed] [Google Scholar]
- Centonze D, Grande C, Saulle E, Martin AB, Gubellini P, Pavón N, Pisani A, Bernardi G, Moratalla R, Calabresi P. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci. 2003;23:8506–8512. doi: 10.1523/JNEUROSCI.23-24-08506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Han DD, Gu HH. A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate. J Neurochem. 2005;94:352–359. doi: 10.1111/j.1471-4159.2005.03199.x. [DOI] [PubMed] [Google Scholar]
- Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, Quan N, Stephens RL, Hill ER, Nottoli T, Han DD, Gu HH. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci U S A. 2006;103:9333–9338. doi: 10.1073/pnas.0600905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mei C, Ramos M, Iitaka C, Borrelli E. Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol. 2009;9:53–58. doi: 10.1016/j.coph.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E, Huston JP, De Souza Silva MA. The pharmacology, neuroanatomy and neurogenetics of one-trial object recognition in rodents. Neurosci Biobehav Rev. 2007;31:673–704. doi: 10.1016/j.neubiorev.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Jons PH, Cohen RM. High midbrain [18F]DOPA accumulation in children with attention deficit hyperactivity disorder. Am J Psychiatry. 1999;156:1209–1215. doi: 10.1176/ajp.156.8.1209. [DOI] [PubMed] [Google Scholar]
- Errico F, Santini E, Migliarini S, Borgkvist A, Centonze D, Nasti V, Carta M, De Chiara V, Prosperetti C, Spano D, Herve D, Pasqualetti M, Di Lauro R, Fisone G, Usiello A. The GTP-binding protein Rhes modulates dopamine signalling in striatal medium spiny neurons. Mol Cell Neurosci. 2008;37:335–345. doi: 10.1016/j.mcn.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Errico F, Nistico R, Napolitano F, Oliva AB, Romano R, Barbieri F, Florio T, Russo C, Mercuri NB, Usiello A. Persistent increase of d-aspartate in d-aspartate oxidase mutant mice induces a precocious hippocampal age-dependent synaptic plasticity and spatial memory decay. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Faraone SV. Genetics of adult attention-deficit/hyperactivity disorder. Psychiatr Clin North Am. 2004;27:303–321. doi: 10.1016/S0193-953X(03)00090-X. [DOI] [PubMed] [Google Scholar]
- Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res. 1992;14:186–195. doi: 10.1016/0169-328x(92)90173-9. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR. Dopamine transporter mutant mice in experimental neuropharmacology. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:301–313. doi: 10.1007/s00210-007-0216-0. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999a;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Caron MG. Functional hyperdopaminergia in dopamine transporter knock-out mice. Biol Psychiatry. 1999b;46:303–311. doi: 10.1016/s0006-3223(99)00122-5. [DOI] [PubMed] [Google Scholar]
- Garris PA, Collins LB, Jones SR, Wightman RM. Evoked extracellular dopamine in vivo in the medial prefrontal cortex. J Neurochem. 1993;61:637–647. doi: 10.1111/j.1471-4159.1993.tb02168.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet. 2009;126:51–90. doi: 10.1007/s00439-009-0694-x. [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill LL. Pharmacologic treatment of attention deficit hyperactivity disorder. Psychiatr Clin North Am. 1992;15:1–27. [PubMed] [Google Scholar]
- Håkansson K, Pozzi L, Usiello A, Haycock J, Borrelli E, Fisone G. Regulation of striatal tyrosine hydroxylase phosphorylation by acute and chronic haloperidol. Eur J Neurosci. 2004;20:1108–1112. doi: 10.1111/j.1460-9568.2004.03547.x. [DOI] [PubMed] [Google Scholar]
- Håkansson K, Galdi S, Hendrick J, Snyder G, Greengard P, Fisone G. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J Neurochem. 2006;96:482–488. doi: 10.1111/j.1471-4159.2005.03558.x. [DOI] [PubMed] [Google Scholar]
- Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Greengard P, Tung HY, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984;310:503–505. doi: 10.1038/310503a0. [DOI] [PubMed] [Google Scholar]
- Hervé D, Le Moine C, Corvol JC, Belluscio L, Ledent C, Fienberg AA, Jaber M, Studler JM, Girault JA. Gαolf levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J Neurosci. 2001;21:4390–4399. doi: 10.1523/JNEUROSCI.21-12-04390.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DM, Westlind-Danielsson A. Dopamine receptors: molecular biology, biochemistry and behavioural aspects. Pharmacol Ther. 1994;64:291–370. doi: 10.1016/0163-7258(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Hu XT, Cooper DC, Wightman RM, White FJ, Caron MG. Loss of autoreceptor functions in mice lacking the dopamine transporter. Nat Neurosci. 1999;2:649–655. doi: 10.1038/10204. [DOI] [PubMed] [Google Scholar]
- Kawagoe KT, Wightman RM. Characterization of amperometry for in vivo measurement of dopamine dynamics in the rat brain. Talanta. 1994;41:865–874. doi: 10.1016/0039-9140(94)e0064-x. [DOI] [PubMed] [Google Scholar]
- Kuhr WG, Bigelow JC, Wightman RM. In vivo comparison of the regulation of releasable dopamine in the caudate nucleus and the nucleus accumbens of the rat brain. J Neurosci. 1986;6:974–982. doi: 10.1523/JNEUROSCI.06-04-00974.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, Hokfelt T, Borrelli E, Fisone G. Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci U S A. 2003;100:4305–4309. doi: 10.1073/pnas.0730708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog M, Svenningsson P, Pozzi L, Kim Y, Fienberg AA, Bibb JA, Fredholm BB, Nairn AC, Greengard P, Fisone G. Involvement of DARPP-32 phosphorylation in the stimulant action of caffeine. Nature. 2002;418:774–778. doi: 10.1038/nature00817. [DOI] [PubMed] [Google Scholar]
- Lynch G, Kessler M, Arai A, Larson J. The nature and causes of hippocampal long-term potentiation. Prog Brain Res. 1990;83:233–250. doi: 10.1016/s0079-6123(08)61253-4. [DOI] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–2349. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stefani A, Bernardi G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. Eur J Neurosci. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Saiardi A, Bonci A, Picetti R, Calabresi P, Bernardi G, Borrelli E. Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience. 1997;79:323–327. doi: 10.1016/s0306-4522(97)00135-8. [DOI] [PubMed] [Google Scholar]
- Morice E, Billard JM, Denis C, Mathieu F, Betancur C, Epelbaum J, Giros B, Nosten-Bertrand M. Parallel loss of hippocampal LTD and cognitive flexibility in a genetic model of hyperdopaminergia. Neuropsychopharmacology. 2007;32:2108–2116. doi: 10.1038/sj.npp.1301354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Nairn AC, Greengard P. Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J Neurochem. 1999;72:2015–2021. doi: 10.1046/j.1471-4159.1999.0722015.x. [DOI] [PubMed] [Google Scholar]
- Nishi A, Bibb JA, Snyder GL, Higashi H, Nairn AC, Greengard P. Amplification of dopaminergic signaling by a positive feedback loop. Proc Natl Acad Sci U S A. 2000;97:12840–12845. doi: 10.1073/pnas.220410397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rouge-Pont F, Usiello A, Benoit-Marand M, Gonon F, Piazza PV, Borrelli E. Changes in extracellular dopamine induced by morphine and cocaine: crucial control by D2 receptors. J Neurosci. 2002;22:3293–3301. doi: 10.1523/JNEUROSCI.22-08-03293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell VA. Reprint of “Neurobiology of animal models of attention-deficit hyperactivity disorder.”. J Neurosci Methods. 2007;166:I–XIV. doi: 10.1016/j.jneumeth.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson's disease prevents l-DOPA-induced dyskinesia. Sci Signal. 2009;2:ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Libert F, Vassart G, Vanderhaeghen JJ. Distribution of adenosine A2 receptor mRNA in the human brain. Neurosci Lett. 1991;130:177–181. doi: 10.1016/0304-3940(91)90391-6. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Shi L, Beuming T, Weinstein H, Javitch JA. A pincer-like configuration of TM2 in the human dopamine transporter is responsible for indirect effects on cocaine binding. Neuropharmacology. 2005;49:780–790. doi: 10.1016/j.neuropharm.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Shekim WO, Masterson A, Cantwell DP, Hanna GL, McCracken JT. Nomifensine maleate in adult attention deficit disorder. J Nerv Ment Dis. 1989;177:296–299. doi: 10.1097/00005053-198905000-00008. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998;94:127–152. doi: 10.1016/s0166-4328(97)00175-7. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Welcome SE, Henkenius AL, Toga AW, Peterson BS. Cortical abnormalities in children and adolescents with attention-deficit hyperactivity disorder. Lancet. 2003;362:1699–1707. doi: 10.1016/S0140-6736(03)14842-8. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Le Moine C, Fisone G, Fredholm BB. Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog Neurobiol. 1999;59:355–396. doi: 10.1016/s0301-0082(99)00011-8. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Lindskog M, Ledent C, Parmentier M, Greengard P, Fredholm BB, Fisone G. Regulation of the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa in vivo by dopamine D1, dopamine D2, and adenosine A2A receptors. Proc Natl Acad Sci U S A. 2000;97:1856–1860. doi: 10.1073/pnas.97.4.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, McKinzie DL, Fienberg AA, Nomikos GG, Greengard P. Diverse psychotomimetics act through a common signaling pathway. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Kinsbourne M, Nigg J, Lanphear B, Stefanatos GA, Volkow N, Taylor E, Casey BJ, Castellanos FX, Wadhwa PD. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev. 2007;17:39–59. doi: 10.1007/s11065-007-9019-9. [DOI] [PubMed] [Google Scholar]
- Thapar A, O'Donovan M, Owen MJ. The genetics of attention deficit hyperactivity disorder. Hum Mol Genet. 2005;14:R275–R282. doi: 10.1093/hmg/ddi263. [DOI] [PubMed] [Google Scholar]
- Tilley MR, Gu HH. Dopamine transporter inhibition is required for cocaine-induced stereotypy. Neuroreport. 2008a;19:1137–1140. doi: 10.1097/WNR.0b013e3283063183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley MR, Gu HH. The effects of methylphenidate on knockin mice with a methylphenidate-resistant dopamine transporter. J Pharmacol Exp Ther. 2008b;327:554–560. doi: 10.1124/jpet.108.141713. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rougé-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Troyer KP, Wightman RM. Response times of carbon fiber microelectrodes to dynamic changes in catecholamine concentration. Anal Chem. 2002;74:539–546. doi: 10.1021/ac010819a. [DOI] [PubMed] [Google Scholar]
- Viggiano D, Ruocco LA, Sadile AG. Dopamine phenotype and behaviour in animal models: in relation to attention deficit hyperactivity disorder. Neurosci Biobehav Rev. 2003;27:623–637. doi: 10.1016/j.neubiorev.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Newcorn J, Fowler JS, Telang F, Solanto MV, Logan J, Wong C, Ma Y, Swanson JM, Schulz K, Pradhan K. Brain dopamine transporter levels in treatment and drug naive adults with ADHD. Neuroimage. 2007;34:1182–1190. doi: 10.1016/j.neuroimage.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Wilens TE. Mechanism of action of agents used in attention- deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(Suppl 8):32–38. [PubMed] [Google Scholar]
- Wilens TE, Haight BR, Horrigan JP, Hudziak JJ, Rosenthal NE, Connor DF, Hampton KD, Richard NE, Modell JG. Bupropion XL in adults with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled study. Biol Psychiatry. 2005;57:793–801. doi: 10.1016/j.biopsych.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Yang L, Wang YF, Li J, Faraone SV. Association of norepinephrine transporter gene with methylphenidate response. J Am Acad Child Adolesc Psychiatry. 2004;43:1154–1158. doi: 10.1097/01.chi.0000131134.63368.46. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Belluscio L, Hen R. GOLFα mediates dopamine D1 receptor signaling. J Neurosci. 2000;20(RC91):1–5. doi: 10.1523/JNEUROSCI.20-16-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]