

Abstract
Intraneuronal tau aggregations are distinctive pathological features of Parkinson's disease (PD) with autosomal-dominant mutations in leucine-rich repeat kinase 2 (LRRK2). The most prevalent LRRK2 mutation, G2019S (glycine to serine substitution at amino acid 2019), causes neurite shrinkage through unclear pathogenetic mechanisms. We found that expression of G2019S mutant in Drosophila dendritic arborization neurons induces mislocalization of the axonal protein tau in dendrites and causes dendrite degeneration. G2019S-induced dendrite degeneration is suppressed by reducing the level of tau protein and aggravated by tau coexpression. Additional genetic analyses suggest that G2019S and tau function synergistically to cause microtubule fragmentation, inclusion formation, and dendrite degeneration. Mechanistically, hyperactivated G2019S promotes tau phosphorylation at the T212 site by the Drosophila glycogen synthase kinase 3β homolog Shaggy (Sgg). G2019S increases the recruitment of autoactivated Sgg, thus inducing hyperphosphorylation and mislocalization of tau with resultant dendrite degeneration.
Introduction
Neurodegenerative disorders are often characterized with abnormal protein aggregates, such as the Lewy body (LB) in Parkinson's disease (PD) (Forno, 1996). Although PD is mostly a sporadic disorder, familial forms of PD have been shown recently to associate with several genetic loci (Lesage and Brice, 2009). Mutations in leucine-rich repeat kinase 2 (LRRK2), identified in sporadic and familial PD, are associated with pleomorphic pathological features ranging from typical α-synuclein-positive LB, nonspecific nigral cell degeneration to, surprisingly, tau depositions in degenerated neurites (Wszolek et al., 2004; Zimprich et al., 2004; Rajput et al., 2006). The interconnection between PD and tau is further strengthened by the identification of mutations in the tau-encoding MAPT (microtubule-associated protein tau) gene that are linked to frontotemporal dementia with parkinsonism (FTDP-17) (Wszolek et al., 2006).
Dominant mutations in LRRK2, by far the most prevalent genetic cause for PD, display indistinguishable clinical features in sporadic and familial cases. Among identified mutations of LRRK2, the amino acid substitution G2019S (substitution of glycine to serine at amino acid 2019) augments Lrrk2 kinase activity, causing neurite degeneration and neuronal cell death (Smith et al., 2005; West et al., 2005; MacLeod et al., 2006). Although several studies suggest cellular pathogenesis ranging from abnormal activations of the cell death pathway and autophagy to perturbed homeostasis of pERM (phosphorylated ezrin/radixin/moesin) and F-actin in sprouting neurites in LRRK2 mutants (Plowey et al., 2008; Ho et al., 2009; Parisiadou et al., 2009), the linkage to tau aggregates identified in LRRK2-inflicted PD patients has not been explored.
The axonal protein tau is involved in the establishment and maintenance of neuronal morphogenesis through the activity to bind microtubules, which are regulated by the levels and sites of phosphorylation (Shahani and Brandt, 2002). Among identified kinases that phosphorylate tau, glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (Cdk5) were copurified with microtubules (Hosoi et al., 1995; Billingsley and Kincaid et al., 1997). The involvement of GSK3β in PD development is substantiated by a recent study that PD risk is associated with genetic polymorphisms of GSK3β (Kwok et al., 2005). Knowing that activities of tau kinases are also regulated by phosphorylation events (Kim and Kimmel, 2006; Timm et al., 2008), LRRK2 disease mutations may affect tau phosphorylation through upstream kinases such as GSK3β, thereby contributing to PD pathogenesis.
In this study, we used the system of Drosophila dendritic arborization (DA) neurons to study the pathogenetic mechanism induced by LRRK2 mutations (Gao et al., 1999; Grueber et al., 2003). We showed that expressions of LRRK2 transgenes cause dendrite degeneration, with the kinase activity-elevated G2019S inducing the most severe defect. The increased kinase activity in the G2019S mutant does not affect dendritic protein distribution, but the localization of axonal proteins, including tau, was found in dendrites. We further studied the pathological consequences of tau dendrite distribution and the genetic interaction with Tau in these pathological processes. Furthermore, we demonstrated the molecular mechanism by which G2019S mutation causes tau phosphorylation, tau dendrite localization, and dendrite degeneration through recruiting autoactivated GSK3β.
Materials and Methods
Fly stocks and generation of LRRK2 transgenics.
Human LRRK2 cDNA or carrying mutations G2019S, R1441C (substitution of arginine to cystine at amino acid 1441), G2385R (substitution of glycine to arginine at amino acid 2385), or G2019S-K1906M (substitutions of lysine to methionine at amino acid 1906 in G2019S) were subcloned into the N-terminally Flag-tagged GateWay pUAST vector, and the resulting constructs were injected into w1118 embryos to generate transgenic flies. Flies lines used in this study are 109(2)80 (Gao et al., 1999), ppk-GAL4, Ig1-1 (Grueber et al., 2003), GAL80ts (Suster et al., 2004), UAS-Dscam(TM1)-GFP (Soba et al., 2007), UAS-tauWT (Steinhilb et al., 2007), UAS-tau175/181(Steinhilb et al., 2007), UAS-tau212 (Steinhilb et al., 2007), UAS-tau214 (Steinhilb et al., 2007), UAS-tau-RNAi (Vienna Drosophila RNAi Center), UAS-tauGFP (Murray et al., 1998), UAS-Syt-GFP (Estes et al., 2000), UAS-sggWT (Jia et al., 2002), UAS-sggDN (Jia et al., 2002), and sggnull (Bloomington Drosophila Stock Center).
Antibodies for Western blots, immunoprecipitation, and immunohistochemistry.
Primary antibodies used are against Flag (M2; Sigma), α-Tubulin [Developmental Studies Hybridoma Bank (DSHB)], AT8 (recognizes phosphorylated tau at serine 202 and threonine 205) (Innogenetics), Tau1 (Millipore Bioscience Research Reagents), AT270 (recognizes phosphorylated tau at threonine 175 and tyrosine 181) (Pierce-Endogen), AT100 (recognizes phosphorylated tau at threonine 212 and serine 214) (Innogenetics), GSK-3β (Abcam), phospho-Y216 (recognizes phosphorylated GSK-3 at tyrosine 216) (BD Biosciences), Cdk5 (Abcam), green fluorescent protein (GFP; Invitrogen), dsRed (BD Biosciences), Futsch (22C10; DSHB), BP104 (DSHB), anti-tyrosine hydroxylase (TH; Pel-Freez), anti-5-HT (Sigma), and phalloidin (Jackson ImmunoResearch). In immunostaining of DA neurons, wandering third instar larvae were dissected in cold 1× PBS solution and then fixed in 4% paraformaldehyde with 0.4% Triton X-100. Fluorescence intensities of AT270 and of GFP immunostaining were monitored by dual-channel confocal microscopy and quantified using MetaMorph imaging system (Universal Imaging). For quantification of fluorescence intensities in Figure 2, a square frame was positioned between two dorsolateral groups of dopaminergic neurons to obtain the total GFP fluorescence intensities. The ratios of the GFP fluorescence intensities of different time points relative to control flies are presented in Figure 2, C and D. For quantification in Figure 3, a square frame was positioned over the area of the axon and primary dendrites to obtain their fluorescence intensities in both channels for AT270 and GFP. The frame was then positioned outside the neuron to obtain background fluorescence intensities for subtraction. The ratios of AT270 to GFP are presented in Figure 3E.
Figure 2.

Dendrite degeneration in G2019S-expressing dopaminergic neurons. A, Whole-mount adult brains show dendrites of dopaminergic neurons marked by ddc-GAL4, UAS-dscam [TM1]-GFP. Projected images show reduced GFP signals (green) in adult brains (costained with Phalloidin, red) of G2019S compared to ddc-GAL4 control at 2 weeks after eclosion. At 4 weeks, GFP signals are markedly reduced in both control and G2019S adult brains compared to respective 2-week-old brains. B, Whole-mount adult brains show axons of dopaminergic neurons marked by ddc-GAL4, UAS-syt-GFP. Projected images show equivalent GFP signals in G2019S and ddc-GAL4 control brains at 2 weeks after eclosion. At 4 weeks, whereas GFP signals are slightly reduced in ddc-GAL4, they are markedly reduced in G2019S transgenic adult brains. Dashed boxes enclose the regions for immunointensity scoring. Scale bar: (in A) A, B, 50 μm. C, D, Quantification of GFP immunoreactivities within dashed boxes for ddc-GAL4 control and transgenic adult brains with denoted genotypes at 2 and 4 weeks after eclosion. Significances are compared by Mann–Whitney test; *p < 0.05; **p < 0.01. Error bars indicate mean ± SEM.
Figure 3.

Dendritic localization of tau induced by G2019S. A–D, Class IV ddaC neurons marked by ppk-GAL4-driven mCD8GFP (A), or also carrying Drosophila tau-RNAi (B), G2019S (C), or G2019S-K1906M (D) are immunostained by AT270 for tau (magenta, left panels), and merged images with GFP are shown in the right panels. The axons (arrowheads), cell bodies (asterisks), and dendrites with tau signals (arrows) are indicated. Scale bar: (in D) A–D, 50 μm. E, Quantifications of immunofluorescence intensities in the axon and dendrites for indicated genotypes (listed at right with numbers of samples scored in parentheses) are shown as ratios of AT270/GFP (mean ± SEM). Significant differences (by Mann–Whitney test) are shown in comparison with the axons (⋀p < 0.05) and dendrites (*p < 0.05; **p < 0.01).
Results
G2019S induces DA dendrite degeneration
To examine the potential impacts on DA dendrites by human disease forms of Lrrk2, we generated transgenic lines expressing Flag-tagged wild-type (WT) Lrrk2, or Lrrk2 carrying amino acid substitutions G2019S (abbreviated as G2019S and thereafter), R1441C, or G2385R. Unlike G2019S and R1441C mutations, which are tightly associated with familial PD and have been shown to elevate the kinase activity of Lrrk2 (Smith et al., 2005; West et al., 2005; MacLeod et al., 2006), G2385R is the Asian-specific polymorphism that enhances the risk of PD development by twofold (Farrer et al., 2007). Transgenic lines with similar expression levels were selected for subsequent experiments (Fig. 1A). Expressions of these LRRK2 transgenes in DA neurons by the GAL4 driver 109(2)80 cause dendrite arborization defects, as shown by the quantification of numbers of dendritic ends in the dorsal field of A6 segments in third-instar larvae. Whereas R1441C and G2385R cause similar levels of defects to wild-type LRRK2, G2019S causes the strongest reduction in the number of dendritic ends among all transgenes (Fig. 1B–D) (for phenotypes of wild-type Lrrk2, R1441C and G2385R, see supplemental Fig. S1A–C, available at www.jneurosci.org as supplemental material). In G2019S-expressing DA neurons, the dendritic field is significantly reduced. The lengths of both lower- and higher-order branches are shortened, and many terminal arbors fail to reach the segmental and dorsal-midline boundaries (Fig. 1D). To examine whether G2019S also affects axonal processes, the class IV DA-neuron-specific ppk-GAL4 was used to drive wild-type Lrrk2 or G2019S expression. Axonal terminals of the class IV DA neurons remain normal, targeting to the ventral nerve cord (Fig. 1E,F), although the complexity of dendrites is severely compromised, displaying shorter and fewer terminal arbors and leaving some receptive fields uncovered (supplemental Fig. S1D,E, available at www.jneurosci.org as supplemental material). By scoring the number of dendrites in each order, we found that higher-order dendrites are more vulnerable to expressions of G2019S or R1441C mutants than lower-order ones (supplemental Fig. S1F, available at www.jneurosci.org as supplemental material). Expressions of these transgenes in simple class I DA neurons by Ig1-1 GAL4, however, cause no observable defect (supplemental Fig. S1G–I, available at www.jneurosci.org as supplemental material). The effect of G2019S to sensory neurons in the peripheral nervous system seems specific, since we failed to observe significant difference in the dendrite complexity of motor neurons expressing G2019S (data not shown).
Figure 1.
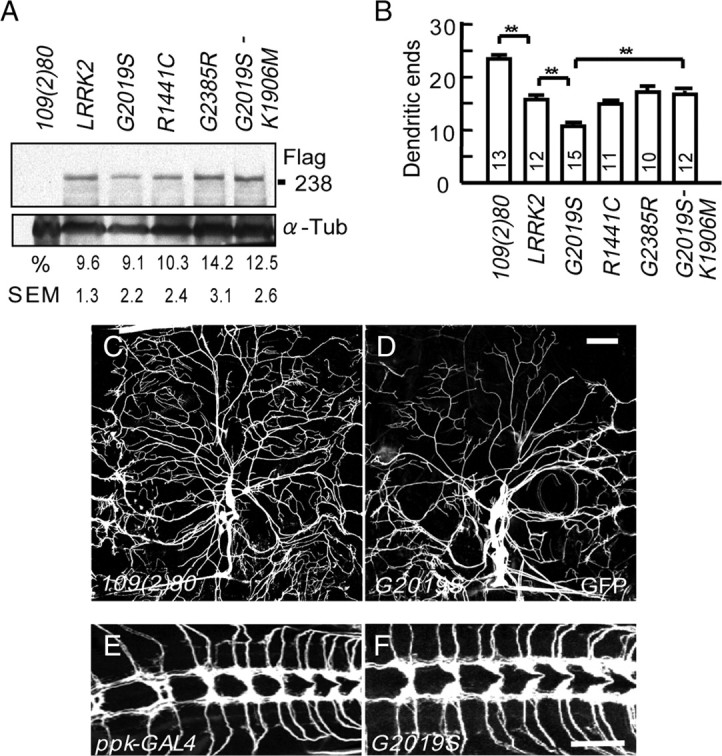
Degeneration of DA neuron dendrites induced by LRRK2 mutants. A, Expression levels of Flag-tagged human LRRK2 transgenes of wild-type or with amino acid substitutions G2019S, R1441C, G2385R, and G2019S-K1906M driven by 109(2)80 in DA neurons of third-instar larvae are shown in Western blots using the Flag antibody (top) or α-Tubulin (α-Tub, bottom). The ratio of Flag to α-Tub immunointensities are averaged from three experiments and shown as mean percentages and SEMs. B, Quantification of dendritic ends in dorsal fields of A6 segments of third instar larvae that express individual LRRK2 transgenes and mCD8GFP by 109(2)80. Averages are the mean ± SEM of dendritic ends per 10,000 μm2 area. Significance is compared by Student's t test with **p < 0.01, and numbers of segments scored are inside columns. C, D, Images of dorsal DA dendrites in the A6 segment of third instar larvae of the 109(2)80 control (C) or the 109(2)80-driven G2019S (D). Dendrites are labeled by the coexpressed mCD8GFP. E, F, Axonal terminals of ppk-GAL4 control (E) and G2019S (F) ddaC neurons. Scale bars: (in D) C, D, 50 μm; (in F) E, F, 10 μm.
Given that G2019S has the highest population-attributable risk among all LRRK2 mutations and induces the most severe defects in dendrite arborization among all LRRK2 transgenes, we decided to focus on the pathogenetic mechanism underlying G2019S-induced dendritic defects (Lesage et al., 2006). To examine whether the kinase activity of G2019S is involved in causing the dendritic defect, Lys1906 that orients the γ-phosphate of ATP onto Lrrk2 was mutated to Met in G2019S to generate the kinase-decreased G2019S-K1906M mutant (Smith et al., 2005; West et al., 2005; MacLeod et al., 2006; Plowey et al., 2008). It has been shown that the K1906M mutation nullifies the increased level of kinase activity in G2019S but preserves its basal level similar to wild-type Lrrk2 (Luzon-Toro et al., 2007). When expressed by either 109(2)80 in all DA neurons or ppk-GAL4 in class IV DA neurons, G2019S-K1906M behaves similarly to the wild-type Lrrk2 in inducing dendritic defects (Fig. 1B; supplemental Fig.S1F, available at www.jneurosci.org as supplemental material), suggesting that the gain-of-function G2019S mutant aggravates dendrite arborization defect by the elevated level of Lrrk2 kinase activity.
The distinct dendrite phenotypes of G2019S DA neurons suggest a possibility of dendrite retraction, leaving some dendritic field uncovered (Fig. 1D; supplemental Fig. S1E, available at www.jneurosci.org as supplemental material). To further confirm the involvement of degeneration, we used the temperature-sensitive form of GAL80 (GAL80ts) that functions as a transcription repressor for GAL4 at the permissive temperature (25°C) but allows GAL4-dependent transcription at 30°C because of the inactivation of GAL80ts, providing a temporal regulation of the GAL4/UAS system (Suster et al., 2004). The G2019S transgenic larvae carrying the GAL4 driver 109(2)80 and tubP-GAL80ts were reared at 25°C after egg laying and shifted to 30°C at day 3 to induce expression of the transgene (see scheme in supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). The control larvae carrying only 109(2)80 and tubP-GAL80ts were performed in parallel. When larvae were continuously reared at 25°C without temperature shift, the numbers of dendritic ends were comparable between G2019S and 109(2)80 control larvae at day 4 or day 6 (supplemental Fig. S2B–E,J, available at www.jneurosci.org as supplemental material), suggesting that G2019S expression is silenced in the presence of functional GAL80 at 25°C. When G2019S larvae were shifted to 30°C at day 3, the DA dendrites grow normally when examined at day 4, compared to the GAL4 control (supplemental Fig. S2F,H,J, available at www.jneurosci.org as supplemental material), indicating that G2019S affects little, if any, dendrite growth in early stages of larval growth. Strikingly, G2019S DA neurons display a sparse dendrite pattern at day 6; the number of dendritic ends is significantly lower than that of G2019S DA neurons at day 4 and that of the GAL4 control at day 6 (supplemental Fig. S2G–J, available at www.jneurosci.org as supplemental material). Therefore, after G2019S expression, the dendrite complexity is reduced in later larval stages compared to that in early stages, an indication of degeneration.
Dendrite degeneration precedes axon degeneration in G2019S dopaminergic neurons
With dopaminergic neurons being the prime pathological target of PD, we examined whether dendrite degeneration is also the prominent feature of dopaminergic neurons expressing LRRK2 mutants. We first determined whether the introduction of LRRK2 transgenes in dopaminergic neurons causes parkinsonism-like phenotypes in adult flies. Using dopa decarboxylase (ddc)-GAL4 to drive G2019S expression in Drosophila dopaminergic neurons, locomotion of adult flies is affected by the third week after eclosion, and the viability shows significant decline at the fourth week compared to ddc-GAL4 control (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material). Adult brains dissected from transgenic flies at the fourth week after eclosion were immunostained with anti-TH antibodies. We found a significant loss of dopaminergic neurons in both dorsomedial and dorsolateral groups of G2019S adult brains at the fourth week after eclosion compared to ddc-GAL4 controls of the same age (supplemental Fig. S3C–E, available at www.jneurosci.org as supplemental material). We further examined whether G2019S affects serotonin (5-HT) neurons that also express ddc-GAL4 by using anti-5-HT immunohistochemical analysis. We found that the brains of G2019S flies at the fourth week after eclosion displayed 5-HT immunoreactivity similar to that of the control flies (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). These results suggest that G2019S specifically induces the loss of dopaminergic neurons, thus compromising life span and motor activity.
We then examined whether dendrite degeneration occurs before neuronal loss in G2019S dopaminergic neurons. As revealed by Dscam (Down's syndrome cell adhesion molecule)-[TM1 (transmembrane 1)]-GFP that labels dendrites (Soba et al., 2007), G2019S expression in ddc-GAL4-neurons results in significant reduction of dendritic structures in 2- and 4-week-old adult brains (Fig. 2A,C). Quantification of GFP immunoreactivity in a defined region shows that dendritic GFP signal in G2019S animals in 2-week-old brains is expressed at 0.6-fold the level in ddc-GAL4 control animals (Fig. 2A,C). With decreasing numbers of neurons in both control and G2019S animals at the fourth week (supplemental Fig. S3D,E, available at www.jneurosci.org as supplemental material), the dendritic GFP immunoreactivity in G2019S animals is decreased to 0.5-fold that in the ddc-GAL4 control of the same age (Fig. 2C). To examine the degeneration of axonal processes, the transgene for synaptotagmin (Syt)-GFP is driven by ddc-GAL4 in dopaminergic neurons. In 2-week-old adults, G2019S neurons retained correct projection and targeting in the ventral nerve cord (data not shown). No significant difference in the levels of GFP immunoreactivity was measured between the ddc-GAL4 control and G2019S adults at this stage (Fig. 2B,D). When advancing to the fourth week after eclosion, the axonal GFP signals decreased in both ddc-GAL4 and G2019S brains. The axonal GFP immunoreactivity in G2019S animals is decreased to 0.85-fold of that in the ddc-GAL4 control (Fig. 2B,D). Together, these analyses inferred from GFP immunoreactivity suggest that dendrite degeneration precedes axon degeneration and cell-body loss in G2019S dopaminergic neurons.
G2019S causes tau localization in DA dendrites
To dissect the molecular pathogenetic mechanism underlying G2019S-induced dendrite degeneration, the larval DA neurons were further used as the system for additional study for the following reasons: First, the two-dimensional elaboration of dendrites is feasible for quantitative analysis of dendrite phenotypes and protein localization. Second, the axon and dendrites can be easily differentiated by their stereotypical projection patterns. Third, several class-specific GAL4 drivers are available for different expression levels.
The axonal protein tau is detected as aggregates in neurites of dopaminergic neurons in the autopsied brains of G2019S-inflicted PD patients and in dorsal striatum and piriform cortex of R1441G transgenic mice (Wszolek et al., 2004; Zimprich et al., 2004; Li et al., 2009). It is not clear whether tau aggregation is a consequence of degeneration or indeed contributes to the LRRK2 mutant-induced degeneration. To test this, G2019S expressing-DA neurons were examined for the distribution of tau protein. We first screened a panel of antibodies (see Materials and Methods) for recognizing endogenous tau proteins in DA neurons, and found that AT270 labels predominantly the axon of ppk-GAL4-driven mCD8GFP-labeled class IV DA neurons (Fig. 3A, arrowhead). Weak punctates are also detected in some but not all primary dendrites (Fig. 3A, arrow). To confirm that AT270 recognizes the endogenous tau protein, the Drosophila tau-RNAi transgene was used to knock down tau expression. Reduction of tau expression by tau-RNAi was detected in the Western blot for larval extracts (data not shown). The immunoreactivity by AT270 in the axon is significantly reduced when tau-RNAi is driven by ppk-GAL4 (Fig. 3B,E), indicating the specificity of AT270 in the recognition of the Drosophila tau protein in DA neurons. Strikingly, in class IV DA neurons that express G2019S, the tau protein localizes in all primary dendrites, and in many cases the localization reaches secondary branches (Fig. 3C, arrows). Axonal distribution of tau, however, is unaltered (Fig. 3C, arrowhead, E, statistics). The mislocalization of tau depends on the elevated kinase activity of G2019S because the tau localization is predominantly limited to the axon of G2019S-K1906M DA neurons (Fig. 3D,E). The expression levels of tau recognized by AT270 are similar in these transgenic lines, as shown by Western blot analyses (see Fig. 6A). In addition to tau, we also observed the mislocalization of the axonal protein Syt-GFP to dendrites in G2019S DA neurons (supplemental Fig. S4A,B, available at www.jneurosci.org as supplemental material). Conversely, the distribution of the dendrite-specific Dscam-[TM1]-GFP fusion protein in DA neurons is not affected (data not shown). These results suggest that G2019S promotes the localization of axonal proteins, including tau, to dendrites in DA neurons.
Figure 6.
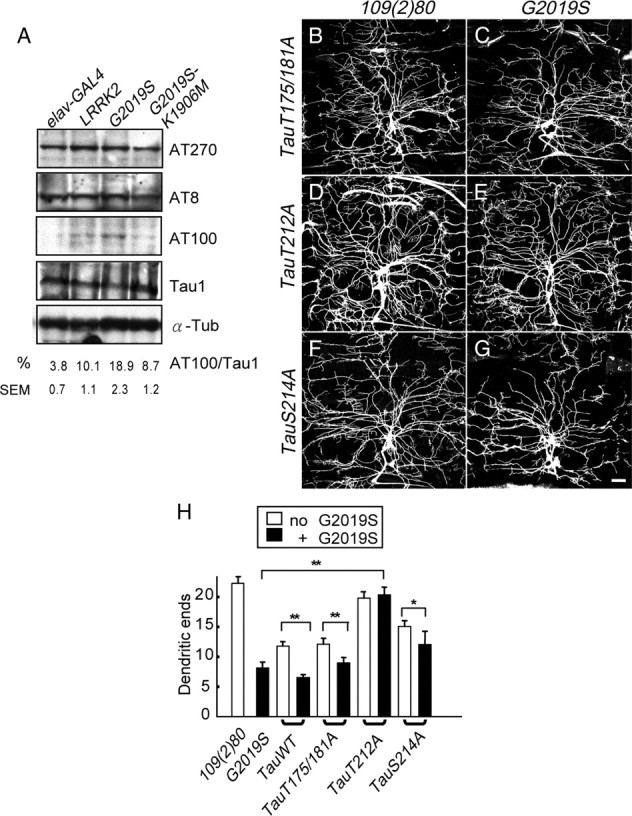
Tau phosphorylation at T212/S214 in G2019S neurons. A, Western blot analysis of adult brain lysates of the elav-GAL4 control or elav-GAL4-driving LRRK2, G2019S, or G2019S-K1096M expressions to detect phospho-tau levels by antibodies AT270 (recognizing pT175/pT181), AT8 (pS202/pT205), and AT100 (pT212/pS214). Tau 1 detects total tau protein and α-Tubulin (α-Tub) as control. The immunoreactivities of AT100 are shown as a percentage relative to those of Tau 1, averaged from three independent experiments. B–G, Images of dorsal DA dendrites in the A6 segment of third instar larvae with 109(2)80 driving transgenes TauT175/181A (B), G2019S + TauT175/181A (C), TauT212A (D), G2019S + TauT212A (E), TauS214A (F), or G2019S + TauS214A (G). Dendrites are marked by coexpressed mCD8GFP. Scale bar: (in G) B–G, 50 μm. H, Quantification of dendritic ends for 109(2)80 (n = 13), G2019S (n = 12), TauWT (n = 15), TauWT + G2019S (n = 8), TauT175/181A (n = 10), TauT175/181A + G2019S (n = 8), TauT212A (n = 10), TauT212A + G2019S (n = 10), TauS214A (n = 10), and TauS214A + G2019S (n = 10). Averages are the mean ± SEM of dendritic ends per 10,000 μm2 area, and significance is compared by Mann–Whitney test with *p < 0.05; **p < 0.01.
G2019S enhances Tau-induced dendrite degeneration, microtubule fragmentation, and inclusion formation
With the mislocalization of axonal proteins in dendrites, it is imperative to assess the contribution of tau in the G2019S-induced dendrite degeneration. To assess this, tau-RNAi that can effectively suppress tau protein expression was introduced into G2019S DA neurons. Although expression of tau-RNAi alone causes no defect on the dendrite morphology of DA neurons (Fig. 4B,I, statistics), coexpression of tau-RNAi alleviates dendritic defects caused by G2019S (Fig. 4C,D). The dorsal dendritic field is almost fully restored and the number of dendritic ends is also markedly increased (Fig. 4I). These results suggest that tau contributes significantly to G2019S-induced dendrite degeneration.
Figure 4.

G2019S and lrrk modify Tau-induced dendrite degeneration. A–H, Images of dorsal DA dendrites in the A6 segment of third instar larvae of the 109(2)80 control (A) or DA dendrites that also expresses transgenes UAS- Drosophila tau RNAi (B), G2019S (C), G2019S + Drosophila tau-RNAi (D), Tau-GFP (E), Tau-GFP in e03680/+ (F), or Tau-GFP + G2019S with typical (G) or severe (H) phenotypes. Asterisks in H mark cell bodies without apparent dendritic processes. I, Quantification for dendritic ends in dorsal fields of A6 segments of third instar larvae with genotypes listed at right. Averages are the mean ± SEM of dendritic ends per 10,000 μm2 area. Significances are compared by Mann–Whitney test; *p < 0.05; **p < 0.01. Numbers of segments scored are in parentheses. Scale bars: G (for A–G), H, 50 μm.
Ectopic expression of the bovine Tau-GFP fusion transgene in DA neurons causes severe dendrite degeneration (Fig. 4E). Such defect is strongly enhanced with the coexpression of G2019S (Fig. 4G,I). In severe cases, only six of the eight DA somas are present in the dorsal field, an indication of neuronal loss that is not detected in the expression of G2019S or tau-GFP alone (Fig. 4H). Thus, G2019S and tau-GFP interact synergistically in inducing dendrite degeneration and, in severe cases, neuronal loss.
The genetic interaction between G2019S and microtubule-binding protein tau prompted us to examine the structure of dendritic microtubules, which can be revealed by immunostaining for the MAP1B-like protein Futsch (Hummel et al., 2000; Roos et al., 2000). In the ppk-GAL4 control, dendrites of class IV DA neurons display a continuous and smooth Futsch-expression pattern along the process (Fig. 5A). Expression of tau-GFP causes a defect in the microtubule structure, as shown by the discontinuous and irregular Futsch staining pattern in dendrites (Fig. 5B, arrowhead). Expression of G2019S also causes discontinuous Futsch staining in dendrites, an indication that G2019S functions in the same pathway as tau in causing dendrite degeneration (Fig. 5D, arrowhead). This effect of G2019S depends on the elevated kinase activity because the Futsch staining pattern is normal in dendrites of DA neurons expressing either wild-type Lrrk2 or G2019S-K1906M (Fig. 5C,E). Furthermore, combination of tau-GFP and G2019S causes severe microtubule destruction in dendrites with large gaps void of Futsch expression (Fig. 5F, arrowhead). Very often, dendrites contain nearly no Futsch signals (Fig. 5F, asterisk). Such microtubule deficiency has not been detected in either G2019S or tau-GFP alone. Therefore, G2019S and tau act synergistically in causing microtubule fragmentation and dendrite degeneration, suggesting that they function closely in the degenerative process.
Figure 5.

Microtubule fragmentation in G2019S- and Tau-expressing dendrites. A–G, Low-order trunks of class IV ddaC neurons visualized by ppk-GAL4-driven mCD8GFP (A), or also carrying transgenes Tau-GFP (B), wild-type LRRK2 (C), G2019S (D), G2019S-K1906M (E), G2019S + Tau-GFP (F), or Tau-GFP in an e03680/+ background (G). Third-instar larvae were dissected and stained for the microtubule-associated protein Futsch (left panels) and GFP (merged in right panels). Arrowheads in B, D, and F indicate dendrites with discontinuous Futsch expression, and asterisks in F mark dendrites containing nearly no Futsch signals. Scale bar: (in A) A–G, 15 μm.
Overexpression of tau proteins is sufficient to induce tau inclusions in neurites (Buee et al., 2000; Lee et al., 2001; Gong et al., 2005). With the effect of G2019S on tau distribution and the enhancement of tau-induced dendrite degeneration and microtubule fragmentation, we then tested whether G2019S also enhances the formation of tau inclusions in dendrites. The tau-GFP fusion protein, when expressed in class IV DA neurons by ppk-GAL4, localizes in dendrites and induces spheroid inclusion-like structures (supplemental Fig. S4C–C″, available at www.jneurosci.org as supplemental material). These inclusions form preferentially at distal dendrites (supplemental Fig. S4C′, arrows, available at www.jneurosci.org as supplemental material), although some are also present at proximal segments, causing local swelling (supplemental Fig. S4C″, arrow, available at www.jneurosci.org as supplemental material). Strikingly, G2019S enhances the formation of spheroid inclusions that appear much more frequent in both distal and proximal dendrites (supplemental Fig. S4D–D″, available at www.jneurosci.org as supplemental material). The spheroid structures along the primary and secondary dendrites with diameters 1.5-fold larger than the neighboring dendritic arbors were counted (supplemental Fig. S4F, available at www.jneurosci.org as supplemental material). The presence of G2019S enhances the formation of spheroid structures by fivefold. Therefore, G2019S is able to enhance tau-induced microtubule fragmentation and inclusion formation, two processes that can lead to dendrite degeneration and neuronal loss.
Drosophila lrrk mutations suppress tau-induced dendrite degeneration and inclusion formation
The Drosophila genome includes a single LRRK2 homolog lrrk (CG5483). The PBac(PB)e03680 insertion in the lrrk locus was regarded as a null allele and has a subtle defect in the loss of dopaminergic neurons in adult flies (Lee et al., 2007). Also, the dendrite arborization of DA neurons is normal in homozygous larvae for the e03680 allele (data not shown). Overexpression of lrrk in DA neurons suppresses dendrite arborization (in end points/1 × 104 μm2: 109(2)80, 23.3 ± 2.3, n = 8; UAS-lrrk, 16.8 ± 2.2, n = 8; p = 0.031 by Mann–Whitney test), with phenotypes indistinguishable from the overexpression of human LRRK2, suggesting that Drosophila lrrk and human LRRK2 function similarly in regulating dendrite arborization.
We then addressed whether the gene dosage of lrrk affects the toxicity of tau in dendrite degeneration. With the introduction of one e03680 allele, dendrites of the tau-GFP-expressing neurons are longer and the number of dendritic ends is increased (Fig. 4F,I). The suppression by the lrrk mutation is also observed in inclusion formation and microtubule fragmentation. The number of tau-GFP inclusions is reduced by more than fourfold in e03680/+ heterozygotes (supplemental Fig. S4E–E″,F, available at www.jneurosci.org as supplemental material). The discontinuous Futsch pattern in tau-GFP dendrites regains the smooth and continuous pattern by the introduction of the e03680 allele (Fig. 5G). Therefore, reduction of the lrrk gene dosage is able to alleviate pathogenic phenotypes induced by tau overexpression, including dendrite degeneration, inclusion formation, and microtubule fragmentation.
The human Tau T212A phosphomutant suppresses G2019S-induced dendrite degeneration
Our results suggest a genetic correlation between G2019S and tau in microtubule fragmentation and inclusion formation, two pathogenic processes involving tau hyperphosphorylation (Blard et al., 2007). It is therefore important to examine whether G2019S affects the phosphorylation status of tau. Antibodies that specifically recognize phosphorylated serine-proline or threonine-proline sites of tau were used, including AT270 (pT175/pT181), AT8 (pS202/pT205), and AT100 (pT212/pS214) (Steinhilb et al., 2007). Western blots show that the phosphorylation levels of tau protein at T175/T181 and S202/T205 sites are unaltered by neuronal expressions of wild-type and mutant LRRK2 transgenes, compared to the elav-GAL4 GAL4 driver alone (Fig. 6A, top two panels). In contrast, the level of phospho-tau at the T212/S214 sites is elevated in G2019S-expressing neurons, compared to wild-type Lrrk2 or G2019S-K1906M (Fig. 6A, third panel, statistics at bottom). Expression levels of total tau proteins measured by the Tau1 antibody are comparable among all samples (Fig. 6A, fourth panel). These results suggest that G2019S, through its elevated kinase activity, directly or indirectly induces tau phosphorylation at the T212/S214 sites.
If phosphorylation at the tau T212/S214 sites is critical to mediate the G2019S activity, mutating T212 or S214 with alanine (TauT212A and TauS214A transgenes) would abrogate G2019S-induced dendrite degeneration. As a control, TauWT and TauT175/181A transgenes were tested in parallel. Expressions of these Tau transgenes, which are at comparable levels (Steinhilb et al., 2007), suppress dendrite arborization of DA neurons with different severities (Fig. 6H). Whereas tauWT and tauT175/181A suppress dendrite arborization strongly, tauS214A confers intermediate suppression, and tauT212A has almost no suppression (Fig. 6B,D,F,H). When coexpressed in DA neurons, G2019S causes further dendrite degeneration in DA neurons that express tauWT, tauT175/181A, or tauS214A (Fig. 6C,G,H) (data not shown). In contrast, G2019S fails to cause dendrite degeneration in tauT212A-expressing neurons, suggesting that T212A blocks the G2019S neuronal toxicity (Fig. 6E,H). Together, the analyses of phospho-tau levels and genetic suppression support the model that the dendritic defect caused by G2019S is mediated through phosphorylation at the tau T212 site.
GSK3β mediates the activity of G2019S in dendrite degeneration
The T212 site of tau may be directly phosphorylated by hyperactivated G2019S, which was not detected in the phosphorylation assay we performed (data not shown). As the tau T212/S214 sites are known targets for GSK3β (Zheng-Fischhofer et al., 1998), we then tested whether Shaggy (Sgg), the Drosophila homolog of GSK3β, mediates G2019S-induced dendrite degeneration. By introducing a single copy of the sggnull allele, dendrite arborization in G2019S DA neurons is significantly restored (Fig. 7A–C), suggesting that Sgg functions in the G2019S pathogenic pathway to cause dendrite degeneration. We also tested the introduction of dominant-negative Sgg (SggDN) in blocking the G2019S effect. Whereas SggDN alone causes significant dendritic defects as compared to SggWT, the dendritic defect is not enhanced by SggDN and G2019S coexpression (supplemental Fig. S5A–E, available at www.jneurosci.org as supplemental material). In supporting that Sgg kinase activity is involved in the G2019S pathogenesis, we found that expression of SggWT has no effect on tau phosphorylation at T212/S214 sites, but SggDN reduces tau phosphorylation by 77% (supplemental Fig. S5F, available at www.jneurosci.org as supplemental material). Together, these results suggest that tau phosphorylation by Sgg at T212/S214 is important for G2019S to cause dendrite degeneration, which can be blocked by SggDN.
Figure 7.
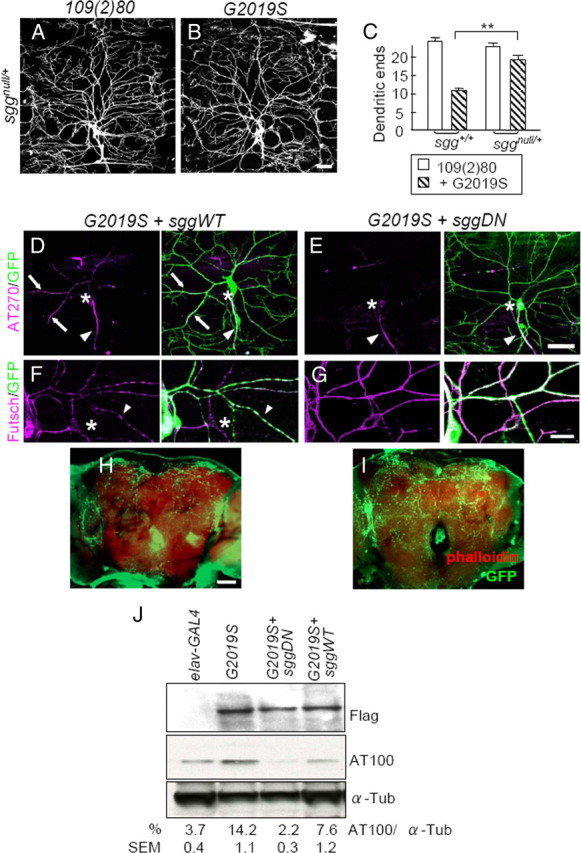
Sgg suppresses G2019S-induced dendrite degeneration. A, B, Images of dorsal DA dendrites in A6 segments of third instar larvae of 109(2)80 (A) or G2019S (B) in an sggnull/+ background. Dendrites are marked by coexpressed mCD8GFP. C, Quantification of dendritic ends for 109(2)80 (n = 7), G2019S (n = 8), sggnull/+ (n = 11), and sggnull/+;G2019S (n = 11). Averages are the mean ± SEM of dendritic ends per 10,000 μm2 area, and significance is compared by Mann–Whitney test with **p < 0.01. D–G, ddaC neurons marked by ppk-GAL4-driven mCD8GFP with coexpression of G2019S + sggWT (D, F) or G2019S + sggDN (E, G). D, E, AT270 (magenta) and GFP (green) staining. The axons (arrowheads), cell bodies (asterisks), and dendrites with tau signals (arrows) are indicated. Scale bar is 50 μm. F, G, Futsch staining (magenta). Arrowheads indicate dendrites with discontinuous Futsch expression, and asterisks mark dendrites containing no Futsch signals. H, I, Whole-mount adult brains shows dendrites of dopaminergic neurons marked by ddc-GAL4, UAS-dscam [TM1]-GFP, as done for Figure 2, A and B. Projected images show reduced GFP signals (green) in adult brains (costained with Phalloidin, red) of G2019S + SggDN compared to G2019S + sggWT control at 2 weeks after eclosion. J, Western blot analysis of adult brain lysates of elav-GAL4 control or elav-GAL4-driven G2019S, G2019S + sggWT, and G2019S + sggDN by Flag, AT100, or α-Tubulin (α-Tub). The immunoreactivities of AT100 are shown as percentages to those of α-Tub, averaged from three independent experiments. Scale bars: B (for A, B), E (for D, E), H (for H, I), 50 μm; F (for F, G), 15 μm.
We then tested whether dendrite phenotypes induced by G2019S can be suppressed by compromising the Sgg activity. Expression of SggWT in G2019S DA neurons maintains the dendrite localization of tau (Fig. 7D), similarly to expression of G2019S alone (Fig. 3C). However, expression of SggDN restricts tau to the axon in G2019S-expressing neurons (Figs. 3E, statistics, 7E). In addition, SggDN suppresses the toxic effects of G2019S on microtubule fragmentation, showing smooth pattern of Futsch staining in dendritic processes (Fig. 7G). As a control, SggWT fails to suppress discontinuous Futsch staining pattern in G2019S-expresing neurons and sometimes enhances such phenotype as revealed by dendrites without Futsch staining (Fig. 7F, asterisk). We next examined whether the level of phospho-T212/S214 tau, recognized by the AT100 antibody in G2019S DA neurons would also be altered by the expression of Sgg. The AT100 level that is upregulated in G2019S neurons is completely suppressed by coexpressed SggDN but not SggWT (Fig. 7J). Therefore, compromising Sgg activities in DA neurons is able to block the detrimental effects induced by G2019S, including upregulated phosphorylation and dendrite localization of tau, microtubule fragmentation, and dendrite degeneration.
We further tested whether Sgg also mediates G2019S in causing dendrite degeneration of dopaminergic neurons. Coexpression of SggWT in G2019S-expressing neurons by ddc-GAL4 maintains the disrupted dendritic structure in 2-week-old adult brains, similar to expression of G2019S alone (compare Figs. 2A, 7H). However, expression of SggDN restores dendritic patterns caused by G2019S (Figs. 2C, statistics, 7I). In addition, SggDN suppresses the effects of G2019S on the life-span-shortening and locomotion defects (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material). Therefore, Sgg is the major executor of G2019S in causing dendrite degeneration in both peripheral DA neurons and central dopaminergic neurons.
G2019S associates with autoactivated Sgg to increase tau phosphorylation
We further examined how G2019S regulates Sgg activities. In neurons expressing LRRK2 transgenes, the protein levels of Sgg are enhanced in Western blot analyses using the GSK3β antibody, compared to the GAL4 control (Fig. 8A, left). The upregulated levels of Sgg are almost identical among all LRRK2 transgenes, which is not sufficient to account for the more aggravated defect caused by G2019S (Fig. 1B). We then tested whether Lrrk2 and Sgg form a protein complex to mediate tau phosphorylation. In immunoprecipitates by the anti-Flag antibody, we found that G2019S is able to associate with Sgg (Fig. 8A, right). Quantitative analysis of the immunointensities indicates that the level of G2019S-associated Sgg is nearly threefold that of G2019S-K1906M-associated Sgg and more than fivefold that of wild-type Lrrk2-associated Sgg (Fig. 8A, ratios below the panel). Activity of GSK3β is regulated by phosphorylation at the Ser 9 site (Papadopoulou et al., 2004). In the immunoprecipitates from G2019S-expressing neurons, no significant changes in the levels of Sgg pS9 were detected (data not shown). However, GSK3β is autoactivated at the Y216 site in the activation loop through a chaperone-assisted process (Lochhead et al., 2006). The Y216 site is conserved at Y214 of Sgg, which is also recognized by the phospho-Y216 antibody. The phospho-level of Sgg pY214 in the Flag-G2019S immunoprecipitates is at least fourfold that precipitated by wild-type Lrrk2 or G2019S-K1906M (Fig. 8B). It thus appears that the kinase-activated G2019S is able to enhance the levels of and associate with autoactivated pY214, rather than pS9 Sgg.
Figure 8.

G2019S associates with Sgg to phosphorylate tau at T212/S214. A, Western blot analysis of adult brain lysates of elav-GAL4 control or elav-GAL4-driven LRRK2, G2019S, or G2019S-K1096M to detect expressions of Lrrk2 proteins (Flag antibody), Sgg (GSK3β antibody), and α-Tub (left panels). Immunoprecipitated wild-type and mutant forms of Lrrk2 are shown by blotting with the Flag antibody, and the coprecipitated Sgg is revealed by GSK3β antibodies (right panels). Percentages of GSK3β to Flag immunointensities of the IP (immunoprecipitation) complex are averages from three experiments (bottom right). B, Similarly, Flag immunoprecipitates are analyzed by Western blots using antibodies against Flag or GSK3β-Y216p antibody. Percentages of Y216p to Flag signals are shown (n = 3). The value of the mean percentage is expressed as a percentage relative to IP Flag controls. C, Flag immunoprecipitates were incubated with recombinant human tau in the presence of 150 mm LiCl (left four lanes) or NaCl (right four lanes). Phosphorylation of tau at T212/S214 is revealed by Western blot using AT100 antibody. α-Tub (bottom) was used as a loading control. The immunoreactivities of AT100 are shown as percentages relative to those of α-Tub, averaged from three independent experiments. D, Western blot analyses fail to detect the association between CDK5 and G2019S, and were performed essentially as for association with Sgg in A, except that the CDK5 antibodies were used to substitute the GSK3β antibodies.
We further tested whether G2019S-associated Sgg is active in tau phosphorylation at T212/S214 sites. The Flag immunoprecipitates were incubated with the recombinant tau substrate, and the phosphorylation at T212/S214 sites was revealed by the AT100 antibody in Western blots. In neurons expressing wild-type Lrrk2, tau phosphorylation at the T212/S214 sites was almost undetectable, identical to the elav-GAL4 control. In G2019S neurons, however, the Flag immunoprecipitate was able to phosphorylate tau, and the level of phosphorylation was dramatically reduced in G2019S-K1906M neurons (Fig. 8C, four lanes from right). To test whether the phosphorylation at the T212/S214 sites of tau is indeed through Sgg, LiCl that blocks GSK3β autoactivation was added during the incubation. With the presence of LiCl but not NaCl in the incubation buffer, all immunoprecipitates failed to phosphorylate the recombinant tau protein at the T212/S214 sites (Fig. 8C, four lanes from left). Thus, Sgg that associates with G2019S appears to be active for the phosphorylation of the tau T212/S214 sites.
CDK5 is also the upstream kinase for tau protein at T212/S214 sites (Liu et al., 2002). We then examined whether G2019S regulates Drosophila CDK5 activity as we have done for GSK3β. In neurons that express either wild-type LRRK2, G2019S, or G2019S-K1906M transgenes, the protein levels of CDK5 are comparable (Fig. 8D, left), and no coprecipitations of CDK5 can be detected with any Lrrk2 immunoprecipitates (Fig. 8D, right). In addition, no significant genetic interaction between G2019S and CDK5 can be detected (data not shown). These analyses rule out a direct relationship between G2019S and CDK5, and further strengthen the specific role of GSK3β in mediating G2019S-induced dendrite degeneration phenotypes.
Discussion
Drosophila melanogaster has been a model organism for studying neurodegenerative diseases (Sang and Jackson, 2005; Lu and Vogel, 2009). The adult compound eye has been used extensively to screen for modifiers of human disease genes, in the attempt to dissect genetic pathways involved in pathogenesis. Equipped with complex but stereotypic dendrite patterns that can be easily visualized and quantified, DA neurons have become an ideal model to study pathogenetic mechanisms causing neurite degeneration characteristic in several neurodegenerative diseases, including G2019S-induced dendrite degeneration (Iseki et al., 2001). With this system, we have studied the underlying mechanism in G2019S-induced neurite shortening (MacLeod et al., 2006). We have observed that dendrites are more vulnerable than the axon to LRRK2 G2019S mutant expressions in both dopaminergic neurons and DA neurons. Neurites of the peripheral sensory nervous system have been shown recently to undergo degeneration in skin biopsy findings of PD patients, suggesting the neurite defect is a common process devastating both central and peripheral nervous systems in PD (Nolano et al., 2008).
In DA neurons, we show that the Lrrk2 G2019S dominant mutant causes several dendrite defects, including tau mislocalization in dendrites, tau hyperphosphorylation at the T212/S214 sites, microtubule fragmentation, and dendrite degeneration, which phenocopy tau overexpression in DA neurons. These phenotypic similarities suggest that tau and G2019S likely function in the same pathway in causing neuronal degeneration. Importantly, our data further show that tau is a prominent mediator for G2019S-induced dendrite degeneration, as reducing endogenous tau protein levels by the RNAi transgene strikingly rescues these dendrite phenotypes. However, the strong genetic interaction from coexpression of G2019S and Tau suggests that they may not function in a simple linear pathway. Coexpression of G2019S and Tau leads severe phenotypes that are not detected in the expression of either one alone, such as neuronal loss and lack of microtubules in dendrites. Thus, the synergistic interaction of G2019S with Tau is consistent with the idea that the G2019S mutation induces and enhances the detrimental activities of tau in neurodegeneration.
How does G2019S induce the detrimental activities of tau? There are >30 phosphorylation sites identified on the tau protein, and phosphorylated tau plays a major role in neuronal degeneration (Kenessey and Yen, 1993; Buee et al., 2000; Lee et al., 2001; Gong et al., 2005). Some of these phosphorylation sites, including T212/S214, T231/S235, and S422, are pathological and not detected in normal aging brains (Hoffmann et al., 1997; Bussiere et al., 1999). We propose that tau phosphorylation at T212 is the primary site responding to G2019S induction, as evidenced by the increased level of phospho-tau at the T212/S214 sites and that the T212A mutant completely suppresses the toxic effects induced by G2019S. How does G2019S induce hyperphosphorylation of tau at the T212 site? One of the upstream kinases that targets the T212/S214 sites of tau is GSK3β (Zheng-Fischhofer et al., 1998). Cotransfection of human tau and GSK3β in mammalian cells is shown to induce tau hyperphosphorylation with the loss of microtubule-binding affinity (Lovestone et al., 1996). Our genetic approaches provide the evidence that GSK3β mediates G2019S activity to regulate detrimental effects on dendrites, because compromising the activity of Sgg by either removing one gene dosage of sgg or expressing SggDN suppresses G2019S-induced hyperphosphorylation and dendritic distribution of tau, microtubule fragmentation, and dendrite degeneration.
In addition, we also found that expression of SggDN alone is sufficient to cause considerable dendritic arborization defect that is likely independent of the G2019S dendritic toxicity (supplemental Fig. S5E, available at www.jneurosci.org as supplemental material). GSK3β has many substrates involved in microtubule binding, like Crmp-2 and adenomatous polyposis coli tumor suppressor protein. Alterations in GSK3β activity could therefore have multiple effects on microtubule dynamics and, consequently, dendrite arborization (Zumbrunn et al., 2001; Yoshimura et al., 2005). Although SggDN fails to modify the effect of G2019S on peripheral DA dendrites, it significantly restores the dendritic reduction of dopaminergic neurons induced by G2019S (Fig. 7H,I). Such tissue specificity implies the more dominant contribution of the G2019S–Sgg–tau pathway in central dopaminergic neurons than in the peripheral DA neurons. Recent studies in human dopaminergic neuronal cell lines and rat striatal pathology sections have shown that activation of dopamine D1 receptors augment GSK3β activity and tau hyperphosphorylation through phosphorylation at tyrosine 216 of GSK3β (Beaulieu et al., 2004; Lebel et al., 2009). Thus, the vulnerability of dopaminergic neurons to the G2019S–Sgg–tau pathway is evident in both mammalian and Drosophila systems.
To further depict the mechanism how G2019S induces tau phosphorylation through Sgg, we found an enhanced association between G2019S and activated Sgg that is capable of phosphorylating tau at T212. With the ability of the ROC (Ras of complex) domain of Lrrk2 to bind microtubules (Gandhi et al., 2008), G2019S might bring Sgg to the proximity of the microtubule-binding protein tau for phosphorylation. Autoactivation of GSK3β is deemed as a protein-folding event after protein synthesis. This step is not coupled to protein translation and is assisted by the molecular chaperone Hsp90 (Lochhead et al., 2006). Interestingly, Lrrk2 and G2019S were shown recently to bind to Hsp90 with equal affinities (Wang et al., 2008). One possible model is that the G2019S mutation promotes the autoactivation of Sgg and facilitates the recruitment of autoactivated Sgg for tau phosphorylation at the T212 site during the pathogenesis of dendrite degeneration.
We also observed mislocalization of tau in the somatodendritic compartment in G2019S-DA neurons. The mislocalization of tau is GSK3β-activity regulated as it is suppressed by SggDN, underscoring the important role of GSK3β in dendritic distribution of tau in addition to tau phosphorylation. The hyperphosphorylated tau protein, unbound to microtubules, is therefore not limited to the axonal compartment and accumulates in dendrites. Phosphorylation of tau by GSK3β can also affect its binding ability to the kinesin-1 light chain, and therefore the transport rate in the axon (Cuchillo-Ibanez et al., 2008). Thus, G2019S might act through GSK3β to impair the binding affinity to kinesin-1 and decreased axonal tau transportation (Scott et al., 1993; Cuchillo-Ibanez et al., 2008). Alternatively, G2019S may cause abnormal intracellular trafficking in DA neurons, causing axonal proteins including tau to localize to dendrites. The function of Lrrk2 in regulating protein trafficking has been highlighted in the study of the Caenorhabditis elegans LRRK2 homolog lrk-1. In the lrk-1 deletion mutant, the presynaptic vesicle proteins are aberrantly localized to dendritic endings in the head sensory neurons (Sakaguchi-Nakashima et al., 2007). It is suggested that LRK-1 functions to exclude synaptic vesicle proteins from sorting into the somatodendritic domain. In addition to tau, we also found that the presynaptic protein Syt-GFP mislocalizes to dendrites of G2019S neurons, suggesting a more profound effect of G2019S in disrupting the integrity of protein sorting or trafficking.
In addition to G2019S, expressions of wild-type LRRK2 or the kinase-activity-lessened G2019S-K1906M mutant in DA neurons also cause dendrite arborization defects, to a lesser extent than G2019S. However, the underlying mechanisms of these defects are very different, as we did not observe tau accumulation or microtubule fragmentation in dendrites of wild-type LRRK2 and G2019S-K1906M DA neurons. In addition, the association with activated GSK3β and the ability of the coimmunoprecipitates to phosphorylate tau at T212/S214 sites are greatly reduced compared to those of G2019S. The effects of wild-type LRRK2 and G2019S-K1906M on the dendrite arborization may be caused by the upregulation of GSK3β levels (Fig. 8A), which have been shown to impede neurite outgrowth by disrupting organelle transport in mammalian cell cultures (Munoz-Montano et al., 1999).
Neurite degeneration represents a prominent feature of neurodegeneration associated with LRRK2 mutations (MacLeod et al., 2006; Plowey et al., 2008). We propose a model in which Lrrk2, GSK3β, and tau function in the same pathogenetic pathway to cause neuronal degeneration in the highly recognized LRRK2-inflicted PD. We also confirm previous observations in PD patients that dopaminergic neurons and peripheral sensory neurons are more sensitive to the LRRK2 neurotoxicity (Nolano et al., 2008). However, the vulnerability of specific neurons, especially nigral dopaminergic neurons, in the pathogenesis of PD is still unclear. Potential clues to the susceptibilities of these neurons include their increasing reliance on Ca2+ channels to maintain autonomous activity with age, which could pose a sustained metabolic stress on mitochondria (Surmeier, 2007). Additionally, neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), with its metabolite MPP+ (1-methyl-4-phenylpyridinium), preferentially binds to neuromelanin in the dopaminergic neurons, contributing to the susceptibility of these neurons in PD pathogenesis (Hirsch et al., 1988). These studies suggest that although genetic factors such as the G2019S mutation can hasten the onset, PD stems from a joint effect of both genetic and environmental factors. Our study will provide a tractable model to facilitate the study of PD pathogenetic mechanisms as well as the development of therapeutic molecules to slow the detrimental process.
Footnotes
This work was supported by National Science Council (NSC) Grant NSC 96-2628-B-002-103-MY2 (C.H.L., R.M.W.) and grants from the NSC and Academia Sinica of Taiwan (C.T.C.). We are grateful to M. B. Feany, M. J. Farrer, J. T. Wu, Bloomington Drosophila Stock Center, Vienna Drosophila RNAi Center, the Exelixis Collection at the Harvard Medical School for fly stocks, and the Developmental Studies Hybridoma Bank at the University of Iowa for antibodies. We also thank M. H. Chen for embryo microinjection, and H. S. Lee, H. W. Pi, H. Y. Sun, and J. T. Wu for comments on the manuscripts.
The authors declare no competing financial interests.
References
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blard O, Feuillette S, Bou J, Chaumette B, Frebourg T, Campion D, Lecourtois M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16:555–566. doi: 10.1093/hmg/ddm011. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Buee L, Delacourte A. Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol. 1999;97:221–230. doi: 10.1007/s004010050978. [DOI] [PubMed] [Google Scholar]
- Cuchillo-Ibanez I, Seereeram A, Byers HL, Leung KY, Ward MA, Anderton BH, Hanger DP. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008;22:3186–3195. doi: 10.1096/fj.08-109181. [DOI] [PubMed] [Google Scholar]
- Estes PS, Ho GL, Narayanan R, Ramaswami M. Synaptic localization and restricted diffusion of a Drosophila neuronal synaptobrevin–green fluorescent protein chimera in vivo. J Neurogenet. 2000;13:233–255. doi: 10.3109/01677060009084496. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, Ross OA, Wu RM. Lrrk2 G2385R is an ancestral risk factor for Parkinson's disease in Asia. Parkinsonism Relat Disord. 2007;13:89–92. doi: 10.1016/j.parkreldis.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res. 2008;86:1711–1720. doi: 10.1002/jnr.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao FB, Brenman JE, Jan LY, Jan YN. Genes regulating dendritic outgrowth, branching, and routing in Drosophila. Genes Dev. 1999;13:2549–2561. doi: 10.1101/gad.13.19.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer's disease. J Neural Transm. 2005;112:813–838. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Different levels of the homeodomain protein cut regulate distinct dendrite branching patterns of Drosophila multidendritic neurons. Cell. 2003;112:805–818. doi: 10.1016/s0092-8674(03)00160-0. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- Ho CC, Rideout HJ, Ribe E, Troy CM, Dauer WT. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J Neurosci. 2009;29:1011–1016. doi: 10.1523/JNEUROSCI.5175-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R, Lee VM, Leight S, Varga I, Otvos L., Jr Unique Alzheimer's disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry. 1997;36:8114–8124. doi: 10.1021/bi970380+. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Uchiyama M, Okumura E, Saito T, Ishiguro K, Uchida T, Okuyama A, Kishimoto T, Hisanaga S. Evidence for cdk5 as a major activity phosphorylating tau protein in porcine brain extract. J Biochem. 1995;117:741–749. doi: 10.1093/oxfordjournals.jbchem.a124771. [DOI] [PubMed] [Google Scholar]
- Hummel T, Krukkert K, Roos J, Davis G, Klambt C. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–370. doi: 10.1016/s0896-6273(00)81169-1. [DOI] [PubMed] [Google Scholar]
- Iseki E, Kato M, Marui W, Ueda K, Kosaka K. A neuropathological study of the disturbance of the nigro-amygdaloid connections in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2001;185:129–134. doi: 10.1016/s0022-510x(01)00481-6. [DOI] [PubMed] [Google Scholar]
- Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature. 2002;416:548–552. doi: 10.1038/nature733. [DOI] [PubMed] [Google Scholar]
- Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993;629:40–46. doi: 10.1016/0006-8993(93)90478-6. [DOI] [PubMed] [Google Scholar]
- Kim L, Kimmel AR. GSK3 at the edge: regulation of developmental specification and cell polarization. Curr Drug Targets. 2006;7:1411–1419. doi: 10.2174/1389450110607011411. [DOI] [PubMed] [Google Scholar]
- Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, Mellick GD, Buchanan DD, Silburn PA, Halliday GM, Schofield PR. GSK3B polymorphisms alter transcription and splicing in Parkinson's disease. Ann Neurol. 2005;58:829–839. doi: 10.1002/ana.20691. [DOI] [PubMed] [Google Scholar]
- Lebel M, Patenaude C, Allyson J, Massicotte G, Cyr M. Dopamine D1 receptor activation induces tau phosphorylation via cdk5 and GSK3 signaling pathways. Neuropharmacology. 2009;57:392–402. doi: 10.1016/j.neuropharm.2009.06.041. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002;530:209–214. doi: 10.1016/s0014-5793(02)03487-7. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V. A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell. 2006;24:627–633. doi: 10.1016/j.molcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Hartley CL, Pearce J, Anderton BH. Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience. 1996;73:1145–1157. doi: 10.1016/0306-4522(96)00126-1. [DOI] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzon-Toro B, Rubio de la Torre E, Delgado A, Perez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet. 2007;16:2031–2039. doi: 10.1093/hmg/ddm151. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Munoz-Montano JR, Lim F, Moreno FJ, Avila J, Diaz-Nido J. Glycogen synthase kinase-3 modulates neurite outgrowth in cultured neurons: possible implications for neurite pathology in Alzheimer's disease. J Alzheimers Dis. 1999;1:361–378. doi: 10.3233/jad-1999-1602. [DOI] [PubMed] [Google Scholar]
- Murray MJ, Merritt DJ, Brand AH, Whitington PM. In vivo dynamics of axon pathfinding in the Drosophila CNS: a time-lapse study of an identified motorneuron. J Neurobiol. 1998;37:607–621. [PubMed] [Google Scholar]
- Nolano M, Provitera V, Estraneo A, Selim MM, Caporaso G, Stancanelli A, Saltalamacchia AM, Lanzillo B, Santoro L. Sensory deficit in Parkinson's disease: evidence of a cutaneous denervation. Brain. 2008;131:1903–1911. doi: 10.1093/brain/awn102. [DOI] [PubMed] [Google Scholar]
- Papadopoulou D, Bianchi MW, Bourouis M. Functional studies of shaggy/glycogen synthase kinase 3 phosphorylation sites in Drosophila melanogaster. Mol Cell Biol. 2004;24:4909–4919. doi: 10.1128/MCB.24.11.4909-4919.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klambt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Sang TK, Jackson GR. Drosophila models of neurodegenerative disease. NeuroRx. 2005;2:438–446. doi: 10.1602/neurorx.2.3.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CW, Spreen RC, Herman JL, Chow FP, Davison MD, Young J, Caputo CB. Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J Biol Chem. 1993;268:1166–1173. [PubMed] [Google Scholar]
- Shahani N, Brandt R. Functions and malfunctions of the tau proteins. Cell Mol Life Sci. 2002;59:1668–1680. doi: 10.1007/PL00012495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soba P, Zhu S, Emoto K, Younger S, Yang SJ, Yu HH, Lee T, Jan LY, Jan YN. Drosophila sensory neurons require Dscam for dendritic self-avoidance and proper dendritic field organization. Neuron. 2007;54:403–416. doi: 10.1016/j.neuron.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell. 2007;18:5060–5068. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ. Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol. 2007;6:933–938. doi: 10.1016/S1474-4422(07)70246-6. [DOI] [PubMed] [Google Scholar]
- Suster ML, Seugnet L, Bate M, Sokolowski MB. Refining GAL4-driven transgene expression in Drosophila with a GAL80 enhancer-trap. Genesis. 2004;39:240–245. doi: 10.1002/gene.20051. [DOI] [PubMed] [Google Scholar]
- Timm T, Balusamy K, Li X, Biernat J, Mandelkow E, Mandelkow EM. Glycogen synthase kinase (GSK) 3beta directly phosphorylates Serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J Biol Chem. 2008;283:18873–18882. doi: 10.1074/jbc.M706596200. [DOI] [PubMed] [Google Scholar]
- Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, Moore DJ, Dawson TM, Dawson VL, Chiosis G, Cookson MR, Cai H. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci. 2008;28:3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, Strongosky AJ, Zimprich A, Muller-Myhsok B, Farrer MJ, Gasser T, Calne DB, Dickson DW. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62:1619–1622. doi: 10.1212/01.wnl.0000125015.06989.db. [DOI] [PubMed] [Google Scholar]
- Wszolek ZK, Tsuboi Y, Ghetti B, Pickering-Brown S, Baba Y, Cheshire WP. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) Orphanet J Rare Dis. 2006;1:30–38. doi: 10.1186/1750-1172-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zumbrunn J, Kinoshita K, Hyman AA, Näthke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11:44–49. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]