

Abstract
GluR2 is a subunit of the AMPA receptor, and the adenosine for the Q/R site of its pre-mRNA is converted to inosine (A-to-I conversion) by the enzyme called adenosine deaminase acting on RNA 2 (ADAR2). Failure of A-to-I conversion at this site affects multiple AMPA receptor properties, including the Ca2+ permeability of the receptor-coupled ion channel, thereby inducing fatal epilepsy in mice (Brusa et al., 1995; Feldmeyer et al., 1999). In addition, inefficient GluR2 Q/R site editing is a disease-specific molecular dysfunction found in the motor neurons of sporadic amyotrophic lateral sclerosis (ALS) patients (Kawahara et al., 2004). Here, we generated genetically modified mice (designated as AR2) in which the ADAR2 gene was conditionally targeted in motor neurons using the Cre/loxP system. These AR2 mice showed a decline in motor function commensurate with the slow death of ADAR2-deficient motor neurons in the spinal cord and cranial motor nerve nuclei. Notably, neurons in nuclei of oculomotor nerves, which often escape degeneration in ALS, were not decreased in number despite a significant decrease in GluR2 Q/R site editing. All cellular and phenotypic changes in AR2 mice were prevented when the mice carried endogenous GluR2 alleles engineered to express edited GluR2 without ADAR2 activity (Higuchi et al., 2000). Thus, loss of ADAR2 activity causes AMPA receptor-mediated death of motor neurons.
Introduction
GluR2 (also known as GluR-B or GluA2) is a subunit of the AMPA receptor. The adenosine within the glutamine codon for the Q/R site of its pre-mRNA is converted to inosine (A-to-I conversion) (Yang et al., 1995) by adenosine deaminase acting on RNA 2 (ADAR2) (Melcher et al., 1996). Because inosine is read as guanosine during translation, the genomic glutamine codon (Q: CAG) is converted to a codon for arginine (R: CIG) at the Q/R site of GluR2 in virtually all neurons in the mammalian brain (Seeburg, 2002). Conversion of Q to R at the Q/R site of GluR2 affects multiple AMPA receptor properties, including the Ca2+ permeability of the receptor-coupled ion channel, receptor trafficking, and assembly of receptor subunits (Sommer et al., 1991; Burnashev et al., 1992; Greger et al., 2002, 2003). Genetically modified mice in which the Q/R site of GluR2 remains unedited displayed fatal status epilepticus at early postnatal stages with exaggerated excitation of neurons (Brusa et al., 1995; Feldmeyer et al., 1999). Systemic ADAR2-null mice exhibit a similar phenotype, which was attributed to the absence of GluR2 Q/R site RNA editing (Higuchi et al., 2000). These findings indicate that the A-to-I conversion of the GluR2 Q/R site by ADAR2 is crucial for survival in mice. However, it has not been demonstrated whether neuronal death occurs in mice lacking GluR2 Q/R site editing or in those lacking ADAR2.
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease. Patients with sporadic ALS account for >90% of all cases, and the majority of them do not carry mutations in the causative genes of familial ALS that have been identified thus far (Schymick et al., 2007; Beleza-Meireles and Al-Chalabi, 2009). There is strong evidence indicating that AMPA receptor-mediated excitotoxic mechanism plays a pathogenic role in ALS and SOD1-associated familial ALS model animals (Rothstein et al., 1992; Carriedo et al., 1996; Van Damme et al., 2005). Recently, we demonstrated that a significant proportion of GluR2 mRNA was unedited at the Q/R site in spinal motor neurons of postmortem patients with sporadic ALS. This is in marked contrast to the fact that all GluR2 mRNA was edited in the motor neurons of control subjects (Takuma et al., 1999; Kawahara et al., 2004) and of patients with motor neuron diseases other than sporadic ALS (Kawahara et al., 2006), as well as in dying neurons in other neurodegenerative diseases, including Purkinje cells of patients with spinocerebellar degeneration (Paschen et al., 1994; Akbarian et al., 1995; Kawahara et al., 2004; Suzuki et al., 2003). The disease specificity of inefficient GluR2 Q/R site editing implies the pathogenic relevance of ADAR2 insufficiency in the death of motor neurons in sporadic ALS but leaves open the possibility that other genes whose products remain unedited by ADAR2 insufficiency might contribute to the demise of motor neurons.
We therefore generated a conditional ADAR2 knock-out mouse strain (designated here as AR2), using the Cre/loxP recombination system, and demonstrated that the loss of ADAR2 activity induces the slow death of motor neurons also in the mouse. Importantly, all motor neuron death in AR2 mice could be prevented by substituting the wild-type GluR2 alleles for alleles point mutated to express Q/R site-edited GluR2 in the absence of ADAR2. Our genetic studies in the mouse clearly demonstrate that the underediting of the GluR2 Q/R site specifically induces death of motor neurons with reduced ADAR2 activity.
Materials and Methods
All studies were performed in accordance with the Declaration of Helsinki, the Guideline of Animal Studies of the University of Tokyo, and National Institutes of Health. The committee of animal handling of the University of Tokyo also approved the experimental procedures used.
ADAR2flox allele and conditional ADAR2 knock-out mice.
DNA for the targeted region was obtained from a mouse strain 129/SvEv genomic library (supplemental Table S1, available at www.jneurosci.org as supplemental material). A LoxP site was inserted into intron 6 and another LoxP site was inserted into intron 9 of the mouse ADAR2 gene (adarb1), along with a selection cassette containing a neomycin resistance gene (Neo) flanked by flippase recognition target (FRT) sites (Fig. 1A). Exons 7–9 encode the majority of the adenosine deaminase motif. Chimeric mice were generated by injection of a targeted embryonic stem cell clone into C57BL/6-derived blastocysts. ADAR2flox/+ intercrosses produced ADAR2flox/flox mice at apparent Mendelian frequencies, and ADAR2flox/flox homozygous mice were phenotypically normal. Determination of the ADAR2flox allele was conducted by genomic PCR (Fig. 1B). Then, to knock-out ADAR2 activity selectively in motor neurons, we crossed ADAR2flox/flox mice with VAChT–Cre.Fast mice to obtain AR2 mice.
Figure 1.

Generation of a conditional ADAR2 knock-out mouse. A, A LoxP site (filled triangle) was inserted into intron 6 and another LoxP site in intron 9 with a selection cassette containing the gene for neomycin resistance (Neo) flanked by FRT sites. Exons are depicted as black bars with numbers. RBDs, RNA binding domains; F1/R1, primer pair (supplemental Table S1, available at www.jneurosci.org as supplemental material) for B; S, SfiI; BI, BglI; BII, BglII; E, ERI. B, Genomic PCR using template DNA obtained from the tails of ADAR2flox/flox mice (lane 1), ADAR2flox/+ mice (lane 2), and ADAR2+/+ mice (lane 3). C, Exons excised by recombination are shown as shaded areas in the mRNA, and a black bar indicates the in situ hybridization probe (supplemental Table S1, available at www.jneurosci.org as supplemental material) for D. F2/R2, Primer pair (supplemental Table S1, available at www.jneurosci.org as supplemental material) used in Figure 2B. D, In situ hybridization using a probe that encompasses the region excised by Cre-mediated recombination. There is a large number of punctate signals in the gray matter (outlined with dotted lines) of control mice (Ctl), whereas nuclei of some large neurons in the anterior horn were devoid of signal in the ADAR2flox/flox/VAChT–Cre.Fast (AR2) mice at 6 months of age (6m; arrowheads in magnified view). The sense probe did not yield a visible signal in the control mice at the same age (Ctl sense). Scale bars: top panels, 200 μm; bottom panels, 25 μm. E, All SMI-32-positive large neurons in the anterior horn (AHCs, brown color in the cytoplasm) of the cervical cord (C5) were ADAR2 positive (dark gray color in the nuclei) in the control mice (Ctl), whereas some of them were devoid of ADAR2 immunoreactivity in AR2 mice at 2 months of age (2m, arrowheads and inset). Sections were counterstained with hematoxylin. Scale bar: 50 μm; inset, 25 μm.
AR2 mice.
Intercrosses of ADAR2flox/+/VAChT–Cre.Fast mice produced ADAR2flox/flox/VAChT–Cre.Fast (AR2) mice, either heterozygous or homozygous for the Cre transgene, which directs restricted Cre expression under the control of the vesicular acetylcholine transporter gene promoter in a subset of cholinergic neurons, including the spinal motor neurons (Misawa et al., 2003). Cre expression levels were found not to differ in mice heterozygous or homozygous for the VAChT–Cre.Fast transgene (Misawa et al., 2003). The same intercrosses also produced, as littermates of AR2, ADAR2flox/flox (Ctl1) and ADAR2+/+/VAChT–Cre.Fast mice (Ctl2), which were used as controls. Both genders of AR2 and control mice were used, but littermates heterozygous for the floxed ADAR2 allele were not used in this study. All genotyping was performed by PCR on DNA from tail biopsies. PCR primers and amplicon sizes for the different alleles are listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material).
AR2/GluR-BR/R mice.
AR2/GluR-BR/R mice were generated by intercrossing ADAR2flox/+/VAChT–Cre.Fast/GluR-BR/+mice, which had been produced by crossbreeding AR2 mice with GluR-BR/R mice. The AR2/GluR-BR/R mice used by us were either heterozygous or homozygous for the Cre transgene (Misawa et al., 2003) and homozygous for the floxed ADAR2 and the GluR-B(R) allele. The desired genotype was found approximately once in every 20 offspring. Other genotypes produced by the intercrosses were not used in this study. All genotyping for the ADAR2 and GluR2 (GluR-B) alleles as well as for the Cre transgene was by PCR on DNA extracted from tail biopsies. PCR primers and amplicon sizes for the different alleles are listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material).
Genomic PCR and reverse transcription-PCR.
Genomic DNA was extracted from mouse tails using the High Pure PCR Template Preparation kit (Roche). Total RNA was isolated from brain and spinal cord tissue, and first-strand cDNA was synthesized and then treated with DNase I (Invitrogen) as described previously (Kawahara et al., 2003b). Primer pairs and the conditions used for PCR are presented in supplemental Table S1 (available at www.jneurosci.org as supplemental material). Positions of primer pairs used for genomic ADAR2 PCR (Fig. 1A, F1/R1) and ADAR2 reverse transcription (RT)-PCR (Fig. 1C, F2/R2) are indicated.
Analysis for editing efficiency at A-to-I sites.
Editing efficiencies at the Q/R sites in GluR2 mRNAs were calculated by quantitative analyses of the digests of RT-PCR products with BbvI as described previously (Takuma et al., 1999; Kawahara et al., 2003a, 2004). In brief, 2 μl of cDNA were subjected to first PCR in duplicate in a reaction mixture of 50 μl containing 200 mm each primer, 1 mm dNTP Mix (Eppendorf), 5 μl of 10× PCR buffer, and 1 μl of Advantage 2 Polymerase mix (Clontech). The PCR amplification began with a 1 min denaturation step at 95°C, followed by 40 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s, and extension at 68°C for 40 s. Nested PCR was conducted on 2 μl of the first PCR product under the same conditions with the exception of the annealing temperature (58°C). Primer pairs used for each PCR were listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material). After gel purification using the Zymoclean Gel DNA Recovery kit according to the protocol of the manufacturer (Zymo Research), an aliquot (0.5 mg) was incubated with BbvI (New England Biolabs) at 37°C for 12 h. The PCR products originating from Q/R site-edited GluR2 mRNA had one intrinsic restriction enzyme recognition site, whereas those originating from unedited mRNA had an additional recognition site. Thus, restriction digestion of the PCR products originating from edited GluR2 mRNA should produce different numbers of fragments (two bands at 219 and 59 bp) from those originating from unedited GluR2 mRNA (three bands at 140, 79, and 59 bp). Because the 59 bp band would originate from both edited and unedited mRNA but the 219 bp band would originate from only edited mRNA, we quantified the molarity of the 219 and 59 bp bands using the 2100 Bioanalyzer (Agilent Technologies) and calculated the editing efficiency as the ratio of the former to the latter for each sample (supplemental Table S1, available at www.jneurosci.org as supplemental material).
With similar methods, we calculated the editing efficiencies at the Q/R sites in GluR5 and GluR6 mRNA and in GluR2 pre-mRNA, the R/G site in GluR2 mRNA, and the I/V site in Kv1.1 mRNA (Paschen et al., 1994; Takuma et al., 1999; Kawahara et al., 2003a, 2004; Nishimoto et al., 2008). The following restriction enzymes were used for restriction digestion of the respective A-to-I sites: BbvI for the Q/R sites, MfeI (New England Biolabs) for the I/V site, and MseI (New England Biolabs) for the R/G site. Primer pairs used for each PCR and sizes of restriction digests of PCR products were indicated in supplemental Table S1 (available at www.jneurosci.org as supplemental material).
Behavioral analyses.
Using a mouse-specific rotarod (SN-445; Neuroscience Corp.), we determined the maximal time before falling at 10 rpm during a 180 s period; each run consisted of three trials. Grip strength was measured with a dynamometer (NS-TRM-M; Neuroscience Corp.). Measurements were conducted weekly by a researcher blind to genotype and age of the mice.
Isolation of single motor neurons and brain tissue.
Single-cell isolation from frozen spinal cord tissue was performed with a laser microdissection system (Leica AS LMD; Leica Microsystems) as described previously (Kawahara et al., 2003b, 2004). All of the large motor neurons (diameter larger than 20 μm) in the anterior horn were dissected from 14-μm-thick cervical cord sections, and three neurons each were collected together into respective single test tubes containing 200 μl of TRIZOL Reagent. In addition, using the same method, nuclei of oculomotor nerve and of facial nerve were dissected from the brainstem sections of AR2 mice and control mice at 12 months of age. The positions of these cranial nerve nuclei were identified using the Paxinos and Franklin mouse brain atlas (Paxinos and Franklin, 2001). All samples were kept at −20°C until use.
Immunohistochemistry.
Under deep anesthesia with isoflurane, mice were transcardially perfused with 3% paraformaldehyde and 1% glutaraldehyde in PBS. The brains and spinal cords were removed and immersed in serially increasing concentrations of a sucrose–PBS solution (final sucrose concentration of 30%). The immunohistochemical procedure was performed on 10-μm-thick sections, which were cut with a cryostat (model HM500 O; Microm). The sections were analyzed with a standard avidin–biotin–immunoperoxidase complex method using the M.O.M. Immunodetection kit (Vector Laboratories) for mouse primary antibodies and Vectastain ABC IgGs (Vector Laboratories) for other primary antibodies. The following primary antibodies were used: mouse anti-nonphosphorylated neurofilament H (SMI-32; dilution at 1:1000; Covance), mouse anti-neuronal nuclei (NeuN) (dilution at 1:500; Millipore Bioscience Research Reagents), sheep anti-rat RED1 (ADAR2) N terminus [dilution at 1:500; a gift from Dr. R. B. Emeson (Sansam et al., 2003)], rabbit anti-glial fibrillary acidic protein (GFAP) (dilution at 1:200; Lab Vision), and rat anti-mouse MAC-2 (dilution at 1:500; Cedarlane). Color was developed with the HRP–DAB System (Vector Laboratories).
Muscles and neuromuscular junctions.
Medial gastrocnemius muscles and medial quadriceps muscles were dissected, pinned in mild stretch, and mounted on cork blocks and were quickly frozen in isopentane–liquid nitrogen. Samples were stored at −80°C until use. Five-micrometer-thick transverse frozen sections were stained with hematoxylin and eosin. Twenty-micrometer-thick frozen longitudinal sections were stained with tetramethylrhodamine–bungarotoxin. The same section was incubated with monoclonal antibodies to neurofilament (NF160; dilution at 1:200; Millipore Bioscience Research Reagents) and synaptophysin (dilution at 1:100; Cell Signaling Technologies) and then with Alexa Fluor 488 rabbit anti-mouse IgG (dilution at 1:100; Invitrogen) as the secondary antibody. Stained sections were examined under an LSM-510 confocal microscope system (Carl Zeiss).
Electrophysiology.
Mice were anesthetized with isoflurane and placed in a prone position on a thermal pad at 37°C for the examination. Electromyogram (EMG) recordings using a Power Lab 26T and EMG machine (AD Instruments) were obtained using a 29-gauge, Teflon-coated, monopolar needle electrode. The recording electrode was inserted into the gastrocnemius muscles, and spontaneous electrical activity was recorded for 120 s using a Lab Chart analysis system (AD Instruments).
Morphological observation and stereology.
Sections of the fifth cervical (C5) and fifth lumbar (L5) spinal cord segments were sequentially immunostained with RED1 and SMI-32 using the HRP–DAB system with and without the addition of NiCl2 for color development. Some sections were immunostained with NeuN. ADAR2-positive and -negative neurons were separately counted among SMI-32-positive neurons with diameters larger than 20 μm in 10 sections for each mouse. The number of NeuN-positive neurons with diameter smaller than 20 μm in the ventral gray matter (ventral to the line running though the ventral edge of the central canal) was counted in 10 C5 sections for each mouse at 12 months of age. None of the NeuN-positive small neurons exhibited SMI-32 or GFAP immunoreactivity. The entire brainstem of each mouse at 12 months of age was cut axially to produce a 10-μm-thick section, and the numbers of all the neurons with nucleoli in the nuclei of cranial motor nerves were counted under a light microscope after cresyl violet staining. The position of each nucleus was stereologically determined using a mouse brain atlas (Paxinos and Franklin, 2001). The positions from the bregma were from −3.80 to −4.24 mm (nucleus of oculomotor nerve), from −4.36 to −4.48 mm (nucleus of trochlear nerve), from −4.84 to −5.34 mm (motor nucleus of trigeminal nerve), from −5.52 to −5.80 mm (nucleus of abducens nerve), from −5.68 to −6.48 mm (nucleus of facial nerve), from −7.08 to −7.92 mm (dorsal nucleus of vagus nerve), and from −7.08 to −8.12 mm (nucleus of hypoglossal nerve). The density of neurons in each nucleus was estimated by dividing the total number of neurons in each nucleus by the volume of the nucleus, which was calculated as the product of the area of the nucleus and the thickness of each section. In addition, transverse, 1-μm-thick, Epon-embedded sections of the anterior horns of the spinal cord, and the ventral roots at the L5 level were prepared and stained with 0.1% toluidine blue. Cell counting was performed by researchers who were blind to the genotype of the mouse.
In situ hybridization.
Anesthetized mice were perfusion fixed with Tissue Fixative (GenoStaff). Dissected cervical cord tissues were sectioned after they were embedded in paraffin. Antisense and sense adarb1 cRNA probes (Fig. 1C) (supplemental Table S1, available at www.jneurosci.org as supplemental material) were generated from the mouse adarb1 open reading frame sequence, which was cloned into the pGEMT-Easy vector (Promega). Digoxigenin-labeled cRNA probes were prepared with the DIG RNA Labeling mix (Roche Applied Science). Color was developed with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate, and tissue sections were counterstained with Kernechtrot stain solution (Muto Pure Chemicals). After mounting, 24-bit color images were acquired by scanning of the sections. Digoxigenin signals were isolated by uniformly subtracting the counterstaining color component using Photoshop version 9.0.2 (Adobe Systems) (Ohmae et al., 2006; Takemoto-Kimura et al., 2007).
Statistics.
Differences in behavior and survival rates between groups were analyzed using log-rank analysis with SPSS software (version 15; SPSS Inc.), and GraphPad Prism version 4 (GraphPad Software), respectively. The differences in neuronal number between each group and the control samples were examined with a repeated-measures ANOVA. The SPSS version 15 software was used for ANOVA, followed by a Tukey–Kramer statistical test.
Results
Generation of the ADAR2flox/flox/VAChT–Cre mouse, designated as AR2 mouse
We constructed the mouse ADAR2flox allele by flanking exons 7–9 of the adarb1 gene (mouse ADAR2 gene) with loxP sites (Fig. 1A) (supplemental Table S1, available at www.jneurosci.org as supplemental material). Exons 7–9 encode the majority of the adenosine deaminase motif in the adarb1 gene (Feng et al., 2006), and Cre-mediated deletion of this region ablates ADAR2 activity. To ablate ADAR2 activity selectively in motor neurons, we crossed ADAR2flox/flox mice with VAChT–Cre.Fast mice. In VAChT–Cre.Fast mice, Cre expression is under the control of the vesicular acetylcholine transporter gene promoter, which is active in cholinergic neurons, including spinal motor neurons (Misawa et al., 2003). In these transgenic mice, Cre expression is developmentally regulated, and ∼50% of motor neurons express Cre by 5 weeks of age, independent of the heterozygous or homozygous state of the transgene (Misawa et al., 2003). The resulting ADAR2flox/flox/VAChT–Cre.Fast mice, referred to here as AR2 mice (for breeding, see Materials and Methods), therefore would lack ADAR2 activity in a subset of motor neurons in the spinal cord and other brain motor nuclei after expression of Cre by 5 weeks of age. In situ hybridization with a probe encompassing the sequence excised by Cre-mediated recombination (Fig. 1C) demonstrated that several large neurons in the anterior horn (AHCs) were devoid of adarb1 gene signal in the AR2 mice, whereas all the AHCs exhibited the signal in control littermates (Fig. 1D). Similarly, a subset of the AHCs were devoid of ADAR2 immunoreactivity in AR2 mice, whereas all AHCs exhibited ADAR2 immunoreactivity in the controls (Fig. 1E). There was no difference in the results on male and female AR2 mice.
ADAR2 activity in ADAR2-null motor neurons
Next we examined the effects of recombination of the ADAR2flox allele on ADAR2 activity. We dissected all large neurons in the anterior horn (AHCs) (for AHC identification, see supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) from frozen sections from 2-month-old AR2 mice (n = 4) using a laser microdissector (Fig. 2A). We verified that these AHCs, but not small neurons in the anterior horn, are the spinal motor neurons by RT-PCR for choline acetyltransferase on a single-cell lysates (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Because RT-PCR of GluR2 mRNA on the lysates of three neurons, but not the lysates of one or two motor neurons, reproducibly yielded amplification products, we analyzed the extent of GluR2 Q/R site editing on RNA extracted from the lysates of three pooled AHCs (designated as a specimen) by quantitative analysis of the BbvI-restriction digests of the RT-PCR products, as described previously (Kawahara et al., 2003b, 2004). Among 116 specimens examined, eight showed 0% and 42 showed 100% Q/R site editing, with the remaining 66 specimens distributed between the ranges of 17 and 98% (Fig. 2A) (supplemental Table S2, available at www.jneurosci.org as supplemental material). Because AHCs of control littermates (these carried wild-type ADAR2 alleles or no Cre transgene; see Materials and Methods) expressed only edited GluR2 mRNA, the presence of samples exhibiting 0% Q/R site editing suggests that ADAR2-expressing neurons expressed only edited GluR2 mRNA, whereas ADAR2-null neurons expressed only unedited GluR2 mRNA. Then, DNA and total RNA from the specimens were collected in four different groups according to the proportions of unedited GluR2 (Fig. 2A). Using PCR, we demonstrated that the samples with 100% editing efficiency (group 0:3) harbored the truncated ADAR2flox gene and Cre transcripts, whereas the samples with 100% editing efficiency (group 3:0) carried the full-length ADAR2flox gene and did not express Cre (Fig. 2B). Those samples with both edited and unedited GluR2 mRNA (groups 1:2 and 2:1) exhibited both full-length and truncated ADAR2 along with the Cre transcript. These qualitative results are consistent with the assumption that recombination of the ADAR2flox alleles occurred in a Cre-dependent manner and that this recombination abolished the editing of the GluR2 Q/R site. Among other A-to-I sites examined, we found a significant reduction in editing efficiency only at the GluR6 Q/R site (supplemental Table S3, available at www.jneurosci.org as supplemental material).
Figure 2.

Cre-dependent targeting of ADAR2 and GluR2 Q/R site-editing in motor neurons. A, Frequency histogram of editing efficiency at the GluR2 Q/R site in specimens (lysates containing 3 motor neurons) obtained from AR2 mice at 2 months of age (2m; n = 4). Neurons were dissected with a laser microdissector (inset). B, Specimens (n = 116) were collected into four groups depending on the predicted number of ADAR2-deficient neurons in each specimen; the groups of specimens containing 3, 2, 1, and 0 unedited GluR2-expressing neurons were designated as groups 0:3, 1:2, 2:1, and 3:0, respectively. The ADAR2flox gene and transcripts of the Cre gene and the ADAR2flox alleles before and after recombination were analyzed for each group by PCR. AHCs expressing unedited GluR2 mRNA (group 0:3) harbored the truncated ADAR2flox gene and Cre transcripts, whereas AHCs expressing edited GluR2 mRNA (group 3:0) carried the full-length ADAR2flox gene and did not express Cre. Ctl1, ADAR2flox/flox mice; Ctl2, VAChT–Cre.Fast mice; AH, anterior horn of the spinal cord.
Behavioral changes
AR2 mice were hypokinetic (supplemental movie, available at www.jneurosci.org as supplemental material) and abnormal in posture (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material), but they displayed no overt paralysis or vesico-urinary disturbances and exhibited a normal withdrawal response to noxious stimuli. They showed a lower rotarod performance than their control littermates after 5 weeks of age (Fig. 3A), when the Cre expression reached the maximum level (∼50% of motor neurons) (Misawa et al., 2003). Their rotarod performance rapidly declined during the initial 5–6 months of life, followed by stable performance until about 18 months of age (Fig. 3A). Control mice exhibited full performance (180 s) until ∼12 months of age, followed by slightly lower performance (>164.5 ± 6.4 s) until 24 months. Grip strength declined with kinetics similar to those of rotarod performance (Fig. 3B). The AR2 mice had slightly lower body weight than the controls (Fig. 3C) and were relatively long-lived (81.5 ± 16.4 weeks; mean ± SEM), although not as long as control mice (105.1 ± 13.5 weeks; p = 0.0262, log-rank analysis) (Fig. 3D).
Figure 3.

Behavioral changes in AR2 mice. A, Rotarod performance presented as latency to fall (at 10 rpm, 180 s at the maximum) began to decline at 5 weeks of age in AR2 mice and rapidly fell to low levels during the initial 5–6 months, remaining stable until 18 months of age. Control mice exhibited full performance (180 s) until ∼12 months of age, followed by slightly lower performance (>164.5 ± 6.4 s) until 24 months. B, Grip strength measured declined with kinetics similar to those of rotarod performance. In A and B, the scores obtained for the AR2 mice (mean ± SEM; n = 28) are indicated as percentage performance of control mice (n = 15). C, AR2 mice exhibited slightly lower body weight than controls (p > 0.05). D, AR2 mice (n = 33) had long lifespans, but the rate of death increased after month 18. The median ± SEM survival was 81.5 ± 16.4 weeks for AR2 mice compared with 105.1 ± 13.5 weeks for control mice (p = 0.0262, log-rank analysis).
Pathological alterations in the spinal cords and muscles
Immunohistochemical examination demonstrated that all the AHCs in the spinal cord that were immunoreactive to anti-phosphorylated neurofilament antibodies (SMI-32) showed intense ADAR2 immunoreactivity in their nuclei in control mice, whereas a fraction of these cells was devoid of ADAR2 immunoreactivity in AR2 mice (Fig. 1E) (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). There were a number of degenerating AHCs with cytoplasmic vacuoles (Fig. 4A) and darkly stained degenerating axons in the ventral roots (Fig. 4B). The number of AHCs in AR2 mice markedly decreased between 1 and 2 months of age and then slowly decreased beyond 1 year of age (Fig. 4C). The number of ADAR2-positive AHCs in the AR2 mice decreased from 83 to 54% of the number of total AHCs in the age-matched control littermates between 1 and 2 months of age. The rapid reduction in the proportion of ADAR2-positive AHCs during this period is likely attributable to the Cre-dependent recombination of the floxed ADAR2 alleles, because the number of Cre-expressing AHCs in VAChT–Cre.Fast mice increases developmentally until 5 weeks of age (Misawa et al., 2003). After 2 months of age, the number of ADAR2-positive AHCs did not change over the course of more than 1 year, whereas that of total AHCs decreased from 80 to 54% of the number of AHCs in the age-matched control mice (Fig. 4C) (Table 1). Consistent with the Cre-dependent recombination, the proportion of ADAR2-lacking AHCs in AR2 mice is in accordance with that of Cre-expressing AHCs presented in the original study of VAChT–Cre mice (Misawa et al., 2003). Concomitant with AHC degeneration, the number of myelinated axons in the ventral roots was significantly decreased (Table 1).
Figure 4.
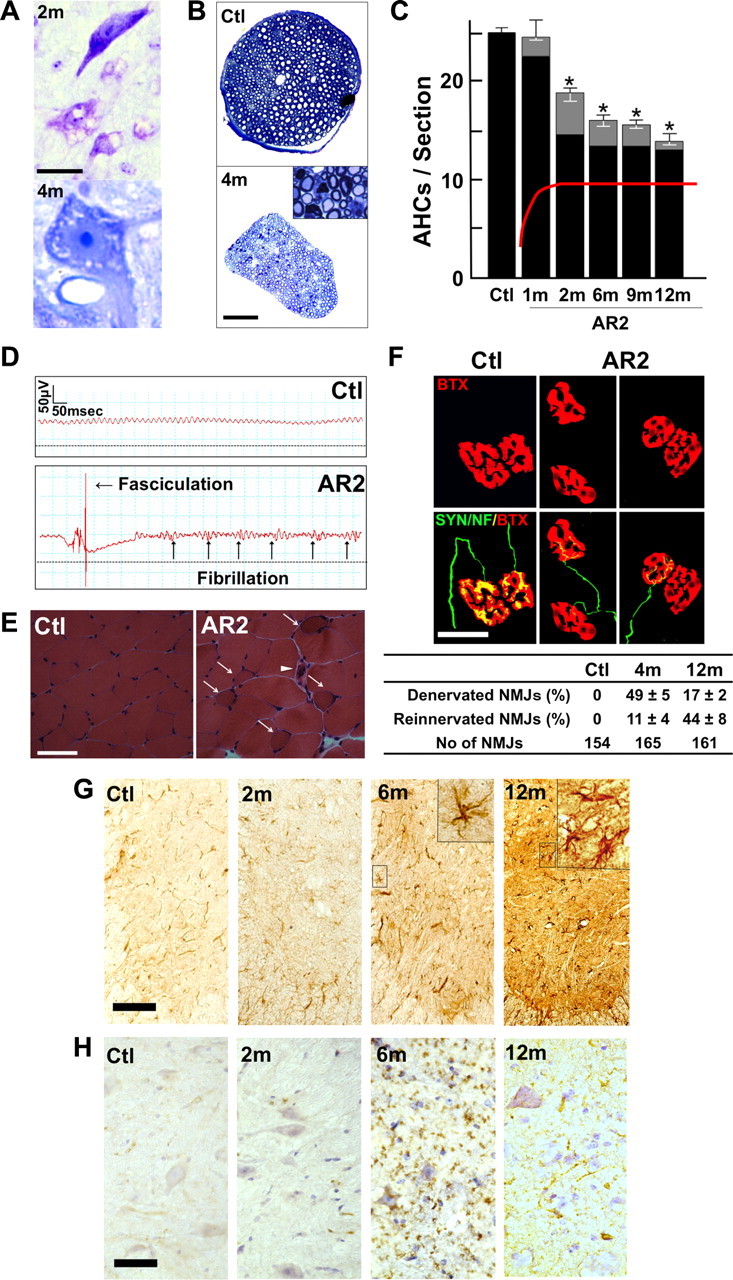
Loss of ADAR2-deficient motor neurons. A, Degenerating AHCs in AR2 mice at 2 months (2m; Nissl staining) and 4 months (4m; toluidine blue staining, 1 μm section) of age. Scale bar: 2m, 25 μm; 4m, 12.5 μm. B, Ventral root (L5) of control (Ctl) and AR2 mice at 4 months of age (4m). Inset, Magnified view of degenerating axons. Scale bar: 100 μm; inset, 20 μm. C, Numbers of AHCs showing ADAR2 immunoreactivity (black columns) and lacking this immunoreactivity (gray columns) (mean ± SEM) in AR2 mice at different ages (1m, 2m, 6m, 9m, 12m). In AR2 mice, Cre expression is developmentally regulated (orange line), and ∼50% of motor neurons express Cre by 5 weeks of age, with recombination of the ADAR2 gene in ∼10% of AHCs at 1 month of age and 40–45% of AHCs after 2 months of age (orange line). The number of ADAR2-lacking AHCs significantly decreased in AR2 mice after 2 months of age as a result of Cre-dependent knock-out of ADAR2 (*p < 0.01, repeated-measures ANOVA). The number of AHCs in the control mice did not change at different ages, and all the AHCs in controls showed ADAR2 immunoreactivity. D, Electrophysiological examination in AR2 mice. Electromyography from an AR2 mouse at 12 months of age showing fibrillations and fasciculations, common findings in ALS indicative of muscle fiber denervation and motor unit degeneration and regeneration. These findings were observed in two other AR2 mice examined but never in control mice (Ctl; n = 2). E, Calf muscles from a wild-type mouse (left) and an AR2 mouse (middle and right) at 12 months of age. Characteristics of denervated muscles, including muscle fiber atrophy (white arrow), centrally placed nuclei, and pyknotic nuclear clumps (white arrowhead) are observed in the AR2 mouse. Hematoxylin and eosin. Scale bar, 60 μm. F, NMJs and distal axons. Quadriceps muscles from a wild-type mouse (Ctl; left) and an AR2 mouse (AR2; middle and right) at 12 months of age are stained with tetramethylrhodamine–bungarotoxin (BTX) (red) and immunostained concomitantly with anti-synaptophysin and neurofilament (SYN/NF) antibodies (green). Endplates (red) were counted as “innervated“ if they were merged with axon terminals (merge; yellow). Each endplate is innervated by a thick axon terminal in the Ctl mouse. In AR2 mice, in addition to the normally innervated NMJs, some NMJs were innervated by axons that simultaneously innervate more than one NMJ (reinnervated NMJs; middle), and other NMJs were devoid of axon terminals (denervated NMJs; right). More than 50 NMJs were counted in each animal in the control group and groups of AR2 mice at 4 and 12 months of age (n = 3 in each group). Proportions of denervated NMJs and reinnervated NMJs among total NMJs in each group are indicated as mean ± SD (percentage). Scale bar, 25 μm. G, H, Immunohistochemistry in the anterior horn (C5). There was a time-dependent increase in GFAP immunoreactivity (G) and an increase in MAC2 immunoreactivity maximal at 6 months of age (H) in the spinal anterior horn of AR2 mice. m, Months of age; inset, activated astroglia. Scale bars: G, 100 μm; insets and H, 50 μm.
Table 1.
Density of neurons in motor nerve nuclei and spinal cord
| Nucleus | Control (n = 3) neurons/mm3 | AR2 (n = 4) neurons/mm3 |
|---|---|---|
| III | 11,253 ± 1783 | 10,441 ± 632 |
| IV | 15,783 ± 1694 | 16,032 ± 658 |
| VI | 10,117 ± 996 | 10,699 ± 195 |
| Vm | 8809 ± 417 | 8623 ± 246 |
| Vm (>25 μm) | 3603 ± 213 | 2767 ± 175** |
| VII | 1041 ± 124 | 1016 ± 96 |
| VII (>20 μm) | 91.1 ± 32.7 | 67.7 ± 13.1** |
| X | 11,442 ± 1932 | 11,652 ± 2387 |
| XII | 11,800 ± 541 | 9834 ± 1530 |
| XII (>20 μm) | 832.7 ± 92.9 | 677.8 ± 116.2** |
| C5 AH (≤20 μm) | 37,147 ± 326 | 37,941 ± 331 |
| C5 AH (>20 μm) | 25.5 ± 0.9a | 13.7 ± 0.7a,** |
| L5 AH (>20 μm) | 29.3 ± 0.32a | 15.9 ± 0.31a,** |
| DH | 476,312 ± 12,623 | 498,816 ± 21,446 |
| VR | 840.0 ± 26.5b | 626.3 ± 31.4b,* |
Numbers are the neuronal density per cubic millimeter (mean ± SEM) in each nucleus from mice at 12 months of age. For Vm, VII, and XII, neurons with large diameter (>20 or 25 μm) were also counted. AR2, ADAR2flox/flox/VAChT–Cre.Fast mice; III, nucleus of oculomotor nerve; IV, nucleus of trochlear nerve; VI, nucleus of abducens nerve; Vm, motor nucleus of trigeminal nerve; VII, nucleus of facial nerve; X, dorsal nucleus of the vagus nerve; XII, nucleus of hypoglossal nerve; C5 AH, anterior horn of the fifth cervical cord; L5 AH, anterior horn of the fifth lumbar cord; DH, zona gelatinosa of the spinal cord; VR, ventral roots (L5).
*p < 0.005;
**p < 0.001 (ANOVA).
aNumber of neurons per section.
bNumber of axons.
The kinetics of neuronal loss (Fig. 4C) were consistent with the kinetics of progressive motor-selective behavioral deficits (Fig. 3A,B). The long survival with hypoactivity beyond 6 months of age indicates that the remaining ADAR2-expressing neurons functioned normally during the remainder of life. The high rate of death after 18 months may reflect the failure of the remaining AHCs to compensate for an age-related decline in skeletal muscle power, including a decline in respiratory muscle strength.
We also examined denervation of skeletal muscles. Electromyography performed on AR2 mice at 12 months of age revealed fibrillation potentials and fasciculations, which are common findings in ALS, indicative of muscle fiber denervation and motor unit degeneration and regeneration (Fig. 4D). We observed characteristics of denervation, including muscle fiber atrophy, centrally placed nuclei, and pyknotic nuclear clumps in the skeletal muscles of AR2 mice (Fig. 4E). Some neuromuscular junctions (NMJs) were not innervated and other NMJs were innervated by ramified axons that innervated more than one NMJ in AR2 mice, indicating reinnervated NMJs (Fig. 4F). In contrast, in control mice, all the NMJs were innervated by a single axon. The proportion of denervated NMJs decreased, whereas reinnervated NMJs increased with age in AR2 mice (Fig. 4F). In addition, proliferation of activated astrocytes with increased GFAP immunoreactivity and of MAC2-positive activated microglial cells was detected in the anterior horns of AR2 mice (Fig. 4G,H). These results suggest that degeneration of ADAR2-lacking AHCs induced degeneration of their axon terminals, and then denervated NMJs were reinnervated by collaterally sprouted axons of ADAR2-expressing AHCs after longer survival.
Neurons in the motor nuclei of cranial nerves
The numbers of large neurons in facial and hypoglossal nerve nuclei in AR2 mice were significantly smaller than those in control mice at 12 months of age, whereas the numbers of neurons in nuclei of oculomotor nerves were not decreased (Table 1). Conversely, GluR2 Q/R site editing was significantly decreased both the in oculomotor nerve nuclei (the efficiency of GluR2 Q/R site editing, mean ± SEM: for AR2 mice, 89.7 ± 5.8%, n = 3; for control mice, 100%, n = 3, p = 0.0048) and in the facial nerve nuclei (for AR2 mice, 82.6 ± 9.1%, n = 3; for control mice, 99.2 ± 0.2%, n = 3, p = 0.0017) of AR2 mice at 12 months of age. These results indicate that ADAR2-lacking motor neurons do not always undergo cell death, and some motor neurons, including those in the oculomotor nerve nucleus, are relatively resistant to cell death mediated by deficient ADAR2. Indeed, motor neurons innervating extraocular muscles are much less vulnerable than those innervating bulbar and limb muscles in ALS patients (Lowe and Leigh, 2002).
GluR-BR alleles prevent motor neuron death in AR2 mice
To investigate by genetic means the role of RNA editing at the GluR2 Q/R site in the death of motor neurons, we exchanged the endogenous GluR2 alleles in AR2 mice with GluR-BR alleles (Kask et al., 1998), which directly encode Q/R site-edited GluR2, thus circumventing the requirement for ADAR2-mediated RNA editing. AR2/GluR-BR/R mice were obtained by ADAR2flox/+/VAChT–Cre.Fast/GluR-BR/+ mice intercrosses to generate ADAR2flox/flox/VAChT–Cre.Fast/GluR-BR/R (AR2/GluR-BR/R) mice (see Materials and Methods). AR2/GluR-BR/R mice (AR2rescue, or AR2res, mice) were phenotypically normal and had full motor function until 6 months of age (Fig. 5A). The AHCs, including the ∼30% AHCs lacking ADAR2 from Cre-mediated recombination, were viable in AR2res mice at 6 months of age, and the total number of AHCs was the same as in age-matched control mice (Fig. 5A,B). Consistent with a lack of AHC loss, there was no detectable increase in GFAP or MAC2 immunoreactivity in the anterior horns (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). These results demonstrate that it is specifically the failure of GluR2 Q/R site editing by which ADAR2 deficiency induces the slow death of motor neurons (Fig. 5C).
Figure 5.

Crucial role of GluR2 Q/R site editing in death of ADAR2-deficient motor neurons. A, AR2/GluR-BR/R mice (AR2res) displayed full rotarod score and normal grip strength at 6 months of age compared with control mice (Ctl). The number of total AHCs, of which a considerable proportion was deficient in ADAR2, did not decrease in AR2res mice. B, At 6 months of age, although only a few AHCs lacking ADAR2 immunoreactivity (arrowheads) were observed in AR2 mice, a considerable number of AHCs lacking ADAR2 immunoreactivity was present in AR2res mice. The density of AHCs in AR2res mice was similar to that in the control mice in which all the AHCs were immunoreactive to ADAR2 in their nuclei. Sections were counterstained with hematoxylin. Scale bar, 100 μm. C, Scheme illustrating that lack of ADAR2 induces slow death of motor neurons in AR2 mice but not in AR2res mice that express Q/R site-edited GluR2 in the absence of ADAR2 activity. The exonic Q codon at the Q/R site of GluR2 was substituted by an R codon in the endogenous GluR2 alleles of GluR-BR/R mice.
Discussion
We generated the AR2 mouse (Hideyama et al, 2008), a conditional ADAR2 knock-out line, which carries gene-targeted floxed ADAR2 alleles that become functionally ablated by Cre recombinase expressed from a transgene (VAChT–Cre.Fast) in ∼50% of motor neurons (Misawa et al., 2003). These displayed progressive motor dysfunctions. The ADAR2-lacking motor neurons expressed only Q/R site-unedited GluR2. Virtually all of the ADAR2-lacking AHCs underwent degeneration, whereas the surviving ADAR2-expressing AHCs remained intact by 12 months of age. The death of ADAR2-lacking AHCs was completely prevented by a point mutation in the endogenous GluR2 alleles of AR2 mice, thus generating Q/R site-edited GluR2 in the absence of ADAR2 (Kask et al., 1998). These findings highlight the crucial role of RNA editing at the GluR2 Q/R site for survival of motor neurons and demonstrate that expression of Q/R site-unedited GluR2 is a cause of slow death of motor neurons. Therefore, it is necessary to investigate the relevance of inefficient GluR2 Q/R site-RNA editing found in the patient's motor neurons to the pathogenesis of sporadic ALS (Kawahara et al., 2004; Kwak and Kawahara, 2005).
Concomitant with the loss of ADAR2-lacking AHCs, proximal and distal axons of AHCs underwent degeneration with resultant neurogenic changes in neuromuscular units. These pathological changes in AHCs and neuromuscular units caused motor dysfunctions in AR2 mice. The prevention of slow neuronal cell death observed in AR2 mice by GluR-BR alleles expressing Q/R site-edited GluR2 in the absence of ADAR2 (Kask et al., 1998) means that, although ADAR2 edits numerous A-to-I positions in many RNAs expressed in the mammalian brain (Levanon et al., 2004; Li et al., 2009), failure of A-to-I conversions at sites other than the GluR2 Q/R site did not play a role in neuronal cell death (Fig. 5C).
When the GluR2 Q/R site is unedited, the Ca2+ permeability of the AMPA receptor is greatly increased, and trafficking of the receptor to synaptic membranes is facilitated (Sommer et al., 1991; Burnashev et al., 1992; Greger et al., 2002). This enhances neuronal excitability by increasing the density of Ca2+-permeable functional AMPA channels, which is typically observed as fatal epilepsy in mice carrying Q/R site-uneditable GluR-B (GluR2) alleles (Brusa et al., 1995; Feldmeyer et al., 1999) and in systemic ADAR2-null mice (Higuchi et al., 2000). The results obtained from AR2 mice indicate that motor neurons expressing only Q/R site-unedited GluR2 undergo slow death when the mice live sufficiently long.
Some ADAR2-lacking AHCs die shortly after recombination, whereas others survive for more than 1 year. These observations indicate that, although all the ADAR2-lacking AHCs undergo neuronal death, the ability to compensate for the increased Ca2+ overload through the functionally altered AMPA receptor differs among AHCs. It is likely that the increased Ca2+ overload might have already led to dysfunction of the ADAR2-lacking AHCs before their death, causing a decline of motor functions at earlier stages. Vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated toxicity was demonstrated in GluR-B(N) transgenic mice, which additionally to wild-type GluR2 express an engineered GluR2 subunit that features asparagine (N) in place of glutamine (Q) at the Q/R site (Kuner et al., 2005). ADAR2 activity is downregulated in the rat after transient forebrain ischemia, resulting in the selective death of hippocampal CA1 pyramidal cells (Peng et al., 2006).
An intriguing observation in AR2 mice was the selective vulnerability among motor neurons in different cranial nerve nuclei. Neurons in facial and hypoglossal nerve nuclei decreased in number, whereas those in the oculomotor nerve nuclei did not, although the extent of GluR2 Q/R site editing was significantly reduced in all these nuclei. These results indicate that motor neurons in the oculomotor nerve nuclei can survive despite the incomplete nature of GluR2 Q/R site editing. Notably, motor neurons in the nuclei of oculomotor nerves are also much less vulnerable in ALS patients; this has been attributed to differential expression levels of Ca2+-binding proteins, particularly parvalbumin, among motor neurons in different cranial nerve nuclei. Expression of parvalbumin is high in oculomotor neurons and low in the facial and spinal motor neurons (Ince et al., 1993). Indeed, overexpression of parvalbumin attenuated kainate-induced Ca2+ transients and protected spinal motor neurons from resultant neurotoxicity in parvalbumin transgenic mice (Van Den Bosch et al., 2002). It is likely that neurons with an efficient Ca2+-buffering system, such as oculomotor neurons, are resistant to Ca2+ overload resulting from Ca2+-permeable AMPA receptors.
The present results indicate that the failure of A-to-I conversion at the Q/R site of GluR2 pre-mRNA in motor neurons of sporadic ALS patients (Takuma et al., 1999; Kawahara et al., 2004; Kwak and Kawahara, 2005) is likely attributable to reduced ADAR2 activity. Indeed, the expression level of ADAR2 mRNA was decreased in the spinal cord of patients with sporadic ALS (Kawahara and Kwak, 2005). Molecular abnormalities found in postmortem tissues of patients with neurodegenerative diseases have shown signs of mechanisms underlying the disease and may represent both the neuronal death process and death-protective reactions arising from the protracted nature of the death process. It is therefore necessary to determine whether these molecular abnormalities are the cause or the result of neuronal cell death by developing an appropriate animal model. Although excitotoxicity has long been implicated in the pathogenesis of neurological diseases including ALS (Vosler et al., 2008; Bezprozvanny, 2009), surprisingly little direct evidence indicating excitotoxic neuronal cell death has been demonstrated in patient-derived materials. Here we demonstrate that the molecular abnormality found in motor neurons of patients with sporadic ALS is a direct cause of neuronal death in mice via a mechanism upregulating Ca2+-permeable AMPA receptors. In addition, the AR2 mice possess certain characteristics found in ALS, including slow progressive death of motor neurons, neuromuscular unit-dependent motor dysfunction and differential low vulnerability of motor neurons of extraocular muscles. Therefore, this mouse model mimicking patient-derived molecular abnormalities may be useful for research on sporadic ALS.
Footnotes
This study was supported in part by Ministry of Education, Culture, Sports, Science, and Technology of Japan Grants-in-Aid for Scientific Research 17390251, 19390235, and 20023008 (S.K.), Ministry of Health, Labor, and Welfare of Japan Grant H18-Kokoro-017 (S.K.), and Amyotrophic Lateral Sclerosis Association Grant 875 (P.H.S.). We thank Dr. R. B. Emeson at Vanderbilt University (Nashville, TN) for antibodies to ADAR2 and D. Kimura, K. Awabayashi, Dr. J. Shimizu, Dr. M. Fukaya, and T. Kakinoki for technical assistance.
References
- Akbarian S, Smith MA, Jones EG. Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res. 1995;699:297–304. doi: 10.1016/0006-8993(95)00922-d. [DOI] [PubMed] [Google Scholar]
- Beleza-Meireles A, Al-Chalabi A. Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph Lateral Scler. 2009;10:1–14. doi: 10.1080/17482960802585469. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- Feng Y, Sansam CL, Singh M, Emeson RB. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol Cell Biol. 2006;26:480–488. doi: 10.1128/MCB.26.2.480-488.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40:763–774. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Tsuji S, Misawa H, Takahashi R, Suzuki T, Kwak S. Slow neuronal death of motor neurons in sporadic ALS mouse model by RNA editing enzyme ADAR2 knockout. Soc Abstr Neurosci. 2008;34:745–17. [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Ince P, Stout N, Shaw P, Slade J, Hunziker W, Heizmann CW, Baimbridge KG. Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol Appl Neurobiol. 1993;19:291–299. doi: 10.1111/j.1365-2990.1993.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Kask K, Zamanillo D, Rozov A, Burnashev N, Sprengel R, Seeburg PH. The AMPA receptor subunit GluR-B in its Q/R site-unedited form is not essential for brain development and function. Proc Natl Acad Sci U S A. 1998;95:13777–13782. doi: 10.1073/pnas.95.23.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Kwak S. Excitotoxicity and ALS: what is unique about the AMPA receptors expressed on spinal motor neurons? Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:131–144. doi: 10.1080/14660820510037872. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Kanazawa I, Kwak S. Low editing efficiency of GluR2 mRNA is associated with a low relative abundance of ADAR2 mRNA in white matter of normal human brain. Eur J Neurosci. 2003a;18:23–33. doi: 10.1046/j.1460-9568.2003.02718.x. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Kwak S, Sun H, Ito K, Hashida H, Aizawa H, Jeong SY, Kanazawa I. Human spinal motoneurons express low relative abundance of GluR2 mRNA: an implication for excitotoxicity in ALS. J Neurochem. 2003b;85:680–689. doi: 10.1046/j.1471-4159.2003.01703.x. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427:801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Sun H, Ito K, Hideyama T, Aoki M, Sobue G, Tsuji S, Kwak S. Underediting of GluR2 mRNA, a neuronal death inducing molecular change in sporadic ALS, does not occur in motor neurons in ALS1 or SBMA. Neurosci Res. 2006;54:11–14. doi: 10.1016/j.neures.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Kuner R, Groom AJ, Bresink I, Kornau HC, Stefovska V, Müller G, Hartmann B, Tschauner K, Waibel S, Ludolph AC, Ikonomidou C, Seeburg PH, Turski L. Late-onset motoneuron disease caused by a functionally modified AMPA receptor subunit. Proc Natl Acad Sci U S A. 2005;102:5826–5831. doi: 10.1073/pnas.0501316102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S, Kawahara Y. Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J Mol Med. 2005;83:110–120. doi: 10.1007/s00109-004-0599-z. [DOI] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- Lowe JS, Leigh N. Motor neuron disease (amyotrophic lateral sclerosis) In: Love S, Louis DN, Ellison DW, editors. The Greenfield's neuropathology. Oxford: Oxford UP; 2002. pp. 372–383. [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- Misawa H, Nakata K, Toda K, Matsuura J, Oda Y, Inoue H, Tateno M, Takahashi R. VAChT–Cre.Fast and VAChT–Cre.Slow: postnatal expression of Cre recombinase in somatomotor neurons with different onset. Genesis. 2003;37:44–50. doi: 10.1002/gene.10224. [DOI] [PubMed] [Google Scholar]
- Nishimoto Y, Yamashita T, Hideyama T, Tsuji S, Suzuki N, Kwak S. Determination of editors at the novel A-to-I editing positions. Neurosci Res. 2008;61:201–206. doi: 10.1016/j.neures.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Ohmae S, Takemoto-Kimura S, Okamura M, Adachi-Morishima A, Nonaka M, Fuse T, Kida S, Tanji M, Furuyashiki T, Arakawa Y, Narumiya S, Okuno H, Bito H. Molecular identification and characterization of a family of kinases with homology to Ca2+/calmodulin-dependent protein kinases I/IV. J Biol Chem. 2006;281:20427–20439. doi: 10.1074/jbc.M513212200. [DOI] [PubMed] [Google Scholar]
- Paschen W, Hedreen JC, Ross CA. RNA editing of the glutamate receptor subunits GluR2 and GluR6 in human brain tissue. J Neurochem. 1994;63:1596–1602. doi: 10.1046/j.1471-4159.1994.63051596.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Peng PL, Zhong X, Tu W, Soundarapandian MM, Molner P, Zhu D, Lau L, Liu S, Liu F, Lu Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;49:719–733. doi: 10.1016/j.neuron.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transporter by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci U S A. 2003;100:14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymick JC, Talbot K, Traynor BJ. Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2007;16:R233–R242. doi: 10.1093/hmg/ddm215. Spec No 2. [DOI] [PubMed] [Google Scholar]
- Seeburg PH. A-to-I editing: new and old sites, functions and speculations. Neuron. 2002;35:17–20. doi: 10.1016/s0896-6273(02)00760-2. [DOI] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Tsuzuki K, Kameyama K, Kwak S. Recent advances in the study of AMPA receptors. Nippon Yakurigaku Zasshi. 2003;122:515–526. doi: 10.1254/fpj.122.515. [DOI] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, Fujii H, Fuse T, Hoshino M, Suzuki S, Kojima M, Mishina M, Okuno H, Bito H. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron. 2007;54:755–770. doi: 10.1016/j.neuron.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Takuma H, Kwak S, Yoshizawa T, Kanazawa I. Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann Neurol. 1999;46:806–815. doi: 10.1002/1531-8249(199912)46:6<806::aid-ana2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Braeken D, Callewaert G, Robberecht W, Van Den Bosch L. GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2005;64:605–612. doi: 10.1097/01.jnen.0000171647.09589.07. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Schwaller B, Vleminckx V, Meijers B, Stork S, Ruehlicke T, Van Houtte E, Klaassen H, Celio MR, Missiaen L, Robberecht W, Berchtold MW. Protective effect of parvalbumin on excitotoxic motor neuron death. Exp Neurol. 2002;174:150–161. doi: 10.1006/exnr.2001.7858. [DOI] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Sklar P, Axel R, Maniatis T. Editing of glutamate receptor subunit B pre-mRNA in vitro by site-specific deamination of adenosine. Nature. 1995;374:77–81. doi: 10.1038/374077a0. [DOI] [PubMed] [Google Scholar]