

Abstract
Tumor necrosis factor α (TNF-α) is a potent cytokine in neurodegenerative disorders, but its precise role in particular brain disorders is ambiguous. In motor neuron (MN) disease of the mouse, exemplified by the model wobbler (WR), TNF-α causes upregulation of the metalloprotease-disintegrin ADAM8 (A8) in affected brain regions, spinal cord, and brainstem. The functional role of A8 during MN degeneration in the wobbler CNS was investigated by crossing WR with A8-deficient mice: a severely aggravated neuropathology was observed for A8-deficient WR compared with WR A8+/− mice, judged by drastically reduced survival [7 vs 81% survival at postnatal day 50 (P50)], accelerated force loss in the forelimbs, and terminal akinesis. In vitro protease assays using soluble A8 indicated specific cleavage of a TNF-α receptor 1 (p55 TNF-R1) but not a TNF-R2 peptide. Cleavage of TNF-R1 was confirmed in situ, because levels of soluble TNF-R1 were increased in spinal cords of standard WR compared with wild-type mice but not in A8-deficient WR mice. In isolated primary neurons and microglia, TNF-α-induced TNF-R1 shedding was dependent on the A8 gene dosage. Furthermore, exogenous TNF-α showed higher toxicity for cultured neurons from A8-deficient than for those from wild-type mice, demonstrating that TNF-R1 shedding by A8 is neuroprotective. Our results indicate an essential role for ADAM8 in modulating TNF-α signaling in CNS diseases: a feedback loop integrating TNF-α, ADAM8, and TNF-R1 shedding as a plausible mechanism for TNF-α mediated neuroprotection in situ and a rationale for therapeutic intervention.
Introduction
Tumor necrosis factor α (TNF-α) is a potent cytokine with beneficial as well as proinflammatory and neurotoxic effects in the CNS (for review, see Sriram and O'Callaghan, 2007). Its pleiotropic activities are mediated by two cell surface receptors, p55 (TNF-R1) and p75 (TNF-R2). These receptors feed into diverse signaling pathways according to differences in their intracellular domains. A death domain in TNF-R1 not present in TNF-R2 leads to apoptotic cell death. However, alternative pathways of TNF-R1 signaling involve activation of MAP kinase (Chen and Goeddel, 2002), ERK kinases (ERK1/2) (Subramaniam and Unsicker, 2010), or signal transducers and activators of transcription (STATs, i.e., STAT3) (Dziennis and Alkayed, 2008) serving neuroprotective functions.
TNF receptors are synthesized as membrane-bound proteins, and their release from the cell surface is mediated by proteolysis, a process known as ectodomain shedding (Peschon et al., 1998). Proteolytic release of TNF-R1 has been described to serve a sequestering function in binding excess TNF-α in the extracellular compartment thereby desensitizing a cell to TNF-α action. Soluble forms of TNF receptors (sTNF-Rs) are constitutively released from cell membranes. After TNF-α stimulation, as observed under disease conditions, TNF receptor release is increased. In cell culture systems, soluble TNF receptors are produced in response to numerous stimuli, such as TNF-α, bacterial lipopolysaccharides (LPS), phorbol esters (tetradecanoylphorbol acetate), and IL-10, or after T-cell and neutrophil activation (Leeuwenberg et al., 1994; Pedron et al., 2003). An aminopeptidase, ARTS-1 (Cui et al., 2002, 2003) and ADAM17/TACE (Black et al., 1997; Moss et al., 1997), a member of the family of ADAM proteins, have so far been identified as proteases responsible for ectodomain shedding of TNF receptors. TACE can cleave pro-TNF-α as well as TNF-R2 and, to a lesser extent, TNF-R1 from the cell surface (Peschon et al., 1998; Reddy et al., 2000), which could account for the level of constitutive shedding in cells expressing TNF-R1. It is generally accepted that neurodegeneration in the CNS causes an inflammatory phenotype, i.e., by activation of innate immunity effectors such as proinflammatory cytokines TNF-α and IL-1 (cf. Gosselin and Rivest, 2007). Analysis of motor neuron degeneration in the mouse mutant wobbler (WR) showed that proinflammatory signaling is essential for neuron–glia signaling (Schlomann et al., 2000). The wobbler gene (wr) is a missense allele of the gene encoding VPS54, a protein involved in retrograde transport (Schmitt-John et al., 2005), and causes a progressive motor neuron degeneration in the cervical spinal cord, leading to forelimb atrophy, similar to disease symptoms observed in human limb gurdle atrophy and amyotrophic lateral sclerosis.
In wobbler mice, TNF-α causes a strong upregulation of ADAM8 in neurons, astrocytes, and microglia in brain regions affected by neurodegeneration, brainstem, and spinal cord (Schlomann et al., 2000). We hypothesized that ADAM8, a protease involved in inflammatory responses (Schlomann et al., 2002; Koller et al., 2009), as a TNF-α target gene might affect disease progression in the wobbler mouse. Therefore, we investigated the role of ADAM8 in WR mice by generating WR/ADAM8-deficient mice and analyzed disease progression.
Materials and Methods
Reagents
Recombinant TNF-α was obtained from Peprotech with a specific activity of 10 U/ng. LPS from Escherichia coli was purchased from Sigma, dissolved in PBS, and used at concentrations indicated.
Mouse breeding and maintenance
Adam8-deficient (Kelly et al., 2005) and wr/+ mice (Kaupmann et al., 1992; Rathke-Hartlieb et al., 1999) were kept on a C57BL/6 background for at least 10 generations before starting the crossing experiments. In our studies, we used a large number of mice by crossing wr/+ with homozygous A8-deficient (Adam8 −/−) mice. The resulting F1 was crossed to obtain the required genotypes. Littermates were taken as controls. Both genders of mice were used for experiments, and no gender differences were observed in the neurological disease phenotype. All animal experiments were performed in accordance with the German law on the protection of Animals (“Tierschutzgesetz”) and with permission by the Local Authorities (Regierungspräsidium Detmold).
PCR diagnosis of the wr allele
The wr allele was diagnosed by a duplex PCR based on the molecular identification of the wr mutation (Schmitt-John et al., 2005). For the wild-type (WT) allele, the primers Vps54–WT forward (5′ cgt tct ctg ttg aag cca ca 3′) and Vps54–WT reverse (5′ ccc aga tct cgg cca tat tta 3′) were used. For the wobbler mutation, the primers Vps54–wr forward (5′ agg cct taa aga tct gga tca 3′) and Vps54–wr reverse 255 (5′ tgc tcc tta ctc agg gat gc 3′) were used. All four primers were mixed with the template DNA, and PCR was performed at 65°C annealing temperature. The PCR products have a size of 413 bp for the wild-type allele and 255 bp for the wr allele of Vps54.
Scoring muscular atrophy
Force measurements were performed on a grid connected to a force meter (Mitsumoto et al., 1994) detecting forces within a range of 0 to 200 cN. Mice were placed on the grid, and, after they have grasped the grid using both paws, a force from the tail was applied to pull the mice from the grid. The force required to pull the mice from the grid was recorded, and the mean value from at least five attempts was taken as one data point.
Morphological analyses
Mice were deeply anesthetized and transcardially perfused with fixative, as described previously (Laage et al., 1988) for preparation of paraffin serial sections of spinal cords. For motor neuron counts, cryosections (10 μm) and paraffin sections from spinal cords were serially cut (5 μm), and every 10th section was processed by counting motor neuron numbers in the ventrolateral motor neuron pool. In this area, motor neurons can be distinguished from interneurons. The raw counts were corrected for double counting of split nuclei as described previously (Rathke-Hartlieb et al., 1999). For each experimental group, four mice were analyzed and 10 sections were evaluated for numbers (n) of healthy motor neurons in the anterior horn on each side of one section. Differences between groups were evaluated with Student's t test (significance level p < 0.05, unpaired), using Microsoft Excel.
Immunohistochemistry
For immunofluorescence, cryosections (10 μm) of CNS tissues were fixed for 5 min in 3% formaldehyde at room temperature or with methanol at −20°C for 6 min. For detection of reactive gliosis, we used a monoclonal cyanine 3 (Cy3)-conjugated anti-glial fibrillary acidic protein (GFAP) (1:800; Sigma), and for microglia detection, a rat anti-CD45 (BD Biosciences) was used in conjunction with a secondary goat anti-rat IgG (1:200; Dianova) conjugated with Cy3. For combined anti-CD45 and Nissl stains, a goat anti-rat HRP antibody with diaminobenzidine (DAB) as a substrate was used. To detect TNF-R1 and ADAM8, the polyclonal antibodies a-TNF-R1nt (1:50, sc-1069; Santa Cruz Biotechnology) and a-ADAM8 cytoplasmic domain antibody (1:50) (Schlomann et al., 2000) were used as primary antibodies. Secondary antibodies were rabbit anti-goat Alexa488 (1:250; Dianova) and goat anti-rabbit Cy3 (1:300; Sigma).
Quantitative reverse transcription-PCR
Total RNA from cultured cells was prepared with Trizol reagent (Invitrogen) according to the instructions of the manufacturer. RNA samples were stored at −80°C pending reverse transcription (RT)-PCR. Three micrograms of total RNA were transcribed using the Bioline RT kit in a volume of 20 μl. Subsequently, 1 μl of RT product was used for PCR in a 20 μl reaction volume. The following primers were used: ADAM8 forward, 5′-ctccagatcccatcatgctt-3′; ADAM8 reverse, 5′-ggtccaggggctactgct-3′ (amplicon 70 nt); ADAM10 forward, 5′-gggaagaaatgcaagctgaa-3′; ADAM10 reverse, 5′-ctgtacagcagggtccttgac-3′ (amplicon 64 nt); ADAM17 forward, 5′-ctttggtgcctttcgtcct-3′; ADAM17 reverse 5′-gagcaaagaatcaagcttctcaa-3′ (amplicon 89 nt). For referencing, the amplicons for Ubiquitin C (UBC) and ribosomal protein L7 (RPL7) were used: UBC forward, 5′-gtctgctgtgtgaggactgc-3′; UBC reverse, 5′-gtcttgcctgtcagggtctt-3′ (amplicon 76 nt); RPL7 forward, 5′-tggaaccatggaggctgt-3′; RPL7 reverse, 5′-cacagcgggaacctttttc-3′ (amplicon 115 nt). The resulting PCR products were initially separated on a 4% agarose gel and subjected to sequencing to verify identities of the PCR products. Real-time PCR was performed in a 20 μl reaction volume with QuantiTect SYBR Green PCR kit (Qiagen); between 2 μl of undiluted and 1 μl of a 1:10 dilution of the RT product was used as template. Samples were run on a LightCycler (Roche). Each sample was analyzed in duplicate in three independent experiments. Differences in ADAM mRNA concentrations were assessed relative to controls, and significance was analyzed by Student's t test. Differences with p < 0.01 were considered significant.
Cell culture
Microglial cells were obtained from neonatal mice (P0–P4). The brain hemispheres were dissected and subsequently homogenized by mincing them with scissors, incubating in papain solution and shearing them through a Pasteur pipette and a syringe with a 21 gauge needle. The cells were spun down, resuspended in DMEM, 10% fetal calf serum (FCS), and 1% glutamine, and seeded on a poly-d-lysine-coated 75 cm2 tissue culture flask (cells from three brains per flask). The medium was removed after 48 h, 96 h, and 1 week, clarified, and stored at −20°C for the microglia cultures. After 12 d, the cells were resuspended by trypsination and subsequently incubated for 20 min on two and 90 min on one bacterial grade plates. Cells attached to the bacterial plates were cultivated in a one-to-one mixture of culture medium and the stored medium. After 12 d, the conditioned medium was replaced by normal cell culture medium to obtain resting microglia that were used for additional experiments. Purity of microglial cultures was assessed by mouse anti-CD11b (clone OX42; 1:1000; MCA275EL; Serotec) and was shown to be >95%. Primary astrocytes and cerebellar neurons were generated as described previously (Schlomann et al., 2000; Naus et al., 2004). Purity of primary cultures was determined by parallel stainings with monoclonal GFAP–Cy3 (Sigma) and neuronal-specific nuclear protein (MAB377; Millipore Bioscience Research Reagents), respectively. Purities of >90% were obtained and further processed. TNF-α or LPS stimulation was performed 2 d after seeding in serum-reduced medium (1% FCS). Cell death rates were determined 5 d after seeding by cell counts and MTT assays.
ELISAs.
Cell culture supernatants were removed, clarified by centrifugation, and directly used for ELISAs. Tissue was extracted by homogenizing in 10 μl/mg tissue in ice-cold PBS containing a protease inhibitor cocktail (EDTA-free; Roche). The homogenate was spun down for 10 min at 4°C, and the supernatant was directly used for ELISAs. ELISA for soluble TNF receptors (TNF-R1 and TNF-R2) were used according to the manual of the supplier (R & D Systems). All ELISA measurements were done in triplets from five mice with identical genotypes (n = 15). Data are expressed as mean ± SD. Values were compared using an unpaired Student's t test, and p values <0.05 were considered significant.
Peptide assays.
Peptides were obtained from CPC Scientific and assayed as described previously (Naus et al., 2006). Briefly, peptides were incubated with either recombinant human (R & D Systems) or mouse ADAM8 ectodomain (Naus et al., 2006) for 16 h at 37°C in a buffer containing 10 mm Tris-HCl, pH 7.5, 10 mm calciumchloride, 1 μm zinc chloride, and 0.015% Brij-35. Peptide fragments were purified by HPLC, and sequence determination was performed by mass spectrometry as described previously (Naus et al., 2006).
Microphotography and image analysis
Fluorescence images were documented using either a Nikon Coolpix camera attached to Leitz Fluorescence microscope or by confocal laser scanning microscopy (TCS SP2; Leica). Digital images with identical sampling fields were transferred to an NIH ImageJ program equipped with a plug-in to quantify the pixel values and numbers above a given threshold value. For each histological sample, sections were obtained from four mice, and 10 serial sections per mouse were quantified (n = 4). Data were averaged for each of the mice and presented as mean values ± SEM (n = 4). For statistical significance, data were compared using an unpaired Student's t test with p < 0.05 considered significant.
Results
Absence of ADAM8 aggravates the wobbler pathology
To assess the contribution of ADAM8 in neurodegeneration, WR mice deficient in ADAM8 were generated. The effect of ADAM8 on the progression of the WR motor neuron disease was examined in four cohorts of wr/+ Adam8 +/+ (“wild type”), wr/+ Adam8 −/− (“ADAM8 deficient”), wr/wr Adam8 +/− (“standard wobbler WR”), and wr/wr Adam8 −/− (“ADAM8-deficient WR”) mice. The survival times of these cohorts (n ≥ 32) of mice within 50 d after birth were determined for each genotype (Fig. 1 A). The survival of ADAM8-deficient WR individuals (wr/wr Adam8 −/−) was down to 0% after 55 d compared with wild-type/ADAM-deficient mice (88% survival after 55 d, respectively) and standard WR mice (wr/wr Adam8 +/−) with 69% survival after 55 d. Control wr/+ Adam8 +/+ and wr/+ Adam8 −/− mice did not differ in survival rates, demonstrating that Adam8 deficiency in heterozygous wr/+ mice has no effect on survival within 50 d and thereafter, which is consistent with a normal life expectancy of ADAM8-deficient mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To monitor the disease onset and progression in these four cohorts of mice, force measurements of forelimb muscles were performed between days 10 and 40 after birth (Fig. 1 B). Wild-type (wr/+ Adam8 +/+) and ADAM8-deficient (wr/+ Adam8 −/−) mice gained significant force during postnatal development ranging from initial 15 to 80 cN, and they were indistinguishable from wild-type C57BL/6 mice. In contrast, wobbler mice, because of their progressive forelimb atrophy, were not able to gain force after disease onset at approximately day 18 after birth (cf. Mitsumoto et al., 1994). After disease onset, the forelimb force in wobbler mice (wr/wr Adam8 +/−) reached a constant value (∼20 cN) and dropped on day 22 after birth. ADAM8-deficient Wobbler mice (wr/wr Adam8 −/−) showed a more dramatic decline in the forelimb force at P18 and complete loss of force at approximately day 24 after birth. Concomitant with the observed muscle atrophy, 80% of the ADAM8-deficient wobbler mice (wr/wr Adam8 −/−) were akinetic from P30 onward. These data suggest an essential role for ADAM8 in the wobbler pathology. With respect to survival times and force development, the control mice with the genotypes wr/+ Adam8 +/+ and wr/+ Adam8 −/− were indistinguishable. Thus, only the genotype wr/+ Adam8 −/− was used as control genotype for additional analyses.
Figure 1.

Absence of ADAM8 aggravates the wobbler (WR) disease. A, Kaplan–Meier survival curves of wr/+ Adam8 +/+ (black line; n = 45), wr/+ Adam8 −/− (dashed black line; n = 34), wr/wr Adam8 +/− (gray line; n = 32), and wr/wr Adam8 −/− (dashed gray line; n = 38) mice for 54 d after birth. Significance was calculated by pairwise log rank tests for survival with p < 0.07. B, Grip strengths of forelimbs of wr/+ Adam8 +/+ (squares), wr/+ Adam8 −/− (circles), wr/wr Adam8 +/− (rhombs), and wr/wr Adam8 −/− (triangles) mice. For all genotypes, n = 15. Mean ± SD values. Note the total loss of force within 24 d after birth in wr/wr Adam8 −/− individuals, leading to complete akinesis and premature death. p.n., Postnatal.
ADAM8 deficiency in wobbler mice leads to changes in neuron numbers, reactive gliosis, and microglia activation
The aggravated disease phenotype of A8-deficient WR mice was assessed at the histological level. Monitoring for neurodegeneration, reactive gliosis, and microglia activation in mice was performed with genotypes wr/+Adam8 −/−, wr/wr Adam8 +/−, and wr/wr Adam8 −/−, using cresyl violet (Nissl) staining, and GFAP and tyrosine phosphatase (CD45) immunostaining, respectively (Fig. 2). Morphological analyses of ventrolateral motor neurons pools in spinal cords revealed all stages of motor neuron degeneration (Fig. 2 B–E), including chromatolytic neurons (Fig. 2 C), vacuolized cell bodies (Fig. 2 E), and faded neurons (Fig. 2 C,D). Motor neuron counts revealed that standard wobbler individuals (wr/wr Adam8 +/−) had decreased numbers of motor neurons (17 ± 4 per section, p < 0.001, n = 4) compared with ADAM8-deficient non-WR individuals (wr/+ Adam8 −/−; 22 ± 2, p < 0.001, n = 4). In accordance with the aggravated disease phenotype of ADAM8-deficient WR mice (wr/wr Adam8 −/−), we observed an additional reduction of motor neuron counts in spinal cords of wr/wr Adam8 −/− mice (8 ± 3 per section, p < 0.001, n = 4) compared with wr/wr Adam8 +/− individuals (Fig. 3 A). There were no apoptosis in spinal cords of WR mice deficient in ADAM8, suggesting that the mode of cell death is not affected by ADAM8 deficiency.
Figure 2.
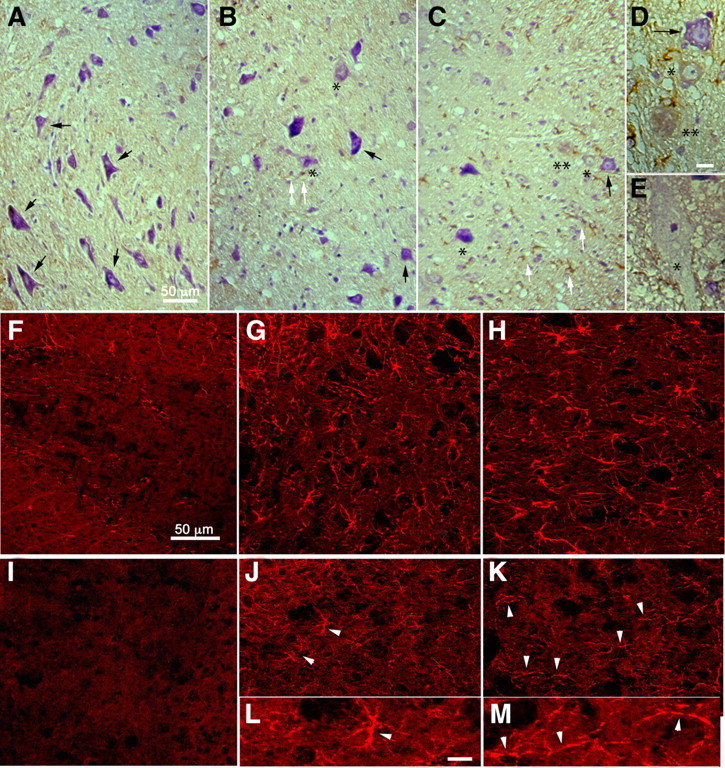
WR histopathology of spinal cord is aggravated by ADAM8 deficiency. A–C, Morphology of spinal cord neurons in the anterior horn of mice with wr/+ Adam8 −/− (A), wr/wr Adam8 +/− (B), and wr/wr Adam8 −/− (C) genotypes, respectively. Spinal cord sections were stained with cresyl violet and immunohistochemistry/DAB staining (brown) for microglia (CD45). The ventrolateral part of the spinal cord is shown (white matter to the left in A and to the right in B and C). Motor neurons in A show normal morphology (black arrows). Motor neurons in wr/wr Adam8 +/− mice (B) display early stages of degeneration, characterized by chromatolysis and fading, characteristic features of neurodegeneration in WR mice (asterisks). In addition, compared with A, increased staining of small CD45-positive microglia is detectable surrounding degenerating motor neurons (white arrows in B). In C, increased numbers of degenerated neurons in the spinal cord of wr/wr A8 −/− compared with wr/wr Adam8 +/− mice (black arrows, unaffected neuron) and degenerating neurons of different stages (* and ** reflect different stages of degeneration, respectively). D, E, More detailed views of degenerating motor neurons in wr/wr Adam8 +/− and wr/wr A8 −/− WR mice with additional vacuolized motor neurons (E). There were no signs of morphological differences between degeneration observed in wr/wr Adam8 +/− and wr/wr A8 −/− spinal cords. In F–H, immunohistochemistry for GFAP (reactive gliosis) and CD45 immunostaining for microglia activation (I–M) for genotypes wr/+ Adam8 −/− (I), wr/wr Adam8 +/− (J, L), and wr/wr Adam8 −/− (K, M) genotypes. L, M, Enlarged view of microglia indicated by arrowheads in J and K, respectively. Note the chain-like appearance of microglia in wr/wr A8 −/− mice, resulting from lesser ramification. Scale bars: (in A) A–C, 50 μm; (in D) D, E, 10 μm; (in F) F–K, 50 μm; (in L) L, M, 10 μm.
Figure 3.

Quantitative evaluation of histopathological data. A, Motor neuron numbers in ventrolateral pool of anterior horns, judged by Nissl staining; B, GFAP immunoreactivity, given as pixel values per spinal cord section; C, quantification of CD45 immunoreactivity. In each bar graph, quantification of image data were performed for wr/+ Adam8 −/− (black bars), wr/wr Adam8 +/− (gray bars), and wr/wr Adam8 −/− (white bars). Pixel values from individual images were acquired using NIH ImageJ analysis software. *p < 0.05, ***p < 0.001.
Aggravated neurodegeneration in double mutants was accompanied by only an additional moderate increase in GFAP-positive astrocytes, suggesting that reactive gliosis in standard wobbler mice is already maximal (Figs. 2, 3 B). Also, no differences in astrocyte morphology was observed between standard wobbler and double mutants (Fig. 2 G,H). In contrast, there is a more significant increase in microglial activation as judged by CD45 staining and quantification (Fig. 3 C). CD45 staining in wr/+ Adam8 −/− control mice was very low with 8 ± 1.2. The relative signals for microglia increased from 150 ± 16 in standard wobbler mice (wr/wr Adam8 +/−, p < 0.001) to 250 ± 13 in ADAM8-deficient Wobbler individuals (wr/wr Adam8 −/−, p < 0.001). Moreover, microglia morphology in wr/wr Adam8 −/− spinal cords appeared less ramified and had a chain-like morphology (Fig. 2). These data show that the absence of ADAM8 has a significant effect on both the primary effect of the wr mutation, neurodegeneration, and its secondary consequences on microglial activation.
ADAM8 mRNA levels are upregulated in the wobbler CNS
We have shown previously that ADAM8 mRNA levels are elevated in affected CNS regions of WR mice and that the ADAM8 mRNA levels and distribution correlate with TNF-α concentration. To test whether this correlation is specific for ADAM8, relative levels of mRNAs for ADAM8, ADAM10, and ADAM17 in WT and in WR CNS regions were determined by quantitative RT-PCR (Fig. 4 A). ADAM8 mRNA levels were significantly increased in WR cerebellum (×3.0 ± 0.5 compared with WT) and, more dramatically, in WR spinal cord (×23 ± 3 compared with WT). No ADAM8 induction was observed in the WR cortex. In contrast to ADAM8, mRNA levels for ADAM10 remained unchanged, whereas a significant induction of ADAM17 (×4 ± 1, p < 0.001) was observed in the WR spinal cord, arguing for a TNF-α inducible expression of ADAM8 and ADAM17 under inflammatory conditions in the CNS.
Figure 4.

ADAM mRNA levels in WR mice and in primary CNS cells. Levels of ADAM8, ADAM10, and ADAM17 mRNA in cortex, cerebellum, and spinal cords of WT and WR mice are elevated by neurodegeneration (wobbler CNS), LPS, and TNF-α in neurons, astrocytes, and microglia. A, Quantitative RT-PCR analysis of Adam genes encoding ADAM8, ADAM10, and ADAM17 in the CNS of WT and WR cortex, cerebellum, and spinal cord. B, Quantitative RT-PCR analysis for Adam8 in primary neurons (cerebellar granular cells), spinal cord astrocytes, and cortical microglia cells from mouse CNS in the presence of recombinant mouse TNF-α (100 ng/ml) or LPS (250 ng/ml). Light cycler data were normalized to reference mRNAs (Ubiquitin C and ribosomal protein L7) and given relative to WT. Mean ± SD values were derived by three independent runs performed in triplets for each sample (n = 9). *p < 0.05, **p < 0.01, ***p < 0.001.
In addition, the induction characteristics of ADAM8 mRNA levels by TNF-α and LPS were evaluated in isolated primary cells from mouse CNS, cerebellar neurons, spinal cord astrocytes, and primary microglia (Fig. 4 B). After administration of 100 U/ml TNF-α, a 7- to 10-fold induction of ADAM8 mRNA was observed, whereas higher concentrations (≥500 U/ml) of TNF-α lead to a decline in ADAM8 mRNA levels. Similarly, the effect of LPS shows an induction optimum at 250 ng/ml and declines at higher LPS concentrations of 1000 ng/ml.
ADAM8 cleaves TNF-R1 in vitro but not TNF-R2
We determined the TNF-R1 cleavage site with soluble recombinant human and mouse ADAM8 comparing four different TNF receptor peptides (Table 1) (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material). In this assay, a species-specific cleavage preference was observed, as recombinant human ADAM8 effectively cleaved human TNF-R1 peptide, whereas mouse recombinant ADAM8 cleaved the respective mouse TNF-R1 peptide much more efficiently than human TNF-R1 peptide. However, for ADAM8 from both species, the position of the cleavage site was identical, arguing for a physiological cleavage site recognized by ADAM8. Peptides with the sequences of the juxtamembraneous cleavage sites of TNF-R2 were cleaved by neither human nor mouse ADAM8, indicating that ADAM8 is not a potential shedding enzyme for TNF-R2.
Table 1.
Cleavage of mouse and human TNF receptor peptides by recombinant mouse and human ADAM8
| Peptide | Origin | mADAM8 | hADAM8 | Cleavage site | Reference |
|---|---|---|---|---|---|
| NPQDSGTAVL | mTNF-R1 | +++ | + | NPQ ⇓ DSGTAVL | Mohan et al., 2002 |
| LPQLENVKGTED | hTNF-R1 | 0 | ++ | LPQLE ⇓ NVKGTED | This work |
| PPLANVTNPQ | mTNF-R2 | 0 | 0 | ND | Pinckard et al., 1997 |
| CMKLCLPPPL | hTNF-R2 | 0 | 0 | ND | Reddy et al., 2000 |
Cleavage sites were analyzed using coupled HPLC/MS. For details, see supplemental data (available at www.jneurosci.org as supplemental material). m, Mouse; h, human; ND, not determined.
TNF-R1 release correlates with the Adam8 gene dosage in CNS tissue
The concentrations of sTNF-R1 were determined in several tissues and cell preparations to confirm that TNF-R1 release and ADAM8 expression correlate in situ under pathological conditions. Brain lysates of ADAM8-deficient mice (wr/+ Adam8 −/−) and standard wobbler mice (wr/wr Adam8 +/−) were analyzed to quantify the levels of sTNF-R1 by ELISA. No differences in the amounts of sTNF-R1 were observed in lysates from cortex, in accordance with the absence of neurodegeneration in these CNS regions (Fig. 5 A). However, in spinal cord lysates of standard wobbler mice (wr/wr Adam8 +/−), the concentration of sTNF-R1 was significantly increased. This increase was not observed in wobbler mice with a homozygous Adam8 deficiency (wr/wr Adam8 −/−), arguing for a role of ADAM8 in the release of TNF-R1 under pathological conditions. In addition, TNF-R1 localization was determined by immunohistochemistry (Fig. 5 B). In WT spinal cords, motor neurons express TNF-R1 weakly, mainly localized to vesicles (Fig. 5 Bi). Accumulations of TNF-R1 in vesicles and on the cell surface were detected in motor neurons of wr/+ Adam8 −/− mice (Fig. 5 Bii), with increased vesicular localization of TNF-R1 in motor neurons of wr/wr Adam8 +/− mice (Fig. 5 Biii) and deficient shedding in wr/wr Adam8 −/− motor neurons (Fig. 5 Biv), resulting in intracellular accumulations of TNF-R1.
Figure 5.

A, Concentrations of soluble TNF-R1 in the spinal cord of wobbler mice determined by ELISA. Lysates from cortex and spinal cord from wild-type Adam8 +/+ wr/+ (white bars), Adam8 +/+ wr/wr (black bars), Adam8 −/− wr/+ (hatched bars), and Adam8 −/− wr/wr (gray bars) were analyzed with an sTNF-R1 ELISA kit. Mean ± SD values from three independent experiments performed in triplets (n = 9). Data were analyzed by t test. *p < 0.05, **p < 0.01, ***p < 0.001. B, Immunohistochemistry of TNF-R1 in spinal cord sections corresponding to the samples analyzed in A: i, wr/+ Adam8 +/+ with large intact motor neurons, weakly stained for TNF-R1; ii, wr/+ Adam8 −/− with vesicular and membrane-associated TNF-R1 signals; iii, wr/wr Adam8 +/− increased vesicular TNF-R1 staining compared with ii; iv, wr/wr Adam8 −/− with strong TNF-R1 staining throughout the cell body; v, negative control (omission of primary antibody). Scale bar, 50 μm. Immunostaining for TNF-R1 is indicated by white arrowheads.
Neuronal survival is impaired in ADAM8-deficient primary neurons
We used a neuronal survival assay as a physiological readout for TNF-α-induced ADAM8-mediated TNF-R1 shedding (Fig. 6). The comparative analysis of wild-type and Adam8-deficient primary neurons, isolated from cerebellum, revealed that TNF-α-induced TNF-R1 shedding was almost nil in the absence of ADAM8 (Fig. 6 A). To determine whether this affected neuronal survival, wild-type and ADAM8-deficient neurons were treated with increasing concentrations of TNF-α. TNF-α at 100 and 500 U/ml caused a 20% loss of wild-type neurons. In comparison, loss of A8-deficient neurons under identical conditions was dramatic, amounting to >90% (Fig. 6 B). TNF-α induction of ADAM8-deficient neurons was performed in the presence of recombinant soluble TNF-R1 (10 ng/ml) (Fig. 6 C). Cell numbers were comparable with those of wild-type neurons, indicating that the observed neuronal loss is attributable to a lack in TNF-R1 shedding. In contrast, no effect on neuronal loss was observed when primary neurons were treated with soluble recombinant CHL1-Fc protein (10 ng/ml), an ADAM8 substrate in the CNS (Naus et al., 2004). Together, these experiments demonstrate that shedding of TNF-R1 has a protective effect on neuronal survival during TNF-α administration (Fig. 6).
Figure 6.

Effect of TNF-α on cultured cerebellar neurons from wild-type and Adam8-deficient mice. A, TNF-α (10 ng/ml = 100 U/ml) induces the release of soluble TNF-R1 from A8+/+ neurons (white bars), which is dependent on ADAM8, because there is no induction in ADAM8-deficient neurons (black bars). In B, ADAM8-deficient neurons show a higher sensitivity to TNF-α-induced cell death. Less than 10% of the cells survive a TNF-α dosage of 500 ng/ml, whereas >80% of wild-type cells survived. In C, TNF-α (100 U/ml)-induced neuronal cell death was prevented by incubation with an excess of soluble TNF-R1 (10 ng/ml) but not with soluble recombinant CHL1 (sCHL1-Fc; 50 ng/ml), a neuronal substrate of ADAM8 that was shown to protect neurons from constitutive cell death. *p < 0.05, **p < 0.01, ***p < 0.001.
ADAM8-mediated TNF-R1 shedding is gene dosage dependent in microglia cells
Having shown that ADAM8 expression correlated with TNF-R1 release, we prepared primary microglia from 4-d-old Adam8 +/+, Adam8 +/−, and Adam8 −/− mice (Fig. 7 A). Microglia cultures were treated with either LPS or TNF-α, and, after 12 h, concentrations of soluble TNF-R1 were determined by ELISA. As with neurons, the extent of constitutive and TNF-α-induced TNF-R1 shedding was positively correlated with the gene dosage of Adam8, whereas TNF-R2 shedding was slightly increased in Adam8 −/− microglia (Fig. 7 B).
Figure 7.
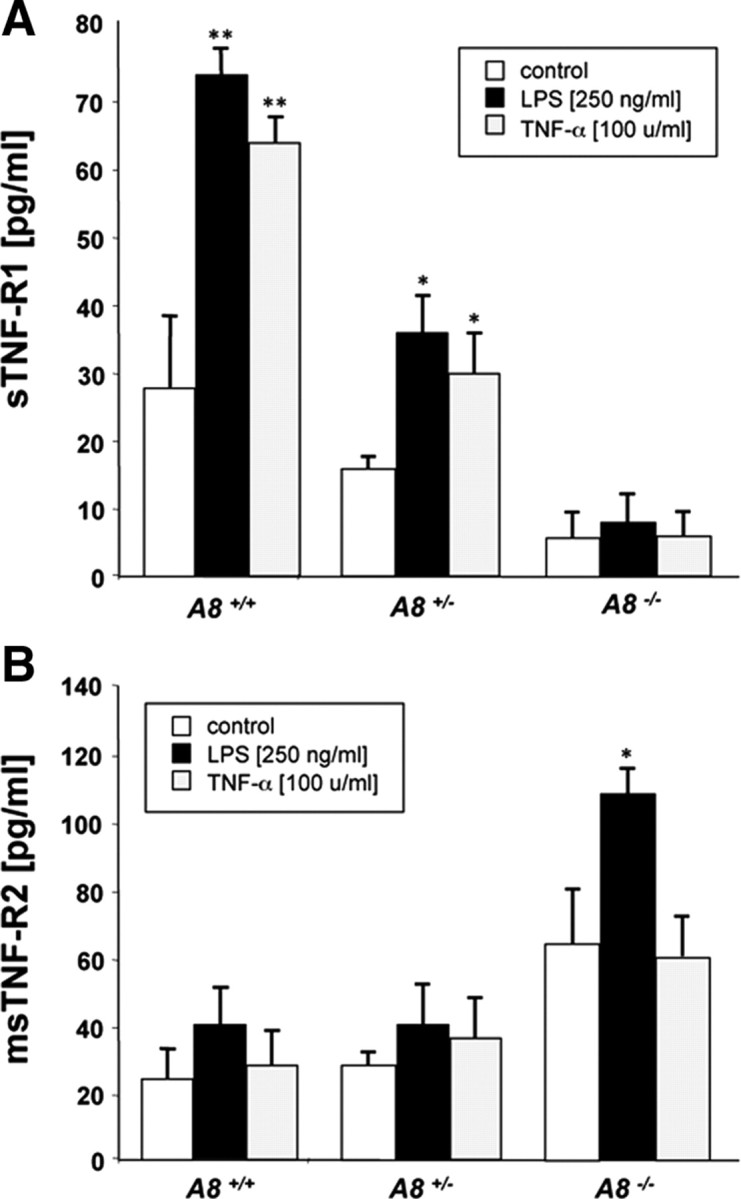
A, B, Release of soluble TNF-R1 (A) and TNF-R2 (B) from primary microglia cells isolated from wild-type, heterozygous, and homozygous Adam8 knock-out mice and induced with indicated concentrations of LPS or TNF-α. ELISA data (in picograms per milliliter medium) were acquired from three independent experiments performed in triplets (n = 9) and were given as mean ± SD. *p < 0.05, **p < 0.01.
Discussion
The data presented here provide experimental evidence for TNF-α-induced shedding of TNF-R1 by the metalloprotease-disintegrin ADAM8 in the mammalian CNS and in cell culture. The resulting feedback loop consists of a dose-dependent induction of ADAM8 by TNF-α, leading to enhanced release of TNF-R1 from the cell membrane. In turn, soluble TNF-R1 can bind extracellular TNF-α, so that overall, ADAM8-dependent TNF-R1 shedding can serve neuroprotective effects (Fig. 8).
Figure 8.

Proposed neuroprotective feedback loop involving TNF-α, TNF-R1, and ADAM8. At low TNF-α concentrations, the transcription rate of the Adam8 gene is low and very low levels of soluble TNF-R1 are found in the extracellular compartment. At high TNF-α concentrations (e.g., 100 U/ml), TNF-R1 activates intracellular pathway(s) strongly stimulating transcription of the Adam8 gene. Its product, the cell-surface-exposed protease ADAM8 cleaves the transmembrane protein TNF-R1 and releases sTNF-R1 from the cell into the intercellular space, in which high concentrations of sTNF-R1 are able to scavenge free TNF-α. This in turn reduces the cellular response to TNF-α, i.e., preventing activation of TNF-dependent genes involved in neuronal cell death.
We demonstrate a significant impact of this feedback loop on the progression of a neurodegenerative disease by analyzing disease progression and survival of wobbler mice. Although there is up to now no human disease known that is genetically homologous to the WR disease of the mouse, we have made use of this stringent phenotypic model because the symptoms develop rapidly and with full penetrance. Double-mutant ADAM8-deficient WR mice obtained by genetic crossing show an aggravated phenotype and drastically reduced survival compared with standard WR mice. From our data, we conclude that ADAM8-dependent TNF-R1 shedding dampens TNF-α-induced motor neuron degeneration in WR, as demonstrated by a neuroprotective effect of ADAM8 in cultured primary neurons.
For a number of CNS diseases, the control of TNF-α function is of crucial importance for neuronal function and survival (Sriram and O'Callaghan, 2000; O'Callaghan et al., 2008). In particular, TNF-R1 signaling is instrumental in mediating neuroprotective effects (Taoufik et al., 2008), i.e., by inducing intracellular neuroprotective pathways (MAP, phosphatidylinositol 3-kinase) or by scavenging extracellular TNF-α through binding to sTNF-R1. The importance of systemic TNF-R1 shedding has been demonstrated in TRAPS (TNF receptor associated periodic syndrome) (McDermott et al., 1999; Hull et al., 2002) patients with mutations in TNF-R1 leading to shedding deficiency (Galon et al., 2000) and in TRAPS model mice expressing a knock-in non-sheddable TNF-R1 (TNF-R1ΔNS) variant (Xanthoulea et al., 2004). These mice show enhanced inflammation in the liver, are sensitive to septic shock (LPS-induced lethality), and display an increased susceptibility to multiple sclerosis-like symptoms in experimental autoimmune encephalitis (EAE) (Xanthoulea et al., 2004). Compared with these cases, the progression of EAE in ADAM8-deficient mice is not significantly affected (A. Pagenstecher and J.W.B., unpublished observations), and ADAM8-deficient mice were not susceptible to LPS-induced lethality to the extent observed in TNF-R1ΔNS mice (supplemental Table 1, available at www.jneurosci.org as supplemental material). As a result of this difference, we conclude that only TNF-α-induced but not constitutive TNF-R1 shedding levels are affected by ADAM8 deficiency, so that rather a reduction but not a complete lack of TNF-R1 shedding (as observed in TNF-R1ΔNS mice) accounts for the aggravated neuropathology of A8-deficient wobbler mice. In the normal CNS with low levels of ADAM8, constitutive TNF-R1 shedding is probably catalyzed by the aminopeptidase ARTS-1 (Cui et al., 2002, 2003) and TACE/ADAM17 (Reddy et al., 2000). Because of its abundance under physiological conditions, ADAM17 is considered the constitutive shedding enzyme for TNF-R1 (Reddy et al., 2000), but, in contrast to ADAM8, transcriptional induction of the Adam17 gene by TNF-α was only moderate. It is likely that ADAM17 activation occurs as an acute response, i.e., by vesicle recruitment to the cell membrane (Black, 2002), whereas the effect of TNF-α on transcription of the Adam8 gene might be a long-term response.
We suspect, however, that activation of a TNF-α–A8–TNF-R1 feedback loop might be a general feature of neurodegeneration in the CNS involving astrogliosis and inflammatory processes. In this respect, the role of TNF-α in other types of motor neuron diseases has been discussed extensively, in particular in the mouse model of familial amyloid lateral sclerosis, superoxide dismutase 1 (SOD1)G93A transgenic (tg) mice. In these mice, an essential role for TNF-α in disease progression has been excluded by crossing TNF-α-deficient with SOD1(G93A) tg mice (Gowing et al., 2006). The minor importance of TNF-α in these mice might be reflected by a relatively low response of glial cells to motor neuron degeneration in the spinal cord, whereas astrogliosis and microglia activation are prominent in WR spinal cord and brainstem (Laage et al., 1988; Rathke-Hartlieb et al., 1999). Nevertheless, ADAM8 is upregulated in spinal cords of SOD1(G93A) tg under disease conditions, most likely by other proinflammatory cytokines such as IL-1β (U. Schlomann and J.W.B., unpublished data). Interestingly, ADAM8 upregulation was also observed after spinal cord injury (Mahoney et al., 2009). Under these conditions, ADAM8 was reported to be involved in angiogenesis by colocalization with PECAM-1 in endothelial cells in the CNS, suggesting that, apart from inflammatory modulation, ADAM8 has additional functions in CNS pathologies.
For these reasons, analyses of other genetic mouse models of neurodegeneration, such as the SOD1(G93A) tg mice, the 3xTg–AD Alzheimer's disease model mice (Oddo et al., 2003), as well as spinal cord injury models, are required to define the functional importance of ADAM8 in these CNS pathologies.
Scavenging excess TNF-α has been predicted to result in beneficial effects on the WR pathology (Naus et al., 2007). Therapeutic application of a recombinant TNF-α binding protein (rh-TBP-1) resulted in a milder disease progression in WR mice (Bigini et al., 2008), thus proving this hypothesis. In all likelihood, TNF-α-induced TNF-R1 release by ADAM8 in the WR motor neuron disease serves neuroprotective functions by balancing physiological levels of free TNF-α through a positive feedback loop. Based on our findings, induction rather than inhibition of ADAM8 might be a promising therapeutic approach to neuronal disorders, characterized by pro-inflammatory cytokine activation.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft SFB549-A4 (J.W.B., H.J.) and BA1303/2-1 (J.W.B.), King's College London (J.W.B.), and the Fonds der Chemischen Industrie (H.J.). We thank Elke Redecker, Marion Knufinke, and Christiane Graebe for excellent technical assistance and mouse care, Dr. Peter Heimann for preparation of perfused tissues, and Angelos Vompras for computer assistance and NIH ImageJ analyses.
References
- Bigini P, Repici M, Cantarella G, Fumagalli E, Barbera S, Cagnotto A, De Luigi A, Tonelli R, Bernardini R, Borsello T, Mennini T. Recombinant human TNF-binding protein-1 (rhTBP-1) treatment delays both symptoms progression and motor neuron loss in the wobbler mouse. Neurobiol Dis. 2008;29:465–476. doi: 10.1016/j.nbd.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Black RA. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. 2002;34:1–5. doi: 10.1016/s1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Cui X, Hawari F, Alsaaty S, Lawrence M, Combs CA, Geng W, Rouhani FN, Miskinis D, Levine SJ. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest. 2002;110:515–526. doi: 10.1172/JCI13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Rouhani FN, Hawari F, Levine SJ. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol. 2003;171:6814–6819. doi: 10.4049/jimmunol.171.12.6814. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Alkayed NJ. Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev Neurosci. 2008;19:341–361. doi: 10.1515/revneuro.2008.19.4-5.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Aksentijevich I, McDermott MF, O'Shea JJ, Kastner DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol. 2000;12:479–486. doi: 10.1016/s0952-7915(00)00124-2. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Rivest S. Role of IL-1 and TNF in the brain: twenty years of progress on a Dr. Jekyll/Mr. Hyde duality of the innate immune system. Brain Behav Immun. 2007;21:281–289. doi: 10.1016/j.bbi.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Gowing G, Dequen F, Soucy G, Julien JP. Absence of tumor necrosis factor-α does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci. 2006;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, Dean J, Powell RJ, Kastner DL. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002;81:349–368. doi: 10.1097/00005792-200209000-00002. [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Simon-Chazottes D, Guénet JL, Jockusch H. Wobbler, a mutation affecting motoneuron survival and gonadal functions in the mouse, maps to proximal chromosome 11. Genomics. 1992;13:39–43. doi: 10.1016/0888-7543(92)90199-3. [DOI] [PubMed] [Google Scholar]
- Kelly K, Hutchinson G, Nebenius-Oosthuizen D, Smith AJ, Bartsch JW, Horiuchi K, Rittger A, Manova K, Docherty AJ, Blobel CP. Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn. 2005;232:221–231. doi: 10.1002/dvdy.20221. [DOI] [PubMed] [Google Scholar]
- Koller G, Schlomann U, Golfi P, Ferdous T, Naus S, Bartsch JW. ADAM8/MS2/CD156, an emerging drug target in the treatment of inflammatory and invasive pathologies. Curr Pharm Des. 2009;15:2272–2281. doi: 10.2174/138161209788682361. [DOI] [PubMed] [Google Scholar]
- Laage S, Zobel G, Jockusch H. Astrocyte overgrowth in the brain stem and spinal cord of mice affected by spinal atrophy, wobbler. Dev Neurosci. 1988;10:190–198. doi: 10.1159/000111969. [DOI] [PubMed] [Google Scholar]
- Leeuwenberg JF, Dentener MA, Buurman WA. Lipopolysaccharide LPS-mediated soluble TNF receptor release and TNF receptor expression by monocytes. Role of CD14, LPS binding protein, and bactericidal/permeability-increasing protein. J Immunol. 1994;152:5070–5076. [PubMed] [Google Scholar]
- Mahoney ET, Benton RL, Maddie MA, Whittemore SR, Hagg T. ADAM8 is selectively up-regulated in endothelial cells and is associated with angiogenesis after spinal cord injury in adult mice. J Comp Neurol. 2009;512:243–255. doi: 10.1002/cne.21902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, Mansfield E, Gadina M, Karenko L, Pettersson T, McCarthy J, Frucht DM, Aringer M, Torosyan Y, Teppo AM, Wilson M, Karaarslan HM, Wan Y, Todd I, Wood G, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, Ikeda K, Klinkosz B, Cedarbaum JM, Wong V, Lindsay RM. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science. 1994;265:1107–1110. doi: 10.1126/science.8066451. [DOI] [PubMed] [Google Scholar]
- Mohan MJ, Seaton T, Mitchell J, Howe A, Blackburn K, Burkhart W, Moyer M, Patel I, Waitt GM, Becherer JD, Moss ML, Milla ME. The tumor necrosis factor-alpha converting enzyme (TACE): a unique metalloproteinase with highly defined substrate selectivity. Biochemistry. 2002;41:9462–9469. doi: 10.1021/bi0260132. [DOI] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Naus S, Richter M, Wildeboer D, Moss M, Schachner M, Bartsch JW. Ectodomain shedding of the neural recognition molecule CHL1 by the metalloprotease-disintegrin ADAM8 promotes neurite outgrowth and suppresses neuronal cell death. J Biol Chem. 2004;279:16083–16090. doi: 10.1074/jbc.M400560200. [DOI] [PubMed] [Google Scholar]
- Naus S, Reipschläger S, Wildeboer D, Lichtenthaler SF, Mitterreiter S, Guan Z, Moss ML, Bartsch JW. Identification of candidate substrates for ectodomain shedding by the metalloprotease-disintegrin ADAM8. Biol Chem. 2006;387:337–346. doi: 10.1515/BC.2006.045. [DOI] [PubMed] [Google Scholar]
- Naus S, Reipschläger S, Wildeboer D, Minai Y, Lichtenthaler SF, Mitterreiter S, Guan Z, Moss ML, Bartsch JW. Identification of ADAM proteinase substrates in neurodegeneration and neuroinflammation. BMC Neurosci. 2007;8(Suppl I):30. [Google Scholar]
- O'Callaghan JP, Sriram K, Miller DB. Defining “neuroinflammation”. Ann N Y Acad Sci. 2008;1139:318–330. doi: 10.1196/annals.1432.032. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Pedron T, Girard R, Chaby R. TLR4-dependent lipopolysaccharide-induced shedding of tumor necrosis factor receptors in mouse bone marrow granulocytes. J Biol Chem. 2003;278:20555–20564. doi: 10.1074/jbc.M203551200. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Pinckard JK, Sheehan KC, Arthur CD, Schreiber RD. Constitutive shedding of both p55 and p75 murine TNF receptors in vivo. J Immunol. 1997;158:3869–3873. [PubMed] [Google Scholar]
- Rathke-Hartlieb S, Schmidt VC, Jockusch H, Schmitt-John T, Bartsch JW. Spatiotemporal progression of neurodegeneration and glia activation in the wobbler neuropathy of the mouse. Neuroreport. 1999;10:3411–3416. doi: 10.1097/00001756-199911080-00028. [DOI] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem. 2000;275:14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor α induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron–glia interactions during neurodegeneration. J Neurosci. 2000;20:7964–7971. doi: 10.1523/JNEUROSCI.20-21-07964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlomann U, Wildeboer D, Webster A, Antropova O, Zeuschner D, Knight CG, Docherty AJ, Lambert M, Skelton L, Jockusch H, Bartsch JW. The metalloprotease disintegrin ADAM8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J Biol Chem. 2002;277:48210–48219. doi: 10.1074/jbc.M203355200. [DOI] [PubMed] [Google Scholar]
- Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM, Meisler MH, Jockusch H. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. 2005;37:1213–1215. doi: 10.1038/ng1661. [DOI] [PubMed] [Google Scholar]
- Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277:22–29. doi: 10.1111/j.1742-4658.2009.07367.x. [DOI] [PubMed] [Google Scholar]
- Taoufik E, Petit E, Divoux D, Tseveleki V, Mengozzi M, Roberts ML, Valable S, Ghezzi P, Quackenbush J, Brines M, Cerami A, Probert L. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc Natl Acad Sci U S A. 2008;105:6185–6190. doi: 10.1073/pnas.0801447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoulea S, Pasparakis M, Kousteni S, Brakebusch C, Wallach D, Bauer J, Lassmann H, Kollias G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J Exp Med. 2004;200:367–376. doi: 10.1084/jem.20040435. [DOI] [PMC free article] [PubMed] [Google Scholar]