

Abstract
Mutations in cyclin-dependent kinase-like 5 (CDKL5), also known as serine/threonine kinase 9 (STK9), have been identified in patients with Rett syndrome (RTT) and X-linked infantile spasm. However, the function of CDKL5 in the brain remains unknown. Here, we report that CDKL5 is a critical regulator of neuronal morphogenesis. We identified a neuron-specific splicing variant of CDKL5 whose expression was markedly induced during postnatal development of the rat brain. Downregulating CDKL5 by RNA interference (RNAi) in cultured cortical neurons inhibited neurite growth and dendritic arborization, whereas overexpressing CDKL5 had opposite effects. Furthermore, knocking down CDKL5 in the rat brain by in utero electroporation resulted in delayed neuronal migration, and severely impaired dendritic arborization. In contrast to its proposed function in the nucleus, we found that CDKL5 regulated dendrite development through a cytoplasmic mechanism. In fibroblasts and in neurons, CDKL5 colocalized and formed a protein complex with Rac1, a critical regulator of actin remodeling and neuronal morphogenesis. Overexpression of Rac1 prevented the inhibition of dendrite growth caused by CDKL5 knockdown, and the growth-promoting effect of ectopically expressed CDKL5 on dendrites was abolished by coexpressing a dominant-negative form of Rac1. Moreover, CDKL5 was required for brain-derived neurotrophic factor (BDNF)-induced activation of Rac1. Together, these results demonstrate a critical role of CDKL5 in neuronal morphogenesis and identify a Rho GTPase signaling pathway which may contribute to CDKL5-related disorders.
Introduction
Rett syndrome (RTT, OMIM 312750) is one of the most common causes of mental retardation (MR) in females (Hagberg, 1985). Patients with classic RTT appear to develop normally until 6–18 months of age, and then start to manifest neurological symptoms including loss of speech and purposeful movements, autistic traits, and stereotypic hand movements (Hagberg et al., 1983). Autopsies of RTT brains reveal smaller and more densely packed neurons with shorter dendrites in the motor and frontal cortex (Jellinger et al., 1988; Armstrong et al., 1995), suggesting that neuronal development is impaired in this disorder. Mutations in the methyl-CpG-binding protein 2 (MECP2) gene account for the majority of classic RTT cases (Cheadle et al., 2000). The MECP2 gene encodes the nuclear protein MeCP2, which selectively binds to CpG dinucleotides in the mammalian genome and regulates gene transcription through the recruitment of corepressor complexes (Jones et al., 1998; Nan et al., 1998). It is generally thought that dysregulation of gene expression caused by mutations in MECP2 underlies the pathology of RTT.
Although mutations in MECP2 are believed to be the major cause of classic RTT, the existence of patients carrying no MECP2 mutation (∼20% of classic RTT and 60–80% of RTT variants) suggests other genetic causes (Buyse et al., 2000; Cheadle et al., 2000; Gill et al., 2003). Recently, mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene have been identified in patients with the Hanefeld variant of RTT and other severe encephalopathies (Tao et al., 2004; Weaving et al., 2004; Evans et al., 2005; Scala et al., 2005; Archer et al., 2006). The common disease features in these patients are early onset seizures and severe mental retardation. CDKL5 mutations known to cause disease include chromosome translocations, deletions, insertions, nonsense mutations causing premature termination of transcription, or mutations within the catalytic domain resulting in decreased catalytic activity (Rusconi et al., 2008). The predicted human CDKL5 protein has 1030 aa. It contains an N terminus serine/threonine kinase domain that shares high homology with both mitogen-activated protein kinase (MAPK) and cyclin-dependent kinase (CDK) families (Tao et al., 2004). CDKL5 mRNA is abundant in adult human and rodent brains (Montini et al., 1998). In the mouse brain, CDKL5 is most abundant in the cerebral cortex and hippocampus (Rusconi et al., 2008). Interestingly, CDKL5 is highly expressed in neurons but is undetectable in glial cells (Rusconi et al., 2008), indicating an important role of CDKL5 in neuronal development and function. However, little is known about the function of CDKL5 in neurons. In the current study, we characterized the function of CDKL5 in the developing rat brain by using RNAi to specifically silence CDKL5 expression in cortical neurons. We found that CDKL5 is required for neuronal morphogenesis. In addition, we present evidence to show that CDKL5 is involved in BDNF-Rac1 signaling. These findings provide novel insight into how CDKL5 mutations contribute to the pathology of RTT and other CDKL5-related diseases.
Materials and Methods
Rat CDKL5 cloning and constructs preparation.
The cDNAs of rat CDKL5 (GenBank accession numbers FJ807484 and GU351881) were cloned from a cDNA library of rat cerebral cortex by RT-PCR in conjunction with 3′ rapid amplification of cDNA ends (3′ RACE). Briefly, PCR products of Gene-Specific Primer (GSP, 5′-GCA TCT TTC TTC GTC AAC CAA TCA CC-3′) and the universal amplification primer were ligated into T-easy vector (Promega) and sequenced. To obtain full-length rat CDKL5, 5′ end primer (5′-CGC GGA TCC ATG AAG ATT CCT AAC ATT GGT AAT-3′) and 3′ end primer (5′-CCG GAA TTC AGC AGA TGC TCG TCA GTG G-3′) were designed to amplify a cDNA library of rat cortex by PCR. The PCR products were cloned into pCMV-tag3B (Stratagene) at BamHI/EcoRI sites and sequenced. pCAG-GFP-CDKL5 was generated by inserting the rat CDKL5a coding sequence into BglII/EcoRI sites of pCAG-EGFP-C1 vector which was constructed by replacing the CMV promoter with a CMV early enhancer/chicken β actin (CAG) promoter in pEGFP-C1 (Clontech). pCAG-GFP-CDKL5* was generated by inserting the RNAi resistant form of rat CDKL5a (CDKL5*, see below) into BglII/EcoRI sites of pCAG-EGFP-C1. To generate pCAG-NES-GFP-CDKL5*, two DNA oligos (5′-CTA GCA TGC CTC TGC AGC TCC CGC CAC TGG AAC GTC TTA CCC TCG ACG AA-3′ and 5′-CCG GTT CGT CGA GGG TAA GAC GTT CCA GTG GCG GGA GCT GCA GAG GCA TG-3′) corresponding to HIV-1 Rev protein nuclear export signal sequence (NES, LQLPPLERLTLD) were synthesized, annealed and inserted into NheI/AgeI sites of pCAG-GFP-CDKL5*. For overexpression of CDKL5 in cultured neurons, wild-type CDKL5a cDNA was cloned into pCAG-IRES-EGFP at BglII/EcoRI sites. CDKL5-K42R, C152F, R175S and ΔC (1-790 aa) mutants were generated by a PCR-based method and cloned into pCAG-IRES-EGFP as wild-type CDKL5. GFP-WT-Rac1, GFP-CA-Rac1 (Q61L) and GFP-DN-Rac1 (T17N) were provided by Dr. Xiaobin Yuan (Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China). All constructs were verified by sequencing.
Generation of CDKL5 shRNAs.
Three DNA oligos targeted to different sequences of rat CDKL5 cDNA and a scrambled control oligo were designed, synthesized and cloned into pSuper-basic (Oligoengine). The 19-nucleotide target sequences are as follows: shRNA-#1, 5′-AGT GAG AGC TAA AGG CAT T-3′; shRNA-#2, 5′-GGA GCC TAT GGA GTT GTA C-3′; shRNA-#3, 5′-CAG CTT GCA GCT GTT ATC A-3′; and shRNA-Scr, 5′-TTC TCC GAA CGT GTC ACG T-3′. The shRNA-#1-resistant form of CDKL5 (CDKL5*) was generated by mutating three nucleotides of shRNA-#1 targeting sequence in CDKL5a cDNA (from AGT GAG AGC TAA AGG CAT T to AGT AAG GGC CAA AGG CAT T) without changing their corresponding amino acids. All constructs were verified by sequencing.
Reagents and antibodies.
The following reagents and antibodies were used in this study: polyclonal anti-CDKL5 and its blocking peptide (Abcam), mouse monoclonal anti-Rac1 (Millipore), mouse monoclonal anti-β-tubulin III (Tuj1, Sigma), mouse monoclonal anti-GFAP (Millipore Bioscience Research Reagents), mouse monoclonal anti-GFP (Santa Cruz Biotechnology), polyclonal anti-GFP (Invitrogen), Polyclonal anti-phospho-threonine (Cell Signaling Technology), Rabbit monoclonal anti-phospho-ERK1/2 (Cell Signaling Technology), Rabbit monoclonal anti-ERK1/2 (Cell Signaling Technology), and phalloidin-TRITC (Sigma).
Cell culture and transfection.
HEK 293T cells, NIH3T3 fibroblasts and COS-7 cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS. Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Primary cortical neurons were prepared from brains of embryonic day 18 (E18) Sprague Dawley (SD) rat as described previously (Zhou et al., 2006). Cortical neurons were transfected by Nucleofector (Amaxa) or Lipofectamine 2000.
In utero electroporation.
Female SD rats were checked for vaginal plugs. Electroporation was performed at E15. Rat embryos were exposed in the uterus, and 1 μl of DNA solution was injected into the lateral ventricle through the uterus wall, followed by electroporation. Plasmids were prepared in 1 × PBS at a concentration of 1 μg/μl for pCAG-GFP, 3 μg/μl for shRNAs and 6 μg/μl for pCAG-GFP-CDKL5*. Electric pulses of 50 V were generated with an ElectroSquirePortator T830 (BTX) and applied to the cerebral wall for 50 ms each, a total of five pulses at an interval of 100 ms.
Virus packaging and infection.
The lentiviral expression system used in the present study consists of three vectors (FUGW, pCMV 8.9, and pVSVG) as described previously (Lois et al., 2002). In brief, RNAi expression cassettes containing U6 promoter (derived from pSicoR vector, Addgene plasmid 11579) and RNAi sequences (shRNA-Scr, 5′-TTC TCC GAA CGT GTC ACG T-3′; shRNA-#1, 5′-AGT GAG AGC TAA AGG CAT T-3′) were subcloned into FUGW vector at XbaI/XhoI sites upstream of EGFP expression cassette which is driven by a ubiquitin promoter. HEK293T cells were used for virus production. To make viral particles, three plasmids (FUGW, pCMV 8.9, and pVSVG) were transiently cotransfected into HEK293T cells by a calcium phosphate method. Two days after transfection, the culture medium was concentrated by ultracentrifugation with SW 41 rotor (Beckman Instruments) at 30,000 rpm for 1.5 h. After removal of the supernatant, pellets with viruses were dissolved in 1× PBS overnight. The viral stocks were stored at −80°C. Lentiviruses were titrated using HEK293 cells with serial dilutions of virus stocks, followed by fluorescence microscopy examination 3 d after infection (2∼3 × 109 infectious units/ml). For Semliki Forest virus (SFV) production, BHK-21 cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) and 100 U/ml penicillin/streptomycin. Cells were maintained at 37°C with a constant 5% CO2. In vitro transcribed RNA from pSFV(pd)-EGFP or pSFV(pd)-CDKL5-EGFP were cotransfected with pSFV-helper2 RNA (a gift from Dr. Kenneth Lundstrom, PanTherapeutics, Lutry, Switzerland) by electroporation (GenePulserII, Bio-Rad). All steps of virus production were performed at 31°C. Two days after electroporation, virus stocks were harvested, filter sterilized and activated with chymotrypsin (Invitrogen). The reaction was terminated with the trypsin inhibitor aprotinin (Invitrogen). Virus was concentrated by centrifugation at 20,000 × g for 4 h at 4°C and then dissolved in PBS. Final virus titers were determined by infection of BHK-21 cells (≥1010 infectious units/ml) followed by fluorescence microscopy examination.
Detection of CDKL5 phosphorylation.
Cultured cortical neurons were plated on poly-d-lysine-coated 10 cm dishes at a density of 7.5 × 106 cells per dish. Cells were infected with SFV expressing GFP-CDKL5. Eight hours after infection, cells were serum starved and stimulated with BDNF (10 ng/ml) for different lengths of time. After stimulation, cells were lysed in a modified RIPA buffer (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mm EDTA, 1 mm PMSF, 1× Protease Inhibitor Cocktail (Sigma), 1 mm Na3VO4, 1 mm NaF). After centrifugation at 12,000 × g for 15 min, the supernatant was incubated with 2 μg of GFP antibody (Santa Cruz Biotechnology) overnight at 4°C. At the end of incubation, protein A/G agarose beads (Santa Cruz Biotechnology) was added and incubated for another 3 h with agitation. Beads were washed 3 times with RIPA buffer and mixed with 30 μl of 2× reduced SDS sample buffer. Samples were resolved by SDS-PAGE and immunoblotted with antibodies against phospho-threonine or GFP.
Purification of growth cone proteins.
We purified growth cone proteins (GCPs) according to the procedure described by Pfenninger et al. (Lohse et al., 1996). All purification steps were performed at 4°C. Rat brains of E16-E18 were homogenized by five strokes with a Teflon-glass homogenizer in ∼8 volumes (w/v) of lysis buffer (1 mm MgCl2, 1 mm N-Tris-[hydroxymethyl]methyl-2-aminoethanesulfonic acid-NaOH, pH 7.3, 1× Protease Inhibitor Cocktail, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, and 1 mm 0.32 m sucrose). The homogenate (H) was spun at 1660 × g for 15 min. The resultant low-speed supernatant (LSS) was loaded onto discontinuous sucrose density gradients consisting of the following three layers: 0.83 m, 1.0 m, and 2.66 m sucrose. The gradients were spun to equilibrium at 242,000 g for 40 min in a Beckman VTi50 rotor. The fraction at the load/0.83 m sucrose interface was collected (GCPs), diluted with 3∼4 volumes of buffered 0.32 m sucrose, and pelleted at 39,000 × g for 30 min. The resulting pellets were resuspended in 0.32 m sucrose for further analysis.
GST pull-down assay.
GST and GST fusion proteins were purified from Escherichia coli. Cells were lysed in lysis buffer [50 mm Tris-HCl, pH7.5, 150 mm NaCl, 1% Triton X-100, 10 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 1× phosphatase (PhoSTOP, Roche), and 1× Protease Inhibitor Cocktail (Sigma)] on ice. The lysate was cleared by centrifugation for 20 min at 14,000 × g and incubated with agarose beads coupled with GST or GST fusion proteins for 45 min. The bound proteins and lysates were analyzed by immunobloting.
Rac1 activation assay.
We measured Rac1 activity using methods described previously (Ren et al., 1999). Briefly, lysates of cortical neurons were incubated with the bacterially produced glutathione S-transferase (GST)-fused Rac/Cdc42-binding domain of Pak (GST–PBD, 20 μg) bound to glutathione-coupled Sepharose beads at 4°C for 45 min with constant rocking. The beads were washed five times with lysis buffer, eluted in reduced SDS sample buffer, and analyzed for bound Rac1 by immunoblotting with Rac1 monoclonal antibody.
Image acquisition and quantitation.
Images were obtained using either Nikon TE2000-S or Zeiss LSM 510 confocal microscope with 40× [numerical aperture (NA) = 1.3] or 63× (NA = 1.4) objectives. Neuronal morphology was traced and analyzed by Neurolucida software (MBF Bioscience).
Statistical analysis.
Values were expressed as mean ± SEM. Student's t test was used to measure significance of differences between two groups. Statistical significance was defined as p < 0.05.
Results
Identification of a neuron-specific CDKL5 isoform in the developing rat brain
To begin our study of CDKL5 function in the developing rat brain, we first cloned rat CDKL5 from a rat brain cDNA library by reverse transcription-PCR (RT-PCR) in conjunction with 3′ RACE. Interestingly, we identified two different splicing isoforms, which we named CDKL5a and CDKL5b (GenBank accession numbers: FJ807484 and GU351881). Sequence alignment revealed that CDKL5b cDNA lacks the CDKL5a exon 17, resulting in a shorter amino acid sequence and a distinct C terminus (Fig. 1A; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The open reading frames of CDKL5a and CDKL5b are 2802 bp and 2631 bp, respectively, corresponding to predicted proteins of 934 aa and 877 aa in length (Fig. 1A; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Cross-species protein comparison showed that both isoforms share high sequence similarity with published human and mouse CDKL5 genes (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). However, the last 100-150 aa in the C terminus differ among the three species (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 1.

Molecular cloning of rat CDKL5 and expression of CDKL5 in the developing rat brain. A, Schematic diagrams showing the mRNA and protein structures of CDKL5a and CDKL5b. The immunogen region for the CDKL5 antibody is marked by a thick black line. B, Western blot analysis of lysates extracted from neuronal, glial, or mixed neuron-glia cultures, as well as 293T cells transfected with plasmids expressing CDKL5a and CDKL5b; using antibodies against CDKL5, Tuj1, and GFAP. C, Western blot analysis of CDKL5 protein levels in brain, heart, liver, spleen, lung, kidney, and muscle lysates (top); and in lung, liver, brain and HEK293T cells expressing CDKL5a or CDKL5b (bottom). D, Western blot analysis of CDKL5 protein levels during cortical development at the indicated time points. E, In situ hybridization of CDKL5 mRNA in the P7 rat brain. CX, cortex; OB, olfactory bulb; STR, striatum; CE, cerebellum; TH, thalamus; HIP, hippocampus. F, In situ hybridization (ISH) and immunohistochemistry (IHC) of CDKL5 mRNA and protein in adjacent sections of P7 rat cerebral cortex. The inset (bottom right) shows a higher-magnification view of the boxed region of the cortex. ISH with Sense probe and IHC with blocking peptide to neutralize CDKL5 antibody are negative controls. Scale bars, 50 μm.
To determine whether CDKL5a and CDKL5b exhibit cell type-specific expression patterns in the brain, we immunoblotted lysates of cultured cortical neurons, glial cells, and mixed neuron-glia cultures using a commercial polyclonal antibody against CDKL5. Based on its immunogen sequence, this antibody was predicted to recognize both CDKL5 isoforms (Fig. 1A). In mixed neuron-glia lysates, the CDKL5 antibody detected two bands between 100 kDa and 130 kDa, which comigrated with ectopically expressed CDKL5a and CDKL5b (Fig. 1B). The expression level of CDKL5b in these lysates was much lower than that of CDKL5a (Fig. 1B). In pure neuronal cultures, almost all the detectable CDKL5 protein was CDKL5a, whereas in pure glial cultures the only form present was CDKL5b (Fig. 1B). The purity of these cultures was confirmed by probing with the neuron-specific marker Tuj1 or the glia-specific marker GFAP (Fig. 1B). Next, we performed immunoblotting experiments to examine the expression of CDKL5 in various body tissues. At postnatal day 14 (P14), CDKL5 was only detectable in the brain, liver and lung of rats (Fig. 1C), with the highest expression level being observed in the brain (Fig. 1C). In lung and liver samples, CDKL5b was the only isoform expressed (Fig. 1C).
We next examined the temporal expression pattern of CDKL5a and CDKL5b in rat brains. Consistent with a report that CDKL5 expression is increased during postnatal development (Rusconi et al., 2008), we found that the expression of CDKL5a was relatively low before E16 and markedly induced during perinatal stages (Fig. 1D). The expression of CDKL5b was relatively low throughout development relative to CDKL5a, but was also induced in the perinatal-postnatal period (Fig. 1D). Together, these results demonstrate that CDKL5a is a neuron-specific isoform that is developmentally regulated in the rat brain. To elucidate the function of CDKL5 in neurons, we thus chose to focus the remainder of our experiments on CDKL5a, and refer to CDKL5a as CDKL5 unless otherwise stated.
CDKL5 regulates neuronal morphogenesis of cultured cortical neurons
To determine the function of CDKL5 in neuronal development, we first examined the spatial distribution of CDKL5 during early postnatal stages. In situ hybridization on P7 rat brain sections revealed that CDKL5 mRNA was expressed throughout the brain, with high levels in the cerebral cortex, hippocampus and cerebellum (Fig. 1E). In the cerebral cortex, both CDKL5 mRNA and protein were enriched in cortical layers (Fig. 1F). Notably, CDKL5-labeled cells in the cortex exhibited neuronal morphological features, with processes extending from their cell bodies (Fig. 1F, inset).
The expression pattern of CDKL5 in the rat brain together with the observed disease phenotype of RTT patients led us to hypothesize that CDKL5 is important for neuronal morphogenesis. This hypothesis was first tested in cultured cortical neurons. Immunoblotting of lysates of neurons prepared from E18 rat brain and cultured for a variable number of days in vitro (DIV) showed that CDKL5 expression increased over time, similar to the dynamics we observed in the rat brain (Fig. 2A). To perform loss-of-function studies, we generated plasmid-based RNAi constructs to specifically knockdown CDKL5 expression. Among the three shRNAs tested, shRNA-#1 suppressed CDKL5 expression most effectively (Fig. 2B,C).
Figure 2.

CDKL5 regulates neuronal morphogenesis in vitro. A, Western blot analysis of CDKL5 expression in cultured cortical neurons at the indicated time points. B, C, Downregulation of CDKL5 in HEK293T cells and cultured cortical neurons. Extracts from HEK293T cells (B) or cortical neurons (C) transfected with (B) or without (C) GFP-CDKL5 together with shRNA-Scr, shRNA-#1, shRNA-#2, or shRNA-#3 were immunoblotted with GFP antibody (B) or CDKL5 antibody (C). − indicates lysates of untransfected cells. D, E, Effects of CDKL5 knockdown on neurite growth. D, Representative images of neurons transfected with GFP together with indicated constructs. CDKL5* is an RNAi resistant form of CDKL5. Scale bar, 20 μm. E, Quantitative analysis of total dendritic length and total axon length of neurons treated as in D. Data represent mean ± SEM, n = 50–70 in each group; **p < 0.001; t test. F–H, Effects of CDKL5 knockdown on dendritic arborization. F, Representative images of neurons transfected with GFP together with indicated constructs. Scale bar, 20 μm. G, Quantitative analysis of total dendritic length of neurons treated as in F. Data represent mean ± SEM; n = 50–53; **p < 0.001; t test. H, Sholl analysis of dendritic arborization of neurons in G.
To determine the effect of CDKL5 knockdown on neurite growth, an early event in neuronal differentiation, we transfected cultured cortical neurons by electroporation at the time of plating with plasmids encoding GFP together with either shRNA-Scr (a non-targeting control shRNA) or shRNA-#1, and analyzed neuronal morphology after 3 d. We found that CDKL5 knockdown resulted in a marked reduction in the total length of both dendrites and axons (Fig. 2D,E). These effects were prevented by cotransfecting a shRNA-resistant form of CDKL5 (CDKL5*) (Fig. 2D,E; supplemental Fig. S3, available at www.jneurosci.org as supplemental material), demonstrating that they were not off-target effects. In addition, shRNA-#2 had a similar but milder effect on neurite growth compared with shRNA-#1, correlating with its lesser knockdown efficiency (Fig. 2B,C; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The development of neuronal polarity was not affected by the expression of CDKL5 shRNA. On DIV 3, the percentages of neurons exhibiting a single axon (SA), multiple axons (MA) or no axon (NA) were similar between neurons expressing shRNA-Scr and shRNA-#1 (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). To examine whether CDKL5 is also required for dendritic arborization, we transfected cortical neurons with GFP together with shRNA-#1 on DIV 5 and analyzed dendritic arbors 3 d later. In neurons expressing shRNA-#1, the total dendrite length and the complexity of arbors, measured by Sholl analysis, were markedly reduced (Fig. 2F–H), suggesting that CDKL5 expression in cortical neurons is required for dendritic arborization.
We next examined whether CDKL5 was sufficient to promote dendrite growth. Cultured cortical neurons were transfected on DIV 4 with plasmids expressing various forms of CDKL5 together with GFP, and dendrite morphology was analyzed 3 d later. Overexpression of wild-type CDKL5 significantly increased total dendrite length, whereas overexpression of a kinase dead version of CDKL5 (CDKL5-K42R) had no effect (Fig. 3A,B). To explore the effects of disease-associated CDKL5 mutations on dendrite growth, we transfected neurons with plasmids expressing CDKL5-C152F, CDKL5-R175S, or CDKL5-ΔC (1-790 aa), three disease-associated mutations (Bertani et al., 2006). CDKL5-ΔC, which lacks part of the C terminus, had no effect on dendrite growth, similar to CDKL5-K42R. And although neurons expressing CDKL5-C152F or CDKL5-R175S exhibited an increase in total dendrite length relative to neurons expressing GFP alone, the effect was significantly less robust than that caused by overexpression of wild-type CDKL5. Together, these data indicate that CDKL5 is sufficient to promote dendrite growth, and that disease-associated mutations impair this ability.
Figure 3.

Effects of CDKL5 and mutated CDKL5 variants on dendrite growth. A, Representative images of neurons transfected with plasmids expressing wild-type or the indicated variants of CDKL5 together with GFP. B, Quantitative analysis of total dendrite length of neurons treated as in A. Data represent mean ± SEM; n = 80–100 in each group; **p < 0.001 relative to the GFP or CDKL5 group, as indicated on the graph; t test.
CDKL5 is required for dendritic arborization in vivo
To explore the importance of CDKL5 in neuronal morphogenesis in vivo, we downregulated CDKL5 expression in the rat brain using in utero electroporation (Saito and Nakatsuji, 2001). Plasmids encoding GFP together with either shRNA-Scr or CDKL5 shRNAs were electroporated into a subpopulation of neural progenitor cells at E15, and the position and morphology of transfected neurons were evaluated postnatally. At P0, neuronal migration defects were seen in neurons expressing shRNA-#1 (Fig. 4A). These defects were rescued by coexpressing CDKL5* (Fig. 4A). Migration was not altogether blocked but delayed in neurons expressing shRNA-#1, since they were able to migrate into cortical layers and extend apical dendrites toward the pial surface by P14 (Fig. 4A). To investigate the role of CDKL5 in dendritic morphogenesis, we analyzed the morphology of layer II/III pyramidal neurons in the P4 rat brain (Fig. 4B). At this time point, most transfected neurons had migrated into cortical layers, and their dendrites were easy to trace. We found that neurons expressing shRNA-#1 exhibited a marked reduction in both the total length of apical dendrites and the number of apical dendrite branches (Fig. 4C,D). This phenotype was partially rescued by coexpression of CDKL5* together with shRNA-#1 (Fig. 4D). Moreover, shRNA-#2, a less effective shRNA construct that had little effect on neuronal migration (data not shown), exerted a similar but less robust effect on dendritic morphogenesis compared with shRNA-#1, indicating that the impairment in dendritic arborization was not a secondary consequence of the migratory defect (Fig. 4D). Thus, neuron morphogenesis in the rat cortex requires CDKL5.
Figure 4.

CDKL5 is required for dendritic arborization in vivo. A, Representative images of coronal slices of P0 and P14 rat brains that were transfected with GFP together with indicated constructs by in utero electroporation at E15. Transfected cells were visualized by staining coronal slices with GFP antibody. Scale bar, 100 μm. B, Representative images of P4 slices of rat brains transfected with GFP together with indicated constructs. Scale bar, 50 μm. C, Representative high-magnification images and Neurolucida tracings of layer II/III pyramidal neurons in B. Scale bar, 50 μm. D, Quantitative analysis of total dendritic length and number of dendritic branches in neurons treated as in B. Data represent mean ± SEM; n = 30–40 in each group; **p < 0.001; t test.
Rac1 mediates the function of CDKL5 in neuronal morphogenesis
In previous studies, CDKL5 was found to function in the nucleus (Lin et al., 2005; Bertani et al., 2006; Ricciardi et al., 2009). However, CDKL5 was also found to be present in the dendrites of cultured neurons (Rusconi et al., 2008), suggesting a cytoplasmic function independent of its role in the nucleus. To explore this possibility, we further analyzed the subcellular localization of CDKL5 in neurons. Anti-CDKL5 immunostaining of cortical neurons on DIV 2 revealed high levels of CDKL5 in neurites (Fig. 5A), consistent with previously published results (Rusconi et al., 2008). A cell fractionation assay of cultured cortical neurons confirmed that the majority of CDKL5 was present in the cytoplasmic fraction (Fig. 5B). Interestingly, CDKL5 was found to colocalize with F-actin in the peripheral domain of growth cones (Fig. 5A). The high level of CDKL5 in the growth cone was confirmed by immunoblotting of biochemically purified GCPs (Fig. 5B). To test whether CDKL5 acts outside the nucleus to regulate neurite growth and dendrite morphogenesis, we expressed GFP-CDKL5* exclusively in the cytoplasm by adding a NES to the N terminus of GFP-CDKL5* (NES-GFP-CDKL5*) (Fig. 5C), and determined whether this cytoplasm-localized, RNAi-resistant form of CDKL5 could rescue the CDKL5 knockdown phenotype. The cytoplasm-exclusive localization of NES-GFP-CDKL5* was confirmed in COS-7 cells and neurons by visualization of GFP (Fig. 5D,E). Revealingly, coexpression of NES-GFP-CDKL5* in cortical neurons from DIV 0 rescued at least partially the effect of shRNA-#1 on neurite growth and branching at DIV 3 (Fig. 5F–H). Thus, CDKL5 functions in the cytoplasm to regulate neuronal morphogenesis.
Figure 5.
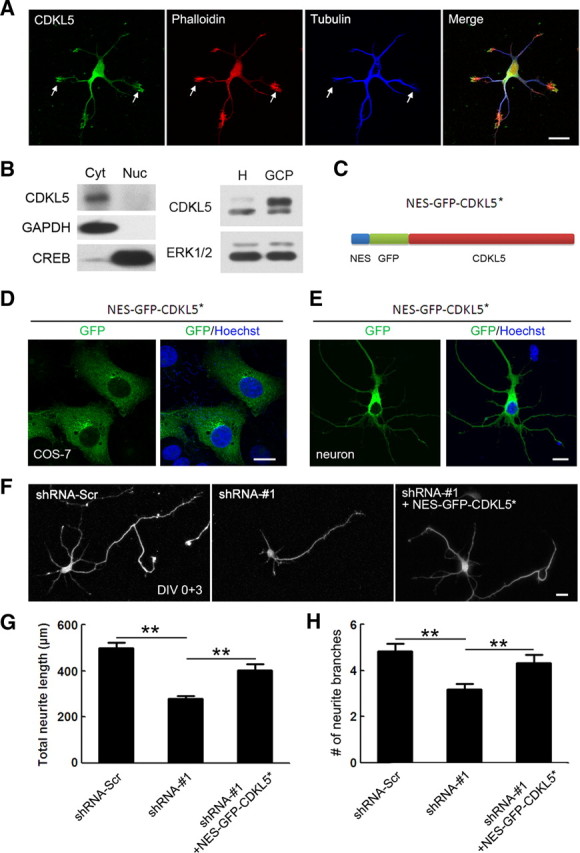
Subcellular localization of CDKL5. A, Representative images of DIV 2 neurons costained with CDKL5 and tubulin antibody. F-actin was visualized by phalloidin staining. Arrows indicate growth cones with abundant CDKL5. Scale bar, 20 μm. B, Left, Western blot showing CDKL5 enrichment in the cytoplasmic fraction, but not the nucleus, of DIV 14 cultured cortical neurons. Cytoplasmic and nuclear lysate were immunoblotted with antibodies against CDKL5, the cytoplasmic factor GAPDH and the nuclear factor CREB. Right, Anti-CDKL5 Western blot analysis of whole-cell homogenate (H) and GCPs isolated from E18 rat cortex. ERK1/2 was used as the loading control. C, Schematic illustration of the protein structure of NES-GFP-CDKL5*. D, E, Representative images showing the localization of NES-GFP-CDKL5* in COS-7 cells (D) and in neurons (E). Scale bar, 10 μm. F, Representative images of neurons transfected with GFP together with the indicated constructs. Scale bar, 20 μm. G, H, Quantitative analysis of total neurite length and total number of dendrite branches in neurons treated as in F. Data represent mean ± SEM; n = 70–80 in each group; **p < 0.001; t test.
The colocalization of CDKL5 with F-actin in growth cones (Fig. 5A) suggested that CDKL5 might be involved in regulating the actin cytoskeleton. Rho GTPases are well known regulators of actin dynamics and neuronal morphogenesis (Hall, 1998; Luo, 2002; Govek et al., 2005). Therefore, we tested the possibility that CDKL5 regulates neuronal morphogenesis through Rho GTPases. We first determined whether CDKL5 colocalizes with Rho GTPases in NIH3T3 fibroblasts, a cell line widely used to study Rho GTPases (Hall, 1998). PDGF stimulation of serum-starved cells, including fibroblasts, resulted in rapid reorganization of actin to form membrane ruffles (Eriksson et al., 1992; Hooshmand-Rad et al., 1997; Ruusala et al., 1998). In serum-starved cells, CDKL5 and Rac1 were diffusely distributed throughout the cells. However, upon PDGF stimulation, CDKL5 was recruited to F-actin-enriched membrane ruffles, where it colocalized with Rac1 (Fig. 6A).
Figure 6.

CDKL5 forms a complex with Rac1. A, Colocalization of CDKL5 and Rac1 in membrane ruffles. To detect endogenous CDKL5, NIH3T3 fibroblasts were serum starved for 48 h, and then left unstimulated or stimulated with PDGF (25 ng/ml) for 5 min. Cells were fixed and costained with CDKL5, Rac1, and phalloidin. Arrows indicate membrane ruffles induced by PDGF. Scale bar, 20 μm. B, Colocalization of CDKL5 and CA-Rac1 in membranes of serum-starved COS-7 cells. Cells transfected with GFP-CA-Rac1 either with or without Myc-CDKL5 were serum starved and stained for CDKL5 or Myc. Arrows indicate membranes where CDKL5 and CA-Rac1 colocalize. Scale bar, 20 μm. C, DN-Rac1 does not colocalize with CDKL5 in membrane regions in response to PDGF. COS-7 cells transfected with GFP-DN-Rac1 either with or without Myc-CDKL5 were serum starved and stimulated with PDGF (25 ng/ml) for 5 min. Cells were fixed and stained for CDKL5 or Myc. Scale bar, 20 μm. D, GST pull-down assay showing the interaction of CDKL5 and Rac1. COS-7 cell lysates were incubated with agarose bead embedded GST or GST-CDKL5Ct. Proteins eluted from the beads and in the lysates were immunoblotted with GFP antibody. GST and GST fusion proteins were visualized by staining the same membrane with Ponceau S. E, F, Western blots showing that the association of CDKL5 with Rac1 is induced by growth factor stimulation. COS-7 cells (E) or cultured cortical neurons (F) were serum starved and stimulated with PDGF (25 ng/ml) or BDNF (25 ng/ml), respectively, for 0, 1, 5, 10, and 20 min. Lysates from the cells were then incubated with GST-CDKL5Ct, and proteins bound to the beads and in the lysates were immunoblotted with Rac1, pERK1/2, and ERK1/2 antibodies.
To determine whether the location or activity of Rac1 influenced CDKL5 targeting we transfected cells with a constitutively active form (CA) or a dominant-negative form (DN) of Rac1. In transfected COS-7 cells, GFP-CA-Rac1 localized to membranes without PDGF stimulation (Fig. 6B). In these cells, both endogenous and Myc-tagged CDKL5 were recruited to membranes, colocalizing with CA-Rac1 (Fig. 6B). In contrast, in cells expressing GFP-DN-Rac1, most of the DN-Rac1 was in the cytoplasm and CDKL5 failed to translocate to membranes even in the presence of PDGF (Fig. 6C). These results indicate that CDKL5 and Rac1 may form a protein complex and translocate together to the membrane region in response to extracellular signals. To test whether CDKL5 and Rac1 forms a protein complex, we performed a GST pull-down assay in COS-7 cells with a GST fusion protein containing part of the C terminus of rat CDKL5 (670-934 aa, GST-CDKL5Ct). We found that Rac1 was pulled down by GST-CDKL5Ct, whereas ERK1/2 was not, suggesting that CDKL5 specifically forms a protein complex with Rac1 (Fig. 6D). The activity of Rac1 is known to be induced by growth factors (Hall, 1998); we therefore tested whether growth factor stimulation could regulate the interaction between CDKL5 and Rac1. Serum-starved COS-7 cells were simulated with PDGF for various lengths of time, and the interaction between CDKL5 and Rac1 was again measured by GST pull-down assay. We found that PDGF treatment increased the amount of Rac1 bound to GST-CDKL5Ct in a time-dependent manner, and the strongest binding was observed after 10 min of stimulation (Fig. 6E). Moreover, in cortical neurons, brain-derived neurotrophic factor (BDNF) stimulation also enhanced the interaction between CDKL5 and Rac1 (Fig. 6F), indicating that this interaction is conserved among different cell types. Together, these data suggest that CDKL5 and Rac1 interact directly or indirectly by formation of a protein complex, and that this interaction is regulated by growth factors.
Next, we functionally tested whether changes in Rac1 activity could mediate the effects of CDKL5 on neuronal morphogenesis, and found that coexpressing Rac1 prevented the inhibition of dendrite growth caused by shRNA-#1 (Fig. 7A). Moreover, the growth-promoting effect of CDKL5 overexpression on dendrites was abolished by coexpressing DN-Rac1 (Fig. 7B). Together, these results suggest that Rac1 acts downstream of CDKL5 in neuronal morphogenesis.
Figure 7.

CDKL5 regulates neuronal morphogenesis through Rac1. A, Left, Representative images of neurons transfected with GFP together with shRNA-Scr, shRNA-#1, or shRNA-#1 plus wild-type Rac1. Scale bar, 20 μm. Right, Quantitative analysis of total dendritic length of transfected neurons. Data represent mean ± SEM; n = 30 in each group; **p < 0.001; t test. B, Left, Representative images of neurons transfected with GFP alone, GFP together with CDKL5, or GFP with CDKL5 plus DN-Rac1. Scale bar, 20 μm. Right, Quantitative analysis of total dendritic length of transfected neurons. Data represent mean ± SEM; n = 132, 110, and 109 from left to right; **p < 0.001; t test. C, Western blot analysis showing the effects of CDKL5 knockdown on BDNF-induced Rac1 activation. DIV 1 cultured cortical neurons were infected with lentivirus expressing shRNA-Scr or shRNA-#1. Four days after infection, cells were stimulated with BDNF (10 ng/ml) for the indicated length of time, and active Rac1 was pulled down by GST-PBD. The activation of Rac1 was determined by normalizing the amount of pulled down Rac1 to total Rac1 in the lysate. Data represent mean ± SEM; n = 4; *p < 0.05 relative to the unstimulated group. D, E, Western blot analysis showing the effects of BDNF on CDKL5 phosphorylation. Neurons infected with SFV-expressing GFP-CDKL5 were serum starved and subsequently stimulated with BDNF (10 ng/ml) for the indicated length of time (D). To inhibit Trk activity, neurons were preincubated with K252a 1 h before BDNF application (E). After BDNF stimulation, GFP-CDKL5 was immunoprecipitated with GFP antibody and probed with phospho-threonine antibody. The same membrane was stripped and reprobed with GFP antibody to confirm equal protein loading. Data represent mean ± SEM; n = 5; *p < 0.05 relative to the unstimulated group; t test.
CDKL5 is involved in BDNF-induced activation of Rac1
To directly test whether CDKL5 regulates Rac1, we first measured the activity of Rac1 in cultured cortical neurons. To knockdown CDKL5 in neurons for biochemical analysis, we infected DIV 1 cortical neurons with lentivirus expressing shRNA-Scr or shRNA-#1 4 d before measurements (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Initially, we assessed the basal activity of Rac1 in neurons expressing shRNA-Scr or shRNA-#1, but failed to obtain consistent results, probably due to low and variable levels of active Rac1 in cultured neurons or to the limited sensitivity of our method. We therefore measured Rac1 activation under growth stimulation conditions. In cultured neurons, Rac1 is activated by BDNF, a critical regulator of neuronal development and function (McAllister et al., 1995; Yuan et al., 2003; Zhou et al., 2007) (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). In control neurons expressing shRNA-Scr, BDNF stimulation induced robust Rac1 activation within 5 min (Fig. 7C). When CDKL5 was downregulated by expressing shRNA-#1, BDNF-induced activation of Rac1 was inhibited (Fig. 7C). This effect was not due to a general silencing of BDNF-induced responsiveness in these cells, because no difference was observed in BDNF-induced ERK1/2 phosphorylation between neurons transfected with control and CDKL5 shRNA constructs (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). Moreover, we found that BDNF also triggered a transient increase in CDKL5 phosphorylation (Fig. 7D), and this effect was blocked by the Trk tyrosine kinase inhibitor K252a (Fig. 7E). Importantly, the time course of BDNF-induced CDKL5 phosphorylation correlated well with the activation of Rac1, supporting a role for CDKL5 in BDNF-dependent Rac1 activation.
Discussion
Mutations in the CDKL5 gene have been identified in patients with RTT variants and other X-linked neurological syndromes (Rusconi et al., 2008). However, understanding the connection between genotype and disease phenotype is hindered by the lack of information regarding the role of CDKL5 in neuronal development and function. We found that CDKL5 was highly expressed in the developing rat brain, and that neurons and glial cells expressed different splice isoforms. Our finding that CDKL5 is essential for neuronal morphogenesis provides the first evidence of a cellular mechanism that may account for the developmental effects caused by CDKL5 mutations, and for the disease phenotypes observed in patients. In fact, abnormal morphology is a consistent feature of neurons in patients with neurodevelopmental disorders such as nonsyndromic mental retardation and RTT (Jellinger et al., 1988; Armstrong et al., 1995; Kaufmann and Moser, 2000). In despite of the clear function of CDKL5a in neuronal development, it is worth noting that CDKL5b, the isoform which is not expressed in neurons but glial cells may have a function in glial development because its expression is also induced during the perinatal-postnatal stages when a rapid onset of massive gliogenesis starts. If this is the case, the function of CDKL5 in non-neuronal cells may also be implicated in the pathogenesis of CDKL5-related diseases.
The majority of CDKL5 mutations have been identified in a form of RTT called the Hanefeld variant. As opposed to classic RTT, in which there is a period of normal development, the Hanefeld variant is characterized by early-onset seizures or infantile spasm (Scala et al., 2005). Neuronal migration defects have been implicated in infantile spasm and seizures. For example, mutations in the human doublecortin gene (DCX) cause profound defects in cortical neuron migration, and patients with DCX mutations have lissencephaly and infantile spasm (Gleeson et al., 1998). Our result showing that CDKL5 knockdown delayed neuronal migration as early as P0 in the rat brain suggests that migratory defects may be involved in the occurrence of early seizures in patients with CDKL5 mutations.
The involvement of CDKL5 in RTT indicates that CDKL5 may act in the same pathway with MeCP2, a protein that is mutated in classic RTT. The finding that CDKL5 interacts with and phosphorylates MeCP2 in vitro strengthened this possibility (Lin et al., 2005; Mari et al., 2005; Bertani et al., 2006). However, CDKL5 mutations exhibit some distinctive phenotypes. For example, in addition to the early-onset seizures mentioned above, patients with CDKL5 mutations often have a better preserved autonomous nervous system compared with patients with MeCP2 mutations (Rusconi et al., 2008). This suggests that CDKL5 may function independently of MeCP2. Our finding that CDKL5 regulated neuronal morphogenesis through a mechanism involving BDNF-Rac1 signaling supports this argument and suggests a novel mechanism underlying the pathology of CDKL5-related diseases. This finding can also explain why CDKL5 mutations that enhance kinase activity cause similar phenotypes in patients as those that decrease kinase activity (Lin et al., 2005). Variants with enhanced kinase activity (R781X and L879X) mislocalize to the cell nucleus (Bertani et al., 2006; Rusconi et al., 2008), which may prevent CDKL5 from acting within the cytoplasm, leading to a loss-of-function phenotype similar to that caused by kinase-inactive mutations. Thus we propose that proper function of CDKL5 requires both kinase activity and correct localization to the cytoplasm. It should be mentioned that although our results demonstrate a cytoplasmic function of CDKL5 in neuronal morphogenesis, we cannot exclude the possibility that nuclear function of CDKL5 also contributes to this process. As shown in Figure 5, the shRNA-#1 phenotype was not fully but partially rescued by overexpressing the cytoplasm-localized CDKL5, suggesting that nuclear CDKL5 may play a role in the growth process, possibly by regulating transcription events.
Neurotrophins and Rho GTPases are important regulators of neuronal development and function (Huang and Reichardt, 2003; Govek et al., 2005; Reichardt, 2006). Dysregulation of these proteins and their signaling pathways are frequently linked to neurodevelopmental disorders. Interestingly, in MeCP2 mutant mice, a mouse model of RTT, BDNF protein level is decreased and disease progression is affected by the level of BDNF expression (Chang et al., 2006). Mutations in a number of regulators and effectors of Rho GTPases have also been implicated in mental retardation (Ramakers, 2002). Thus, given the importance of neurotrophin and Rho GTPase signaling in cognitive development and function, our finding that CDKL5 is involved in BDNF-Rac1 signaling provides a reasonable explanation of why CDKL5 mutations cause severe defects in cognitive functions.
Although our data suggest that CDKL5 may complex with Rac1, it is not clear how CDKL5 affects the activity of Rac1 in neurons. Rho GTPase activity is temporally and spatially regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). It has been demonstrated that the activity of GEFs and GAPs is regulated by phosphorylation events (Miyamoto et al., 2006; Ohta et al., 2006). Therefore, it is possible that CDKL5 regulates the activity of Rac GEFs or GAPs by phosphorylation, and thereby affects Rac1 activity. The activity of Rac1 is also regulated by endocytosis (Palamidessi et al., 2008). Thus, regulation of endocytic pathway activity might be another possible mechanism by which CDKL5 controls Rac1 activation. Identification and characterization of substrates and interacting partners of CDKL5 in neurons will provide further understanding of the mechanisms underlying the function of CDKL5 in neuronal development.
Footnotes
This work was supported by grants from the National Basic Research Program of China (2006CB806600), the Key State Research Program of China (2006CB943900), The National Natural Science Foundation of China (30721004, 30925016) to Z.-Q.X., the National Natural Science Foundation of China (30500292), the “Shu Guang” project of the Shanghai Municipal Education Commission, and the Shanghai Education Development Foundation (06SG21) to J.T. We thank Dr. Mu-Ming Poo for critical reading of this manuscript.
References
- Archer HL, Evans J, Edwards S, Colley J, Newbury-Ecob R, O'Callaghan F, Huyton M, O'Regan M, Tolmie J, Sampson J, Clarke A, Osborne J. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet. 2006;43:729–734. doi: 10.1136/jmg.2006.041467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Bertani I, Rusconi L, Bolognese F, Forlani G, Conca B, De Monte L, Badaracco G, Landsberger N, Kilstrup-Nielsen C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J Biol Chem. 2006;281:32048–32056. doi: 10.1074/jbc.M606325200. [DOI] [PubMed] [Google Scholar]
- Buyse IM, Fang P, Hoon KT, Amir RE, Zoghbi HY, Roa BB. Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms. Am J Hum Genet. 2000;67:1428–1436. doi: 10.1086/316913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression mutant mice is affected of Mecp2 by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Cheadle JP, Gill H, Fleming N, Maynard J, Kerr A, Leonard H, Krawczak M, Cooper DN, Lynch S, Thomas N, Hughes H, Hulten M, Ravine D, Sampson JR, Clarke A. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum Mol Genet. 2000;9:1119–1129. doi: 10.1093/hmg/9.7.1119. [DOI] [PubMed] [Google Scholar]
- Eriksson A, Siegbahn A, Westermark B, Heldin CH, Claesson-Welsh L. PDGF alpha- and beta-receptors activate unique and common signal transduction pathways. EMBO J. 1992;11:543–550. doi: 10.1002/j.1460-2075.1992.tb05085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JC, Archer HL, Colley JP, Ravn K, Nielsen JB, Kerr A, Williams E, Christodoulou J, Gécz J, Jardine PE, Wright MJ, Pilz DT, Lazarou L, Cooper DN, Sampson JR, Butler R, Whatley SD, Clarke AJ. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet. 2005;13:1113–1120. doi: 10.1038/sj.ejhg.5201451. [DOI] [PubMed] [Google Scholar]
- Gill H, Cheadle JP, Maynard J, Fleming N, Whatley S, Cranston T, Thompson EM, Leonard H, Davis M, Christodoulou J, Skjeldal O, Hanefeld F, Kerr A, Tandy A, Ravine D, Clarke A. Mutation analysis in the MECP2 gene and genetic counselling for Rett syndrome. J Med Genet. 2003;40:380–384. doi: 10.1136/jmg.40.5.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Rett's syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr Scand. 1985;74:405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hooshmand-Rad R, Claesson-Welsh L, Wennström S, Yokote K, Siegbahn A, Heldin CH. Involvement of phosphatidylinositide 3′-kinase and Rac in platelet-derived growth factor-induced actin reorganization and chemotaxis. Exp Cell Res. 1997;234:434–441. doi: 10.1006/excr.1997.3636. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol. 1988;76:142–158. doi: 10.1007/BF00688098. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Lin C, Franco B, Rosner MR. CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet. 2005;14:3775–3786. doi: 10.1093/hmg/ddi391. [DOI] [PubMed] [Google Scholar]
- Lohse K, Helmke SM, Wood MR, Quiroga S, de la Houssaye BA, Miller VE, Negre-Aminou P, Pfenninger KH. Axonal origin and purity of growth cones isolated from fetal rat brain. Brain Res Dev Brain Res. 1996;96:83–96. doi: 10.1016/0165-3806(96)00076-4. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C, Ariani F, Hayek G, Balestri P, Bergo A, Badaracco G, Zappella M, Broccoli V, Renieri A, Kilstrup-Nielsen C, Landsberger N. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–1946. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Yamauchi J, Tanoue A, Wu C, Mobley WC. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci U S A. 2006;103:10444–10449. doi: 10.1073/pnas.0603914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M, Consalez G, Trump D, Ballabio A, Franco B. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics. 1998;51:427–433. doi: 10.1006/geno.1998.5391. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biology. 2006;8:803–U835. doi: 10.1038/ncb1437. [DOI] [PubMed] [Google Scholar]
- Palamidessi A, Frittoli E, Garré M, Faretta M, Mione M, Testa I, Diaspro A, Lanzetti L, Scita G, Di Fiore PP. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell. 2008;134:135–147. doi: 10.1016/j.cell.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Kilstrup-Nielsen C, Bienvenu T, Jacquette A, Landsberger N, Broccoli V. CDKL5 influences RNA splicing activity by its association to the nuclear speckle molecular machinery. Human Molecular Genetics. 2009;18:4590–4602. doi: 10.1093/hmg/ddp426. [DOI] [PubMed] [Google Scholar]
- Rusconi L, Salvatoni L, Giudici L, Bertani I, Kilstrup-Nielsen C, Broccoli V, Landsberger N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J Biol Chem. 2008;283:30101–30111. doi: 10.1074/jbc.M804613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruusala A, Sundberg C, Arvidsson AK, Rupp-Thuresson E, Heldin CH, Claesson-Welsh L. Platelet-derived growth factor (PDGF)-induced actin rearrangement is deregulated in cells expressing a mutant Y778F PDGF beta-receptor. J Cell Sci. 1998;111:111–120. doi: 10.1242/jcs.111.1.111. [DOI] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Scala E, Ariani F, Mari F, Caselli R, Pescucci C, Longo I, Meloni I, Giachino D, Bruttini M, Hayek G, Zappella M, Renieri A. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet. 2005;42:103–107. doi: 10.1136/jmg.2004.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffmann K, Moser B, Raynaud M, Sperner J, Fryns JP, Schwinger E, Gécz J, Ropers HH, Kalscheuer VM. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 2004;75:1149–1154. doi: 10.1086/426460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, Evans J, Clarke A, Pelka GJ, Tam PP, Watson C, Lahooti H, Ellaway CJ, Bennetts B, Leonard H, Gécz J. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004;75:1079–1093. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol. 2003;5:38–45. doi: 10.1038/ncb895. [DOI] [PubMed] [Google Scholar]
- Zhou P, Porcionatto M, Pilapil M, Chen Y, Choi Y, Tolias KF, Bikoff JB, Hong EJ, Greenberg ME, Segal RA. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55:53–68. doi: 10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, Takemori H, Xiong ZQ. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]