

Abstract
The lateral amygdala (LA) plays a key role in emotional learning and is the main site for sensory input into the amygdala. Within the LA, pyramidal neurons comprise the major cell population with plasticity of inputs to these neurons thought to underlie fear learning. Pyramidal neuron activity is tightly controlled by local interneurons, and GABAergic modulation strongly influences amygdala-dependent learning. Synaptic inputs to some interneurons in the LA can also undergo synaptic plasticity, but the identity of these cells and the mechanisms that underlie this plasticity are not known. Here we show that long-term potentiation (LTP) in LA interneurons is restricted to a specific type of interneuron that is defined by the lack of expression of synaptic NR2B subunits. We find that LTP is only present at cortical inputs to these cells and is initiated by calcium influx via calcium-permeable AMPA receptors. LTP is maintained by trafficking of GluR2-lacking AMPA receptors that require an interaction with SAP97 and the actin cytoskeleton. Our results define a novel population of interneurons in the LA that control principal neuron excitability by feed-forward inhibition of cortical origin. This selective enhanced inhibition may contribute to reducing the activity of principal neurons engaged during extinction of conditioned fear.
Introduction
The amygdaloid complex plays a key role in emotional learning and behavior (LeDoux, 2000; Davis and Whalen, 2001). The lateral amygdala (LA), the main site for sensory input to the amygdala, contains two types of neuron: glutamatergic principal neurons and GABAergic local interneurons (McDonald, 1992; Smith et al., 1998; Sah et al., 2003). Although both types of neuron receive glutamatergic inputs from cortical and subcortical regions (Mahanty and Sah, 1998, 1999; Weisskopf and LeDoux, 1999; Szinyei et al., 2000), distinct postsynaptic receptors are present at the two cell types.
Glutamate activates two types of ionotropic receptors, AMPA and NMDA receptors (NMDARs). AMPA receptors are tetramers assembled from the products of four genes (GluR1–GluR4) (Mansour et al., 2001). Whereas AMPA receptors assembled with GluR2 show linear current–voltage (I–V) relationships and are calcium impermeable, receptors that lack GluR2 subunits are inwardly rectifying and calcium permeable (Jonas et al., 1994; Jonas and Burnashev, 1995; Washburn et al., 1997). NMDARs are also heteromultimers with two obligatory NR1 subunits and two of four different NR2 subunits (NR2A–NR2D) (Cull-Candy et al., 2001).
Glutamatergic afferents to principal neurons make dual-component synapses with both AMPA and NMDA receptors (Mahanty and Sah, 1999; Weisskopf and LeDoux, 1999). AMPA receptors at these synapses have linear I–V relationships, consistent with the presence of GluR2 subunits (Mahanty and Sah, 1999), and NMDARs containing both NR2A and NR2B subunits are present at all synapses (Lopez de Armentia and Sah, 2003). NMDAR-dependent synaptic plasticity of inputs to these neurons is thought to underlie the acquisition and storage of fear memories (Walker and Davis, 2002; Rodrigues et al., 2004; Sah and Westbrook, 2008). In contrast, synaptic AMPA receptors on interneurons lack GluR2 subunits and have inwardly rectifying I–V relationships (Mahanty and Sah, 1998). Although synapses on some interneurons lack detectable NMDARs (Mahanty and Sah, 1998), others express synaptic NMDARs that contain only NR2B subunits (Szinyei et al., 2000, 2003).
Interneurons maintain a tight control over principal neuron activity (Lang and Pare, 1997; Woodruff and Sah, 2007b; Ehrlich et al., 2009), and modulation of inhibition strongly influences amygdala-dependent learning (Harris and Westbrook, 1998b; Shaban et al., 2006). Glutamatergic inputs to interneurons in the LA also show long-term potentiation (LTP) (Mahanty and Sah, 1998; Szinyei et al., 2000). However, although different populations of interneurons have been described in the LA (McDonald, 1992; McDonald and Betette, 2001), whether interneurons that display plasticity comprise a distinct population or the mechanisms that underlie this plasticity remain unknown. Here we show that LTP in LA interneurons is restricted to a specific class of interneuron that lacks NR2B-containing synaptic NMDARs and is mediated by calcium influx via calcium-permeable AMPA receptors. LTP is restricted to cortical inputs and maintained by postsynaptic trafficking of GluR1-containing AMPA receptors. LTP of these inputs potentiates feedforward inhibition to principal neurons, effectively reducing the number of action potentials evoked by cortical stimulation. These results define a distinct population of interneurons that may contribute to reducing the activity of principal neurons engaged during fear extinction.
Materials and Methods
Coronal brain slices containing basolateral amygdala were cut from 18- to 28-d-old GAD67-GFP knock-in mice expressing enhanced green fluorescent protein (EGFP) under the control of the GAD67 promoter (Tamamaki et al., 2003). After halothane anesthesia, animals were decapitated, and the brain was rapidly removed and placed in ice-cold, oxygenated artificial CSF containing (in mm) 118 NaCl, 2.5 KCl, 2.5 NaHCO3, 10 glucose, 1.3 MgCl2, 2.5 CaCl2, and 1.2 NaH2PO4. Slices were cut (300 μm) and incubated at 32°C for 30 min before being allowed to equilibrate at room temperature for at least an additional 30 min. All procedures were approved by the University of Queensland Animal Ethics Committee.
During recording, slices were perfused with heated artificial CSF (34 ± 2°C). Recording pipettes (3–5 MΩ) fabricated from borosilicate glass were filled with a solution containing (in mm) 135 CsMeSO4, 8 NaCl, 10 HEPES, 2 Mg2ATP, 0.3 Na3GTP, 0.1 spermine, 7 phosphocreatine, and 0.3 EGTA. Picrotoxin (100 μm) was added to the artificial CSF during LTP experiments. Visualization of GAD67-EGFP neurons was achieved using an upright microscope (BX50WI; Olympus) equipped with a fluorescence attachment. Whole-cell recordings were made using MultiClamp 700B (Molecular Devices), filtered at 4 kHz, and digitized at 8 kHz using an ITC-16 board (Instru-Tech). Analysis was performed using Axograph 4.9. Ifenprodil, d-APV, nicardipine, and cyclopiazoic acid were obtained from Sigma-Aldrich. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline-2,3-dione (NBQX) Pep1-TGL, and Pep2-EVKI were obtained from Tocris. The NR2B-selective antagonist CP323-488 was a gift from Pfizer. During LTP experiments, after obtaining a whole-cell recording, EPSCs were tested at −60 and +40 mV to determine the kinetics of the NMDAR-mediated EPSCs. Baseline recordings were then initiated at a holding potential of −60 mV. For current-clamp recordings, the CsMeSO4 was replaced with KMeSO4. Bipolar stimulating electrodes were placed on the external capsule to stimulate cortical inputs and were placed medial to the LA to stimulate thalamic inputs (Mahanty and Sah, 1999). Synaptic inputs were evoked at 0.1 Hz using a bipolar stimulator (DS2A; Digitimer). For comparison of deactivation kinetics of the NMDAR EPSCs, 5–10 evoked EPSCs were recorded at +40 mV and averaged, and the current decays were fitted to a double-exponential equation of the form the following:
where If and Is are the amplitudes of the fast and slow and decay components and τf and τs are their respective decay time constants. The weighted time constant was calculated as follows:
This was used for comparisons of the decay times of the EPSCs. LTP was evoked with a tetanic stimulus at −60 mV (100 Hz, 300 ms) repeated three times at an interval of 10 s, at no more than 10 min after gaining whole-cell access. The rectification index (RI) of AMPA receptor-mediated EPSCs was calculated as the peak amplitude at +40 mV divided by the peak amplitude at −60 mV. The AMPA/NMDA ratio of the EPSCs was calculated as the peak AMPA receptor-mediated EPSC at −60 mV divided by NMDA current at 30 ms after the peak of the dual-component EPSC at +40 mV. Paired-pulse ratios were calculated as a ratio of EPSC2 and EPSC1 separated by 50 ms. Spontaneous events recorded at +40 mV were detected and analyzed using variable amplitude templates. Three different templates were created with an amplitude threshold of 5 pA and decay time constants of 50, 100, and 150 ms. Only events >2.5× SD of the noise were detected.
Green fluorescent protein (GFP)-positive GAD67 mice (n = 4) were anesthetized with sodium pentobarbital (150 mg/kg) and perfused transcardially with 2% sodium nitrite solution (in 0.1 m phosphate buffer (PB), pH 7.4) followed by 50 ml of 4% paraformaldehyde containing 0.5% glutaraldehyde (in 0.1 m phosphate buffer, pH 7.4). Brains were then removed and postfixed overnight in the same fixative at 4°C and washed in 0.1 m PBS, after which 50-μM-thick coronal sections containing the amygdala complex were cut using a Vibratome (VT1000S; Leica). All sections were washed in PBS, incubated in 3% H2O2 in 0.1 m PBS for 30 min, and blocked in a solution of 5% BSA/0.05% saponin/0.1 m PBS for 1 h. Sections were incubated in a primary antibody solution containing mouse monoclonal anti-synapse-associated protein 97 (SAP97) antibody (1:500; Stressgen Bioreagents) and chicken polyclonal anti-GFP antibody (1:4000; Abcam) in a blocking solution containing 0.5% BSA/0.05% saponin in 0.1 m PBS with 0.05% sodium azide, at 23°C for 56 h. Sections were then washed in PBS and incubated for 5 h in a solution of biotinylated donkey anti-mouse antibody (Jackson ImmunoResearch) at 1:500 in blocking solution. After additional PBS washes, sections were placed into a solution of avidin–biotin–horseradish peroxidase complex (1:5000; Invitrogen) in blocking solution for 16 h. This was followed by PBS washes (two times for 5 min) and a sodium acetate buffer, pH 6.0, wash for 5 min. Sections were exposed for 15 min to a 2% NiSO4 solution (in sodium acetate buffer) containing 2 mg/ml d-glucose, 0.4 mg/ml NH4Cl, and 0.025% 3,3-diaminobenzidine (DAB). This was followed by another incubation in the same solution but with the addition of glucose oxidase (0.2 μl/ml) to allow the slow production of H2O2, resulting in the deposition of nickel–DAB (black precipitate). Sections were washed in sodium acetate buffer to stop the reaction and then washed thoroughly in PBS. The sections were then incubated for 5 h in a solution of biotinylated donkey anti-chicken antibody (Jackson ImmunoResearch) at 1:500 in blocking solution with 0.05% sodium azide. Sections were washed in PBS and incubated in a solution of avidin–biotin–horseradish peroxidase complex (1:5000) in a blocking solution for 16 h. Sections were washed in PBS, and labeling revealed using the same DAB protocol as described above, except for the omission of nickel in the DAB solutions to allow the production of a brown precipitate. Control sections were processed as above, except that primary or secondary antibodies were omitted. Sections were washed in sodium acetate buffer and mounted onto glass slides and coverslips containing Depex mounting medium. Images were acquired using an Axio Imager Z1 microscope equipped with an AxioCam HR camera (Zeiss). We used Adobe Illustrator to sharpen images, adjust brightness and contrast levels, and compose the final plates.
Results
Principal neurons in the LA are the major input elements and receive direct sensory inputs from thalamic nuclei as well as processed sensory input from a number of cortical areas (McDonald, 1998; Sah et al., 2003). Local circuit GABAergic interneurons in the LA provide substantial feedforward and feedback inhibition that not only controls their activity (Li et al., 1996; Lang and Pare, 1997; Woodruff and Sah, 2007a,b) but also acts as a brake on plasticity of glutamatergic inputs to principal neurons (Bissiere et al., 2003). Cortical inputs to some interneurons in the rat LA also show synaptic plasticity (Mahanty and Sah, 1998; Bauer and LeDoux, 2004). However, neither the identity of these interneurons nor the mechanisms that underlie this LTP is known.
Different interneurons express different NMDARs
Interneurons in the LA were identified by expression of GFP under the control of the GAD67 promoter (Tamamaki et al., 2003). As in the rat (Mahanty and Sah, 1998; Szinyei et al., 2000; Bauer and LeDoux, 2004), interneurons in the mouse amygdala received independent excitatory inputs from both cortical and thalamic regions (Figs. 1, 2; see Fig. 4). Consistent with the relatively low expression of GluR2 subunits in interneurons (McDonald, 1996), stimulation of these inputs evoked AMPA receptor-mediated EPSCs that showed inward rectification (Fig. 1A,B), with a mean RI (see Materials and Methods) of 0.16 ± 0.06 (n = 18) compared with principal neurons, that had linear current–voltage relationships with a rectification ratio of 1.3 ± 0.4 (n = 8; p < 0.01 vs interneuron RI) (Fig. 1C). The contribution of NMDARs at these synapses was highly variable with some interneurons showing little or no NMDAR-mediated synaptic current (Mahanty and Sah, 1998), whereas others clearly had a large postsynaptic NMDAR component (Szinyei et al., 2003) (Fig. 1A). When present, the decay of the NMDAR EPSC could be fit with a sum of two exponentials, and the weighted time constant of decay (τw; see Materials and Methods) revealed two populations of cells. One population had a relatively fast decay time constant with a mean of 53 ± 2 ms (n = 43), whereas the other had significantly slower kinetics with a mean weighted time constant of 109 ± 11 ms (Fig. 1D) (n = 10; p < 0.05). Overall, excitatory synapses on 10 of 63 interneurons (16%) had no detectable NMDARs, whereas 68% (43 of 63) had NMDAR EPSCs with fast kinetics and 16% (10 of 63) had NMDAR EPSCs with slow kinetics (Fig. 1E). When present, the relative contribution of NMDARs to the EPSC was similar in both types of interneuron with the mean AMPA/NMDAR ratio being 5.8 ± 1.1 and 5.7 ± 0.9 for cells with slow and fast NMDAR kinetics, respectively. All glutamatergic synapses on an individual interneuron showed the same properties with respect to both AMPA and NMDA receptors. Thus, all synapses had rectifying AMPA receptors and, when voltage clamped at +40 mV, stimulation of either cortical or thalamic inputs (Fig. 2A–C) as well as the averaged spontaneous synaptic events (Fig. 2D) had NMDARs with identical kinetics. To determine the anatomical distribution of different interneurons, cells were filled with neurobiotin and recovered post hoc (see Materials and Methods), and their location was determined as shown in Figure 2E. Cells with different types of NMDARs were distributed throughout the LA, although cells that lacked NMDARs appear to be somewhat concentrated in the dorsal LA (Fig. 2F).
Figure 1.

Interneurons can be differentiated based on the presence and kinetics of synaptic NMDARs. A, EPSCs evoked at holding potentials of −60 mV and +40 V normalized to the peak EPSC at −60 mV, from representative neurons with synapses that lack NMDARs (left traces), have NMDARs with fast kinetics (middle traces), or have NMDARs with slow kinetics (right traces). The inset shows the NMDAR EPSC at +40 mV with the fit time constant of decay. B, Current–voltage relationships measured at the peak of the AMPA receptor EPSC (filled squares) and 50 ms after the peak (filled circles) at a cell without (left) and with (right) synaptic NMDARs. C, Summary data of the RI of the AMPA receptor-mediated EPSC, calculated as the ratio of peak EPSC at +40 mV to the peak EPSC at −60 mV, in interneurons that have synapses with no NMDARs (NMDA−) and those with NMDARs (NMDA+). For comparison, cortical inputs to principal neurons express AMPA receptors with a rectification ratio near 1. D, Summary of NMDAR EPSC decay time constants in the LA for fast and slow NMDAR-mediated EPSC. E, Summary plot of percentages of interneurons without/with and different types of NMDARs in the LA. Sixteen percent (n = 10 of 63) of cells lacked NMDARs, 67% (n = 43 of 63) have NMDARs with fast kinetics, and 16% (n = 10 of 63) of cells have NMDARs with slow kinetics.
Figure 2.
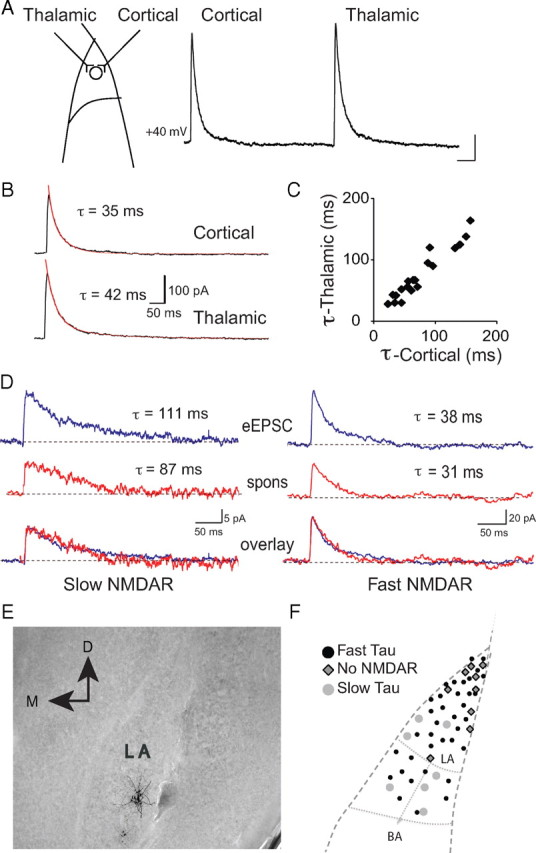
All synapses on LA interneurons have similar postsynaptic NMDARs. A, Diagrammatic representation of the innervation of a single LA interneuron by thalamic and cortical afferents. Traces on the right show EPSCs evoked at +40 mV for cortical stimulation followed by thalamic stimulation in a LA interneuron. Calibration: 100 ms, 50 pA. B, Average EPSCs fit with a weighted time constant (τw). C, Decay time constants for NMDAR-mediated EPSCs at cortical synapses plotted against decay time constants for EPSCs at thalamic synapses on interneurons (n = 19). The straight line indicates a linear fit to the data (r = 0.96) showing that decay time constants for cortical and thalamic inputs are highly correlated. D, Sample traces of evoked (eEPSC) and spontaneous (spons) NMDAR-mediated EPSCs at +40 mV in two cells with fast and slow NMDAR kinetics, respectively. In cells with slow evoked NMDAR EPSCs (top trace; τw = 111 ms). τw for spontaneous EPSCs was 87 ms (middle trace). In cells with fast evoked NMDA EPSCs (top trace; τw = 38 ms), the decay time constant of the spontaneous EPSC was 31 ms (middle trace, red). Normalized evoked and spontaneous EPSCs overlaid showing similar NMDAR kinetics (bottom trace). E, Shown is one interneuron filled with neurobiotin and recovered after electrophysiological recording. F, The distribution of all neurons in the LA with the three types of synaptic NMDAR present.
Figure 4.

Cortical inputs to interneurons with slow synaptic NMDARs do not show LTP. A, Recording from a representative interneuron with synapses that express NMDARs with fast kinetics (τw = 42 ms). Tetanic stimulation leads to potentiation of the AMPA receptor-mediated EPSC. EPSCs recorded at the indicated time points are shown above. Inset, NMDAR-mediated EPSC recorded at +40 mV with the weighted time constant. The average response from interneurons with fast NMDAR-mediated EPSCs is shown in the graph below (n = 8). B, Recording from a representative interneuron with synapses that express NMDARs with slow kinetics (τw = 126 ms). Tetanic stimulation has no effect on EPSC amplitude. EPSCs recorded at the indicated time points are shown above. Inset, NMDAR-mediated EPSC recorded at +40 mV with the weighted time constant. The averaged response from interneurons with slow NMDAR-mediated EPSCs is shown in the graph below (n = 9). C, Scatter plot showing the amount of LTP evoked in each cell plotted against the weighted time constant of the NMDAR-mediated EPSC. D, Facilitation and charge transfer during the high-frequency stimulation are similar in both cell types containing fast and slow NMDAR kinetics. Normalized example trace (normalized to the first EPSC in the train) of HFS from interneurons containing fast NMDAR kinetics and slow NMDAR kinetics, respectively. C, Cortical input; IN, interneuron.
NMDARs are assembled as tetramers with two obligatory NR1 subunits and two of four NR2 subunits, NR2A–NR2D (Cull-Candy et al., 2001; Paoletti and Neyton, 2007). The presence of different NR2 subunits confers distinct pharmacological and kinetic properties on the receptor (Dingledine et al., 1999; Paoletti and Neyton, 2007). Of these, NR2A and NR2B subunits are the most prominent subunits present in most forebrain structures including the basolateral amygdala (Cull-Candy et al., 2001; Lopez de Armentia and Sah, 2003). In both expression systems and cultured neurons, NMDARs composed of NR1/NR2A heteromers have rapid channel kinetics, with offset rate constants of 30–50 ms in response to brief applications of glutamate, whereas NR1/NR2B heteromers are significantly slower with offset kinetics of ∼200 ms (Stocca and Vicini, 1998; Tovar et al., 2000) (room temperature). The significantly slower kinetics of NMDARs at some interneurons in the LA suggests the presence of NR2B subunits at these synapses. Consistent with this, NMDAR-mediated EPSCs with slow kinetics were significantly more sensitive to blockade by NR2B selective antagonists CP323488 (5 μm), Ro25-9681 (1 μm), and ifenprodil (5 μm) (Fig. 3). Together, these results show that all interneurons in the LA express inwardly rectifying AMPA receptors but can be separated into three populations based on the receptor composition of NMDARs they express at excitatory synapses. One type of interneuron expresses little or no synaptic NMDARs. The second type expresses NMDARs that contain mostly NR2B subunits, and the third type express receptors that contain primarily NR2A subunits.
Figure 3.

NMDAR EPSCs with slow kinetics are attributable to the presence of NR2B subunits. NMDARs containing NR1/NR2A and NR1/NR2B can be distinguished pharmacologically. We have used three antagonists that are more specific for NR1/NR2B receptors compared with NR1/NR2A receptors: CP323488, ifenprodil, and Ro25-9681 (Chizh et al., 2001). A, B, NMDAR EPSCs recorded at +40 mV in the presence of 10 μm NBQX and 50 μm picrotoxin from interneurons with slow (A) and fast (B) kinetics. Shown are average traces for control, CP323488 (5 μm, gray), and d-AP5 (30 μm). The time course of block of EPSCs corresponding to A and B are shown below. C, Mean data for percentage block of the slow and fast NMDA currents by CP-323488 (5 μm), Ro 25-9681 (1 μm), and ifenprodil (5 μm). CP-323488, Ro 25-9681, and ifenprodil produced 62 ± 8% (n = 9), 54 ± 1% (n = 4), and 52 ± 2% (n = 3) block in the peak EPSC for cells with slow NMDAR kinetics and 25 ± 5% (n = 4), 19 ± 3% (n = 3), and 24 ± 4% (n = 3) block of EPSCs with fast kinetics.
LTP is restricted to cortical inputs at one type of interneuron
Most glutamatergic synapses in the mammalian brain form dual-component excitatory synapses that express both AMPA and NMDA receptors with basal synaptic transmission being essentially mediated by AMPA receptors, whereas NMDARs are recruited during repetitive stimulation to initiate different forms of synaptic plasticity (Malenka and Nicoll, 1993). As previously described (Mahanty and Sah, 1998), repetitive stimulation of inputs to interneurons at which excitatory synapses did not express NMDARs evoked LTP (168 ± 12%; p < 0.001 vs baseline; n = 9; data not shown). However, for interneurons where synaptic NMDARs were present, repetitive stimulation of cortical afferents revealed two populations. In interneurons in which synaptic NMDARs had fast decay kinetics (τw = 54 ± 2 ms), repetitive stimulation evoked LTP (to 150 ± 10%; p < 0.001 vs baseline; n = 8) (Fig. 4A,C). In contrast, interneurons that expressed NMDARs with significantly slower kinetics (τw = 118 ± 7 ms) failed to show LTP (to 101 ± 15%; p > 0.1 vs baseline; n = 9) (Fig. 4B,C). There was no significant difference in the normalized charge transfer during LTP induction between cell types (0.13 ± 0.02 vs 0.16 ± 0.04; n = 6; p > 0.5) (Fig. 4D) showing that activation of AMPA/kainate receptors during tetanus was similar in the two cell types.
Interneurons in the LA receive both cortical and thalamic input (Fig. 2), and we next tested whether LTP was input specific. We first confirmed that these inputs did not share fibers by testing if there was any cross-facilitation between the two inputs (Fig. 5A,B). Cortical and thalamic inputs were then independently stimulated, and, after obtaining a baseline, cortical inputs were selectively tetanized. In cells with fast NMDAR EPSCs (cortical input, τW = 50 ± 6 ms; thalamic input, τW = 47 ± 7 ms; n = 5), tetanic stimulation resulted in LTP (to 190 ± 20% of control; p < 0.001 vs baseline) but had no effect on thalamic inputs (to 96 ± 3% of control) (Fig. 5C), showing that LTP is input specific. Costimulation of thalamic and cortical inputs also failed to potentiate thalamic inputs (n = 5) (Fig. 5D). Moreover, when thalamic inputs were tetanized alone, they failed to show LTP (99 ± 16% of control; n = 10) (Fig. 5E). This inability to potentiate thalamic inputs was selective for interneurons as stimulation of thalamic inputs to principal neurons (interleaved recordings) led to clear LTP (to 157 ± 13%; p < 0.001 vs baseline; n = 10) (Fig. 5F).
Figure 5.

LTP is input specific and only present at cortical synapses. A, Interneurons receive independent cortical and thalamic inputs. Left, EPSCs evoked by either cortical (top traces) or thalamic (bottom traces) stimulation at a holding potential of −60 mV. Middle, Response to paired pulses for cortical or thalamic stimulation. Right, Primed pulses for thalamic followed by cortical (Thal-Cort) and cortical followed by thalamic (Cort-Thal) stimulations at an interval of 50 ms. Paired pulses for either Cort-Cort or Thal-Thal stimulations result in PPF, whereas primed pulses for either Cort-Thal or Thal-Cort do not show cross-facilitation. B, Cortical and thalamic inputs stimulated simultaneously sum together showing there are no shared fibers between the two inputs. Plotted is the algebraic sum of separately evoked EPSCs in response to cortical and thalamic stimulation ([Cort] + [Thal]) against the EPSC evoked by simultaneous stimulation of cortical and thalamic inputs ([Cort + Thal]). C, LTP of cortical inputs is homosynaptic. Average data from neurons with fast NMDAR-mediated synaptic currents in which cortical and thalamic inputs were independently stimulated (τw cortical input, 50 ± 6 ms; thalamic input, 47 ± 7 ms; n = 5). Tetanic stimulation of cortical inputs leads to potentiation with no effect on thalamic inputs. D, Cortical and thalamic inputs evoked as in C were co-tetanized at time 0. Only cortical inputs are potentiated after paired stimulation of both inputs. τw for cortical input was 43 ± 4 ms and for thalamic input was 44 ± 7 ms (n = 5). E, Thalamic inputs to interneurons do not show LTP. The left panel shows averaged data from interneurons in which thalamic inputs (τw = 54 ± 2 ms; n = 10) were tetanized at time 0. F, Average data of interleaved recordings from principal neurons in which the same thalamic input was stimulated (n = 10). EPSCs recorded at the indicated times are shown above the graph. C, Cortical input; T, thalamic input; IN, interneuron; PN, principal neuron.
LTP requires trafficking of AMPA receptors and SAP97
These experiments show that LTP of excitatory inputs to interneurons is restricted to cortical inputs to a distinct population of interneurons that do not express NR2B subunits. We next asked whether calcium influx via NMDARs is required to trigger LTP at these synapses. Application of the specific antagonist (d-APV, 30 μm) had no effect on the EPSC at −60 mV (data not shown) but fully blocked the NMDAR EPSC (Fig. 2B). However, d-APV had no effect on induction of LTP in these interneurons (τw = 39 ± 4 ms; n = 5) (Fig. 6A). Low-frequency stimulation of inputs at a holding potential of 0 mV or delivering tetanic stimulation at a holding potential of −30 mV to enhance calcium influx via NMDARs (Laezza et al., 1999; Laezza and Dingledine, 2004) did not reveal any dependence of LTP on NMDARs. Thus, low-frequency pairing at 0 mV did not evoke LTP, whereas tetanic stimulation at −30 mV did evoke LTP but was not blocked by d-APV (data not shown). Thus, LTP at these inputs does not require activation of NMDARs. As the presence of a slow NMDAR-mediated EPSC would have significantly larger summation (Lei and McBain, 2002; Lopez de Armentia and Sah, 2003), we wondered whether the lack of LTP in these interneurons resulted from the presence of a large NMDAR-mediated calcium influx. However, d-AP-5 (30 μm) failed to rescue LTP in cells with NR2B subunits (Fig. 6B). LTP induction did, however, require a rise in postsynaptic calcium as it was blocked when neurons were loaded with the calcium chelator BAPTA (τw = 47 ± 5 ms; 102 ± 3% of baseline; p > 0.05; n = 5) (Fig. 6C). What, then, is the source of the calcium rise that triggers LTP? Blockade of L-type voltage-dependent calcium channels with nicardipine (10 μm) did not alter LTP (150 ± 10% of baseline; p < 0.001; n = 4; τw = 58 ± 10 ms) (Fig. 6D), and neither did emptying intracellular calcium stores with cyclopiazionic acid (140 ± 9%; p < 0.001 vs baseline; n = 5) (Fig. 6E). Thus, the rise in cytoplasmic calcium that initiates LTP at interneuron synapses does not result from calcium influx via NMDARs or L-type calcium channels. Moreover, the calcium rise is not attributable to release from intracellular stores, ruling out the involvement of metabotropic receptors.
Figure 6.

LTP at cortical inputs requires calcium influx via AMPA/kainate receptors. A, LTP in interneurons does not require NMDAR activation. d-APV (30 μm) was applied from the start of recording in a cell that had fast NMDAR-mediated kinetics. A representative NMDAR EPSC is shown in the inset (τw = 39 ms). Tetanic stimulation leads to LTP. B, Blocking NMDARs with d-APV does not rescue LTP in interneurons with slow decaying NMDARs. d-APV (30 μm) was applied from the start of recording, and a representative NMDAR EPSC is shown in the inset (τw = 142 ms). Tetanic stimulation did not evoke LTP (n = 5). C, LTP is blocked in interneurons loaded with the calcium chelator BAPTA (10 mm; n = 5). A representative NMDAR EPSC with fast kinetics is shown in the inset. D, L-type voltage-dependent calcium channels are not required for LTP. EPSCs were recorded with 10 μm nicardipine; tetanic stimulation had no effect on LTP (n = 4). E, Store release of calcium is not required for LTP. Application of SERCA (sarco/endoplasmic reticulum Ca2+-ATPase) pump antagonist cyclopiazoic acid (CPA; 30 μm; n = 5) had no effect on LTP. F, AMPA receptor activation is required for LTP. Cortical and thalamic inputs were independently stimulated, and, after obtaining a baseline, kynurenic acid (5 mm) was applied. After EPSCs were blocked, tetanic stimulation was delivered to cortical inputs (time 0). Subsequent washout of kynurenic acid restored the EPSC to baseline equally in the both the tetanized and nontetanized pathways, showing that LTP was blocked. C, Cortical input; IN, interneuron.
AMPA receptors that mediate transmission at interneuron synapses show inward rectification, indicating that they are calcium permeable (Jonas et al., 1994; Jonas and Burnashev, 1995; Washburn et al., 1997). To test whether activation of AMPA receptors is required for LTP, we blocked synaptic transmission with the reversible ionotropic receptor antagonist kynurenic acid (5 mm) and tetanized cortical afferents when AMPA receptors were blocked (Fig. 6F). Subsequent washout of kynurenic acid restored EPSC amplitude to its control levels in both the tetanized cortical afferents and the nontetanized thalamic afferents equally (98 ± 5% for cortical, 93 ± 8%, p > 0.1; τw = 43 ± 4 ms; n = 4), showing that AMPA receptor activation is necessary for LTP induction.
These results demonstrate that in interneurons that express synaptic NMDARs, tetanic stimulation leads to LTP that is initiated by calcium influx via calcium-permeable AMPA/kainate glutamate receptors. As induction of LTP to interneurons is clearly postsynaptic, we next asked whether expression of LTP requires a presynaptic or postsynaptic change in synaptic transmission. We first tested the effects of tetanic stimulation on paired-pulse facilitation (PPF), an index of presynaptic release probability (Zucker, 1989). There was no change in release probability with baseline PPF being 1.14 ± 0.08 before and 1.15 ± 0.13 (n = 8) after LTP, suggesting that LTP at interneuron synapses has a postsynaptic locus. In both hippocampal (Hayashi et al., 2000) and LA (Rumpel et al., 2005) principal neurons, expression of postsynaptic LTP results from trafficking of AMPA receptors to the synapse. To test whether a similar mechanism underlies LTP in LA interneurons, we first tested the role of the actin cytoskeleton by disrupting it with latrunculin B (Spector et al., 1989). Loading neurons with latrunculin B (20 μm) had no effect on basal synaptic transmission (Fig. 7A) but completely blocked LTP (103 ± 3%; p > 0.5 vs baseline; NMDA EPSC, τw = 53 ± 8 ms; n = 6) (Fig. 7B). These recordings were interleaved with those from neurons not loaded with latrunculin B, which displayed normal LTP (170 ± 20%; p < 0.001 vs baseline; n = 4). Thus, LTP at interneuron synapses require an intact actin cytoskeleton.
Figure 7.

LTP requires a functional actin cytoskeleton and SAP97. A, Lactrunculin B had no effect on basal synaptic transmission. Shown are average data of recordings from neurons with fast NMDAR-mediated synaptic currents in interneurons loaded with latrunculin B (20 μm; n = 4). B, Loading neurons with latrunculin B blocks LTP. Average data of recordings from neurons with fast NMDAR-mediated synaptic currents in interneurons loaded with latrunculin B (20 μm; n = 4; open diamonds). For comparison, interleaved recordings from neurons that were not loaded with latrunculin B (n = 4; filled diamonds). Inset, Representative NMDAR-mediated EPSCs in control and latrunculin-loaded neurons. C, AMPA receptors inserted after LTP lack GluR2 subunits. Shown is the RI of AMPA receptor EPSCs recorded before and after induction of LTP. Average RI is not significantly changed after tetanic stimulation, showing that receptors inserted after LTP also lack GluR2 subunits. D, LTP in interneurons requires interaction of GluR1 with its PDZ binding partners. Average data of recordings from neurons with fast NMDAR-mediated synaptic currents in interneurons loaded with pep1-TGL (50 μm; n = 6; open squares). For comparison, interleaved recordings from neurons that were loaded with pep2-EVKI (n = 5; gray squares) and neurons that were not loaded with any peptide (filled squares; n = 4) are shown. Inset, Representative NMDAR-mediated EPSCs in pep2-EVKI- and pep1-TGL-loaded neurons.
At some interneuron synapses, AMPA receptors have been found to change their subunit composition after repetitive activity (Liu and Cull-Candy, 2000). However, in amygdala interneurons, control RI was unchanged after tetanic stimulation and was 0.15 ± 0.04 before and 0.08 ± 0.09 after LTP (p > 0.1; n = 6) (Fig. 7C). Although this difference was not significantly different, it is, if anything, showing greater rectification after LTP, consistent with the view that new AMPA receptors inserted after LTP do not contain GluR2 subunits. As interneurons in the basolateral amygdala express high levels of GluR1 (McDonald, 1996; Kondo et al., 2000) with little or no GluR2/3 (McDonald, 1996), it is likely that AMPA receptors at interneuron synapses consist mainly of GluR1 homomers. Trafficking of GluR1-containing AMPA receptors in neurons has been found to require interactions with the synapse-associated protein SAP97 (Leonard et al., 1998; Rumbaugh et al., 2003), and SAP97 has been associated with synapses that express GluR2 lacking AMPA receptors (Valtschanoff et al., 2000). Consistent with this, most EGFP-positive interneurons in the LA also expressed SAP97; however, a small subset of interneurons did not express SAP97 (Fig. 8). To disrupt interactions of GluA1 with SAP97, we loaded neurons with the 11-amino peptide pep1-TGL (50 μm) corresponding to the C terminus of GluA1 that binds SAP-97 (Hayashi et al., 2000). In pep1-TGL-loaded neurons that had fast NMDAR-mediated EPSCs, basal synaptic transmission was unaffected (data not shown); however, LTP was completely abolished (95 ± 1%; p > 0.1 vs baseline; τw = 52 ± 6 ms; n = 6) (Fig. 7D). In contrast, loading the cells with the peptide Pep2-EVKI that disrupts the interaction of GluR2 with its postsynaptic density-95/Discs large/zona occludens-1 (PDZ) binding partners did not have any effect on LTP (Fig. 7D). These results show that at glutamatergic synapses on interneurons in the LA, tetanic stimulation of cortical afferents leads to trafficking of GluR1-containing AMPA receptors to these synapses by a mechanism that requires SAP97.
Figure 8.
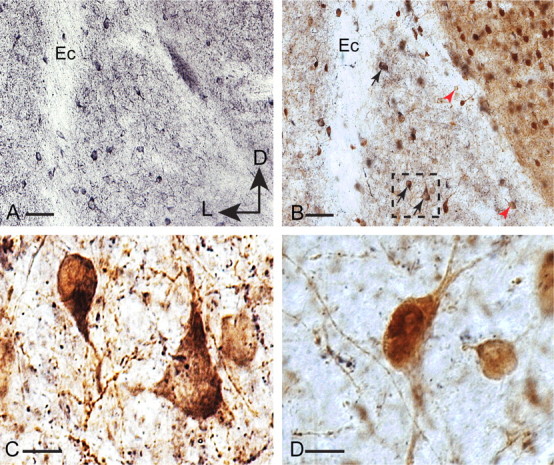
Single (A) and double (B–D) immunoperoxidase labeling of SAP97 (black) and GFP (brown) in coronal sections of LA of GAD-GFP mouse brains. A, SAP97-immunoreactive neurons are present in the LA. B, Double labeling for SAP97 and GFP revealed the majority of GFP-immunoreactive interneurons also exhibited SAP97 immunoreactivity (black arrows). Some GFP-immunoreactive interneurons indicated by red arrowheads lacked SAP97 immunoreactivity. C, Higher magnification of the dotted inset in B illustrates double-labeled interneurons. D, High-magnification image of two GFP-positive interneurons lacking SAP97 immunoreactivity. External capsule (Ec) is marked in A and B for orientation purposes. Scale bars: A, B, 50 μm; C, D, 10 μm. D, Dorsal; L, lateral.
Interneuron LTP dampens the excitability of LA principal cells
To assess the functional impact of LTP on interneurons, we made whole-cell recordings from projection neurons, evoking biphasic currents for both cortical and thalamic stimulation. As described previously (Mahanty and Sah, 1998), tetanic stimulation of cortical inputs led to potentiation of the evoked IPSC (142 ± 17%; p < 0.001 vs baseline; n = 6). LTP was restricted to the tetanized cortical inputs showing that it is synapse specific (Fig. 9A,B). Potentiation of the cortically evoked IPSC was caused by a disynaptic IPSC as it was fully blocked by the AMPA receptor antagonist NBQX (10 μm) (Fig. 9A). The change in evoked EPSC after LTP induction was only 14 ± 12% (vs baseline, p > 0.1), resulting in a shift of balance between inhibition and excitation, and the IPSC/EPSC ratio increased from 1.38 ± 0.44 to 2.80 ± 0.9 (p < 0.01) after LTP (Fig. 9C). As shown previously for thalamic inputs to these neurons (Bissiere et al., 2003), when inhibition was blocked (100 μm picrotoxin), tetanic stimulation led to robust LTP of cortical glutamatergic inputs (158 ± 11%; n = 5; data not shown).
Figure 9.
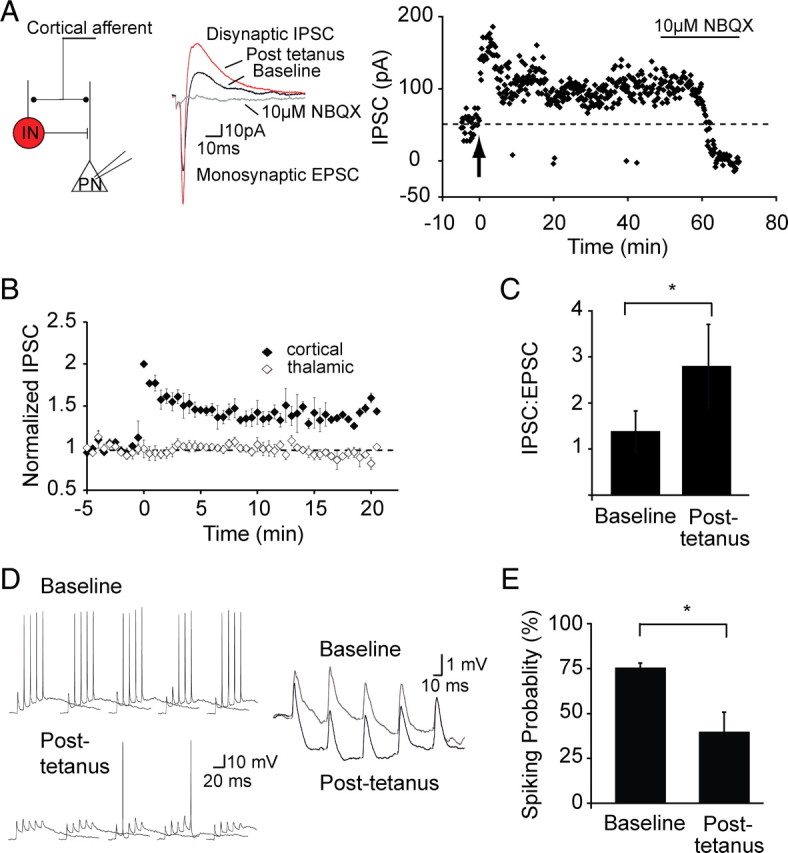
Inhibitory LTP leads to reduction of activity in principal neurons. A, Schematic showing the recording configuration. Whole-cell recordings were obtained from principal neurons (PN) that were voltage clamped at −40 mV. Stimulation of cortical inputs evokes a monosynaptic EPSC followed by a disynaptic IPSC as labeled. The graph on the right plots the peak IPSC amplitude over time in response to cortical stimulation in one principal neuron. Tetanic stimulation (arrow) results in potentiation of the IPSC. Application of the AMPA receptor antagonist NBQX blocks the IPSC, confirming it is disynaptic. B, Mean data (n = 6) showing that tetanic stimulation of cortical inputs leads to an input-specific potentiation of cortical disynaptic IPSC. C, Potentiation of the IPSC shifts the balance toward greater excitation, plotted in the mean peak IPSC/EPSC ratio at baseline (1.38 ± 0.44) and after LTP (2.80 ± 0.9, *p < 0.01). D, Current-clamp recording from a principal neuron. Repetitive stimulation of cortical input (50 Hz, 5 stimuli) generates an EPSP followed by an IPSP that summates and occasionally generates action potentials. An expanded version of the response to stimulation when no action potential evoked is shown on the right. After tetanic stimulation, the resulting disynaptic IPSP is larger and, as a result, fewer spikes are evoked. E, Mean data showing the reduction in probability of spiking after tetanic stimulation. *p < 0.05.
To test the impact of feedforward inhibition on principal cell excitability, current-clamp recordings were made from principal neurons, and cortical stimulation (five stimuli at 20 Hz) led to summation of both the EPSP and the IPSP and evoked action potentials with a spiking probability of 76 ± 2% (Fig. 9D,E). After tetanic stimulation, inhibition is enhanced, and the summating EPSP is shunted to a greater degree, reducing the ability of this input to drive neurons to threshold (Fig. 9D). As a result of this enhanced inhibition, the number of action potentials evoked was reduced, with the probability of spiking now at 40 ± 10% (p < 0.05 vs baseline; n = 4) (Fig. 9E).
Discussion
The amygdala, a temporal lobe structure, is intimately associated with the acquisition and storage of fear memory (LeDoux, 2000; Davis and Whalen, 2001). It is divided into a number of functionally distinct nuclei (Sah et al., 2003), of which the LA is the main input station for incoming sensory information (McDonald, 1998). Glutamatergic projection neurons are the major type of neuron found in the LA (McDonald, 1982), and plasticity (LTP) of inputs to these neurons is thought to underlie the acquisition and perhaps storage of fear memory (Pape and Pare, 2010). Locally projecting GABAergic interneurons form about 20% of the neuronal population within the LA (Sah et al., 2003), and their activity maintains a tight control over principal cell excitability (Li et al., 1996; Lang and Pare, 1997; Bissiere et al., 2003; Woodruff and Sah, 2007b). Interneurons receive excitatory inputs from the same sources as principal neurons and also show LTP (Mahanty and Sah, 1998; Bauer and LeDoux, 2004). Here we have shown LTP to interneurons is restricted to cortical inputs to a specific set of interneurons within the LA. This LTP is initiated by calcium influx through calcium-permeable AMPA receptors and is maintained by trafficking of GluR2-lacking AMPA receptors to the postsynaptic membrane. These interneurons provide feedforward inhibition to principal cells in the LA, and potentiation of excitatory input to these cells acts to limit the excitability of principal neurons.
As in the cortex and hippocampus (Markram et al., 2004), interneurons in the LA comprise diverse populations that can be separated by immunohistochemical markers, firing properties, and the connections they make (McDonald and Mascagni, 2001; Woodruff and Sah, 2007a). Our results show that interneurons in the LA can also be separated based on the types of NMDARs present at excitatory synapses they receive. We have shown three classes of interneuron: those that lack NMDARs, those that express NMDARs that primarily contain NR2A subunits, and those that express NMDARs primarily containing NR2B subunits. All interneurons receive both cortical and thalamic input, but LTP is restricted to cortical inputs to interneurons that do not contain NR2B subunits.
LTP of glutamatergic inputs has been studied extensively at Schaffer collateral synapses onto pyramidal neurons in area CA1 of the hippocampus. At these synapses, calcium influx via NMDARs triggers activation of CAMKII (calcium/calmodulin-dependent protein kinase II) and a cascade of second-messenger systems that leads to trafficking and insertion of new AMPA receptors into the postsynaptic density resulting in an increase in the EPSP (Malenka and Bear, 2004). This trafficking of AMPA receptors requires the presence of proteins that interact via PDZ-binding domains in the C-terminal tails of AMPA receptors (Malinow, 2003). Similar mechanisms have also been demonstrated for LTP of inputs to principal neurons in the LA (Rumpel et al., 2005). LTP has also been demonstrated at excitatory synapses on hippocampal interneurons (Kullmann and Lamsa, 2007). However, the mechanisms that underlie this potentiation are not well understood. For interneurons in the LA, we have shown that its induction requires a rise in postsynaptic calcium as it is blocked by the inclusion of the calcium buffer BAPTA in the recording solution. This rise in postsynaptic calcium does not require activation of NMDARs or release from intracellular stores. However, it is blocked when tetanic stimulation is delivered in the presence of the AMPA/kainate receptor antagonist kynurenic acid indicating that, as in several types of interneurons in the hippocampus, LTP is initiated by calcium influx through calcium-permeable AMPA receptors (Lei and McBain, 2002; Kullmann and Lamsa, 2007; Oren et al., 2009; Nissen et al., 2010).
We find that LTP on LA interneurons requires an interaction with a PDZ binding partner of GluR1, SAP97. SAP97 is a MAGUK (membrane associated guanylate kinase) known to specifically bind to the GluR1 subunit of AMPA receptors (Leonard et al., 1998), and its localization at synapses is maintained by interactions with the actin cytoskeleton (Rumbaugh et al., 2003). Consistent with this, LTP was absent when the actin cytoskeleton was disrupted with latrunculin B. Together, these results show that LTP at cortical inputs is initiated by a rise in postsynaptic calcium that leads to trafficking of GluR2-lacking AMPA receptors to synapses undergoing plasticity. These newly inserted AMPA receptors are likely maintained by an interaction with SAP97 and the actin cytoskeleton.
Three types of interneurons could be separated in the LA based on the contribution of NMDARs at excitatory synapses. One type of cell appears to lack postsynaptic NMDARs (Fig. 1) (Mahanty and Sah, 1998). The other two types of interneuron clearly have postsynaptic NMDARs but can be separated by the presence of NR2B subunits. We have shown that LTP is restricted to cortical inputs on cells that do not express NR2B subunits. LTP is input specific and requires a rise in postsynaptic calcium because of influx via calcium-permeable AMPA receptors that provides spatially confined rise in postsynaptic calcium (Goldberg et al., 2003). Why calcium influx via NMDARs at these synapses does not contribute to this process remains unknown. Cortical and thalamic inputs make synapses that appear to be very similar: they both express inwardly rectifying AMPA receptors, and the complement of NMDARs at the two inputs is identical. However, thalamic inputs to interneurons do not appear to show LTP. One possibility is that SAP97 expression may be restricted to synapses made by cortical inputs.
In the hippocampal formation, mossy fiber inputs to interneurons in CA3 stratum lucidum activate synapses that contain a mixture of calcium-permeable and calcium-impermeable AMPA receptors (Toth and McBain, 1998). Moreover, as for interneurons in the LA, these synapses display two types of NMDARs, those that mostly contain NR2A subunits and those that contain NR2B subunits (Lei and McBain, 2002). However, unlike in the LA, the distribution of different NMDAR subunits depends in part on the type of AMPA receptors present, with NR2B selectively present at synapses that are calcium permeable (Lei and McBain, 2002). Moreover, in CA3 interneurons, it has been suggested that both types of synapses may be mixed in the same interneuron. Interestingly, tetanic stimulation of synapses expressing calcium-permeable AMPA receptors leads to long-term depression that requires a rise in postsynaptic calcium but is NMDAR independent (Lei and McBain, 2002).
Functionally, LTP of cortical inputs to interneurons enhances feedforward IPSPs in local principal neurons, effectively reducing their output. The finding that cortically evoked disynaptic IPSPs also show LTP indicates that at least some of the interneurons showing LTP must act as feedforward interneurons. Fear memories are, at least in part, acquired by strengthening of synaptic inputs to principal neurons in the basolateral amygdala (LeDoux, 2000; Davis and Whalen, 2001), resulting in the increased response of these neurons to the conditioned stimulus (Quirk et al., 1997; Herry et al., 2008). An emerging body of evidence indicates that inhibitory circuits are engaged during both fear learning and extinction. Thus, enhancers of GABAergic inhibition interfere with fear learning and expression (Davis, 1979; Harris and Westbrook, 1998a), whereas reduction in GABAergic inhibition has an opposite effect (Tang et al., 2007). Extinction of conditioned fear had been studied extensively and has been proposed to result from potentiation of excitatory inputs to intercalated neurons with resultant inhibition of output from the central nucleus (Quirk and Mueller, 2008). However, unit recordings in vivo have shown that after extinction there is a concomitant reduction in the activity of neurons in the basolateral amygdala (Herry et al., 2008). Moreover, extinction of conditioned fear is impaired by reduction in GABAergic transmission in the basolateral amygdala (Harris and Westbrook, 1998b). These reports, together with the present findings, suggest that reduction in the activity of BLA neurons after fear extinction may be mediated by potentiation of cortical feedforward inhibition.
Footnotes
This work was supported by grants from the Australian National Health and Medical Research Council and the Australian Research Council. We thank members of the Sah laboratory, Rowan Tweedale, and Luli Faber for comments on this manuscript.
References
- Bauer EP, LeDoux JE. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci. 2004;24:9507–9512. doi: 10.1523/JNEUROSCI.3567-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissiere S, Humeau Y, Luthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Headley PM, Tzschentke TM. NMDA receptor antagonists as analgesics: focus on the NR2B subtype. Trends Pharmacol Sci. 2001;22:636–642. doi: 10.1016/s0165-6147(00)01863-0. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Davis M. Diazepam and flurazepam: effects on conditioned fear as measured with the potentiated startle paradigm. Psychopharmacology (Berl) 1979;62:1–7. doi: 10.1007/BF00426027. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Goldberg JH, Tamas G, Aronov D, Yuste R. Calcium microdomains in aspiny dendrites. Neuron. 2003;40:807–821. doi: 10.1016/s0896-6273(03)00714-1. [DOI] [PubMed] [Google Scholar]
- Harris JA, Westbrook RF. Evidence that GABA transmission mediates context-specific extinction of learned fear. Psychopharmacology (Berl) 1998a;140:105–115. doi: 10.1007/s002130050745. [DOI] [PubMed] [Google Scholar]
- Harris JA, Westbrook RF. Benzodiazepine-induced amnesia in rats: reinstatement of conditioned performance by noxious stimulation on test. Behav Neurosci. 1998b;112:183–192. doi: 10.1037//0735-7044.112.1.183. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Jonas P, Burnashev N. Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron. 1995;15:987–990. doi: 10.1016/0896-6273(95)90087-x. [DOI] [PubMed] [Google Scholar]
- Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- Kondo M, Okabe S, Sumino R, Okado H. A high GluR1:GluR2 expression ratio is correlated with expression of Ca2+-binding proteins in rat forebrain neurons. Eur J Neurosci. 2000;12:2812–2822. doi: 10.1046/j.1460-9568.2000.00167.x. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–699. doi: 10.1038/nrn2207. [DOI] [PubMed] [Google Scholar]
- Laezza F, Dingledine R. Voltage-controlled plasticity at GluR2-deficient synapses onto hippocampal interneurons. J Neurophysiol. 2004;92:3575–3581. doi: 10.1152/jn.00425.2004. [DOI] [PubMed] [Google Scholar]
- Laezza F, Doherty JJ, Dingledine R. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- Lang EJ, Pare D. Similar inhibitory processes dominate the responses of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ. Distinct NMDA receptors provide differential modes of transmission at mossy fiber-interneuron synapses. Neuron. 2002;33:921–933. doi: 10.1016/s0896-6273(02)00608-6. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Li XF, Armony JL, LeDoux JE. GABAA and GABAB receptors differentially regulate synaptic transmission in the auditory thalamo-amygdala pathway: an in vivo microiontophoretic study and a model. Synapse. 1996;24:115–124. doi: 10.1002/(SICI)1098-2396(199610)24:2<115::AID-SYN3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci. 2003;23:6876–6883. doi: 10.1523/JNEUROSCI.23-17-06876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Excitatory synaptic inputs to pyramidal neurons of the lateral amygdala. Eur J Neurosci. 1999;11:1217–1222. doi: 10.1046/j.1460-9568.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Malinow R. AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:707–714. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour M, Nagarajan N, Nehring RB, Clements JD, Rosenmund C. Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron. 2001;32:841–853. doi: 10.1016/s0896-6273(01)00520-7. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Neurons of the lateral and basolateral amygdaloid nuclei: a golgi study in the rat. J Comp Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cell types and intrinsic connections of the amygdala. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory and mental dysfunction. New York: Wiley; 1992. pp. 67–96. [Google Scholar]
- McDonald AJ. Localization of AMPA glutamate receptor subunits in subpopulations of non-pyramidal neurons in the rat basolateral amygdala. Neurosci Lett. 1996;208:175–178. doi: 10.1016/0304-3940(96)12585-4. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Brain Res. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Betette RL. Parvalbumin-containing neurons in the rat basolateral amygdala: morphology and co-localization of Calbindin-D(28k) Neuroscience. 2001;102:413–425. doi: 10.1016/s0306-4522(00)00481-4. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F. Colocalization of calcium-binding proteins and GABA in neurons of the rat basolateral amygdala. Neuroscience. 2001;105:681–693. doi: 10.1016/s0306-4522(01)00214-7. [DOI] [PubMed] [Google Scholar]
- Nissen W, Szabo A, Somogyi J, Somogyi P, Lamsa KP. Cell type-specific long-term plasticity at glutamatergic synapses onto hippocampal interneurons expressing either parvalbumin or CB1 cannabinoid receptor. J Neurosci. 2010;30:1337–1347. doi: 10.1523/JNEUROSCI.3481-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren I, Nissen W, Kullmann DM, Somogyi P, Lamsa KP. Role of ionotropic glutamate receptors in long-term potentiation in rat hippocampal CA1 oriens-lacunosum moleculare interneurons. J Neurosci. 2009;29:939–950. doi: 10.1523/JNEUROSCI.3251-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Sia GM, Garner CC, Huganir RL. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci. 2003;23:4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sah P, Westbrook RF. Behavioural neuroscience: the circuit of fear. Nature. 2008;454:589–590. doi: 10.1038/454589a. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, Barbieri S, van der Putten H, Kaupmann K, Bettler B, Luthi A. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat Neurosci. 2006;9:1028–1035. doi: 10.1038/nn1732. [DOI] [PubMed] [Google Scholar]
- Smith Y, Pare JF, Pare D. Cat intraamygdaloid inhibitory network: ultrastructural organization of parvalbumin-immunoreactive elements. J Comp Neurol. 1998;391:164–179. doi: 10.1002/(sici)1096-9861(19980209)391:2<164::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins–novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szinyei C, Heinbockel T, Montagne J, Pape HC. Putative cortical and thalamic inputs elicit convergent excitation in a population of GABAergic interneurons of the lateral amygdala. J Neurosci. 2000;20:8909–8915. doi: 10.1523/JNEUROSCI.20-23-08909.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szinyei C, Stork O, Pape HC. Contribution of NR2B subunits to synaptic transmission in amygdaloid interneurons. J Neurosci. 2003;23:2549–2556. doi: 10.1523/JNEUROSCI.23-07-02549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tang HH, McNally GP, Richardson R. The effects of FG7142 on two types of forgetting in 18-day-old rats. Behav Neurosci. 2007;121:1421–1425. doi: 10.1037/0735-7044.121.6.1421. [DOI] [PubMed] [Google Scholar]
- Toth K, McBain CJ. Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat Neurosci. 1998;1:572–578. doi: 10.1038/2807. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Sprouffske K, Westbrook GL. Fast NMDA receptor-mediated synaptic currents in neurons from mice lacking the epsilon2 (NR2B) subunit. J Neurophysiol. 2000;83:616–620. doi: 10.1152/jn.2000.83.1.616. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW, Weinberg RJ. SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur J Neurosci. 2000;12:3605–3614. doi: 10.1046/j.1460-9568.2000.00256.x. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol Biochem Behav. 2002;71:379–392. doi: 10.1016/s0091-3057(01)00698-0. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Numberger M, Zhang S, Dingledine R. Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J Neurosci. 1997;17:9393–9406. doi: 10.1523/JNEUROSCI.17-24-09393.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- Woodruff AR, Sah P. Networks of parvalbumin-positive interneurons in the basolateral amygdala. J Neurosci. 2007a;27:553–563. doi: 10.1523/JNEUROSCI.3686-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, Sah P. Inhibition and synchronization of basal amygdala principal neuron spiking by parvalbumin-positive interneurons. J Neurophysiol. 2007b;98:2956–2961. doi: 10.1152/jn.00739.2007. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]