

Abstract
Plasmalemmal repair is necessary for survival of damaged eukaryotic cells. Ca2+ influx through plasmalemmal disruptions activates calpain, vesicle accumulation at lesion sites, and membrane fusion proteins; Ca2+ influx also initiates competing apoptotic pathways. Using the formation of a dye barrier (seal) to assess plasmalemmal repair, we now report that B104 hippocampal cells with neurites transected nearer (<50 μm) to the soma seal at a lower frequency and slower rate compared to cells with neurites transected farther (>50 μm) from the soma. Analogs of cAMP, including protein kinase A (PKA)-specific and Epac-specific cAMP, each increase the frequency and rate of sealing and can even initiate sealing in the absence of Ca2+ influx at both transection distances. Furthermore, Epac activates a cAMP-dependent, PKA-independent, pathway involved in plasmalemmal sealing. The frequency and rate of plasmalemmal sealing are decreased by a small molecule inhibitor of PKA targeted to its catalytic subunit (KT5720), a peptide inhibitor targeted to its regulatory subunits (PKI), an inhibitor of a novel PKC (an nPKCη pseudosubstrate fragment), and an antioxidant (melatonin). Given these and other data, we propose a model for redundant parallel pathways of Ca2+-dependent plasmalemmal sealing of injured neurons mediated in part by nPKCs, cytosolic oxidation, and cAMP activation of PKA and Epac. We also propose that the evolutionary origin of these pathways and substances was to repair plasmalemmal damage in eukaryotic cells. Greater understanding of vesicle interactions, proteins, and pathways involved in plasmalemmal sealing should suggest novel neuroprotective treatments for traumatic nerve injuries and neurodegenerative disorders.
Introduction
Traumatic injury to any eukaryotic cell, including neurons, inevitably produces plasmalemmal damage, and rapid repair (sealing) of this damage is necessary for cell survival (Schlaepfer, 1973; Fishman and Bittner, 2003). Ca2+ influx through plasmalemmal disruptions initiates processes that lead to cell death, often via apoptosis (Schlaepfer and Bunge, 1973; Nguyen et al., 2005). However, such Ca2+ influx also leads to competing processes that activate plasmalemmal sealing pathways (Fishman and Bittner, 2003), sometimes before the internal Ca2+ concentration becomes toxic (Nguyen et al., 2005). Sealing is especially important for adult neurons, since they typically cannot proliferate. Thus, increasing the rate of sealing may increase the probability of cell survival following plasmalemmal damage and, for neurons, may increase the likelihood of axonal regeneration.
Confocal and EM images show that giant invertebrate axons seal plasmalemmal damage by Ca2+-dependent production of vesicles that form a plug, often at a partially constricted cut end (Krause et al., 1994). After plasmalemmal disruption, vesicles from nearby undamaged membrane (Eddleman et al., 1997, 1998), lysosomes (Reddy et al., 2001), and/or myelin delamination (Ballinger et al., 1997) migrate, accumulate, and pack tightly at the damage site. These vesicles interact with each other and undamaged membrane to reduce the influx of extracellular Ca2+ and other ions until a seal is restored. Complete plasmalemmal repair takes approximately 24 h for earthworm giant axons, at which time vesicles are no longer observed at the original lesion site and a continuous plasmalemma is restored (Lichstein et al., 2000). Similar micromorphological images are difficult to obtain from small-diameter unmyelinated or myelinated mammalian axons or other mammalian cell types. Although many proteins and molecules influence plasmalemmal sealing, to date no study has described possible molecular pathways involved in plasmalemmal sealing.
To investigate pathways of plasmalemmal sealing, we transected neurites of individual B104 hippocampal cells and assessed repair (sealing) of the damaged plasmalemma by formation of a barrier to extracellular dye. Cells transected <50 μm (nearer) from the soma always sealed at a slower rate compared to cells transected >50 μm (farther) from the soma. Increasing cAMP concentration or specifically activating protein kinase A (PKA) or Epac via target-specific cAMP analogs all produced similar increases in plasmalemmal sealing. Furthermore, all three cAMP analogs increased sealing in the absence of Ca2+, suggesting that cAMP, PKA, and Epac act downstream of Ca2+ to induce plasmalemmal sealing. Plasmalemmal sealing is decreased by inhibiting PKA or novel protein kinase Cη (nPKCη) or by an antioxidant (melatonin). These and other data have enabled us to develop a model of plasmalemmal sealing that involves redundant, parallel pathways initiated by Ca2+ influx. Because in vivo, ex vivo, and in vitro biochemical and dye exclusion data from many different preparations and many diverse phyla, including B104 cells, show that isomers of the same proteins and processes are likely involved in plasmalemmal sealing (see supplemental list, available at www.jneurosci.org as supplemental material), we suggest that the evolutionary origin of such proteins in eukaryotes was to repair plasmalemmal damage. Therefore, identifying plasmalemmal sealing pathways in neurons may provide insights into sealing in other cell types.
Materials and Methods
B104 cells.
B104 cells derived from a CNS neuroblastoma (Bottenstein and Sato, 1979) have often been used as a model system to study neuronal function in vitro (Toda et al., 1999; Tan et al., 2003; Yoo et al., 2003, 2004; Nguyen et al., 2005; Miller et al., 2006). B104 cells extend neurites that have properties typical of nerve axons such as generation of action potentials, smooth (but not rough) endoplasmic reticulum, release of neurotransmitter, and regeneration of severed neurites. These cells have easily identifiable cell bodies and neurites, allowing precise identification of individually damaged cells at their injury site (Detrait et al., 2000b; Yoo et al., 2003, 2004). Unlike some other model neuronal cell lines (e.g., PC12 cells), B104 cells do not require growth factor supplements to fetal bovine serum to proliferate (Detrait et al., 2000b; Yoo et al., 2003). Data on sealing of B104 cells are consistent with similar data on sealing from at least 20 other preparations from many phyla and different cell types (see supplemental list, available at www.jneurosci.org as supplemental material).
Cell culture.
As described previously (Nguyen et al., 2005), B104 cells were grown in 75 cm2 vented cap flasks (BD Falcon; BD Biosciences) in a humidified incubator at 37°C in 5% CO2 and in 4 ml of “cell growth medium: consisting of a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 (DMEM:F-12, HyClone) supplemented for growth with 10% heat-inactivated fetal bovine serum (FBS; HyClone) and 1% antibiotics (10,000 U of penicillin/ml and 10 mg/ml streptomycin; Sigma-Aldrich). The cell growth medium was changed every 2 d. Cultures were passaged at 80% confluency, and cells were then either subcultured in a vented cap flask or seeded at ∼2000 cells/cm2 in cell growth media on Petri dishes coated with poly-d-lysine (Sigma-Aldrich) to prevent cells from detaching during solution changes and/or neurite transection. After 24 h, the growth media was replaced with serum-free DMEM:F12 (Hyclone) to allow the B104 cells to differentiate. B104 neurites were transected 24–48 h after replacing the cell growth media with serum-free DMEM:F12.
Transection of neurites of B104 cells.
Before transecting neurites, the solution (DMEM:F12) in the Petri dish was washed out twice with a Ca2+-free phosphate- buffered saline (referred to as “Ca2+-free saline,” PBS−/−; HyClone). All neurites were transected in Ca2+-free saline using a broken, pulled glass, microcapillary tube (“micro-knife”) that was placed on a micromanipulator (Narishige Instruments) and quickly drawn across the surface of the Petri dish, etching a score line that showed the path of the knife. We were able to uniquely and individually identify each transected cell by the relation of the transected neurite to its soma and to the score mark on the plate (see Fig. 1). In some experiments, 3 kDa fluorescein dextran (Invitrogen) was added to the Ca2+-free saline before transecting neurites to confirm that the score line could be reliably used to uniquely identify individual cells.
Figure 1.

A–F, Bright-field (A, D) and fluorescence (B, C, E, F) images typical of those used to assess dye uptake or exclusion by B104 cells having a transected neurite. Bright-field images (A, D) were taken after transecting a neurite, marked by a score line [transection line (T- - - -L)], in Ca2+-free saline containing fluorescein dextran. After 10 min in Ca2+-free saline, the cells were bathed in Ca2+-saline for 5 min, at which time Texas Red dextran was added to the Ca2+-saline. The cells were then imaged for fluorescein (B, E) and Texas Red (C, F) emission. The presence of fluorescein emission in B and E (cells marked with asterisk) shows that these cells took up fluorescein dye through a damaged plasmalemma (transected neurite). The presence of Texas Red emission (C) shows that this cell (marked with asterisk) did not exclude dye and therefore did not form a plasmalemmal seal. The absence of Texas Red emission (F) shows that this cell (location of cell marked with asterisk) excluded dye and therefore had formed a plasmalemmal seal. The uninjured cell (indicated by arrows, A–C) did not take up either dye and therefore shows no emission. Scale bars in all panels represent 50 μm.
Microscopy and imaging.
For all experiments, B104 cells were observed under an inverted Zeiss ICM-405 fluorescent microscope with a 40× long focal distance lens. Transected cells were imaged through a hole cut out of the bottom of a plastic dish covered by a thin glass coverslip using a Leica DM IRBE outfitted with a 40× lens and a Leica DFC350 FX fluorescence camera at the University of Texas core microscopy facility. Cells with neurites obviously transected <50 μm from the soma were counted as transected “nearer to” the soma, and those with neurites transected >50 μm from the soma were counted as transected “farther from” the soma. No further observations were made on any cell whose transection distance was not clearly definable.
Assessment of plasmalemmal sealing.
We used the most reliable measure of plasmalemmal repair (dye exclusion) to assay plasmalemmal sealing (Blanchette et al., 1999; Detrait et al., 2000a,b). Other measures of sealing, such as intracellular recordings of membrane potential or input resistance (Krause et al., 1995), vibrating probe measurements of cell currents (Eddleman et al., 2000), preloading cells with Ca2+-sensitive dyes, or fluorescence extinction of membrane-bound dyes (Togo et al., 2003), all have much more ambiguous interpretations as measures of plasmalemmal sealing; electron microscopic images in any preparation have insufficient resolution to assess the status of plasmalemmal repair (Fishman and Bittner, 2003).
To assess plasmalemmal repair, we first transected 10–130 cells in Ca2+-free saline within 10 min. The time elapsed after a neurite is transected in Ca2+-free saline is defined as the post-transection time (PT time). The Ca2+-free saline was then replaced with a phosphate-buffered saline containing 1 mm Ca2+ (“Ca2+-saline.” PBS+/+; HyClone) to initiate the sealing process (Detrait et al., 2000b; Yoo et al., 2003). The time elapsed after exposing cells to Ca2+-saline is defined as the “post-Ca2+ addition time” (PC time). Thus, unless otherwise noted, all cells on a given Petri dish have the same PC time, but each cell has a different PT time.
At various PC times (0–60 min PC), 3 kDa Texas Red dextran (Invitrogen) was added to the Ca2+-saline to assess the formation of a plasmalemmal seal. At 0 min PC, Texas Red dextran was typically added at the same time as Ca2+-saline to investigate whether cells with transected neurites excluded dye before Ca2+ addition. For all experiments, the dye was thoroughly washed out with Ca2+-saline after a 10 min exposure to Texas Red dextran, and was sealing assessed. Transected cells that excluded Texas Red dextran were counted as “sealed” (Fig. 1C). Cells that did not exclude Texas Red dextran were counted as “not sealed” (Fig. 1F). Nearby undamaged cells did not take up Texas Red dextran (Fig. 1A,C, arrows).
Previous studies of sealing using dye exclusion in many preparations have used fluorescent dyes of various molecular weights (600 Da–10 kDa) and showed that higher molecular weight dyes are excluded faster than lower molecular weight dyes (Detrait et al., 2000a; Yoo et al., 2003), which are excluded faster than ions having even lower molecular weights than the smallest dye (Krause et al., 1994; Eddleman et al., 2000). We confirmed that Texas Red dyes of higher molecular weights (10 kDa) are excluded from B104 cells with transected neurites faster than dyes of lower molecular weight. For example, cells transected nearer to the soma and assessed for sealing at 5 min PC with 10 kDa Texas Red dextran (108 cells, 2 Petri dishes) sealed at a significantly [p < 0.001, Cochran-Mantel-Haenszel (CMH) χ2 test)] higher frequency (80%) than cells assessed for sealing with 3 kDa Texas Red dextran (272 cells, 15 Petri dishes; 45%). To avoid variation in observed sealing frequency or time due to the molecular weight of the dye, we consistently used 3 kDa Texas Red dextran to assess sealing in all other experiments reported herein.
Pharmacological reagents.
We used pharmacological agents to quickly inhibit or activate a target protein to avoid complications of compensatory pathways that may result from gene knock-outs or chronic applications of pharmacological inhibitors (Steinberg, 2008). All pharmacological reagents were typically dissolved in distilled water and added to Ca2+-free saline immediately before neurites were transected to backload cells with each reagent during transection. Dibutyryl-cAMP (db-cAMP, 1 mm, 491.37 Da; Sigma-Aldrich) was used to increase intracellular cAMP concentration. Two structurally different, small molecule inhibitors of PKA were used to verify PKA inhibition: KT5720 (1 mm, 537.6 Da; AG Scientific) and PKI (50 μm, 1868 Da; Sigma-Aldrich; a generous gift from Drs. Michael Markham and Harold Zakon, University of Texas, Austin, TX). PKI is very specific for PKA (Dalton and Dewey, 2006). Sp-5,6-Dichloro-1–d-ribofuranosylbenzimidazole-3′,5′-monophosphorothioate (8 μm), referred to as cBiMPS (419.2 Da; Biomol), was used to specifically activate PKA and not other cAMP targets (Christensen et al., 2003). 8-(4-Chlorophenylthio)-2′-O-methyladenosine-cAMP (8 μm), referred to as Epac-cAMP (507.82 Da; Tocris Biosciences), was used to activate Epac and not other cAMP targets (Enserink et al., 2002; Christensen et al., 2003). Melatonin (2 mm; Sigma-Aldrich) was used to decrease cytosolic oxidation following plasmalemmal damage. An nPKCη pseudosubstrate fragment (7 μm, 2 kDa; EMD Chemicals) was used to specifically inhibit nPKCη (Liu et al., 2006; Barros et al., 2009).
Statistical analyses.
We obtained data from individual, uniquely identified B104 cells that all received similar, well-specified, neurite transections as plasmalemmal injuries in a tightly controlled environment. [Other studies of plasmalemmal sealing based on the averaged response of a population of different cell types that receive unknown or variable injuries almost always present statistical measures of a population of indistinguishable cells, each with an unspecified injury, to obtain data that often have more ambiguous interpretations (Mellgren et al., 2007).] For each experimental treatment group, at a given PC time the data were pooled for all cells (n) from all Petri dishes (N). See supplemental Table 1, A and B (available at www.jneurosci.org as supplemental material), for n and N values for each data set at each PC time point in all figures presented in this paper. “Sealing frequency” is defined as the percentage of a set of individually-transected and uniquely identified cells that exclude 3 kDa Texas Red dye (sealed) at a given PC time. The CMH χ2 test for independence was used to determine whether the differences between the sealing frequency at a given PC time for different experimental treatments was statistically significant (p < 0.05), as described previously (Agresti, 1996; Detrait et al., 2000b; Yoo et al., 2003, 2004).
GraphPad Prism was used to fit the sealing frequency for all PC times of a given control or test substance to a one-phase exponential equation,
where y(t) is the sealing frequency (%) of cells examined at a given PC time (t), Ymax is the maximum plateau sealing frequency (%) of cells for that control or test substance, e is Euler's constant, t is a given PC time (min), and k is the rate constant (min−1) of a given exponential equation. The rate constant k is the reciprocal of the time constant (τ, min) defined as the elapsed PC time needed to achieve 63.2% (reciprocal of Euler's constant) of the observed maximum (plateau) sealing.
The solid and dashed lines on all graphs were calculated by GraphPad Prism according to Eq. 1 and represent the exponential equations fitted to the sealing frequencies at all PC times for all data sets. The time constants were calculated according to Eq. 1 and τ = 1/k. The maximum sealing frequency (plateau) was always reached within 60 min PC.
Sealing time constants or rate constants for two experimental conditions were normalized and compared by using Fisher's Z transformation (FZT). Two sealing constants were considered significantly different if p was <0.05, using a Z table. We also calculated R2 values for the exponential equation defining each time constant to determine how closely the exponential equations modeled the observed data (see Table 2).
Table 2.
Effect of test substances on the time constant (τ) and rate constant (k) of sealing
| Test substance | <50 μm τ (min) | <50 μm k (min−1) | R2 < 50 μm | > 50 μm τ (min) | >50 μm k (min−1) | R2 > 50 μm |
|---|---|---|---|---|---|---|
| Control | 5.95 | 0.168 | 0.9865 | 2.16 | 0.461 | 0.9970 |
| db-cAMP | 1.98** | 0.503** | 0.9877 | 0.622** | 1.61** | 0.9976 |
| db-cAMP (no Ca2+) # | 11.62*** | 0.086*** | 0.9455 | 3.98** | 0.251** | 0.9877 |
| cBiMPS | 1.24** | 0.805** | 0.9413 | 0.497** | 2.01** | 0.8231 |
| Epac-cAMP | 2.89** | 0.345** | 0.9141 | 0.628** | 1.59** | 0.7933 |
| DMSO | 7.59 | 0.131 | 0.9798 | 2.15 | 0.461 | 0.9856 |
| KT5720 | 20.1*** | 0.049*** | 0.9769 | 9.91*** | 0.10*** | 0.9769 |
| PKI | 25.9*** | 0.038*** | 0.9972 | 10.7*** | 0.093*** | 0.9676 |
| Epac-cAMP plus PKI | 7.37 | 0.138 | 0.9795 | 3.35 | 0.298 | 0.9404 |
| nPKCη PSF | 6.59** | 0.152*** | 0.9618 |
Statistical comparisons of rate constants (k, min−1) or exponential time constants (τ, min) for sealing of B104 cells transected nearer to (<50 μm) or farther from (>50 μm) the soma. Cells transected nearer to the soma and bathed in a given test substance always sealed at a slower rate compared to cells transected farther from the soma and bathed in the same test substance. R2 < 50 μm or R2 > 50 μm represents the goodness of fit values obtained from a single exponential model to fit the sealing frequency at all PC times for control sealing or for sealing in a given test substance. Asterisks indicate the level of significant difference between sealing time constants or rate constants for a given test substance compared to control sealing. Significance levels for this and all other tables and figures are indicated beside each value as follows: no asterisk = p > 0.05 and p < 0.95; single asterisk (*) = p < 0.05; double asterisk (**) = p < 0.01; and triple asterisk (***) = p < 0.001.
#Initial increase in sealing frequency.
Validation of our single dye assessment method for plasmalemmal sealing.
We confirmed that we can reliably identify individual B104 cells with transected neurites by their relationship to the score mark on a Petri dish by adding 3 kDa fluorescein dextran to the Ca2+-free saline before transecting a set of neurites. We then added 3 kDa Texas Red dextran at 0 min PC (i.e., after neurite transection) to assess sealing. As reported previously (Yoo et al., 2003, 2004; Nguyen et al., 2005), cells with transected neurites identified by their relationship to the score mark were always filled with fluorescein dextran (Fig. 1B,E), whether or not they excluded Texas Red dextran. Nearby undamaged cells did not take up fluorescein dextran or Texas Red dextran (i.e., these cells had an undamaged plasmalemma) (Fig. 1A–C, arrows).
We observed no significant (p > 0.05, CMH χ2) difference in the percentage of cells that excluded Texas Red dextran (sealing frequency) identified as transected because they contained fluorescein dextran at 0 min PC (0%, 45 cells from 2 dishes) compared to the percentage of cells that excluded dye identified as transected solely by their relation to the score mark (0%, 131 cells from 4 dishes). That is, we could reliably identify transected cells by their relationship to the score mark without having to use a second fluorescent dye (fluorescein dextran) to confirm that those neurites were indeed transected.
As further confirmation of the validity of our single-dye assessment technique, when transection distance was not taken into account, the percentage of cells that sealed in the present study at 5, 20, and 60 min PC was not significantly (p > 0.05, CMH χ2) different than that in previous studies in which fluorescein dextran was used as an additional indicator of cells that were transected, but transection distance was not noted (Yoo et al., 2003; 2004; Nguyen et al., 2005). Hence, in all protocols described below, we used a single dye (Texas Red dextran) to assess dye exclusion as a measure of plasmalemmal sealing of transected B104 neurites. This single dye assessment technique allowed us to transect the neurites of a greater number of uniquely identified cells within 10 min on a single Petri dish and to more rapidly assess their ability to seal.
Results
In all experiments, cells were transected in Ca2+-free saline for 10 min, at which time the solution in the dish (Ca2+-free saline) was replaced with Ca2+-saline (1 mm Ca2+). Any experimental compounds were added to the Ca2+-free saline during neurite transection. At various times post-Ca2+ addition (PC time), dye was added and sealing was typically assessed after 10 min of dye exposure.
PC time determines plasmalemmal sealing
The time at which a cell is transected during the 10 min transection period, PT time, does not affect the sealing frequency of B104 cells, while the time elapsed since Ca2+ addition determines sealing frequency. For a given transection distance, cells with longer PT times, up to 10 min PT, did not seal at a significantly (p > 0.05, CMH χ2) different frequency compared to cells with shorter PT times (Table 1). These and all other data in Table 1 confirmed that the frequency and rate of sealing in B104 cells was determined by PC time and not the time of neurite transection, as was previously reported for this and other preparations (Blanchette et al., 1999; Detrait et al., 2000b; Yoo et al., 2004). Hence, unless otherwise stated, all measures of sealing by B104 cells in this article are given in PC times.
Table 1.
PC time, not PT time or dye exposure time, determines sealing time of B104 cells
| PT time (min) | PC time (min) | Dye exposure time (min) | Sealing frequency <50 μm (n, N) | Sealing frequency >50 μm (n, N) |
|---|---|---|---|---|
| 1–2 | 5 | 10 | 48% (38, 2) | 92% (25, 2) |
| 9–10 | 5 | 10 | 52% (50, 2) | 88% (26, 2) |
| 1–2 | 5 | 2 | 48% (42, 2) | 94% (18, 2) |
| 9–10 | 5 | 2 | 46% (46, 2) | 91% (22, 2) |
PT time is post-transection time (min). PC time is the time (min), after adding Ca2+-saline, when Texas Red dextran was added to the bath solution. Dye exposure time is time (min) when B104 cells were bathed in Ca2+-saline that contained Texas Red dextran. Sealing frequency is percentage of individually damaged, uniquely identifiable B104 cells with neurites transected <50 μm or >50 μm from the cell body that excluded Texas Red dextran. Numbers in parentheses (n,N) are the total number (n) of B104 cells with a transected neurite examined in a set (N) of identically treated Petri dishes. The sealing frequency of B104 cells was significantly different when transection distance or PC time was varied, but not when PT time or dye exposure time was varied.
The data in Table 1 also show that the sealing frequency does not depend on the length of time cells are exposed to Texas Red dextran in Ca2+-saline before sealing is assessed. For example, bathing cells in Texas Red dextran for 2 or 10 min did not affect the sealing frequency at 5 min PC, suggesting that Texas Red dextran very rapidly entered and filled all transected cells after the dye was added to Ca2+-saline. Thus, if a cell excludes dye at a given PC time, it is a reliable indicator that the cell had sealed before dye addition. Since dye exposure time did not affect the observed sealing frequency, in all other experiments below we exposed cells to Texas Red dextran for 10 min to more brightly label the score marks on the plastic Petri dishes.
We also examined whether sealing frequency was affected by the time at which cells were imaged after the dye was washed out. We transected cells as described above and added Texas Red dextran at 5 min PC. We then maintained the cells in Ca2+-saline-containing dye for 10 min on one Petri dish, after which cells were immediately imaged and the sealing frequency (48%, 30 cells) was noted. On another Petri dish, we imaged the cells at 60 min after the dye was washed out and the sealing frequency was again noted (49%, 25 cells). There was no significant (p > 0.95, CMH χ2) difference in the sealing frequency between cells on these two Petri dishes. Furthermore, when we reexamined the first Petri dish after 60 min, the same cells continued to exclude dye and the percentage of cells that excluded dye remained unchanged (48%, 30 cells). Therefore, the time at which cells are imaged after dye is washed out (up to 60 min PC) does not affect sealing frequency.
To confirm that sealing was initiated by Ca2+ influx at 0 min PC rather than by any residual Ca2+ remaining in the Ca2+-free saline or released from nearby cells, we transected B104 cells in 0.5 mm EGTA. This EGTA concentration reduces extracellular Ca2+ to <10−6 m, which is too low to initiate plasmalemmal sealing (Yoo et al., 2003) but not low enough to produce plasmalemmal damage for up to 10 min, since cells without transected neurites (undamaged cells) did not take up Texas Red dextran after 10 min of exposure to 0.5 mm EGTA. Cells transected nearer to or farther from the soma in 0.5 mm EGTA did not have a significantly different sealing frequency compared to control sealing at 5 min PC (p > 0.05, CMH χ2, data not shown). Considering all our data, when neurites are transected in Ca2+-free saline and subsequently exposed to Ca2+-saline-containing dye, control sealing frequency is determined by PC time and not by PT time, the time of dye exposure, the time at which cells are observed following dye washout, or by the residual Ca2+ remaining in the Petri dish following solution changes.
Plasmalemmal sealing depends upon transection distance from the soma
We observed a significantly lower control sealing frequency for cells with neurites transected nearer to the soma (<50 μm) compared to cells transected farther from the soma (>50 μm) at all PC times between 1 and 10 min PC (p < 0.01 to p < 0.001) (Fig. 2A). Control sealing was complete between 5 and 10 min PC for cells transected farther from the soma and by 20 min PC for cells transected nearer to the soma. Furthermore, control sealing of cells transected nearer to the soma had a significantly slower rate (longer time constant) compared to cells transected farther from the soma (p < 0.001, FZT) (Table 2). As described in subsequent sections, cells transected nearer to the soma always sealed at a significantly lower frequency and a significantly slower rate compared to cells transected farther from the soma for all test substances used in this study (Table 2). R2 values in Table 2 also show that a single exponential equation very closely models the observed sealing data for control or test substances.
Figure 2.
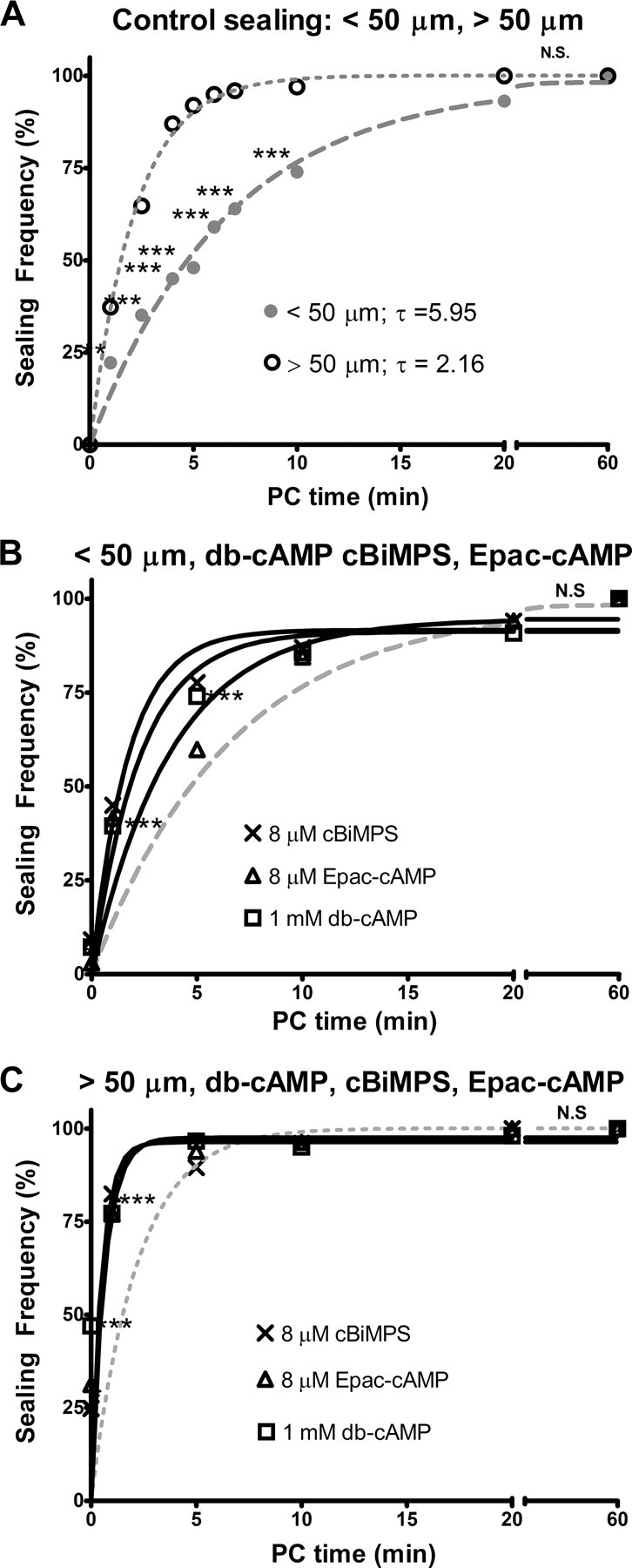
A–C, Sealing frequency (%) plotted versus PC time (min). Sealing frequency increases as PC time increases following neurite transection nearer to (<50 μm; A, B) or farther from (>50 μm; A, C), the soma. In this and subsequent figures, each line represents the sealing frequency for control sealing (dotted or dashed lines) or test substances (solid lines) fitted to Eq. 1 by GraphPad Prism (see Materials and Methods). A, Control sealing for neurites transected nearer to (dashed gray line, filled circles) or farther from (dotted gray line, open circles) the soma in Ca2+-free saline and then exposed to Ca2+ at 0 min PC. B, C, Neurites transected in 1 mm db-cAMP (open squares), 8 μm cBiMPS (crosses), or 8 μm Epac-cAMP (open triangles) in Ca2+-free saline and then exposed to Ca2+at 0 min PC. In A–C, the x-axis is interrupted between 20 and 60 min PC so that differences in sealing frequencies from 0 to 20 min PC can more easily be observed. In B and C and subsequent figures, control sealing curves from Fig. 3A are replotted as dashed (<50 μm) or dotted (>50 μm) lines without data points to compare the effects of test substances (e.g., db-cAMP) to control sealing. The total number of cells transected to obtain each data point (n) varied from 40 to 403 on a total number (N) of Petri dishes (2–15). These numbers (n, N) are given in supplemental Table 1, A and B, available at www.jneurosci.org as supplemental material for all figures. N.S., No significant difference, p > 0.95.
Increasing cAMP, PKA activity, or Epac activity increases frequency and rate of sealing
Because cAMP, PKA, and Epac are involved in vesicle mediated processes (Hatakeyama et al., 2007) and axon regeneration (Cai et al., 2001; Chierzi et al., 2005; Murray and Shewan, 2008), we hypothesized that Ca2+ influx through a damaged plasmalemma increases cAMP concentration, which activates PKA and Epac and leads to repair of plasmalemmal damage. (This hypothesis implies that PKA and Epac act downstream of cAMP, which acts downstream of Ca2+ during plasmalemmal sealing.)
We first examined the effects of increased cAMP concentration by transecting cells in 1 mm db-cAMP, a membrane-permeable analog of cAMP (Qiu et al., 2002). The sealing frequency following neurite transection nearer to the soma at 0, 1, and 5 min PC was significantly increased by 1 mm db-cAMP compared to control sealing (p < 0.01, CMH χ2) (Fig. 2B). Furthermore, cells with neurites transected nearer to the soma in 1 mm db-cAMP sealed at a significantly faster rate compared to control sealing (p < 0.01, FZT) (Table 2). At 10 and 20 min PC, 1 mm db-cAMP did not significantly increase the sealing frequency of cells compared to control sealing, because sealing was almost complete by 10 min PC (p > 0.05, CMH χ2). Similarly, 1 mm db-cAMP significantly increased the sealing frequency (at 0, 1, and 2.5 min PC) and increased the rate of sealing for cells with neurites transected farther from the soma compared to control sealing. Cells transected nearer to or farther from the soma in 50 μm db-cAMP sealed at a significantly greater frequency and faster rate compared to control sealing at 5 and 20 min PC (p < 0.01, CMH χ2; data not shown), indicating that 1 mm db-cAMP likely saturated all relevant cAMP targets.
We next examined whether increased PKA or Epac activity facilitated plasmalemmal sealing. With the exception of cells transected farther from the soma at 0 min PC (see below), cAMP analogs that activate PKA (cBiMPS) or Epac (Epac-cAMP) had nearly the same effect as db-cAMP. Specifically, 8 μm cBiMPS or 8 μm Epac-cAMP significantly increased the sealing frequency (p < 0.05, CMH χ2) (Fig. 2B,C) and rate (p < 0.01, FZT) (Table 2) compared to control sealing for cells transected nearer to and farther from the soma. These data are consistent with our hypothesis that cAMP activates PKA and Epac during plasmalemmal sealing.
Ca2+ independent sealing produced by cAMP analogs
In all previous studies and in our current studies presented above (Table 1, Fig. 2A), Ca2+ influx has been necessary to initiate sealing of plasmalemmal damage (see list in supplemental list, available at www.jneurosci.org as supplemental material). In contrast, Figure 2, B and C, show that this Ca2+ requirement can be bypassed by 1 mm db-cAMP, 8 μm cBiMPS, or 8 μm Epac-cAMP. That is, when cells are transected in Ca2+-free saline containing one of these three cAMP analogs, some cells seal before Ca2+ addition (i.e., at 0 min PC).
To further examine whether cAMP analogs can initiate plasmalemmal sealing in the absence of Ca2+, we transected cells in Ca2+-free saline containing 0.5 mm EGTA and one of these cAMP analogs and evaluated sealing without ever exposing these cells to Ca2+ (i.e., after transecting neurites in Ca2+-free saline, dye was added to the Ca2+-free saline). We observed no significant (p > 0.05, CMH χ2) difference in the sealing frequency between cells transected in a cAMP analog and never exposed to Ca2+ compared to cells transected in a cAMP analog and exposed to Ca2+ at 0 min PC (Fig. 3A, compare gray bars to black bars). Furthermore, B104 cells sealed at a significantly (p < 0.05, CMH χ2) higher frequency at 0 min PC in 1 mm db-cAMP compared to the other two cAMP analogs, whether or not cells were exposed to Ca2+ (Fig. 3A). All these data are consistent with an ability of cAMP-activated proteins to initiate neuroprotective plasmalemmal sealing in the absence of Ca2+ and are consistent with our hypotheses that PKA and Epac act downstream of Ca2+ during plasmalemmal sealing.
Figure 3.

A, Sealing frequency (%) of B104 cells transected >50 μm from the soma in Ca2+-free saline or Ca2+-free saline containing one of the cAMP analogs and assessed for sealing at 0 min PC in either Ca2+ saline (black bars) or Ca2+-free saline with 0.5 mm EGTA (gray bars). B, Sealing frequency (%) versus post-cAMP addition time (min) for cells transected nearer to or farther from the soma (solid lines). Cells were transected in Ca2+ free saline, after which db-cAMP without Ca2+ (squares) was added to the Petri dish. Cells that were bathed in 1 mm db-cAMP following neurite transection were never exposed to extracellular Ca2+. Control sealing frequency (%) versus PC time (min) plotted following neurite transection nearer to (dashed line) or farther from the soma (dotted line) as described for Figure 2A. C, D, Sealing frequency (%) versus PC time (min) following neurite transection nearer to (C) or farther from (D) the soma in 1 mm KT5720 (diamonds) or 50 μm PKI (triangles).
cAMP cannot fully substitute for Ca2+ in plasmalemmal sealing
We examined whether cAMP can completely substitute for Ca2+ by comparing the sealing frequency and rate initiated by 1 mm db-cAMP at various “post-cAMP times” in cells never exposed to Ca2+ to sealing initiated by Ca2+ saline (control sealing) at various PC times. The sealing frequency and rate in 1 mm db-cAMP without Ca2+ was significantly less than the frequency (p < 0.01, CMH χ2) and rate (p < 0.01, FZT) of control sealing (Fig. 3B, compare post-cAMP curves to PC curves). When cells were deprived of Ca2+ for >20 min, even in the presence of 1 mm db-cAMP, we observed membrane blebbing (plasmalemmal breakdown) and a decrease in sealing frequency at ∼20 min post-cAMP time compared to control sealing. These data suggest that cAMP cannot completely substitute for Ca2+ in plasmalemmal sealing and that exposure to Ca2+-free solutions for >15 min has deleterious effects on membrane integrity and cell viability.
Inhibiting PKA activity decreases frequency and rate of plasmalemmal sealing
Because activating PKA increased the ability of B104 cells to seal (Fig. 2B,C), we hypothesized that inhibiting PKA would decrease the ability of cells to seal. We therefore inhibited PKA with KT5720, a membrane-permeant, small molecule, PKA inhibitor that blocks the ATP binding site of PKA (Davies et al., 2000). Since KT5720 is soluble in DMSO but not in distilled water or Ca2+-free saline, we first transected B104 cells in 312 mm DMSO as a vehicle control. Except for one data point at 20 min PC for cells transected farther from the soma, the sealing frequency of cells following transection in 312 mm DMSO was not significantly different from control sealing in which cells are transected in Ca2+-free saline (p > 0.05, CMH χ2; data not shown). Likewise, cells transected nearer t, or farther from the soma in 312 mm DMSO did not seal at a significantly different rate compared to control sealing (p > 0.05, FZT; Table 2).
To examine whether inhibiting PKA decreases sealing, we transected cells in 1 mm KT5720 dissolved in DMSO. Specifically, 1 mm KT5720 significantly decreased the sealing frequency of cells following transection nearer to the soma at all PC times compared to control sealing (p < 0.001, CMH χ2) (Fig. 3C). Furthermore, 1 mm KT5720 significantly decreased the sealing rate compared to control sealing (p < 0.001, FZT) (Table 2). We obtained similar results for neurites transected farther from the soma in 1 mm KT5720 (Fig. 3D).
Since KT5720 has been reported to inhibit protein kinases other than PKA in cell lysates (Davies et al., 2000), we examined the effect of PKI, a structurally dissimilar peptide inhibitor that binds to the regulatory subunit and more specifically inhibits PKA (Dalton and Dewey, 2006). Compared to control sealing, both 50 μm PKI and 1 mm KT5720 decreased the frequency and the rate of sealing to a similar extent for neurites transected nearer to or farther from the soma at all PC times investigated, suggesting that the primary effect of both compounds is to inhibit PKA (Fig. 3C,D). When cells were transected in greater (2×) concentrations of either inhibitor or in both inhibitors at the same time, we did not observe a further decrease in sealing frequency, suggesting that both inhibitors are inhibiting PKA to the maximum possible extent at the concentrations tested (data not shown).
Epac activation partly overcomes PKA inhibition
To further investigate the role of Epac in plasmalemmal sealing, we inhibited PKA with 50 μm PKI while simultaneously increasing Epac activity with 8 μm Epac-cAMP (Fig. 4C,D). Despite a consistent, significant decrease in the sealing frequency at all PC times compared to control sealing (Fig. 4C), the rate of sealing for cells transected nearer to the soma in 8 μm Epac-cAMP and 50 μm PKI was not significantly different from the rate of control sealing (p > 0.05, FZT) (Table 2). Similar results for the frequency and rate of sealing were observed for cells transected farther from the soma in 8 μm Epac-cAMP and 50 μm PKI compared to control sealing (Fig. 4D, Table 2). Thus, when PKA is inhibited, Epac activation is able to restore the rate but not the frequency of sealing to control levels.
Figure 4.

A–C, Sealing frequency (%) versus PC time (min) following neurite transection nearer to (A) or farther from (B, C) the soma in 8 μm Epac-cAMP and 50 μm PKI (unfilled diamonds), 50 μm PKI (filled triangles) or nPKCη pseudosubstrate fragment (PSF; solid line, filled squares). D, Sealing frequency of cells transected nearer to (black bars) or farther from (gray bars) the soma in 2 mm melatonin (Mel) or Ca2+-free solution (control) and assessed for sealing at 5 min PC.
Cells transected in 8 μm Epac-cAMP with 50 μm PKI sealed at a significantly higher frequency (p < 0.01, CMH χ2) and significantly faster rate (p < 0.01, FZT) compared to cells transected nearer to or farther from the soma in 50 μm PKI (Fig. 4C,D). Thus, Epac activation is able to partially bypass PKA inhibition, suggesting that plasmalemmal sealing has an Epac-dependent, PKA-independent component.
nPKCη inhibition decreases frequency and rate of plasmalemmal sealing
PKC isozymes are involved in vesicle exocytosis and plasmalemmal sealing in invertebrate and non-neuronal preparations (Togo et al., 2003), and novel PKC (nPKC) isozymes are involved in cell wall maintenance in yeast (Arellano et al., 1999). To investigate whether nPKCs might affect plasmalemmal sealing, we inhibited an nPKCη isozyme using an nPKCη pseudosubtrate fragment (Steinberg, 2008). We examined cells transected farther from the soma, because this transection distance is a more sensitive measure of sealing (Fig. 4). At 5, 10, and 20 min PC, cells transected farther from the soma in 7 μm pseudosubstrate fragment sealed at a significantly (p < 0.001) lower frequency (CMH χ2) and a significantly slower rate (p < 0.01, FZT) compared to control sealing (Fig. 4C). These data suggest that nPKCη facilitates plasmalemmal sealing.
Melatonin decreases frequency of plasmalemmal sealing
Cytosolic oxidation after traumatic injury enhances plasmalemmal sealing in muscle cells (Cai et al., 2009a,b), and melatonin acts as an antioxidant by scavenging free oxygen radicals (Millán-Plano et al., 2010). To investigate whether plasmalemmal sealing might be affected by an antioxidant, we transected neurites of B104 cells in 2 mm melatonin, the maximum concentration that could be dissolved in distilled water. As seen for other test substances (i.e., KT5720, or PKI), melatonin significantly (p < 0.01, CMH χ2) (Fig. 4D) reduced the sealing frequency for cells transected nearer to or farther from the soma at 5 min PC compared to control sealing. Lower concentrations of melatonin (0.48 mm; data not shown) showed similar effects nearer to (p < 0.05, CMH χ2) and farther from (p < 0.01, CMH χ2) the soma compared to control sealing.
Discussion
Vesicles, plasmalemmal sealing, and cell survival
Before 1994, authors of textbooks (e.g., Kandel et al., 1991) and research publications (Spira et al., 1993) assumed that minor plasmalemmal damage was sealed by spreading of plasmalemmal lipids and that complete cellular or axonal transection was sealed by collapse and fusion of plasmalemmal leaflets. In 1994, we (Krause et al., 1994) showed that in several invertebrate neuronal preparations, either lesion type sealed by an accumulation of membrane-bound structures (mostly vesicles); Steinhardt et al. (1994) also reported a similar sealing mechanism in sea urchin eggs and mammalian epithelial cells. Damage-induced vesicles may continuously pack more densely and/or fuse with each other and nearby undamaged membrane to form a plug that retards the influx/efflux of smaller and smaller particles (Fishman and Bittner, 2003). Alternatively, these vesicles may interact in the vicinity of the damage site to form a “wound vesicle” or “membrane patch” that suddenly seals the membrane disruption (McNeil, 2009). Data from at least 20 different preparations from many phyla (see supplemental references list, available at www.jneurosci.org as supplemental material) consistently show that plasmalemmal sealing requires (isomers of) the same proteins, many of which are Ca2+ dependent and associated with cytoskeletal elements and/or vesicle trafficking/fusion in synapses and/or the Golgi apparatus (for reviews, see Fishman and Bittner, 2003; McNeil, 2009).
The consistent decrease in sealing frequency and rate observed for B104 cells transected nearer to the soma compared to cells transected farther from the soma (Figs. 2–4) may be partly because the diameter of a neurite (or axon) of a B104 (or other neuronal) cell measured nearer to the soma is often larger than the diameter of that same neurite measured farther from the soma (Lucas et al., 1985; 1990). Thus, vesicles probably require more time to accumulate to seal plasmalemmal damage of a larger diameter neurite or axon. Additionally, the potentially greater Ca2+ influx through a damage site nearer to the soma has a shorter diffusion distance to reach the soma and so may produce greater increases in somal Ca2+, thus increasing the probability of cell death (Nguyen et al., 2005).
Model of plasmalemmal sealing
Because rapid repair of plasmalemmal damage is necessary for their survival, all eukaryotic cells may have evolved multiple parallel pathways to ensure rapid plasmalemmal sealing, as shown in Figure 5 and as discussed below.
Figure 5.

Model of plasmalemmal sealing. Ca2+ flowing inward through the damage site activates multiple parallel (redundant) pathways that increase cAMP concentration (Figs. 2B,C, 3A,B) by activating Ca2+-dependent adenylate cyclases (Dunn et al., 2009). Ca2+ influx also activates calpain (Eddleman et al., 1997), whose protease action rearranges the cytoskeleton, partially constricting the cut ends of a transected axon or neurite, and also increases DAG availability by activating phospholipases. Ca2+ influx at sites of plasmalemmal damage also leads to increased cytosolic oxidation, facilitating plasmalemmal sealing (McNeil, 2009). Production of membrane-bound structures, usually vesicles, is stimulated by cAMP activation of PKA and Epac (Hatakeyama et al., 2007), DAG activation of nPKCs (Steinberg, 2008), and cytosolic oxidation (Cai et al., 2009a,b). These vesicles are targeted to and fuse with the damage site by: (1) PKA phosphorylation of SNARE and SNARE-related proteins important for vesicle traffic (Deák et al., 2006; Menegon et al., 2006; Bonanomi et al., 2007) (Figs. 2B,C, 3A,C,D); (2) Epac, which activates proteins important for vesicle motility (Fujimoto et al., 2002) (Figs. 2B,C, 4A,B); (3) nPKC phosphorylation (Uberall et al., 1997; Tsai et al., 2007) (Fig. 4C); and (4) oxidative activation of vesicle motility proteins (van Diepen et al., 2005; Cai et al., 2009a,b) (Fig. 4D).
cAMP increases plasmalemmal sealing (Fig. 2B,C), but cannot completely substitute for Ca2+ (Fig. 3B)
cAMP increases vesicular interactions in Golgi trafficking, growth cone extension, and transmitter release at synapses (Yoshihara et al., 2000; Sedej et al., 2005; for review see Hannila and Filbin, 2008). cAMP and cAMP activated proteins increase plasmalemmal sealing at PC times up to 20 min, including at 0 min PC, even when the bathing solution contains no Ca2+ (Fig. 2B,C). That is, increased cAMP concentration or increased PKA or Epac activity can partly bypass the requirement for Ca2+ in plasmalemmal sealing. A similar result has been reported for Drosophila neurons, where increased cAMP concentration can partly bypass the Ca2+ requirements of membrane fusion proteins for vesicle exocytosis at presynaptic release sites (Yoshihara et al., 1999, 2000). However, cAMP cannot completely substitute for Ca2+ during plasmalemmal sealing, since Ca2+ produces higher sealing frequencies than cAMP or cAMP analogs at all PC times (Fig. 3A). These data suggest the existence of other redundant Ca2+-dependent pathways parallel to cAMP pathways, such as diacylglycerol (DAG) activation of nPKCs (see below) (Fig. 5).
PKA activation increases sealing (Fig. 2B,C); PKA inhibition decreases sealing (Fig. 4A,B)
PKA activates many proteins important for membrane fusion and vesicle trafficking, such as SNAP-25 (Bronk et al., 2007), syntaxin (Nagy et al., 2004), synaptobrevin isoforms (Yoshihara et al., 1999, 2000), NSF (Garcia et al., 1995), and synapsin (Menegon et al., 2006). Most of these membrane fusion proteins are more abundant along axolemmal walls compared to nerve terminals (Tao-Cheng et al., 2000), suggesting that they may play a role in plasmalemmal repair. PKA may increase sealing by activating these and other membrane fusion and vesicle trafficking proteins, whereas PKA inhibition likely prevents activation of these proteins.
Epac activation increases sealing (Fig. 2B,C), partly independent of PKA (Fig. 4A,B)
Epac isoforms are involved in vesicle formation (Hatakeyama et al., 2007), exocytosis (Sedej et al., 2005), and neuronal survival after damage (Murray and Shewan, 2008). Epac may increase plasmalemmal sealing by stimulating vesicle traffic by binding to Rim2 and piccolo (Fujimoto et al., 2002) and exchanging guanine nucleotides for Rab3 (Branham et al., 2009). Epac increases membrane fusion by activating Sec1/Munc13 (Kwan et al., 2007) or SNARE proteins (Sedej et al., 2005). Inhibition of trans-Golgi traffic using the bacterial toxin brefeldin A decreases the influence of Epac on synaptic transmission, suggesting that Epac isoforms activate Rab3A exocytosis in both synaptic and Golgi trafficking pathways (Ster et al., 2009). That is, Epac pathways likely activate proteins in the PKA pathway (e.g., Rab3A, mUNC proteins, SNAREs) (Hochbaum et al., 2008), but also may activate parallel (redundant) Ca2+-dependent pathways important for plasmalemmal sealing (Fig. 5).
nPKCη inhibition decreases plasmalemmal sealing (Fig. 4C)
nPKCs activate many vesicle-mediated processes, such as cell wall maintenance and repair during yeast fission (Arellano et al., 1999), axonal growth cone turning, transport of β1 integrins, and neurite extension through phosphorylation of MARCKS and GAP43 (Sivasankaran et al., 2004; Gatlin et al., 2006; Sisková et al., 2006; Korshunova et al., 2007; Tsai et al., 2007). Inhibition of nPKC isozymes inhibits such vesicle-mediated processes and thus likely decreases the ability of vesicles to seal a damaged plasmalemma. nPKC isozymes are activated by binding of DAG to their regulatory subunits. Intracellular DAG concentration is increased by damage-induced Ca2+ influx that activates calpain to cleave and activate phospholipase C. [Calpains are Ca2+-dependent proteases necessary for plasmalemmal sealing in many preparations (Godell et al., 1997; Mellgren et al., 2007).]
Melatonin decreases sealing (Fig. 4D)
The diffusion of Ca2+ and other substances into or out of a cell at a site of plasmalemmal damage oxidizes the cytosol (McNeil, 2009). Cytosolic oxidation activates proteins (Sod family, glutathiones, thioredoxins) responsible for maintaining the intracellular redox environment and cell survival (Circu et al., 2009). In muscle, cytoplasmic oxidation activates the TRIM protein mitsigumin 53 (MG53), which stimulates vesicle accumulation (Cai et al., 2009a,b). These vesicles likely undergo Ca2+-dependent membrane fusion. Vesicle accumulation by damage-induced TRIM protein activation may also occur in neurons, since neuronal TRIM proteins increase vesicle accumulation in growth cones (van Diepen et al., 2005). Thus, the antioxidant melatonin may decrease sealing by reducing the formation of oxidation-induced vesicles.
Because melatonin decreases plasmalemmal sealing at 5 min PC, we hypothesize that other antioxidants or reducing agents may also impair sealing, whereas oxidizing agents may facilitate plasmalemmal sealing. Since cytosolic oxidation plays an important role in neurodegenerative disorders such as amyotrophic lateral sclerosis (Circu et al., 2009), a more complete understanding of how oxidation affects neurons, especially following plasmalemmal damage, may provide key insights into the etiology of neurodegenerative disorders.
Evolutionary origin of membrane fusion proteins was to seal plasmalemmal damage
Similar cellular/molecular mechanisms of plasmalemmal sealing occur in all eukaryotic cells and use protein isomers that have likely undergone a conservative evolution (see supplemental list, available at www.jneurosci.org as supplemental material). Since eukaryotic cells likely evolved a plasmalemmal membrane before membrane-enclosed organelles (Gerhart and Kirchner, 1997), we suggest that the first evolved role of membrane fusion proteins activated by PKA, Epac, PKC, or cytosolic oxidation was likely to seal plasmalemmal damage. Membrane fusion proteins were likely then co-opted in eukaryotic evolution for use in Golgi trafficking and subsequently, as suggested by Südhof and Rothman (2009), for transmitter release. Therefore, a better understanding of the molecular pathways of plasmalemmal sealing is likely to increase our understanding of vesicle interactions and membrane fusion in the Golgi apparatus, exocytosis, and synaptic transmission.
Clinical importance of plasmalemmal sealing
Repair of plasmalemmal damage is necessary but not sufficient for survival of eukaryotic cells, including B104 cells (Schlaepfer, 1973; Bittner and Fishman, 2000; Nguyen et al., 2005). For example, neurons that seal plasmalemmal disruptions at sites nearer to the soma are less likely to survive compared to neurons that seal damage at sites farther from the soma (Ramon y Cajal, 1928; Loewy and Shader, 1977; Lucas et al., 1985, 1990; Yoo et al., 2004; Nguyen et al., 2005). Limiting Ca2+ influx by increasing the rate of plasmalemmal sealing could increase the survival rate of injured neurons (i.e., provide neuroprotection), potentially increasing behavioral recovery following traumatic injury to the CNS or PNS neurons (Britt et al., 2010; Nehrt et al., 2010). Increasing plasmalemmal sealing also decreases the adverse affects of treadmill climbing in normal mice and in a mouse model of muscular dystrophy (Bansal and Campbell, 2004). A more complete understanding of plasmalemmal repair may lead to new clinical treatments to better treat conditions involving plasmalemmal damage, including traumatic injury, neurodegenerative diseases, stroke or other ischemic conditions, and muscular degenerative disorders.
Footnotes
This work was funded by grants from the Lone Star Paralysis Foundation. We thank Josh Britt, Dr. Jennifer Morgan, Eva Tang, Robert Wilcott, and Dr. Cathy Yang for help with cell maintenance and advice on the manuscript. PKI was a generous gift from Dr. Michael Markham and Dr. Harold Zakon.
References
- Agresti A. An introduction to categorical data analysis. New York: Wiley; 1996. pp. 60–64. [Google Scholar]
- Arellano M, Valdivieso MH, Calonge TM, Coll PM, Duran A, Perez P. Schizosaccharomyces pombe protein kinase C homologues, pck1p and pck2p, are targets of rho1p and rho2p and differentially regulate cell integrity. J Cell Sci. 1999;112:3569–3578. doi: 10.1242/jcs.112.20.3569. [DOI] [PubMed] [Google Scholar]
- Ballinger ML, Blanchette AR, Krause TL, Smyers ME, Fishman HM, Bittner GD. Delaminating myelin membranes help seal the cut ends of severed earthworm giant axons. J Neurobiol. 1997;33:945–960. doi: 10.1002/(sici)1097-4695(199712)33:7<945::aid-neu6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Bansal D, Campbell KP. Dysferlin and the plasma membrane repair pathway in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Barros F, Gómez-Varela D, Viloria CG, Palomero T, Giráldez T, de la Peña P. Modulation of human erg K+ channel gating by activation of a G protein-coupled receptor and protein kinase C. J Physiol. 2009;511:333–346. doi: 10.1111/j.1469-7793.1998.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner GD, Fishman HM. Axonal sealing following injury. In: Ingoglia N, Murray M, editors. Axonal regeneration in the central nervous system. New York: Dekker; 2000. p. 337.p. 370. [Google Scholar]
- Blanchette AR, Ballinger ML, Fishman HM, Bittner GD. Calcium entry initiates processes that restore a barrier to dye entry in severed earthworm giant axons. Neurosci Lett. 1999;272:147–150. doi: 10.1016/s0304-3940(99)00544-3. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, Rusconi L, Colombo CA, Benfenati F, Valtorta F. Synaptophysin I selectively specifies the exocytic pathway of synaptobrevin 2/VAMP2. Biochem J. 2007;404:525–534. doi: 10.1042/BJ20061907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci U S A. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham MT, Bustos MA, De Blas GA, Rehmann H, Zarelli VE, Treviño CL, Darszon A, Mayorga LS, Tomes CN. Epac activates the small G proteins Rap1 and Rab3A to achieve exocytosis. J Biol Chem. 2009;284:24825–24839. doi: 10.1074/jbc.M109.015362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt JM, Kane JR, Spaeth CS, Zuzek A, Robinson GL, Gbanaglo MY, Estler CJ, Boydston EA, Schallert T, Bittner GD. Polyethylene glycol rapidly restores axonal integrity and improves the rate of motor behavior recovery after sciatic crush injury. J Neurophysiol. 2010;104:695–703. doi: 10.1152/jn.01051.2009. [DOI] [PubMed] [Google Scholar]
- Bronk P, Deák F, Wilson MC, Liu X, Südhof TC, Kavalali ET. Differential effects of SNAP-25 deletion on Ca2+-dependent and Ca2+-independent neurotransmission. J Neurophysiol. 2007;98:794–806. doi: 10.1152/jn.00226.2007. [DOI] [PubMed] [Google Scholar]
- Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, Ko JK, Lin P, Thornton A, Zhao X, Pan Z, Komazaki S, Brotto M, Takeshima H, Ma J. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009a;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Weisleder N, Ko JK, Komazaki S, Sunada Y, Nishi M, Takeshima H, Ma J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3 and dysferlin. J Biol Chem. 2009b;284:15894–15902. doi: 10.1074/jbc.M109.009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chierzi S, Ratto GM, Verma P, Fawcett JW. The ability of axons to regenerate their growth cones depends on axonal type and age, and is regulated by calcium, cAMP and ERK. Eur J Neurosci. 2005;21:2051–2062. doi: 10.1111/j.1460-9568.2005.04066.x. [DOI] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Døskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. J Biol Chem. 2003;278:35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- Circu ML, Moyer MP, Harrison L, Aw TY. Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Rad Biol Med. 2009;47:1190–1198. doi: 10.1016/j.freeradbiomed.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Dewey WL. Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides. 2006;40:23–34. doi: 10.1016/j.npep.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Ok-Ho Shin OH, Tang J, Hanson P, Ubach J, Jahn R, Rizo J, Kavalali ET, Südhof TS. Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. 2006;25:2856–2866. doi: 10.1038/sj.emboj.7601165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrait E, Eddleman CS, Yoo S, Fukuda M, Nguyen MP, Bittner GD, Fishman HM. Axolemmal repair requires proteins that mediate synaptic vesicle fusion. J Neurobiol. 2000a;44:382–391. doi: 10.1002/1097-4695(20000915)44:4<382::aid-neu2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Detrait ER, Yoo S, Eddleman CS, Fukuda M, Bittner GD, Fishman HM. Plasmalemmal repair of severed neurites of PC12 cells requires Ca2+ and synaptotagmin. J Neurosci Res. 2000b;62:566–573. doi: 10.1002/1097-4547(20001115)62:4<566::AID-JNR11>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Dunn TA, Storm DR, Feller MB. Calcium-dependent increases in protein kinase-A activity in mouse retinal ganglion cells are mediated by multiple adenylate cyclases. PLoS One. 2009;4:e7877. doi: 10.1371/journal.pone.0007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleman CS, Ballinger ML, Smyers ME, Godell CM, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci U S A. 1997;94:4745–4750. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleman CS, Ballinger ML, Smyers ME, Fishman HM, Bittner GD. Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury. J Neurosci. 1998;18:4029–4041. doi: 10.1523/JNEUROSCI.18-11-04029.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleman CS, Bittner GD, Fishman HM. Barrier permeability at cut axonal ends progressively decreases until an ionic seal is formed. Biophys J. 2000;79:1883–1890. doi: 10.1016/S0006-3495(00)76438-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analog demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Fishman HM, Bittner GD. Vesicle-mediated restoration of a plasmalemmal barrier after axonal injury. News Physiol Sci. 2003;18:115–118. doi: 10.1152/nips.01429.2002. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Shibasaki T, Yokoi N, Kashima Y, Matsumoto M, Sasaki T, Tajima N, Iwanaga T, Seino S. Piccolo, a Ca2+ sensor in pancreatic beta-cells: involvement of cAMP-GEFII·Rim2·Piccolo complex in cAMP-dependent exocytosis. J Biol Chem. 2002;277:50497–50502. doi: 10.1074/jbc.M210146200. [DOI] [PubMed] [Google Scholar]
- Garcia EP, McPherson PS, Chilcote TJ, Takei K, De Camilli P. rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J Cell Biol. 1995;129:105–120. doi: 10.1083/jcb.129.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatlin JC, Estrada-Bernal A, Sanford SD, Pfenninger KH. Myristoylated, alanine-rich C-kinase substrate phosphorylation regulates growth cone adhesion and pathfinding. Mol Biol Cell. 2006;17:5115–5130. doi: 10.1091/mbc.E05-12-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J, Kirchner M. Cells, embryos and evolution. Malden, MA: Blackwell Science; 1997. p. 642. [Google Scholar]
- Godell CM, Smyers ME, Eddleman CS, Ballinger ML, Fishman HM, Bittner GD. Calpain activity promotes the sealing of severed giant axons. Proc Natl Acad Sci U S A. 1997;94:4751–4756. doi: 10.1073/pnas.94.9.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannila SS, Filbin MT. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp Neurol. 2008;209:321–332. doi: 10.1016/j.expneurol.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama H, Takahashi N, Kishimoto T, Nemoto T, Kasai H. Two cAMP pathways differentially regulate exocytosis of large dense core and small vesicles in mouse β cells. J Physiol. 2007;582:1087–1098. doi: 10.1113/jphysiol.2007.135228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochbaum D, Hong K, Barila G, Ribeiro-Neto F, Altschuler DL. Epac, in synergy with cAMP-dependent protein kinase (PKA), is required for cAMP-mediated mitogenesis. J Biol Chem. 2008;283:4464–4468. doi: 10.1074/jbc.C700171200. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessel T. Principles of neural science. Ed 3. New York: McGraw-Hill; 1991. [Google Scholar]
- Korshunova I, Novitskaya V, Kiryushko D, Pedersen N, Kolkova K, Kropotova E, Mosevitsky M, Rayko M, Morrow JS, Ginzburg I, Berezin V, Bock E. GAP-43 regulates NCAM-180 mediated neurite outgrowth. J Neurochem. 2007;100:1599–1612. doi: 10.1111/j.1471-4159.2006.04316.x. [DOI] [PubMed] [Google Scholar]
- Krause TL, Fishman HM, Ballinger ML, Bittner GD. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–6651. doi: 10.1523/JNEUROSCI.14-11-06638.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan EP, Xie L, Sheu L, Ohtsuka T, Gaisano HY. Interaction between Munc13–1 and RIM is critical for glucagon-like peptide-1 mediated rescue of exocytotic defects in Munc13–1 deficient pancreatic beta-cells. Diabetes. 2007;56:2579–2588. doi: 10.2337/db06-1207. [DOI] [PubMed] [Google Scholar]
- Lichstein JW, Ballinger ML, Blanchette AR, Fishman HM, Bittner GD. Structural changes at cut ends of earthworm giant axons in the interval between dye barrier formation and neuritic outgrowth. J Comp Neurol. 2000;416:143–157. doi: 10.1002/(sici)1096-9861(20000110)416:2<143::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Liu XF, Xie X, Miki T. Inhibition of protein kinase C zeta blocks the attachment of stable microtubules to kinetochores leading to abnormal chromosome alignment. Cell Signal. 2006;18:2314–2323. doi: 10.1016/j.cellsig.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Loewy AD, Schader RE. A quantitative study of retrograde neuronal changes in Clarke's column. J Comp Neurol. 1977;171:65–81. doi: 10.1002/cne.901710106. [DOI] [PubMed] [Google Scholar]
- Lucas JH, Gross GW, Emery DG, Gardner CR. Neuronal survival or death after dendrite transection close to the perikaryon: correlation with electrophysiologic, morphologic, and ultrastructural changes. Cent Nerv Syst Trauma. 1985;2:231–255. doi: 10.1089/cns.1985.2.231. [DOI] [PubMed] [Google Scholar]
- Lucas JH, Emery DG, Higgins ML, Gross GW. Neuronal survival and dynamics of ultrastructural damage after dendrotomy in low calcium. J Neurotrauma. 1990;7:169–192. doi: 10.1089/neu.1990.7.169. [DOI] [PubMed] [Google Scholar]
- McNeil P. Membrane repair redux: redox of MG53. Nat Cell Biol. 2009;11:7–9. doi: 10.1038/ncb0109-7. [DOI] [PubMed] [Google Scholar]
- Mellgren RL, Zhang W, Miyake K, McNeil PL. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem. 2007;282:2567–2575. doi: 10.1074/jbc.M604560200. [DOI] [PubMed] [Google Scholar]
- Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao HT, Benfenati F, Baldelli P, Valtorta F. Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci. 2006;26:11670–11681. doi: 10.1523/JNEUROSCI.3321-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán-Plano S, Piedrafita E, Miana-Mena FJ, Fuentes-Broto L, Martínez-Ballarín E, López-Pingarrón L, Sáenz MA, García JJ. Melatonin and structurally-related compounds protect synaptosomal membranes from free radical damage. Int J Mol Sci. 2010;11:312–328. doi: 10.3390/ijms11010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, Mooney SM, Middleton FA. Transforming growth factor β1 and ethanol affect transcription and translation of genes and proteins required for cell adhesion molecules in B104 neuroblastoma cells. J Neurochem. 2006;97:1182–1190. doi: 10.1111/j.1471-4159.2006.03858.x. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cell Neurosci. 2008;38:578–588. doi: 10.1016/j.mcn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sørensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:417–429. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Nehrt A, Haman K, Ouyang H, Shi Polyethylene glycol enhances axolemmal resealing following transection in cultured cells and in ex vivo spinal cord. J Neurotrauma. 2010;27:151–161. doi: 10.1089/neu.2009.0993. [DOI] [PubMed] [Google Scholar]
- Nguyen MP, Bittner GD, Fishman HM. Critical interval of somal calcium transient after neurite transection determines B104 cell survival. J Neurosci Res. 2005;81:805–816. doi: 10.1002/jnr.20606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S. Degeneration and regeneration in the nervous system. New York: Hoffner; 1928. [Google Scholar]
- Schlaepfer WW. Effects of nerve constriction on oxygenated excised segments of rat peripheral nerve. J Neuropathol Exp Neurol. 1973;32:203–217. doi: 10.1097/00005072-197304000-00003. [DOI] [PubMed] [Google Scholar]
- Schlaepfer WW, Bunge RP. Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J Cell Biol. 1973;59:456–470. doi: 10.1083/jcb.59.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedej S, Rose T, Rupnik M. cAMP increase Ca2+-dependent exocytosis through both PKA and Epac2 in mouse melanotrophs from pituitary slices. J Physiol. 2005;567:799–813. doi: 10.1113/jphysiol.2005.090381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisková Z, Baron W, de Vries H, Hoekstra D. Fibronectin impedes “myelin” sheet-directed flow in oligodendrocytes: a role for a beta 1 integrin-mediated PKC signaling pathway in vesicular trafficking. Mol Cell Neurosci. 2006;33:150–159. doi: 10.1016/j.mcn.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Sivasankaran R, Pei J, Wang KC, Zhang YP, Shields CB, Xu XM, He Z. PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat Neurosci. 2004;7:261–268. doi: 10.1038/nn1193. [DOI] [PubMed] [Google Scholar]
- Spira ME, Benbassat D, Dormann A. Resealing of the proximal and distal cut ends of transected axons: electrophysiological and ultrastructural analysis. J Neurobiol. 1993;24:300–316. doi: 10.1002/neu.480240304. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Ster J, de Bock F, Bertaso F, Abitbol K, Daniel H, Bockaert J, Fagni L. Epac mediates PACAP-dependent long-term depression in the hippocampus. J Physiol. 2009;587:101–113. doi: 10.1113/jphysiol.2008.157461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ZY, Chen J, Shun HY, Feng XH, Ji YH. Modulation of BmK AS, a scorpion neurotoxic polypeptide, on voltage-gated Na+ channels in B104 neuronal cell line. Neurosci Lett. 2003;340:123–126. doi: 10.1016/s0304-3940(03)00094-6. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Du J, McBain CJ. Snap-25 is polarized to axons and abundant along the axolemma: an immunogold study of intact neurons. J Neurocytol. 2000;29:67–77. doi: 10.1023/a:1007168231323. [DOI] [PubMed] [Google Scholar]
- Toda M, Shirao T, Uyemura K. Suppression of an actin-binding protein, drebrin, by antisense transfection attenuates neurite outgrowth in neuroblastoma B104 cells. Brain Res Dev Brain Res. 1999;114:193–200. doi: 10.1016/s0165-3806(99)00030-9. [DOI] [PubMed] [Google Scholar]
- Togo T, Alderton JM, Steinhardt RA. Long-term potentiation of exocytosis and cell membrane repair in fibroblasts. Mol Biol Cell. 2003;14:93–106. doi: 10.1091/mbc.E02-01-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SY, Yang LY, Wu CH, Chang SF, Hsu CY, Wei CP, Leu SJ, Liaw J, Lee YH, Tsai MD. Injury-induced Janus kinase/protein kinase C-dependent phosphorylation of growth-associated protein 43 and signal transducer and activator of transcription 3 for neurite growth in dorsal root ganglion. J Neurosci Res. 2007;85:321–331. doi: 10.1002/jnr.21119. [DOI] [PubMed] [Google Scholar]
- Uberall F, Giselbrecht S, Hellbert K, Fresser F, Bauer B, Gschwendt M, Grunicke HH, Baier G. Conventional PKC-alpha, novel PKC-epsilon and PKC-theta, but not atypical PKC-lambda are MARCKS kinases in intact NIH 3T3 fibroblasts. J Biol Chem. 1997;272:4072–4078. doi: 10.1074/jbc.272.7.4072. [DOI] [PubMed] [Google Scholar]
- van Diepen MT, Spencer GE, van Minnen J, Gouwenberg Y, Bouwman J, Smit AB, van Kesteren RE. The molluscan RING-finger protein L-TRIM is essential for neuronal outgrowth. Mol Cell Neurosci. 2005;29:74–81. doi: 10.1016/j.mcn.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Yoo S, Nguyen MP, Fukuda M, Bittner GD, Fishman HM. Plasmalemmal sealing of transected mammalian neurites is a gradual process mediated by Ca2+-regulated proteins. J Neurosci Res. 2003;74:541–551. doi: 10.1002/jnr.10771. [DOI] [PubMed] [Google Scholar]
- Yoo S, Bottenstein JE, Bittner GD, Fishman HM. Survival of mammalian B104 cells following neurite transection at different locations depends on somal Ca2+ concentration. J Neurobiol. 2004;60:137–153. doi: 10.1002/neu.20005. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Ueda A, Zhang D, Deitcher DL, Schwarz TL, Kidokoro Y. Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. J Neurosci. 1999;19:2432–2441. doi: 10.1523/JNEUROSCI.19-07-02432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Suzuki K, Kidokoro Y. Two independent pathways mediated by cAMP and protein kinase A enhance spontaneous transmitter release at Drosophila neuromuscular junctions. J Neurosci. 2000;20:8315–8322. doi: 10.1523/JNEUROSCI.20-22-08315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]