

Abstract
A large body of evidence suggests that the neurotransmitter GABA undergoes a developmental switch from being predominantly depolarizing–excitatory to predominantly hyperpolarizing–inhibitory. Recently published data, however, point to the possibility that the presumed depolarizing mode of GABA action during early development might represent an artifact due to an insufficient energy supply of the in vitro preparations used. Specifically, addition of the ketone body dl-β-hydroxybutyrate (βHB) to the extracellular medium was shown to prevent GABA from exerting excitatory effects. Applying a complementary set of minimally invasive optical and electrophysiological techniques in brain slices from neonatal mice, we investigated the effects of βHB on GABA actions in immature cells of the upper cortical plate. Fluorescence imaging revealed that GABA-mediated somatic [Ca2+] transients, that required activation of GABAA receptors and voltage-gated Ca2+ channels, remained unaffected by βHB. Cell-attached current-clamp recordings showed that, in the presence of βHB, GABA still induced a membrane potential depolarization. To estimate membrane potential changes quantitatively, we used cell-attached recordings of voltage-gated potassium currents and demonstrated that the GABA-mediated depolarization was independent of supplementation of the extracellular solution with βHB. We conclude that, in vitro, GABA depolarizes immature cells of the upper cortical plate in the presence of the ketone body βHB. Our data thereby support the general concept of an excitatory-to-inhibitory switch of GABA action during early development.
Introduction
A widely accepted hypothesis in developmental neurobiology states that the maturation of neuronal networks is mainly genetically determined in the initial phase, but critically relies on neuronal activity during later development (Katz and Shatz, 1996). GABA acts as the main inhibitory neurotransmitter in the adult brain. However, GABA is assumed to provide the main excitatory drive for the neuronal network at an early time of maturation when GABAergic synapses clearly outnumber glutamatergic contacts (Ben-Ari et al., 2007). GABA-mediated excitation is regarded as a consequence of an intracellular chloride accumulation by a differential expression of chloride cotransporters (Blaesse et al., 2009) and has been documented using various in vitro approaches, e.g., in the neocortex. These approaches include intracellular recordings (Luhmann and Prince, 1991), perforated-patch measurements (Owens et al., 1996; Yamada et al., 2004), cell-attached measurements (Wang et al., 2003; Rheims et al., 2008), recordings of GABA-evoked action potential firing (Rheims et al., 2008), and (semi-)quantitative fluorescence microscopy using Ca2+-sensitive (Yuste and Katz, 1991; Owens et al., 1996; Yamada et al., 2004; Kirmse and Kirischuk, 2006) and Cl−-sensitive (Glykys et al., 2009) fluorophores.
Despite the wealth of data supporting the hypothesis of a depolarizing/excitatory mode of GABA action during early development, this concept has been seriously questioned. Recently, it was reported that GABA-mediated depolarization in immature neocortical pyramidal cells could be due to an inadequate energy supply of the in vitro preparation (Rheims et al., 2009; Holmgren et al., 2010). More precisely, Rheims et al. (2009) found that addition of the ketone body dl-β-hydroxybutyrate to the extracellular medium abolished excitatory effects of GABA. Since the extracellular media in previous studies were generally not supplemented with ketone bodies, the study by Rheims et al. (2009) points to the possibility that excitatory effects of GABA might represent an artifact of the specific in vitro conditions. Interestingly, the rodent brain displays high ketone body utilization during early postnatal life that is supported by a comparably high concentration of ketone bodies in the plasma and a peak expression of ketone body-transporting proteins in the blood–brain barrier during suckling (Nehlig, 2004).
In the present study, a complementary set of Ca2+ imaging and noninvasive electrophysiological techniques were applied to evaluate whether addition of dl-β-hydroxybutyrate to the extracellular medium was able to prevent the depolarizing action of GABA in immature cells of the upper cortical plate. Our data suggest that, even in the presence of dl-β-hydroxybutyrate, GABA maintains a primarily depolarizing effect. They thereby support the hypothesis that GABA could shape neuronal circuit development by providing excitation for the immature network.
Materials and Methods
Preparation of brain slices.
All experimental procedures were performed with approval from the local government and complied with international and European Union norms. Experiments were performed with acute brain slices prepared from C57BL/6J mice at postnatal day (P) 1–4 (P0, day of birth). Animals were decapitated under deep isoflurane anesthesia. The brain was removed quickly and transferred into ice-cold saline containing the following (in mm): 125 NaCl, 4 KCl, 10 glucose, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, and 2.5 MgCl2, bubbled with 5% CO2/95% O2, pH 7.4. Sagittal slices (300 μm) comprising the occipital cortex were cut on a vibratome and stored for at least 1 h before their use at room temperature in artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 4 KCl, 10 glucose, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, and 1 MgCl2, bubbled with 5% CO2/95% O2, pH 7.4. In subsets of experiments, an energy substrate-enriched ACSF (eACSF) was used containing (in mm): 126 NaCl, 3.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 1.3 MgCl2, 2 CaCl2, 5 choline chloride, 5 glucose, 2 dl-sodium β-hydroxybutyrate, 5 sodium pyruvate, pH 7.4 (Holmgren et al., 2010).
For recordings, slices were placed into a submerged-type recording chamber on the microscope stage (Eclipse FN1, Nikon Instruments) equipped with near-infrared differential interference contrast video microscopy (ACSF flow rate, 2–3 ml/min). All experiments were done at 33–35°C.
Confocal Ca2+ imaging.
Cells were loaded with the AM-ester of the Ca2+-indicator Oregon Green 488 BAPTA-1 (OGB1) using the multicell bolus-loading procedure (Stosiek et al., 2003). Fluorescence signals were acquired at a frame rate of 125 Hz using a CSU10 Nipkow-disc scanning unit in combination with a NeuroCCD-SM camera and the software Neuroplex 9.1 (Redshirt Imaging). Alternatively and for higher spatial resolution, a RoleraXR camera driven by the software QCapture Pro 6 (QImaging) was used at 10 Hz. Excitation light at 488 nm was provided by a single wavelength solid-state laser Sapphire CDRH-LP (Coherent) (Fig. 1A). Fluorescence signals were corrected for background fluorescence and expressed as relative changes from prestimulus levels (ΔF/F).
Figure 1.

GABA-mediated Ca2+ transients persist in the presence of dl-β-hydroxybutyrate. A, Schematic drawing of the experimental arrangement. B, Raw fluorescence image displaying OGB1-stained cells in the upper cortical plate. C, ΔF image to illustrate GABA-responsive cells (field of view as in B). D, Single-cell somatic intracellular [Ca2+] responses to puff application of GABA (100 μm, 200 ms) (arrowhead). E, βHB (4 mm) did not affect GABA-induced [Ca2+] transients in the presence of TTX. F, GABA-induced [Ca2+] transients were sensitive to antagonists of GABAA receptors (BMI, 100 μm) and voltage-gated Ca2+ channels (CdCl2, 100 μm). Examples in E and F represent averages of eight to ten cells. G–I, Quantification of results. G, Pooled data from unpaired and paired experiments. H, Significance levels for each group were calculated using one-sided paired t tests (BMI/−βHB, n = 5; BMI/+βHB, n = 5; Cd2+/−βHB, n = 4; Cd2+/+βHB, n = 5; bumetanide/−βHB, n = 7; bumetanide/+βHB, n = 5). Baclo, Baclofen; Musci, muscimol; Bume, bumetanide. ns, Not significant; result of a two-way ANOVA in H and result of a paired t test in I. *p < 0.05, **p < 0.01, ***p < 0.001.
Electrophysiological recordings.
Electrophysiological signals were acquired using an Axopatch 200B amplifier, a 16-bit AD/DA board (Digidata 1440A), and pClamp 10.2 (Molecular Devices). Signals were low-pass filtered at 5 kHz and sampled at 20–50 kHz.
To evaluate the polarity of the GABA-induced membrane potential (Vm) alteration, cell-attached current-clamp recordings were performed (I = 0 mode of the amplifier) (Mason et al., 2005; Perkins, 2006). Recording pipettes (3–8 MΩ) were filled with one of the following solutions (in mm): (1) 120 KCl, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose, pH-adjusted to 7.25 with KOH (KCl-based); or (2) 135 K+ gluconate, 5 KCl, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 HEPES, pH-adjusted to 7.25 with KOH (or modified by omitting EGTA, CaCl2 and MgCl2, both termed “K+ gluconate-based”). The total resistance measured in cell-attached mode was 10.3 ± 0.97 GΩ (n = 43).
To estimate Vm quantitatively, we applied the method described by Verheugen et al. (1999) using the reversal potential of voltage-dependent K+ currents measured in the cell-attached configuration. In this set of experiments, the KCl-based intrapipette solution was used (final [K+] ≈ 152 mm). Assuming symmetrical [K+], the holding potential at which the K+ current reverses gives an estimate of Vm. To activate voltage-gated K+ currents, depolarizing voltage ramps (from −100 mV to +200 mV) were applied at a frequency of 0.5 Hz. Between stimulations, the patch was held at −60 mV (with respect to Vm) to attenuate a possible steady-state inactivation of voltage-gated K+ currents. For analysis, a correction was made for the leak component by linear extrapolation to the closed level below the activation threshold. No correction for liquid junction potentials was made (<5 mV). On average, ∼10–20% of the cells tested displayed discernible K+ currents. Because, in most cases, the amplitude of the initial inward current was small, averaged responses (usually five traces) were analyzed. In those cases where the current reversal was prominent in single trials, no prior averaging was performed. Similar current profiles were obtained with K+ gluconate-based intrapipette solution (data not shown). No significant correlation between the estimated Vm and seal resistance was found (p > 0.6).
Loose-patch (seal resistance <1 GΩ, n = 12) or tight-seal cell-attached recordings (>1 GΩ, n = 6) were performed in voltage-clamp mode at a holding potential of 0 mV using recording pipettes filled with ACSF. GABA application was performed three times every 2 min and cells were classified as excited by GABA if ≥1 action current was detected.
Puff application.
Puff application of agonists or ACSF at a nominal pressure of 4–8 psi was performed via a patch pipette (tip diameter, ∼2.5 μm) positioned ∼10–20 μm above the slice. Substances for application were dissolved in extracellular solution of the respective control condition.
Chemicals.
Chemicals were obtained from Biotrend [tetrodotoxin (TTX)], Invitrogen (OGB1-AM), Tocris Bioscience (baclofen), and Sigma-Aldrich [muscimol, bicuculline methiodide (BMI), bumetanide, dl-sodium β-hydroxybutyrate (βHB), d-sodium βHB (d-βHB), sodium pyruvate].
Data evaluation and statistics.
Data were evaluated off-line using Neuroplex 9.1 (Redshirt Imaging), ImageJ (http://rsbweb.nih.gov/ij/index.html), pClamp 10.2 (Molecular Devices), Microsoft Excel, OriginPro 8 (OriginLab) and PSAW Statistics 17 (SPSS). For a subset of Ca2+ imaging experiments, ANOVA suggested that cells within a given slice could not be regarded as statistically independent. We therefore selected 10 cells per slice that exhibited stable responses to two or three agonist applications during the control and used the average of their responses for quantification. In this case, the statistical parameter n refers to the number of slices, in contrast to electrophysiological experiments, where n indicates the number of cells. All results are presented as means ± SEM.
Results
GABA-induced Ca2+ transients in the absence and presence of dl-β-hydroxybutyrate
Previous studies on immature neocortical cells showed that GABA could evoke a depolarization-dependent rise in intracellular calcium concentration ([Ca2+]) (Yuste and Katz, 1991; Owens et al., 1996; Yamada et al., 2004). To examine whether a similar mechanism is operative in our preparation, we puff-applied GABA (100 μm, 200 ms) and recorded somatic [Ca2+] in immature cells of the upper cortical plate at P1–P4. Indeed, distinct somatic [Ca2+] transients were consistently detected in ∼10–40 cells within the field of view, both in the absence of dl-β-hydroxybutyrate (−βHB, n = 11) (Fig. 1B–D) and in its presence (+βHB, n = 12; slices were exposed to 4 mm βHB for >1 h before the experiment) (Fig. 1F). Under both conditions, these [Ca2+] transients were mimicked by application of the specific GABAA receptor (GABAAR) agonist muscimol (20 μm; −βHB, n = 9; +βHB, n = 7), but not by the specific GABAB receptor (GABABR) agonist baclofen (10 μm; −βHB, n = 4; +βHB, n = 5) nor by puff application of ACSF alone (−βHB, n = 4; +βHB, n = 4), arguing against pure mechanical stimulation (Fig. 1G). Moreover, puff-application of GABA did not trigger similar [Ca2+] transients in slices from adult mice (n = 3) (data not shown). GABA-mediated [Ca2+] transients with similar amplitudes were also observed when TTX (0.5 μm) was added to the superfusion medium to block voltage-dependent sodium channels (−βHB, n = 12; +βHB, n = 9) (Fig. 1G). Dependence on GABAAR activation was further corroborated by the observation that bath-application of BMI (100 μm), a specific GABAAR antagonist, completely abolished the GABA-induced rise in intracellular [Ca2+]. In the presence of BMI, the normalized amplitude of the GABA-induced relative change in fluorescence (ΔF/F) was reduced to −0.1 ± 0.5% (−βHB, p < 0.01, n = 5) and −1.9 ± 0.9% (+βHB, p < 0.001, n = 5; one-sided paired Student's t test) (Fig. 1F,H), respectively. Additionally, these GABA-induced [Ca2+] transients were dependent on the activation of voltage-gated Ca2+ channels (VGCCs), since they were strongly reduced in amplitude by the broad-spectrum VGCC antagonist CdCl2 (100 μm) (−βHB, to 6.8 ± 2.6% of the control, p < 0.05, n = 4; +βHB, to 5.1 ± 0.7% of the control, p < 0.05, n = 5; one-sided paired Student's t test) (Fig. 1F,H).
The sodium/potassium/chloride cotransporter NKCC1 is assumed to represent the dominant Cl− accumulating transporter in immature neocortical neurons (Yamada et al., 2004). Indeed, bath-application of the NKCC1 antagonist bumetanide (20 μm) strongly attenuated the amplitude of the GABA-induced somatic [Ca2+] transients (−βHB, to 40.6 ± 11.3% of the control, p < 0.01, n = 7; +βHB, to 45.3 ± 6.1% of the control, p < 0.05, n = 5; one-sided paired Student's t test) (Fig. 1H). The latter experiments were conducted in the presence of TTX (0.5 μm) to reduce contributions from recurrent excitation. Furthermore, two-way ANOVA on normalized data presented in Figure 1H suggested that the effects of BMI, Cd2+, and bumetanide on the amplitude of GABA-induced [Ca2+] transients did not depend on the absence/presence of βHB (p > 0.8).
Next, in paired experiments, we compared peak amplitudes of somatic [Ca2+] transients in the presence of TTX with those after prolonged (i.e., >30 min) wash-in of βHB (4 mm). It was found that βHB did not lead to a significant alteration of the amplitude of [Ca2+] transients evoked by either GABA (94.1 ± 6.2% of the control, p > 0.4, n = 4) or muscimol (127.6 ± 23.2% of the control, p > 0.3, n = 5; two-sided paired Student's t test) (Fig. 1E,I).
We conclude that, in immature neocortical cells, GABAAR-mediated somatic [Ca2+] transients remain unaffected in amplitude and pharmacological characteristics by supplementation of the ACSF with dl-β-hydroxybutyrate (4 mm).
GABA-mediated depolarization in the absence and presence of dl-β-hydroxybutyrate
Conclusions about the change in Vm derived from Ca2+ imaging experiments are limited insofar as an increase in intracellular [Ca2+] might only serve as an indirect correlate of membrane depolarization. To substantiate our finding, we used current-clamp recordings in the cell-attached configuration, which allows recording a significant fraction of the change in membrane potential if the ratio of seal versus patch resistance is sufficiently high (Mason et al., 2005; Perkins, 2006). Importantly, this method correctly depicts the polarity of a change in Vm without disturbing the composition of the cytoplasm. Experiments were conducted in the presence of TTX. In the absence of βHB, all cells tested (n = 8) displayed a significant positive shift of the measured potential (V) in response to puff application of GABA (100 μm, 5 s), indicating a depolarization of Vm (Fig. 2A,C). Similar results were obtained with a KCl-based (ΔV = 23.5 ± 2.9 mV, n = 4) and K+ gluconate-based (ΔV = 19.5 ± 5.5 mV, n = 4) (Fig. 2C) intrapipette solution, arguing against the suggestion that an artificial Cl− loading via the patch pipette might explain this finding. Pure mechanical stimulation was not responsible for the observed effect, since puff application of ACSF alone did not significantly affect V (data not shown). Moreover, the GABA-induced ΔV was completely blocked by BMI (100 μm) (data not shown). When βHB (4 mm) was added to the ACSF, GABA application also induced a positive shift of the measured potential in all tested cells (n = 11) (Fig. 2B,C). We obtained similar results with both a KCl-based (ΔV = 17.9 ± 3.0 mV, n = 7) and a K+ gluconate-based (ΔV = 24.5 ± 5.0 mV, n = 4) (Fig. 2C) intrapipette solution, as well as with a solution containing only NaCl (154 mm; ΔV = 21.5 ± 2.3 mV, n = 4) (data not shown). We conclude that the vast majority of neocortical cells are depolarized by GABA.
Figure 2.

GABA mediates depolarization in the presence of dl-β-hydroxybutyrate. A, B, Cell-attached current-clamp recordings illustrate that puff application of GABA (100 μm)-induced depolarization in the absence (A) and presence (B) of βHB (4 mm). Action potentials were blocked by TTX. C, Quantification of results. Open and filled symbols indicate cells in the absence and presence of βHB, respectively. K-gluc (K-glucose) and KCl refer to a low- and high-chloride intrapipette solution, respectively. D, Example of a cell-attached recording used to estimate Vm. Leak component was corrected for by linear extrapolation (diagonal dotted line; R2 > 0.99) to the closed level below the activation threshold. The voltage command is shown in the upper part (−Vpipette). The intersection of the linear fit with the current trace indicates the point of K+ current reversal and is projected to the voltage command (vertical dashed line). The current trace represents an average of five responses and was low-pass filtered at 1 kHz. E, Leak-corrected current traces (during voltage ramp) in the absence (top) and presence (bottom) of βHB during control (black) and during puff application of GABA (100 μm; gray). In both cases, note the shift of K+ current reversal to the left (corresponding to a more depolarized membrane potential). Upper traces are from the cell shown in D and represent averages of five responses, lower traces represent single-trial responses (action potentials blocked by TTX). F, Quantification of results (−βHB, n = 7; +βHB, n = 5). **p < 0.01, ***p < 0.001.
Therefore, it is of interest whether GABA is able to induce action potential firing. Loose-patch/cell-attached recordings of action currents showed that ∼50% of the cells were excited by puff application of GABA (100 μm, 200 ms; −βHB, 5 of 9 cells; +βHB, 5 of 9 cells) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). A nonsignificant tendency toward a higher number of actions currents per trial in the absence of βHB was noted, but not further explored (p > 0.25, Mann–Whitney test) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
We next examined the GABA-induced change in membrane potential in a quantitative manner using cell-attached recordings of the reversal potential of voltage-gated K+ currents (see Materials and Methods) (Fig. 2D,E) (Verheugen et al., 1999). The calculated resting Vm under control conditions (action potentials blocked by TTX) did not significantly differ between cells maintained either in the absence or presence of βHB (4 mm; −βHB, −84.0 ± 4.0 mV, n = 7; +βHB, −78.4 ± 7.9 mV, n = 5; p > 0.4; two-sided unpaired t test) (Fig. 2F). In the absence of βHB, puff application of GABA (100 μm, 20 s) was found to strongly depolarize all cortical plate cells tested (to −41.0 ± 3.6 mV, p < 0.001, n = 7; one-way repeated-measures ANOVA followed by pairwise comparison using Bonferroni correction; ηp2>0.92) (Fig. 2E,F). This effect was completely reversible (Fig. 2F). Similar results were obtained in the continuous presence of βHB, where application of GABA reversibly depolarized all cells examined (to −34.7 ± 4.4 mV, p < 0.01, n = 5; one-way repeated-measures ANOVA followed by pairwise comparison using Bonferroni correction; ηp2>0.93) (Fig. 2E,F). Furthermore, mixed-model ANOVA indicated that the main effect for presence versus absence of βHB was not significant (p > 0.3).
Robustness of GABA-induced Ca2+ transients and depolarization
Using Ca2+ imaging and cell-attached current-clamp recordings (K+ gluconate-based intrapipette solution), we next examined whether specific alterations of the experimental conditions might influence our conclusions made so far. First, we tested whether energy substrates other than glucose/βHB might affect the response to GABA. To this end, brain slices were superfused with eACSF (see Materials and Methods) that contained pyruvate as potential energy substrate. GABA-induced somatic [Ca2+] transients could be readily evoked (Fig. 3A–D) and they were largely attenuated by CdCl2 (100 μm) (to 5.5 ± 2.1% of the control, p < 0.01, n = 4; one-sided paired Student's t test), indicating dependence on VGCC activation. In cell-attached current-clamp recordings, GABA-mediated depolarization was evident in all cells tested (ΔV = 26.1 ± 3.4 mV, n = 6) (Fig. 3E,F). Second, to address a potential impurity of the racemic mixture of βHB, ACSF was supplemented with the metabolically active stereoisomer d-βHB (2 mm) instead of βHB. Again, both GABA-induced somatic [Ca2+] transients and depolarization detected in cell-attached mode appeared to be unaffected (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Third, depolarizing GABA-mediated responses were also observed in the somatosensory/motor cortex, arguing against the possibility that different cortical areas respond differentially to βHB (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 3.
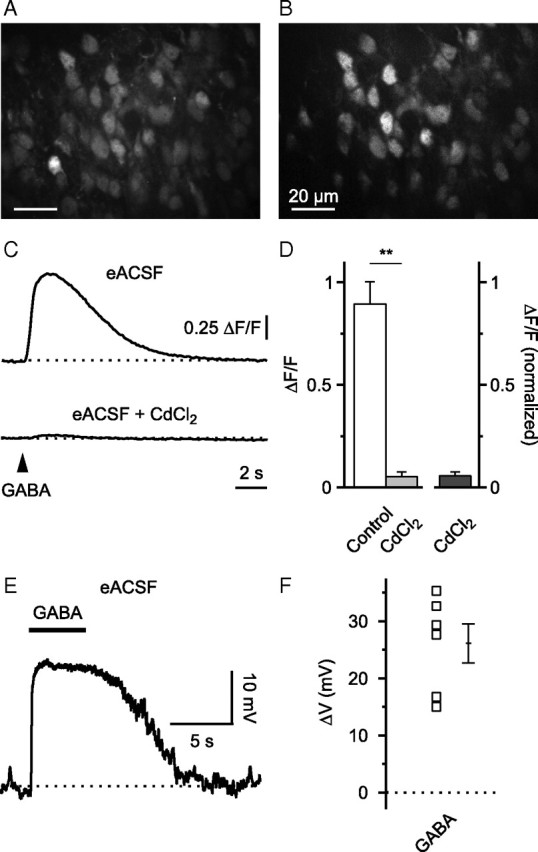
GABA-mediated somatic Ca2+ transients and depolarization persist in energy-substrate enriched ACSF. A, Raw fluorescence image displaying OGB1-stained cells in the upper cortical plate. B, ΔF image to illustrate GABA-responsive cells. C, In eACSF, GABA-mediated somatic [Ca2+] transients were sensitive to CdCl2 (100 μm). Sample traces are averages of five cells. D, Quantification of results. E, Cell-attached current-clamp recording. Puff application of GABA (100 μm) induced membrane depolarization (action potentials blocked by TTX). F, Quantification of results. **p < 0.01.
We conclude that the mode of GABA action in immature neocortical cells at P1–P4 is predominantly depolarizing.
Discussion
To elucidate whether βHB (4 mm) influences the action of GABA on immature cells of the upper cortical plate, we applied a set of techniques that minimally interfere with intrinsic chloride homeostasis. We showed that, in the presence of βHB, GABAAR activation could lead to a rise in intracellular [Ca2+] and that GABA could induce membrane potential depolarization.
The activity-dependent construction of neuronal circuits relies on sources of excitation (Katz and Shatz, 1996). GABA-mediated excitation during early development is a central component of this concept because GABAergic synapses are formed before glutamatergic contacts (Ben-Ari et al., 2007) and the expression profile of Cl− cotransporters favors depolarizing GABAergic responses (Blaesse et al., 2009). Though subject to alternative interpretations, this is exemplified by several studies showing that an interference with intracellular Cl− accumulation at an immature stage perturbs both the proper morphological maturation of neurons and their functional synapse development (Cancedda et al., 2007; Pfeffer et al., 2009). Therefore, an absence of GABA-mediated excitation (Rheims et al., 2009; Holmgren et al., 2010) could have major implications for a central hypothesis of developmental neurobiology.
GABA-evoked [Ca2+] transients
We used somatic [Ca2+] transients of immature neocortical cells evoked by puff application of GABAAR agonists as a correlate of membrane depolarization (Yuste and Katz, 1991; Owens et al., 1996; Yamada et al., 2004). This seems to be plausible, since a broad-spectrum antagonist of VGCCs abolished the responses almost completely (Fig. 1F,H). GABAAR-mediated [Ca2+] transients were not reduced in amplitude by βHB (Fig. 1E,I). In disagreement with previous observations (Rheims et al., 2009), our results therefore point to a persistence of GABA-mediated membrane depolarization in the presence of βHB. Although the quantitative relationship between ΔF/F and ΔVm is unknown, a pronounced hyperpolarizing shift of EGABA, as reported by Rheims et al. (2009), appears to be hardly reconcilable with our data, because GABA-mediated depolarization in the presence of βHB was obviously strong enough to activate VGCCs and blocking NKCC1 strongly attenuated the GABA-induced somatic [Ca2+] transients.
Electrophysiological evidence supporting a depolarizing mode of GABA action
Theoretically, a transient GABA-induced hyperpolarization might activate an excitatory conductance (e.g., low-voltage-activated Ca2+ channels, hyperpolarization-activated Ih channels) that in turn could result in depolarization (Aizenman and Linden, 1999). However, this possibility could be largely ruled out by using cell-attached current-clamp recordings that revealed monophasic GABA-mediated depolarization without initial hyperpolarization (Fig. 2A–C). GABA-mediated depolarization was sufficiently strong to initiate action potential discharge in ∼50% of cells, suggesting that GABA can act as an excitatory neurotransmitter in the presence of βHB. Cell-attached recordings of voltage-dependent K+ currents (Verheugen et al., 1999) showed that the resting membrane potential was, on average, close to −80 mV and did not significantly differ between cells that were continuously exposed to βHB and those that were not (Fig. 2F). These Vm estimates are in agreement with values derived from cell-attached recordings in neocortex (Rheims et al., 2008) and hippocampus (Tyzio et al., 2003). In line with our previous data, GABA was found to considerably depolarize Vm of all cells tested—the amplitude of depolarization being not statistically different between naive and βHB-exposed cells (Fig. 2E,F).
In our hands, GABA-mediated depolarization appears to be a stable phenomenon in vitro, since it was observed in the presence of the metabolically active stereoisomer d-βHB as well as in energy-substrate enriched ACSF and was found to be not cortical-region specific.
In summary, our data demonstrate that the mode of GABA action in immature neocortical cells maintained in vitro is primarily depolarizing in nature, independent of whether the extracellular medium is supplemented with dl-β-hydroxybutyrate or not. Data from in vivo studies are necessary to clarify whether this observation also extends to living neonates.
Footnotes
This study was supported by grants from the Interdisciplinary Centre for Clinical Research Jena to K.K. and the Federal Ministry of Education and Research to K.H. and O.W.W. We thank Christian A. Hübner for valuable comments on the manuscript, Sindy Beck for excellent technical assistance, and Birgitte Berentsen for language editing.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren CD, Mukhtarov M, Malkov AE, Popova IY, Bregestovski P, Zilberter Y. Energy substrate availability as a determinant of neuronal resting potential, GABA signaling and spontaneous network activity in the neonatal cortex in vitro. J Neurochem. 2010;112:900–912. doi: 10.1111/j.1471-4159.2009.06506.x. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kirmse K, Kirischuk S. Ambient GABA constrains the strength of GABAergic synapses at Cajal-Retzius cells in the developing visual cortex. J Neurosci. 2006;26:4216–4227. doi: 10.1523/JNEUROSCI.0589-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ, Prince DA. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol. 1991;65:247–263. doi: 10.1152/jn.1991.65.2.247. [DOI] [PubMed] [Google Scholar]
- Mason MJ, Simpson AK, Mahaut-Smith MP, Robinson HP. The interpretation of current-clamp recordings in the cell-attached patch-clamp configuration. Biophys J. 2005;88:739–750. doi: 10.1529/biophysj.104.049866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehlig A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot Essent Fatty Acids. 2004;70:265–275. doi: 10.1016/j.plefa.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Stein V, Keating DJ, Maier H, Rinke I, Rudhard Y, Hentschke M, Rune GM, Jentsch TJ, Hübner CA. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheims S, Minlebaev M, Ivanov A, Represa A, Khazipov R, Holmes GL, Ben-Ari Y, Zilberter Y. Excitatory GABA in rodent developing neocortex in vitro. J Neurophysiol. 2008;100:609–619. doi: 10.1152/jn.90402.2008. [DOI] [PubMed] [Google Scholar]
- Rheims S, Holmgren CD, Chazal G, Mulder J, Harkany T, Zilberter T, Zilberter Y. GABA action in immature neocortical neurons directly depends on the availability of ketone bodies. J Neurochem. 2009;110:1330–1338. doi: 10.1111/j.1471-4159.2009.06230.x. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Ivanov A, Bernard C, Holmes GL, Ben-Ari Y, Khazipov R. Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J Neurophysiol. 2003;90:2964–2972. doi: 10.1152/jn.00172.2003. [DOI] [PubMed] [Google Scholar]
- Verheugen JA, Fricker D, Miles R. Noninvasive measurements of the membrane potential and GABAergic action in hippocampal interneurons. J Neurosci. 1999;19:2546–2555. doi: 10.1523/JNEUROSCI.19-07-02546.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Krueger DD, Bordey A. GABA depolarizes neuronal progenitors of the postnatal subventricular zone via GABAA receptor activation. J Physiol. 2003;550:785–800. doi: 10.1113/jphysiol.2003.042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Katz LC. Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron. 1991;6:333–344. doi: 10.1016/0896-6273(91)90243-s. [DOI] [PubMed] [Google Scholar]