

Abstract
Rapid saltatory nerve conduction is facilitated by myelin structure, which is composed of Schwann cells in the peripheral nervous system. Schwann cells drastically change their phenotype following peripheral nerve injury. These phenotypic changes are required for efficient degeneration/regeneration. We previously identified ZNRF1 as an E3 ubiquitin ligase containing a RING finger motif, whose expression is upregulated in the Schwann cells following nerve injury. This suggested that posttranscriptional regulation of protein expression in Schwann cells may be involved in their phenotypic changes during nerve degeneration/regeneration. Here we report the identification of glutamine synthetase (GS), an enzyme that synthesizes glutamine using glutamate and ammonia, as a substrate for E3 activity of ZNRF1 in Schwann cells. GS is known to be highly expressed in differentiated Schwann cells, but its functional significance has remained unclear. We found that during nerve degeneration/regeneration, GS expression is controlled mostly by ZNRF1-dependent proteasomal degradation. We also found that Schwann cells increase oxidative stress upon initiation of nerve degeneration, which promotes carbonylation and subsequent degradation of GS. Surprisingly, we discovered that GS expression regulates Schwann cell differentiation; i.e., increased GS expression promotes myelination via its enzymatic activity. Among the substrates and products of GS, increased glutamate concentration inhibited myelination and yet promoted Schwann cell proliferation by activating metabotropic glutamate receptor signaling. This would suggest that GS may exert its effect on Schwann cell differentiation by regulating glutamate concentration. These results indicate that the ZNRF1-GS system may play an important role in correlating Schwann cell metabolism with its differentiation.
Introduction
Rapid saltatory conduction of electrical signals along the distance of an axon is facilitated by myelin, a lipid-rich ionic insulator that wraps around the axon (Poliak and Peles, 2003; Sherman and Brophy, 2005). In the CNS, myelination is performed by oligodendrocytes, whereas in the peripheral nervous system (PNS), the analogous role is fulfilled by Schwann cells. The malfunction of Schwann cells is associated with a number of human diseases, such as Charcot–Marie–Tooth disease and diabetic neuropathy (Warner et al., 1999). A detailed understanding of Schwann cell biology is crucial for the development of treatments for diseases linked to Schwann cell deficits.
Schwann cells drastically change their phenotype during development, as well as during degeneration/regeneration of the peripheral nerves (Scherer and Salzer, 2001). Schwann cells are derived from neural crest cells during embryonic development, and they differentiate into myelinating or nonmyelinating Schwann cells through a series of intermediate differentiation stages (Jessen and Mirsky, 2005). This phenotypic change is enabled by a tightly orchestrated series of transcriptional regulation events (Salzer, 2008). Some of the key transcriptional factors in Schwann cells have been identified, such as Krox20 (Nagarajan et al., 2001) and Oct6 (Jaegle et al., 1996; Ghislain et al., 2002) that promote myelination; and Sox2 which maintains the undifferentiated phenotype (Le et al., 2005). Such transcriptional factors regulate the expression of molecules that define the differentiation status, while other transcriptional factors promote further differentiation/de-differentiation (Ghislain and Charnay, 2006; Parkinson et al., 2008).
During our efforts to identify molecules in Schwann cells whose expression is induced by peripheral nerve injury, we identified a novel RING finger protein, which we termed ZNRF1 (Araki et al., 2001). ZNRF1, together with ZNRF2, which we identified later, form a family of molecules containing a unique zinc finger–RING finger combination at the C-terminal domain, which is conserved in a wide range of species, including Caenorhabditis elegans and Drosophila. We identified that the ZNRF family of proteins function as an E3 ubiquitin ligase (Araki and Milbrandt, 2003). This suggests that the mechanism of posttranscriptional regulation in Schwann cells also could play a role in controlling their phenotypic changes.
Here we report the identification of glutamine synthetase (GS) as a target for ZNRF1-dependent proteasomal degradation in Schwann cells. GS is the enzyme that synthesizes glutamine using glutamate and ammonia as substrates. In the CNS, GS in astrocytes degrades glutamate released from neurons for synaptic transmission or from neuronal damage, and thereby terminates neurotransmission or decreases excitotoxicity caused by excess glutamate. In the PNS, however, GS is known to be highly expressed in differentiated Schwann cells (Miller et al., 2002), while the role of GS continues to be poorly understood. We demonstrate here that GS expression in Schwann cells is controlled largely by posttranscriptional regulation through ZNRF1-dependent degradation. GS is not only a metabolically relevant enzyme, but it also regulates Schwann cell differentiation by regulating the glutamate concentration. Therefore, the ZNRF1-GS system may play a key role in linking cellular metabolism to differentiation in Schwann cells.
Materials and Methods
Purification and identification of ZNRF1 target proteins.
To generate recombinant ZNRF1 with a hexahistidine tag at the C terminus (His-ZNRF1) in bacteria, a full-length coding sequence of ZNRF1 (amplified from pCMV-ZNRF1-EGFP, which we generated previously) was cloned into pET-30a (Novagen) at the BamHI site. The integrity of the clone was verified by nucleotide sequence analysis. Escherichia coli strain BL21 (DE3) cells were transformed with pET-30a-His-ZNRF1, and protein expression was induced by growth in 1 mm IPTG, according to the manufacturer's protocol (Stratagene). His-ZNRF1 was purified using Ni-NTA beads (Promega). For the affinity chromatography assay, adult mouse cerebrum was homogenized in 10-fold volume of a buffer containing 0.32 m sucrose, 4 mm HEPES-KOH at pH 7.5 and a protease inhibitor cocktail (Nacalai). Cerebrum lysate was subsequently solubilized by incubation with a binding/wash buffer (100 mm HEPES-KOH, pH 7.5, 100 mm imidazole, 60 μm ZnSO4, 1% Triton X-100) containing a protease inhibitor cocktail on a rocking platform at 4°C for 1 h. After removing debris by centrifugation at 15,000 × g for 30 min, the supernatant was incubated at 4°C for 2 h with 50 μl of His-ZNRF1 protein-coupled beads. The beads were then washed four times with the binding/wash buffer, and the bound proteins were eluted with 250 mm imidazole, 100 mm HEPES-KOH, pH 7.5 (500 μl). The eluate was concentrated using Microcon YM-30 (Millipore) and subject to SDS-PAGE separation. Proteins were stained using CBB on a SDS-PAGE gel, and the identity of the major band (indicated in Fig. 1) was determined by peptide mass fingerprinting analysis on a MALDI-TOF mass spectrometer.
Figure 1.

Glutamine synthetase binds to ZNRF1. A, Proteins that bound to His-ZNRF1 from mouse brain extract with or without a Zn2+ chelator, TPEN, were purified and separated by SDS-PAGE. Note that ∼40 kDa ZNRF1-binding protein was detected only in the absence of zinc chelator. B, Neuro-2a cells were transfected with expression plasmids for GS-Myc and/or ZNRF1-Flag, and the cell lysate was subjected to immunoprecipitation. Immunoblot analysis showed that anti-Myc immunoprecipitates contained ZNRF1-Flag and anti-Flag immunoprecipitates contained GS-Myc.
Animals and surgical procedures.
Surgical procedures to obtain mouse sciatic nerve samples were approved by the Committee for Animal Resources in the National Center of Neurology and Psychiatry. Sciatic nerves of 8- to 11-week-old mice were subject to crush injury, as previously described (Araki et al., 2001). Injured nerves were collected at indicated time points after injury. Contralateral nerves were used as uninjured control.
Real-time quantitative RT-PCR analysis and data processing.
Total RNA was isolated from indicated mouse tissues or cells using Trizol (Invitrogen), and first-strand cDNA was obtained with Super Script III, according to the manufacturer's protocol (Invitrogen). Real-time quantitative PCR (qRT-PCR) was performed on the ABI Prism 7300 System (Applied Biosystems). The primers and dye-conjugated probe sets of ZNRF1 (Mm00460000_m1), GS (Mm00725701_s1), and rodent GAPDH were purchased from Applied Biosystems. The sequences of the PCR primers using the SYBR green method were as follows: MBP forward, 5′-ACTCACACACAAGAACTACCCA-3′, and reverse, 5′-AGCTAAATCTGCTGAGGGACA-3′. In both TaqMan and the SYBR green RT-PCR methods, the fluorescence data were quantitatively analyzed using serial dilutions of the control samples included in each reaction to produce a standard curve.
Construction of expression plasmids and mutagenesis.
To generate Flag-tagged expression vectors, ZNRF1-Flag, ZNRF1 (C184A) mutant-Flag, and ZNRF2-Flag, full-length coding sequences of ZNRF1, ZNRF1 bearing C184A mutation, and full-length coding sequence of ZNRF2 were excised from pCMV-ZNRF1-EGFP, pCMV-ZNRF1 (C184A) mutant-EGFP, and pCMV-ZNRF2-EGFP, respectively (Araki and Milbrandt, 2003), with BamHI and ligated in frame into pCMV-Tag1 (Stratagene) at the BamHI site.
GS and Myc-tagged GS (GS-Myc) expression vectors were generated by amplifying the full-length coding region of the GS cDNA (GenBank accession number NM_008131) and cloned into pcDNA3 and pCMV-TAG1, respectively. GS bearing P240S and R341C mutations was generated by PCR-mediated site-direct mutagenesis, as previously described (Araki and Milbrandt, 2003). The mutation sites were designed based on a previously reported mutation (R341C) that causes human congenital glutamine deficiency (Häberle et al., 2005), with an additional mutation (P240S) around the enzymatic activity center. We observed that both mutations together abolished the glutamine synthesizing activity of GS (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Myc-tagged ubiquitin (Myc-Ub) expression vector was generated by amplifying the coding region of the ubiquitin cDNA (GenBank accession number D16554, nucleotides 292-519) and cloned into pcDNA3.
The integrity of the clones was verified by nucleotide sequence analysis.
Lentiviral vector constructs and infection.
The flag-tagged ZNRF1, the myc-tagged wild-type GS, and mutant GS were cloned into the FUGW vector (Lois et al., 2002). Control and ZNRF1-specific short hairpin RNAs in the pLKO.1 puromycin-resistant vector were purchased from Sigma. The following clones were used: ZNRF1 (MISSION shRNA: TRCN0000040745, TRCN0000040747), and nontarget control (MISSION shRNA: SHC002). Lentiviral packaging was performed using HEK293T cells as previously described (Araki et al., 2004). The infection efficiency (GFP-positive cells/total cells) routinely reached >95%.
Antibody generation.
Rabbit anti-ZNRF1 antiserum was raised against a peptide containing 14 aa of the ZNRF1 protein (SYNDDVLTKDAGEC) as an antigen. Specificity of the antiserum was confirmed by immunoblot analysis.
Immunoblot and immunoprecipitation.
Neuro-2a and HEK293 cells were cultured at 80% confluence and transfected using LipofectAMINE 2000 (Invitrogen), according to the manufacturer's protocol. The efficiency of transient transfection in these studies was consistently 80% as assessed by fluorescence derived from a control plasmid expressing EGFP. Cells were treated with a proteasome inhibitor, MG132 (Sigma) or lactacystin (Calbiochem), after transfection when necessary. For immunoblot analysis, cultured cells or tissues were lysed with RIPA buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS. The lysate was subject to SDS-PAGE and immunoblot analysis with a standard procedure (Saitoh et al., 2004) using antibodies against Flag (Cell Signaling), Myc (MBL), actin (BioLegend), GS (Sigma), and myelin basic protein (MBP; Covance).
For detecting protein carbonylation, sciatic nerve samples were lysed with RIPA buffer containing 50 mm DTT. Resultant protein lysates were denatured by the addition of SDS (final concentration 6%), and carbonylated proteins were detected using the OxyBlot-protein oxidation detection kit (Millipore Bioscience Research Reagents), per the manufacturer's protocol.
For immunoprecipitation (IP), cells were lysed in a buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 100 mm ZnSO4, 1% Triton X-100. After centrifugation at 15,000 × g for 30 min, the supernatant was incubated at 4°C for 2 h with anti-Flag M2 (Sigma) or anti-Myc 9E10 (Santa Cruz Biotechnology)-coupled beads. After four washes with the IP wash buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 100 mm ZnSO4, proteins were eluted by boiling in 1× Laemmli buffer before immunoblotting using antibodies against Flag, Myc, or GS.
Primary Schwann cell culture.
Primary Schwann cell cultures were prepared as previously described (Brockes et al., 1979) with minor modifications. Briefly, Schwann cells were obtained from sciatic nerves of postnatal day 2 Sprague Dawley rat pups. Contaminating fibroblasts were removed from culture by treating the cells with 10 μm cytosine arabinoside for 48 h and by complement-mediated cytolysis using anti-Thy1.1 (Serotec) and rabbit complement (Cappel). Schwann cells were propagated on poly-l-lysine (PLL)-coated plates in DMEM supplemented with 10% FBS, 2 μm forskolin, and 20 ng/ml rh-HRG-β1 (Sigma).
In vitro myelination assay.
Dorsal root ganglia (DRGs) were dissected from embryonic day 15 Sprague Dawley rat pups and plated on 24-well plates coated with poly-l-lysine (PLL) and laminin at 2 × 105 cells per well in Neurobasal medium supplemented with B27 and 100 ng/ml 2.5S NGF. Non-neuronal cells were removed by treating the cultures with media containing 5-fluorodeoxyuridine for 10 d. Schwann cells (3 × 105 cells per well) were then plated onto the established neuronal cultures in DMEM supplemented with 10% FBS and 100 ng/ml 2.5S NGF. Ten days later, the mixed culture of Schwann cells and neurons were treated with 50 μg/ml ascorbic acid to induce myelination. For some experiments, myelination was induced with varying concentrations of l-glutamine (0.5–2 mm) or 1 mm l-glutamine with varying concentrations glutamate (0–2 mm). For these experiments using defined concentrations of glutamine and glutamate, DMEM lacking glutamine was used.
Ten days after initiation of myelination, the culture was fixed with 4% paraformaldehyde/PBS for 5 min to prepare the cells for immunocytochemistry. Myelination profiles in the culture were visualized by immunocytochemical detection of myelin basic protein (MBP) using anti-MBP antibody followed by Alexa 594-conjugated anti-mouse antibody (Invitrogen). For quantitative analysis, myelination profiles in five randomly chosen fields using a 20× objective lens were counted, and the number of myelinated nerve fibers per arbitrary unit area was calculated. Statistical analysis was performed by Student's t test. All data are reported as mean ± SD; p value of <0.05 was considered significant.
GS enzyme activity assay.
GS enzymatic activity was assessed using a previously described method to measure γ-glutamylhydroxamate synthesized from glutamine and hydroxylamine (Häberle et al., 2005). Briefly, Neuro-2a cells were transfected with GS-Myc or GS-mutant-Myc. After 24 h, the cells were lysed with a buffer containing 20 mm imidazole-HCl, pH 6.8, 150 mm KCl, and 0.1 mm EDTA. After removing debris by centrifugation at 15,000 × g for 30 min, the protein was incubated with an equal amount of buffer containing 60 mm imidazole-HCl, 100 mm l-glutamine, 15 mm hydroxylamine, pH 6.8, 20 mm sodium arsenate, 0.5 mm MnCl2, and 0.2 mm ADP, for 15 min at 37°C. The reaction was terminated by the addition of a buffer containing 0.37 m FeCl3, 0.3 m trichloroacetic acid, and 0.6 m HCl. The solution was cleared by centrifugation at 15,000 × g at 4°C, and the glutamylhydroxamate was measured in the supernatant at 505 nm.
Cell proliferation assay.
Schwann cells were seeded on collagen-coated 24 well plates at 5 × 104 cells per well in DMEM supplemented with 10% FBS and 4 mm l-glutamine. Twenty-four hours later, the medium was changed to DMEM containing varying concentrations of l-glutamine (0.5–2 mm) or 1 mm l-glutamine and varying concentrations of glutamate (0–2 mm). For glutamate receptors or transporters blockade experiments, the medium was changed to DMEM containing 1 mm l-glutamine and 1 mm glutamate with one of the following reagents at indicated concentrations: mGluR receptor antagonist (±)-α-methyl-4-carboxyphenylglycine (MCPG, 100 μm), AMPA receptor antagonist 6-cyano-7-nitroquinoxaline- 2,3-dione (CNQX, 25 μm), NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,b]cyclohepten-5,10-imine hydrogen maleate (MK-801, 20 μm), EAAT2 inhibitor dihydrokainate (DHK, 100 μm) or EAAT1/3 inhibitor l-serine-O-sulfate (l-SOS, 50 μm). For experiments using trans-(1S,3R)-(±)1-amino-1,3-cyclopentanedicarboxylic acid (t-ACPD, an mGluR agonist), the medium was changed to DMEM containing 1 mm l-glutamine with various concentrations of t-ACPD. All antagonists, agonists, or inhibitors were purchased from Sigma.
The proliferation assay was initiated 20 h after the medium change by adding BrdU (20 μm) to the medium. Cells were maintained for another 4 h and fixed as described above. The cells were incubated at 4°C overnight with primary antibodies against anti-BrdU (Sigma), followed by incubation with Alexa 594-conjugated anti-mouse antibody (Invitrogen) for 1 h at room temperature for visualization.
Results
Identification of glutamine synthetase as a ZNRF1-associated protein
We previously identified ZNRF1, an E3 ubiquitin ligase, whose expression is induced by peripheral nerve injury in Schwann cells of the distal nerve stump (Araki et al., 2001). The expression profile of ZNRF1 in peripheral nerves indicates that ZNRF1 belongs to a family of molecules that define promyelinating Schwann cells. We also surveyed the expression profile of ZNRF1 to uncover that ZNRF1 is constitutively expressed in neurons throughout the nervous system (Araki and Milbrandt, 2003). To understand the biological role of ZNRF1 expression, we sought to identify targets for ZNRF1-dependent proteasomal degradation in the nervous system. For this purpose, we isolated proteins from mouse brain lysate that bind to the full-length recombinant ZNRF1 generated in E. coli. The biological activity of zinc finger-containing proteins critically depends on the presence of Zn2+ for the formation of properly folded and active forms (Klug and Schwabe, 1995). And so, we sought to isolate proteins that bind to ZNRF1 in the presence of Zn2+, but do not in the presence of selective Zn2+ chelator, N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN) (Fig. 1 A). The identity of a candidate molecule at ∼40 kDa was determined to be GS by peptide mass fingerprinting analysis. To confirm the affinity between ZNRF1 and GS in mammalian cells, we performed immunoprecipitation of GS and ZNRF1. Expression constructs for GS and ZNRF1 were cotransfected in Neuro-2a cells. We found that ZNRF1 coimmunoprecipitated with GS. In a reciprocal immunoprecipitation, GS was present in ZNRF1 immunoprecipitates (Fig. 1 B). Together, these results indicate that GS is associated with ZNRF1.
GS degradation is promoted by a ZNRF1-dependent proteasomal pathway
The association of ZNRF1 with GS suggests that GS is a substrate for ZNRF1 ubiquitin ligase. To demonstrate ZNRF1-dependent ubiquitination of GS, we transfected expression plasmids for ZNRF1, GS, and ubiquitin, and performed immunoprecipitation of ubiquitinated proteins from the transfected cell lysate. We detected high-molecular-weight molecules immunoreactive for GS in ubiquitin immunoprecipitates (Fig. 2 A). This indicates that GS is ubiquitinated in a ZNRF1-dependent manner. To demonstrate ZNRF1-dependent degradation of GS, we cotransfected expression plasmids for the full-length GS together with wild-type ZNRF1, ZNRF1 carrying C184A mutation, or ZNRF2. ZNRF1 (C184A) is a mutation that disrupts the RING domain of ZNRF1, and thereby inactivates ZNRF1's E3 activity, likely by disrupting ZNRF1's interaction with E2 enzymes (Araki and Milbrandt, 2003). We found that expression of wild-type ZNRF1 specifically downregulated GS expression, and that this effect was blocked by MG132 and lactacystin the same way, both of which serve as proteasome inhibitors (Fig. 2 C). Mutated ZNRF1 or ZNRF2 showed no effect on GS expression (Fig. 2 B). Together, these results indicate that GS serves as a specific target for the E3 ubiquitin ligase function of ZNRF1, and is thereby degraded in proteasome.
Figure 2.

GS is a substrate for ZNRF1-dependent ubiquitination and proteasomal degradation. A, HEK293 cells were transfected with expression plasmids for GS, ZNRF1-Flag, and/or Myc-tagged ubiquitin, and treated with MG132 (20 μm) for 6 h. Cell lysates were subject to immunoprecipitation with anti-Myc, and ubiquitinated GS in the resultant immunoprecipitates was analyzed by immunoblot using an anti-GS antibody. Note that ubiquitinated GS was detected as a smear in the high-molecular-weight range as indicated. B–D, Neuro-2a cells were transfected with expression plasmids for GS-Myc together with either ZNRF1-Flag, ZNRF1(C184A)-Flag, ZNRF2-Flag, or an empty vector (MOCK), and the cell lysates were subjected to immunoblot analysis to detect GS-Myc expression. Actin served as the loading control. Note that GS immunoreactivity became significantly lower only when wild-type ZNRF1 was present (B). When the cells were treated with 10 μm MG132 or 10 μm lactacystin after transfection for 24 h before lysis, ZNRF1-dependent decrease in GS was inhibited (C). In D, ZNRF1-dependent degradation of GS increased its turnover rate. Neuro-2a cells were transfected with GS-Myc plasmid together with wild-type ZNRF1-Flag or ZNRF1 (C184A)-Flag plasmid. Cell lysates were generated at indicated hours after the addition of cycloheximide and subject to immunoblot analysis to detect GS. Actin served as the loading control. Representative immunoblot and quantitative analysis of GS expression normalized to GS levels at 0 h are shown.
Proteasomal degradation of a cellular protein generally leads to its increased turnover rate. To determine whether ZNRF1-mediated degradation of GS affects its turnover, we performed cycloheximide-chase experiments in cultured cells. Wild-type ZNRF1 or ZNRF1 (C184A) was cotransfected with GS, and the expression level of GS was determined up to 3 h after the addition of cycloheximide. We found that degradation of GS was significantly facilitated by the presence of wild-type ZNRF1 (Fig. 2 D). This suggests that ZNRF1-dependent GS degradation increases its turnover rate, and thus involved in the posttranslational regulation of GS expression.
GS expression is regulated posttranslationally in Schwann cells
GS is known to be expressed in differentiated Schwann cells (Miller et al., 2002). Given the induced expression of ZNRF1 in Schwann cells after nerve injury as we previously reported, the GS expression in Schwann cell suggests that GS may be degraded in injured nerve Schwann cells. To examine whether GS serves as a physiological substrate for ZNRF1 in Schwann cells, we first examined the expression profile of GS after mouse peripheral nerve injury. We used a previously established sciatic nerve crush model, and examined expression of ZNRF1 and GS at mRNA and protein levels in a nerve segment distal to the site of injury. As we described previously, expression of ZNRF1 mRNA is highly induced after nerve injury, and comes back down as nerve regeneration ensues. ZNRF1 protein expression also changes, accordingly. In contrast, we found that GS expression is inversely correlated to ZNRF1 expression, just as we had demonstrated in cultured cells. Surprisingly, GS mRNA was slightly downregulated (around 50% of the control level), which is likely to contribute in part the initial decline of GS protein after nerve injury, but constantly expressed during nerve degeneration and regeneration in Schwann cells. GS protein expression was significantly downregulated after nerve injury to an undetectable level (Fig. 3). These results support the hypothesis that GS is a physiological substrate of ZNRF1 in Schwann cells.
Figure 3.
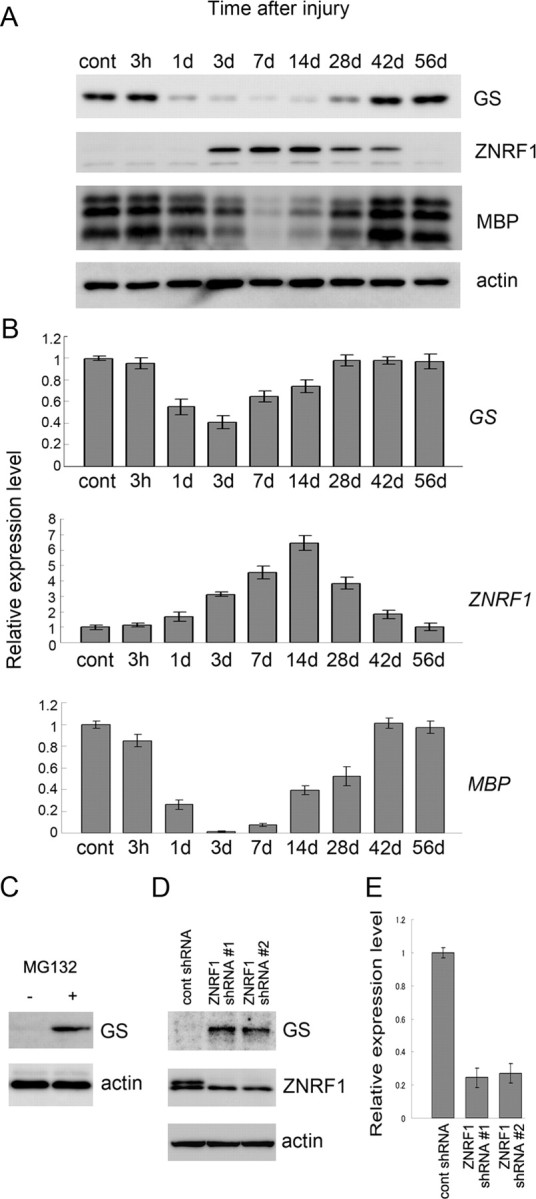
GS expression in Schwann cell is regulated mainly by ZNRF1-dependent proteasomal degradation. A, B, Adult mouse sciatic nerves were crushed and the distal nerve stump was harvested at the indicated time points after crush for immunoblot analysis (A) or quantitative RT-PCR (B) to detect expression of GS, ZNRF1, or MBP. Actin served as the loading control in A. In B, each PCR was normalized to the amount of GAPDH expression. Quantitative changes of expression are shown relative to the expression level in the uninjured nerve control. All reactions were performed in triplicate. Mean expression levels normalized to control ± SD are indicated for each data point. C–E, Primary Schwann cells treated with or without MG132 for 24 h (C) or infected with lentiviral vector for ZNRF1 shRNA (#1 and #2 represent two independent shRNA sequences for ZNRF1) (D) were analyzed for GS protein expression and harvested for immunoblot analysis. shRNA-mediated downregulation of ZNRF1 expression used in this study was confirmed by immunoblot (D) as well as qRT-PCR (E). Actin served as the loading control (C, D). qRT-PCRs were performed in triplicate. Mean expression levels normalized to control ± SD are indicated for each data point.
To show more directly that ZNRF1 induces proteasomal degradation of GS in Schwann cells, we modulated proteasomal activity and ZNRF1 expression in Schwann cells to examine whether GS expression level was affected by these treatments. We found that GS protein expression level in primary cultured Schwann cells is clearly increased in the presence of a proteasome inhibitor, MG132 (Fig. 3 C). Furthermore, we observed that Schwann cells infected with lentiviral vector for ZNRF1 shRNA significantly increased GS protein levels (Fig. 3 D,E). We obtained similar increase in GS protein expression using two independent shRNA sequences. Collectively, these results strongly suggest that GS is a physiological substrate of ZNRF1 in Schwann cells; and that unlike molecules that define the myelinating Schwann cell phenotype which are rapidly downregulated after nerve injury, such as P0 and MAG, GS expression in Schwann cells is primarily controlled posttranslationally by ZNRF1-dependent proteasomal degradation.
GS degradation is induced by increased oxidative stress in Schwann cells after injury
GS is known to be vulnerable to oxidization-induced inactivation under cellular oxidative stress, and is regarded as a well established example of proteins that are degraded in proteasome after oxidization (Stadtman, 1992; Adamo et al., 1999; Labow et al., 2001). The oxidization-induced proteasomal degradation of GS is observed in a wide range of organisms, but the E3 ubiquitin ligase for GS degradation has not yet been identified. Our current finding of proteasomal degradation of GS in Schwann cells suggests that oxidative stress may increase in Schwann cells after injury. To examine whether GS degradation is induced by oxidative stress in Schwann cells, we analyzed protein oxidization status in injured mouse sciatic nerve. Protein oxidization was determined by detecting the carbonylated protein side chain. We found that oxidized proteins were at an almost undetectable level in control nerves and increased significantly after nerve injury (Fig. 4 A; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). The expression profile of oxidized proteins is similar to that of ZNRF1 after injury. To confirm GS oxidization after nerve injury, we performed immunoprecipitation of GS from injured mouse sciatic nerve lysate. GS oxidization was determined by detecting the carbonylated protein side chain. We found that oxidized GS were at an almost undetectable level in control nerves and increased significantly after nerve injury (Fig. 4 B). To examine whether GS oxidization increases ZNRF1-mediated GS degradation, we cotransfected expression plasmids for GS together with wild-type ZNRF1 or empty vector (MOCK) to Neuro-2A cells and treated the cells with hydrogen peroxide to increase cellular oxidative stress. We found that ZNRF1-dependent proteasomal degradation of GS is significantly facilitated by the presence of hydrogen peroxide (Fig. 4 C). These results suggest that GS is oxidized under the increased levels of oxidative stress in degenerating nerve Schwann cells and subsequently subject to proteasomal degradation.
Figure 4.
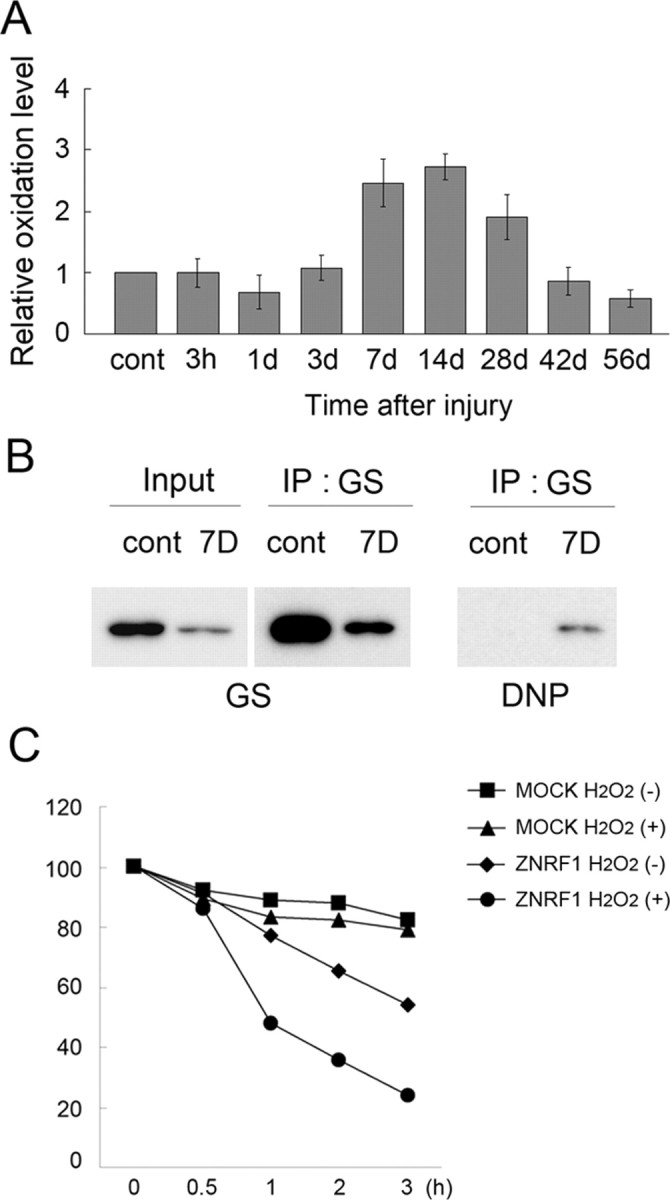
Increased oxidative stress in Schwann cells after nerve injury induces GS oxidization and degradation. Adult mouse sciatic nerves were crushed, and the distal nerve stump was collected at the indicated time after crush. A, Tissue lysates were analyzed for oxidized protein using OxyBlot-protein oxidation detection kit, in which carbonyl groups in the oxidized protein side chains were derivatized to 2,4-dinitrophenylhydrazone (DNP) by reacting with 2,4-dinitrophenylhydrazine (DNPH). The DNP-derivatized protein samples were subjected to immunoblot analysis using an anti-DNP antibody. Carbonylated protein expression level at each indicated time point after injury was normalized to actin and shown relative to the expression level in uninjured control nerves. B, Lysates of nerve tissues (uninjured control and injured nerve 7 d after crush) were subject to immunoprecipitation to detect DNP-derivatized GS. C, ZNRF1-dependent GS degradation in Schwann cells is induced by oxidative stress. Neuro-2a cells transfected with expression plasmids for GS-Myc together with ZNRF1-Flag or an empty vector (MOCK) were treated with H2O2 for 4 h. Both H2O2-treated and untreated cell lysates were collected at indicated hours after the addition of cycloheximide and subjected to immunoblot analysis to detect GS. Quantitative analysis of GS expression normalized to expression levels at 0 h is shown. Note that GS decrease in the presence of ZNRF1 is significantly accelerated by H2O2, suggesting that oxidized GS is preferentially subject to ZNRF1-dependent proteasomal degradation.
Posttranscriptional regulation of GS expression regulates differentiation of myelinating Schwann cells
We demonstrated here that regulation of GS expression via ZNRF1-dependent proteasomal degradation is observed in Schwann cells during nerve degeneration/regeneration. To establish the causal relationship between regulation of GS expression and differentiation of myelinating Schwann cells, we examined how ZNRF1-mediated control of GS expression affects Schwann cell myelination by using in vitro myelination experiments. Schwann cells lentivirally overexpressing GS were cocultured with DRG neurons, and myelination was induced by adding ascorbic acid in culture. We observed increased myelination by overexpressing GS in Schwann cells (Fig. 5 A,B). To examine whether ZNRF1-mediated control of GS expression affects myelination in vitro, we examined myelination by using Schwann cells expressing ZNRF1 shRNA. We found that decreased expression of ZNRF1 also induced myelination, similar to the effect seen with GS overexpression (Fig. 5 C,D). Together these results suggest that ZNRF1-mediated regulation of GS protein expression levels affects differentiation of myelinating Schwann cells, and increased GS expression enabled by decreased expression of ZNRF1 promotes myelination.
Figure 5.
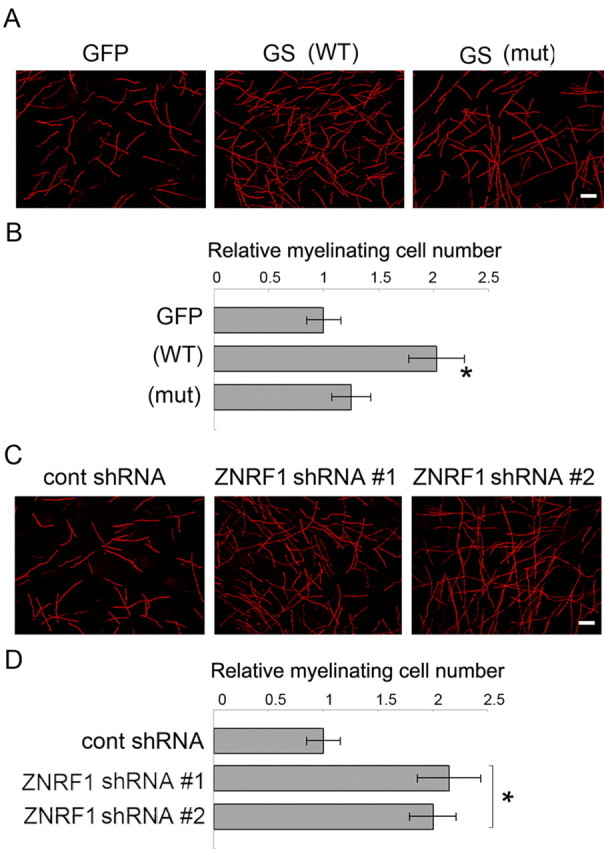
Increased GS expression in Schwann cells promotes myelination in vitro. In vitro myelination was performed using a coculture of DRG neurons and Schwann cells overexpressing wild-type GS, mutated GS bearing P240S and R341C mutations, or GFP-expressing only (A, B). In another set of experiments, in vitro myelination was performed using a coculture of DRG neurons and Schwann cells expressing ZNRF1 shRNA (#1 and #2 represent two independent shRNA sequences for ZNRF1) or control (C, D). In both experiments, myelination was visualized by MBP immunocytochemistry. Representative images of myelination for each condition are shown in A and C. Myelinating profiles in each experimental condition were counted in five randomly selected fields under a microscope using a 20× objective lens. The number of profiles relative to GFP-expressing control (A) or control shRNA-expressing control (C) is shown (mean of 5 independent experiments ± SD) in B and D. Scale bar, 100 μm.
GS likely promotes myelination via degrading glutamate
We found that the promotion of the differentiated phenotype of Schwann cells by increased GS expression is mediated by increased GS enzymatic activity in Schwann cells, as a result of witnessing that the mutated GS lacking its enzymatic activity did not promote myelination (Fig. 5; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The GS enzymatic activity-induced enhancement of myelination suggested that an increased level of GS enzymatic product and/or a decreased level of GS substrate mediates the myelination-promoting effect. To analyze the mechanism of GS expression-mediated promotion of myelination, we examined the effect of glutamine and glutamate on myelination when they are added to the mixed culture of DRG neurons and Schwann cells (Fig. 6). We first examined the effect of differential concentrations of glutamine (the product of GS) on in vitro myelination. We found that increased glutamine concentrations do not enhance myelination. We then examined the effect of glutamate (a GS substrate) on myelination in vitro (Fig. 6), and found that myelination was inhibited by increased concentrations of glutamate in the medium. This tendency is in agreement with the effect of GS overexpression in Schwann cells. Together, these results suggest that GS expression promotes myelination through a reduction of GS substrates.
Figure 6.

Glutamate levels affect the phenotype of myelinating Schwann cells. A–D, In vitro myelination was performed using a coculture of DRG neurons and Schwann cells with indicated concentrations of glutamine (A, B) or 1 mm glutamine together with indicated concentrations of glutamate (C, D). In both experiments, myelination was visualized by MBP immunocytochemistry. Representative images of myelination for each condition are shown in A and C, and quantitative analyses are shown in B, D (performed as described in Fig. 5; normalized to myelination in 0.5 mm glutamine in B, and no glutamate condition in D). E–H, Schwann cell proliferation was monitored with different concentrations of glutamine and glutamate in the culture medium by BrdU pulse labeling followed by immunocytochemistry using an anti-BrdU antibody. Representative images of BrdU-labeled Schwann cells with indicated concentrations of glutamine (E) or 1 mm glutamine together with indicated concentrations of glutamate (G) are shown with overlaid nuclear staining by DAPI. In F and H, quantification data are shown. Mean percentages of BrdU-positive Schwann cells in five independent experiments in each condition ± SD are shown. Statistical analysis was performed by Student's t test. Asterisks and “n.s.” indicate significant (p < 0.05) and nonsignificant differences, respectively. Scale bar, 100 μm.
To further correlate the effect of GS substrates/product with Schwann cell differentiation status, we tested whether GS substrates/product change Schwann cell proliferation, which is an undifferentiated Schwann cell phenotype. We found that Schwann cell proliferation was not affected by varying concentrations of glutamine (Fig. 6). On the other hand, Schwann cell proliferation was increased under higher glutamate concentrations, which proves inhibitory to myelination (Fig. 6). Increased ammonium concentrations seemed toxic to Schwann cells (data not shown). Collectively, these results demonstrate that the concentration of the GS product, glutamine, is unrelated to Schwann cell differentiation status, while differential concentrations of the GS substrate, glutamate, are correlated to the differentiation status of myelinating Schwann cells.
It is possible that extracellular glutamate can affect Schwann cells via a variety of pathways. To determine which types of glutamate receptors and/or transporters mediated the effect of glutamate-induced Schwann cell proliferation, we designed a receptor or transporter blockade experiment using specific glutamate receptor antagonists or inhibitors of glutamate transporters. Schwann cells were cultured in a medium containing glutamate and one of the following reagents: MCPG (a metabotropic receptor antagonist), MK-801 (an NMDA receptor antagonist), CNQX (a kainate–AMPA receptor antagonist), or DHK and l-SOS (blockers of glutamate transporter). We observed that the glutamate-induced increase of Schwann cell proliferation was blocked by MCPG, but not by any other compound. These results suggest that glutamate-induced proliferation of Schwann cells is mediated by mGluRs on Schwann cells. To further substantiate the role of mGluRs on Schwann cells, we examined Schwann cell proliferation in response to an mGluR agonist, t-ACPD. We found that Schwann cells proliferation was induced by t-ACPD in a dose-dependent manner (Fig. 7). Together, these results suggest that glutamate potentiates Schwann cell proliferation via mGluR-mediated signaling, and that this signaling may play an important role in de-differentiation of myelinating Schwann cells after nerve injury.
Figure 7.
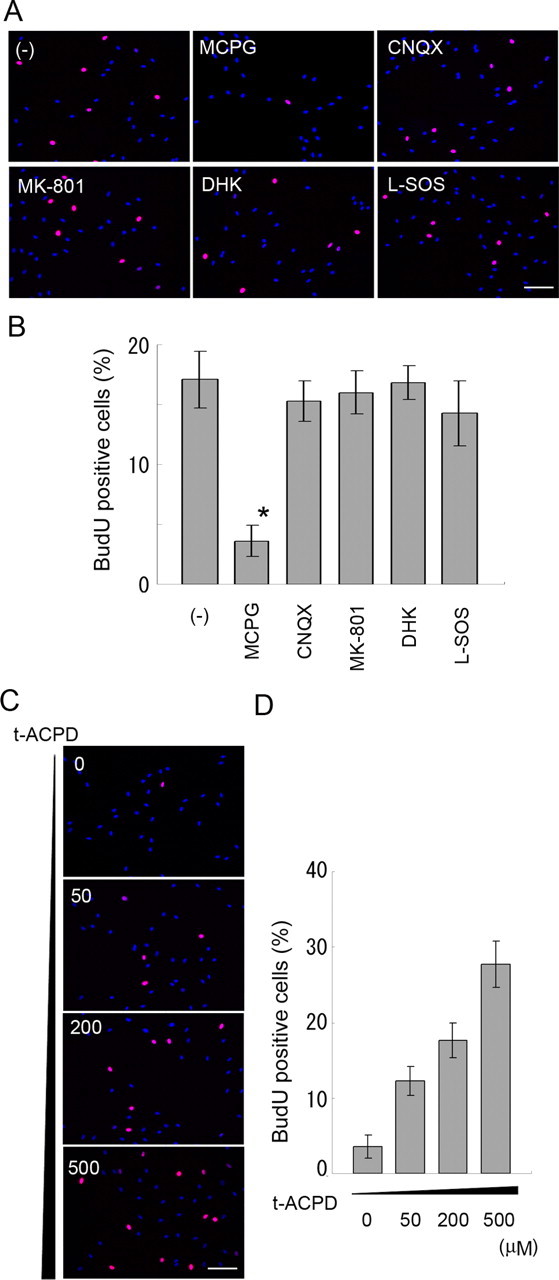
mGluR signaling is involved in the effect of glutamate. Schwann cell proliferation was monitored under presence of 1 mm glutamine together with inhibitors of glutamate receptors or transporters (A, B) or different concentrations of t-ACPD (C, D) by BrdU pulse labeling followed by immunocytochemistry using an anti-BrdU antibody. Results from quantitative analyses were performed as described in Fig. 6 and are shown in B and D. Scale bar, 100 μm.
Discussion
We previously identified that the ZNRF family of E3 ubiquitin ligases, comprised of ZNRF1 and 2, is expressed in the nervous system. ZNRF1 was originally identified as a molecule whose expression is induced in Schwann cells after nerve injury (Araki et al., 2001; Araki and Milbrandt, 2003). Here we report the identification of GS as a physiological substrate of ZNRF1. We found that GS protein expression shows a pattern similar to molecules designating myelinating Schwann cells, such as P0 and MBP (Gupta et al., 1988), but GS expression is controlled mainly posttranscriptionally by ZNRF1-dependent proteasomal degradation. Degradation of GS is induced by oxidative stress generated during the de-differentiating of Schwann cells. Finally, we found that GS expression promotes myelination by degrading its substrates.
Transcriptional and posttranscriptional regulation of GS expression has been described in a variety of organs, and different means of regulation seem to be required specific to tissue type, as well as kinds of stimulation (van Straaten et al., 2006). Induction of GS mRNA by glucocorticoid has been described for many cell types, including L6 and 3T3 L1 cells; as well as for many organs, such as skeletal muscle, lung, and heart (Abcouwer et al., 1995). Posttranscriptional regulation of GS expression is reported in various settings involving metabolic changes, such as metabolic acidosis and changes in tissue glutamine levels (Labow et al., 2001). Induction of GS enzyme activity under low glutamine conditions by posttranscriptional mechanisms, for example, has been commonly observed in a variety of organs such as skeletal muscle, kidney, and lung (Feng et al., 1990). Among them, proteasomal degradation of GS has been shown in only some cases, and the underlying molecular details that control the posttranscriptional regulation of GS expression have remained largely unclear. Here we suggest that GS degradation in Schwann cells is mediated by the ZNRF1-dependent ubiquitin-proteasome system. We previously showed that ZNRF1 expression in normal tissues is restricted to only the nervous system and a few other organs (Araki et al., 2001), and therefore, it is possible that mechanisms other than ZNRF1-dependent proteasomal degradation may be involved in the posttranscriptional regulation of GS expression in some organs. Another possible scenario is that ZNRF1 is induced only on demand. ZNRF1 expression may be undetectable in a steady state and induced in response to stimulation as observed in Schwann cells. Further experiments are necessary to clarify the entire regulatory mechanism of GS expression by protein degradation.
Protein oxidization is observed during normal aging (Agarwal and Sohal, 1993; Sohal et al., 1993), as well as in neuronal disorders such as ischemia/reperfusion injuries (Oliver et al., 1990). Therefore, protein oxidization has often been described in association with oxidative damage of cellular functions. Here we demonstrate that increased protein oxidization induces GS degradation during the de-differentiation of Schwann cells. This suggests that changes in protein oxidization status may affect cellular amino acid metabolism and result in modification of cellular differentiation. A variety of cellular differentiation and fate determination mechanisms have been associated with energy metabolism and redox status (Naujokat and Sarić, 2007). For example, changing levels of protein oxidization are detected in mouse embryonic stem cells during differentiation (Hernebring et al., 2006). Regulation of enzymatic function by inactivation and/or degradation in such situations may also be initiated by increased protein oxidization, and thereby affect the cellular differentiation status, as we showed here using Schwann cells as an example.
We described here that the regulation of GS expression and resultant changes of glutamate metabolism play a role in Schwann cell differentiation. It seems that this ZNRF1-GS-mediated regulation of Schwann cell differentiation needs to be orchestrated by the redox status of Schwann cells during nerve degeneration/regeneration. We showed here that GS upregulation by ZNRF1 downregulation or that GS overexpression in Schwann cells results in increased myelination. On the other hand, we found no detectable reduction of myelination when we used ZNRF1-overexpressing Schwann cells (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). We presume that this is because myelination needs to be induced by the addition of ascorbic acid, a reducing agent, to the culture medium in in vitro myelination experiments, while ZNRF1-induced GS degradation requires oxidative cellular condition. These results may further suggest that cellular metabolic status (such as redox status) of Schwann cells needs to be coregulated with transcription of critical genes to achieve Schwann cell phenotypic changes during nerve degeneration/regeneration.
We report here that de-differentiated Schwann cells proliferate in response to glutamate. Schwann cells are a source of glutamate in themselves, since previous reports showed that cultured Schwann cells secrete glutamate (Wu et al., 2005), although the significance of glutamate secretion is unknown. In the nervous system, the importance of glutamate metabolism has previously been discussed mostly in relation to excitotoxicity and/or glutamatergic neurotransmission (Rosenberg et al., 1992). In the CNS, glutamate is metabolized to glutamine only in astrocytes (Levy, 2005). This activity of GS plays an important role in the cessation of neural transmission and the reduction of glutamate toxicity against neurons. Thus, the role of glutamate in the nervous system has been discussed only in neurons, and glutamate metabolism in astrocytes is thought to be required for support to neurons. In the PNS, the significance of GS expression in Schwann cells has remained unclear, since the glutamate–glutamine shuttle has not been clearly described. Further investigation may reveal functional significance of glutamate on metabolism of other types of glial cells as well during development and degeneration of the nervous system.
In conclusion, we found that GS is a substrate for ZNRF1 E3 ubiquitin ligase. Through analysis of ZNRF1-dependent regulation of GS expression, we identified the function of GS on Schwann cell differentiation and proliferation via regulation of the glutamate level. Further experiments are needed to clarify the significance of specific metabolic conditions that are correlated to Schwann cell differentiation.
Footnotes
T.A. is supported by grants from the Ministry of Health, Labour, and Welfare, National Institute of Biomedical Innovation (Program for Promotion of Fundamental Studies in Health Sciences), Grant-in-Aid for Scientific Research (KAKENHI; 18022051), Japan Health Sciences Foundation, Ichiro Kanehara Foundation, and Novartis Foundation for the Promotion of Science (Japan). We thank Dr. Shuji Wakatsuki for his technical advice, and all the members of our laboratory for many helpful discussions.
References
- Abcouwer SF, Bode BP, Souba WW. Glucocorticoids regulate rat glutamine synthetase expression in a tissue-specific manner. J Surg Res. 1995;59:59–65. doi: 10.1006/jsre.1995.1132. [DOI] [PubMed] [Google Scholar]
- Adamo AM, Pasquini LA, Moreno MB, Oteiza PI, Soto EF, Pasquini JM. Effect of oxidant systems on the ubiquitylation of proteins in the central nervous system. J Neurosci Res. 1999;55:523–531. doi: 10.1002/(SICI)1097-4547(19990215)55:4<523::AID-JNR12>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Sohal RS. Relationship between aging and susceptibility to protein oxidative damage. Biochem Biophys Res Commun. 1993;194:1203–1206. doi: 10.1006/bbrc.1993.1950. [DOI] [PubMed] [Google Scholar]
- Araki T, Milbrandt J. ZNRF proteins constitute a family of presynaptic E3 ubiquitin ligases. J Neurosci. 2003;23:9385–9394. doi: 10.1523/JNEUROSCI.23-28-09385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Nagarajan R, Milbrandt J. Identification of genes induced in peripheral nerve after injury. Expression profiling and novel gene discovery. J Biol Chem. 2001;276:34131–34141. doi: 10.1074/jbc.M104271200. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Brockes JP, Fields KL, Raff MC. Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res. 1979;165:105–118. doi: 10.1016/0006-8993(79)90048-9. [DOI] [PubMed] [Google Scholar]
- Feng B, Shiber SK, Max SR. Glutamine regulates glutamine synthetase expression in skeletal muscle cells in culture. J Cell Physiol. 1990;145:376–380. doi: 10.1002/jcp.1041450224. [DOI] [PubMed] [Google Scholar]
- Ghislain J, Charnay P. Control of myelination in Schwann cells: a Krox20 cis-regulatory element integrates Oct6, Brn2 and Sox10 activities. EMBO Rep. 2006;7:52–58. doi: 10.1038/sj.embor.7400573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain J, Desmarquet-Trin-Dinh C, Jaegle M, Meijer D, Charnay P, Frain M. Characterisation of cis-acting sequences reveals a biphasic, axon-dependent regulation of Krox20 during Schwann cell development. Development. 2002;129:155–166. doi: 10.1242/dev.129.1.155. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Poduslo JF, Mezei C. Temporal changes in PO and MBP gene expression after crush-injury of the adult peripheral nerve. Brain Res. 1988;464:133–141. doi: 10.1016/0169-328x(88)90005-8. [DOI] [PubMed] [Google Scholar]
- Häberle J, Görg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, Gelot A, Suc AL, Höhne W, Schliess F, Häussinger D, Koch HG. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med. 2005;353:1926–1933. doi: 10.1056/NEJMoa050456. [DOI] [PubMed] [Google Scholar]
- Hernebring M, Brolén G, Aguilaniu H, Semb H, Nyström T. Elimination of damaged proteins during differentiation of embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103:7700–7705. doi: 10.1073/pnas.0510944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaegle M, Mandemakers W, Broos L, Zwart R, Karis A, Visser P, Grosveld F, Meijer D. The POU factor Oct-6 and Schwann cell differentiation. Science. 1996;273:507–510. doi: 10.1126/science.273.5274.507. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Klug A, Schwabe JW. Protein motifs 5. Zinc fingers. FASEB J. 1995;9:597–604. [PubMed] [Google Scholar]
- Labow BI, Souba WW, Abcouwer SF. Mechanisms governing the expression of the enzymes of glutamine metabolism—glutaminase and glutamine synthetase. J Nutr. 2001;131:2467S–2474S. doi: 10.1093/jn/131.9.2467S. discussion 2486S–2487S. [DOI] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci U S A. 2005;102:2596–2601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy HL. Metabolic disorders in the center of genetic medicine. N Engl J Med. 2005;353:1968–1970. doi: 10.1056/NEJMe058227. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Miller KE, Richards BA, Kriebel RM. Glutamine-, glutamine synthetase-, glutamate dehydrogenase- and pyruvate carboxylase-immunoreactivities in the rat dorsal root ganglion and peripheral nerve. Brain Res. 2002;945:202–211. doi: 10.1016/s0006-8993(02)02802-0. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron. 2001;30:355–368. doi: 10.1016/s0896-6273(01)00282-3. [DOI] [PubMed] [Google Scholar]
- Naujokat C, Sarić T. Concise review: role and function of the ubiquitin-proteasome system in mammalian stem and progenitor cells. Stem Cells. 2007;25:2408–2418. doi: 10.1634/stemcells.2007-0255. [DOI] [PubMed] [Google Scholar]
- Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci U S A. 1990;87:5144–5147. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Amin S, Leitner M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci. 1992;12:56–61. doi: 10.1523/JNEUROSCI.12-01-00056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh F, Tian QB, Okano A, Sakagami H, Kondo H, Suzuki T. NIDD, a novel DHHC-containing protein, targets neuronal nitric-oxide synthase (nNOS) to the synaptic membrane through a PDZ-dependent interaction and regulates nNOS activity. J Biol Chem. 2004;279:29461–29468. doi: 10.1074/jbc.M401471200. [DOI] [PubMed] [Google Scholar]
- Salzer JL. Switching myelination on and off. J Cell Biol. 2008;181:575–577. doi: 10.1083/jcb.200804136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Salzer JL. Axon-Schwann cell interactions during peripheral nerve degeneration and regeneration. In: Jessen KR, Richardson WD, editors. Glial cell development. Ed 2. London: Oxford UP; 2001. pp. 299–330. [Google Scholar]
- Sherman DL, Brophy PJ. Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci. 2005;6:683–690. doi: 10.1038/nrn1743. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Agarwal S, Dubey A, Orr WC. Protein oxidative damage is associated with life expectancy of houseflies. Proc Natl Acad Sci U S A. 1993;90:7255–7259. doi: 10.1073/pnas.90.15.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- van Straaten HW, He Y, van Duist MM, Labruyère WT, Vermeulen JL, van Dijk PJ, Ruijter JM, Lamers WH, Hakvoort TB. Cellular concentrations of glutamine synthetase in murine organs. Biochem Cell Biol. 2006;84:215–231. doi: 10.1139/o05-170. [DOI] [PubMed] [Google Scholar]
- Warner LE, Garcia CA, Lupski JR. Hereditary peripheral neuropathies: clinical forms, genetics, and molecular mechanisms. Annu Rev Med. 1999;50:263–275. doi: 10.1146/annurev.med.50.1.263. [DOI] [PubMed] [Google Scholar]
- Wu SZ, Jiang S, Sims TJ, Barger SW. Schwann cells exhibit excitotoxicity consistent with release of NMDA receptor agonists. J Neurosci Res. 2005;79:638–643. doi: 10.1002/jnr.20401. [DOI] [PubMed] [Google Scholar]