

Abstract
The prostaglandin E2 receptor, EP2, plays an important role in physiology and in a variety of pathological conditions. Studies indicate that EP2 is pro-inflammatory in chronic peripheral and central nervous system disease and cancer models. Thus, targeting the EP2 receptor with small molecules could be a therapeutic strategy for treating inflammatory diseases and cancer. We recently reported a novel class of competitive antagonists of the EP2 receptor. However earlier leads displayed low selectivity against the DP1 prostanoid receptor, moderate plasma half-life and low aqueous solubility, which renders them sub-optimal for testing in animal models of disease. We now report a novel compound TG8–69, which has suitable drug-like properties. We present synthesis, lead-optimization studies, pharmacological characterization, and anti-inflammatory properties of this compound that support its use in chronic peripheral inflammatory diseases including rheumatoid arthritis, endometriosis and cancer, in which EP2 appears to play a pathogenic role.
Keywords: EP2 antagonist, aqueous-solubility, cytokines, tetrazole, lead-optimization, anti-inflammatory
INTRODUCTION:
Cyclooxygenase-2 (COX-2) and microsomal prostaglandin-E- synthase-1 (mPGES-1) play a key role in several acute and chronic peripheral disease conditions. For example, COX-2 and mPGES-1 are induced in the synovium of rheumatoid arthritis (RA) patients.1, 2 Mice deficient in either COX-2 or mPGES-1 showed a reduced arthritis score in the collagen-induced arthritis (CIA) model.3, 4 Prostaglandin-E2 (PGE2) is synthesized by COX-2 and mPGES-1, and is found in synovial fluid of RA patients supporting its role in inflammatory pathology.5 PGE2 exacerbates CIA in mice through modulation of the inflammatory cytokine IL-23 / IL-17 axis.6
PGE2 exerts downstream signaling through activation of four membrane bound G protein-coupled receptors, EP1, EP2, EP3 and EP4. EP2 and EP4 receptors promote cAMP signaling, EP1 mediates Ca+2 mediated signaling, and EP3 typically inhibits cAMP signaling. Two other prostanoid receptors, DP1 and IP, also promote cAMP mediated signaling, like EP2.7
COX-2 selective inhibitors have been developed to reduce pain and inflammation in arthritis patients. However chronic use of COX-2 drugs (e.g. Rofecoxib and Valdecoxib) has resulted in cardiovascular complications in patients, as a result they were withdrawn from the USA market.8 The less selective COX-2 drug celecoxib (Celebrex) remains on the market with a black-box warning for cardiovascular thrombotic events. The adverse effects of COX-2 drugs have been attributed to inhibition of PGI2 synthesis, thus the IP receptor, which plays a cardioprotective role.9, 10 Future anti-inflammatory strategies could involve inhibition of specific prostanoid receptors or prostanoid synthases, rather than generic block of the entire COX-2 cascade. We found that the EP2 receptor mediates a majority of COX-2 pro-inflammatory effects in the brain.11–13 Although the EP4 receptor seems to exert anti-inflammatory actions in CNS and other diseases,14 it promotes inflammation and exacerbates osteoarthritis disease.15, 16 Therefore, targeting EP2 or EP4 receptors should avoid adverse effects mediated by COX-2 inhibition.8 Recently, an EP4 receptor antagonist, Grapiprant, has been approved to treat dogs to reduce the pain and inflammation associated with osteoarthritis, supporting the notion that prostanoid receptors are potential targets for development of anti-inflammatory therapy.17, 18
Recently Pfizer,19 Amgen,20 and we21, 22 identified distinct classes of EP2 antagonists with good potency. However, these compounds displayed sub-optimal selectivity or in vivo plasma half-life to use in preclinical models of chronic diseases. We initially identified a cinnamic amide class of EP2 antagonists by high-throughput screening23 and conducted structure activity relationship studies to generate a second generation lead EP2 antagonist TG8–15 (3a, Fig 1), which displayed about 600–1000 fold selectivity for EP2 over EP4 and IP receptors, but only had 29-fold selectivity to DP1 receptors.21 Moreover, compound TG8–15 displayed plasma half-life of only 0.3 h after intraperitoneal administration in mice. Therefore, improvement in plasma half-life while maintaining EP2 potency, selectivity against prostanoid receptors and improved aqueous solubility were the main goals for the present study before performing a preclinical evaluation of EP2 antagonist in an animal model of chronic inflammatory disease such as arthritis. In this report, we present lead optimization studies that led to discovery of TG8–69 (Fig 1), which displays high potency, selectivity, aqueous solubility, and plasma half-life. Furthermore, we demonstrate that TG8–69 dampens the EP2 mediated induction of pro-inflammatory genes in a novel BV2 microglial cell line overexpressing the human EP2 receptor.
Figure 1.

Earlier lead compound TG8–15 led to the synthesis of preclinical lead compound TG8–69.
EXPERIMENTAL SECTION
Cell Culture.
The rat C6 glioma (C6G) cells stably expressing human DP1, EP2, EP4, or IP receptors were created in the laboratory22, 23 and grown in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS) (Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen), and 0.8 μg/ml G418 (Invitrogen).
Cell-Based cAMP Assay.
Intracellular cAMP was measured with a cell-based homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) method (Cisbio Bioassays), as previously described.23 The assay is based on generation of a strong FRET signal upon the interaction of two molecules, an anti-cAMP antibody coupled to a FRET donor (Cryptate) and cAMP coupled to a FRET acceptor (d2). Endogenous cAMP produced by cells competes with labeled cAMP for binding to the cAMP antibody and thus reduces the FRET signal. Cells stably expressing human DP1, EP2, EP4, or IP receptors were seeded into 384-well plates in 40 μl complete medium (4,000 cells/well) and grown overnight. The medium was carefully withdrawn and 10 μl Hanks’ Buffered Salt Solution (HBSS) (Hyclone) containing 20 μM rolipram was added into the wells to block phosphodiesterases. The cells were incubated at room temperature for 0.5 h and then treated with 10 μl vehicle or test compound for 10 or 50 min before addition of increasing concentrations of appropriate agonist: BW245C for DP1, PGE2 for EP2 and EP4, or iloprost for IP. The cells were incubated at room temperature for 40 min, then lysed in 10 μl lysis buffer containing the FRET acceptor cAMP-d2 and 1 min later another 10 μl lysis buffer with anti-cAMP-Cryptate was added. After 60–90 min incubation at room temperature, the FRET signal was measured by an Envision 2103 Multilabel Plate Reader (PerkinElmer Life Sciences) with a laser excitation at 337 nm and dual emissions at 665 nm and 590 nm for d2 and Cryptate (50 μs delay), respectively. The FRET signal was expressed as: F665/F590 × 104.
Anti-inflammatory assay using BV2-hEP2 cells
200,000 BV2-hEP2 cells/well (passage 16–20) were grown overnight in Poly-D-lysine (Sigma, USA) coated 12 well plates in duplicate in DMEM-F12 (Gibco, USA) media with 5% FBS (Gibco, USA) and 800 μg/ml G418 (Sigma, USA). The cells were treated with either 0.3 μM or 1 μM TG8–69 for 1 hr, followed by 30 nM ONO-AE1-259-01 (provided by ONO Pharmaceutical Co, Osaka, Japan) for 1 hr and then 100 ng/ml LPS (Sigma, USA) for 2 hrs. For vehicle treatment, a similar dilution of compound vehicle (DMSO) in complete media was used. After all treatments, the entire media was removed from all the wells and RNAs were extracted from the treated cells using RNA extraction kit (Zymo Research, USA) as per the manufacturer’s protocol. RNA samples were converted into cDNA using cDNA conversion kit (Quanta, USA) and further used for qPCR analysis. SYBR Green super mix (Quanta, USA) and primers for GAPDH, COX-2, ILβ, IL6 and hEP2 genes (IDT, USA, see SI Table-2 for the primer sequences) were used for PCR reaction carried out in CFX96 Real Time System (Bio Rad, USA). PCR data was presented as mean fold changes of gene expression in all the treated samples normalized to vehicle treated cells.
Statistics
For the anti-inflammatory assay ΔΔCT values were used for statistical analysis as they were normally distributed compared to their fold changes. ANOVA-with Holm-Sidak multiple comparisons test for post-hoc analysis was used. P values were considered significant at *<0.05.
RESULTS AND DISCUSSION:
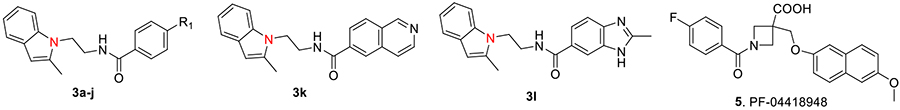
The earlier lead compound TG8–15 (3a) contains three structural moieties - the 2-methyl-1H-indol-1-yl moiety, a middle benzamide ring and a third tetrazole ring (Fig 1). Previous structure activity relationship (SAR) studies that led to identification of 3a indicated that the right side tetrazole ring is good for high EP2 potency, aqueous solubility and mouse liver microsomal stability.21 We also showed that high activity can be achieved with an isomeric replacement of the left hand indole ring.21 In this paper, we expand on the SAR by reporting additional heterocyclic replacements for the tetrazole ring of TG8–15 and the application of the tetrazole ring to the previously reported isomeric indole left hand part.
The novel derivatives with modification on the right side hetero aromatic tetrazole ring are synthesized by following Scheme 1 (see supporting information (SI) for synthesis procedures, and chemical characterization data of the derivatives). As shown in Table 1, we synthesized a N-methylated tetrazole derivative 3b (TG8–185), which displayed 25-fold less potency than 3a. The pyrazole derivatives 3c and 3d (TG8–130 and TG8–168) are found to be > 3-fold more potent than 3a. A triazole derivative 3e was 2-fold less potent than 3a, but imidazole derivatives 3f and 3g (TG8–237 and TG8–239) displayed similar potency to 3a. However, pyrimidine derivatives 3h & 3i (TG8–186, and TG8–238), and a thiazole derivative 3j (TG8–280) were 1.5–2-fold less potent than 3a. Interestingly an isoquinoline derivative 3k (TG8–258) displayed similar potency to 3a, but a benzimidazole derivative 31 (TG8–246) displayed 13-fold less potency than 3a. These SAR data reinforce that notion that modification on the right side ring maintains the high EP2 potency and aqueous-solubility (Table 1). Several of these novel derivatives displayed a modest <100-fold selectivity (see below for a discussion on selectivity) to EP2 over the DP1 and IP prostanoid receptors, and two selective compounds 3e and 3i showed low stability in mouse liver microsomes (Table 1). Replacement of the left hand indole of TG8–15 with an isomeric indole resulted in TG8–69 (Fig 1 & Scheme 1), which, gratifyingly, displayed suitable pharmacology and pharmaceutical properties as described in detail below.
Scheme 1.

Synthesis of EP2 antagonists (Detailed procedures are provided in SI).
Table 1:
EP2 Potency, selectivity, aqueous-solubility and mouse liver microsomal stability of novel EP2 antagonists.a
| Entry | Compd. ID | R1 | EP2 KB (nM) | DP1 KB (μM) | SI (DP1KB/EP2 KB) | IP KB (μM) | SI (IP KB/EP2 KB) | Aq. solb. (nM) | MLMc t1/2 (min) |
|---|---|---|---|---|---|---|---|---|---|
| 3a | TG8-15 | 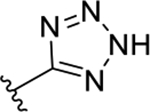 |
22.3 | 0.66 | 29 | >10 | >450 | 257 | 25 |
| 3b | TG8-185 | 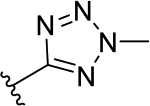 |
560 | nd | nd | nd | nd | nd | nd |
| 3c | TG8-130 | 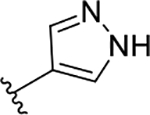 |
6.6 | 5.3 | 803 | 0.34 | 51 | 100 | 3.5 |
| 3d | TG8-168 | 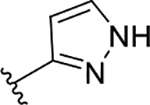 |
4.4 | 0.27 | 61 | nd | nd | 100 | nd |
| 3e | TG8-184 | 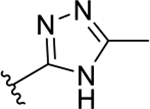 |
50 | >10 | >200 | >10 | >200 | 223 | 7.7 |
| 3f | TG8-237 | 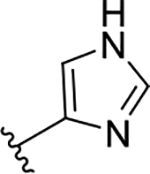 |
19.5 | 0.87 | 45 | nd | nd | 130 | nd |
| 3g | TG8-239 | 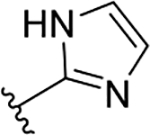 |
31.4 | >10 | >318 | 2.8 | 90 | 75 | nd |
| 3h | TG8-186 | 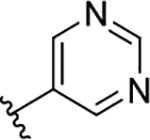 |
35 | >10 | >285 | 0.55 | 16 | 127 | 5.2 |
| 3i | TG8-238 | 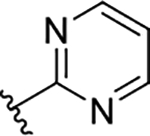 |
46.6 | 4.9 | 105 | >10 | 215 | 75 | 1.8 |
| 3j | TG8-280 | 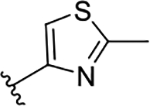 |
51 | 1.4 | 28 | 0.34 | 6.6 | 25 | nd |
| 3k | TG8-258 | see top of the table | 30.5 | 1.0 | 32 | 0.44 | 14 | 76 | nd |
| 31 | TG8-246 | see top of the table | 305 | nd | nd | nd | nd | nd | nd |
| 4 | TG8-69 | see Fig 1 | 48.5 | 18.0 | 370 | 130 | 2680 | 500 | >60 |
| 5 | PF-04418948 | see top of the table | 147 | 48.3 | 330 | 1.7 | 12 | nd | nd |
Schild KB values are calculated using the form ula log (dr-1) = log XB −log KB, where dr (dose ratio) = fold shift in EC50 of PG E2 by the test compound, XB is antagonist concentration (1 μM). KB value indicates a concentration required to produce a 2-fold rightward shift of PGE2 concentration response curve. KB values are average of 2–3 m easurem ents run in duplicate.
The aqueous-solubility of the compounds is measured in PBS buffer (pH 7.4) with 1% DMSO by nephelom etry.26 nd = not determined.
MLM = mouse liver microsomes. Compounds 3b and 3l were low potency compounds, therefore they were not tested in selectivity and solubility assays.
Using a cAMP mediated TR-FRET based functional EP2 assay,27 we first demonstrated that TG8–69 inhibits PGE2 induced EP2 receptor activation in a concentration-dependent manner (Fig 2A) in a C6-glioma cell line overexpressing human EP2 receptors. In this assay, TG8–69 displayed a competitive mechanism of antagonism of EP2 receptors as shown by Schild regression analysis with mean KB 48.5 nM and mean slope of 1.2 (n = 4) (Fig 2B). Schild KB values represent the concentration required to cause a 2-fold rightward shift of EC50 of a full agonist. Ideally, a perfect competitive antagonist will display a slope of unity. To compare the potency of TG8–69 with a known EP2 antagonist PF-04418948 (a Pfizer compound, purchased from Cayman chemical), we tested PF-04418948 in parallel (Fig 2C–D). Previously the Pfizer compound was reported to have a Schild KB value of 1.8 nM in a cAMP mediated functional assay using Chinese hamster ovary (CHO) cells overexpressing human EP2 receptors.19 Surprisingly, in our hEP2 overexpressing C6-glioma cell line, the Pfizer compound displayed 80-fold lower potency with mean KB = 147 nM, and mean slope 1.5 (n = 3) (Fig 2C–D). To understand whether the reduced potency is due to equilibrium not being obtained during the bioassay, we tested these two compounds head-to-head with longer pre-incubation time (50 min, vs. 10 min we normally use; thus the total incubation time is 90 vs 50 min). However, neither the Schild slope nor the KB were much affected for either of the two compounds by extending assay incubation time from 50 to 90 min (see SI Figure 1). To determine whether the cell line plays a role in the relative potency of these two compounds, we tested both in a novel microglia cell line, BV2-hEP2, which expresses human EP2 receptors (Rojas et al., submitted). As shown in the SI Figure 2, the Pfizer compound showed Schild KB 105 nM, which is 5-times less potency than TG8–69, and 60-times lower potency than previously reported value.19 These data suggest that potency does not depend on the cell line, therefore, the observed difference in potency of the Pfizer compound might be attributed to the DiscoveRX assay method used by Pfizer.19 However, both of these compounds, while displaying slope values > 1.0, did not impact the maximum cAMP response elicited by the full agonist PGE2, suggesting they are competitive antagonists. Nonetheless, TG8–69 is about 3-times more potent than the Pfizer compound in the same assay. Furthermore, we also tested TG8–69 in a binding assay, in which it inhibited the binding of radiolabeled H3-PGE2 to EP2 receptors with Ki of 135 nM (Fig 3A).
Figure 2.

EP2 potency of TG8–69 and PF-04418948. (A) TG8–69 inhibited PGE2-induced EP2 receptor activation in a concentration-dependent manner. Data were normalized to percent maximum response to PGE2 in the absence of antagonist; data points represent mean ± SEM from one experiment run in triplicate, the concentration-response experiment was repeated four times in triplicate. (B) TG8–69 displayed competitive antagonism of the EP2 receptor as shown by Schild regression analysis with mean KB = 48.5 nM and slope = 1.2. The Schild plots from four independent experiments are shown. (C) PF-04418948 similarly inhibited PGE2-induced EP2 receptor activation in a concentration-dependent manner and this experiment was repeated three times (D). The Schild plots from three independent experiments are shown. The mean Schild KB = 147 nM and slope = 1.5 was found for PF-04419848.
Figure 3.

(A) Inhibition of H3-PGE2 binding to EP2 receptors by TG8–69. (B) TG8–69 showed >10,000 nM Schild potency in the TR-FRET assay for other Gas-coupled prostanoid receptors, DP1, EP4, and IP. The KBs are estimated from fold changes to agonist EC50 caused by 10 uM compound. Agonists were PGE2 for EP2 and EP4, BW245C for DP1, and iloprost for IP (n = 3–4 repeats in duplicate).
EP2, DPI, EP4 and IP are Gas coupled prostanoid receptors. Among these DPI has the closest sequence homology to EP2, followed by IP, then EP4, although both EP2 and EP4 share a common endogenous ligand PGE2 for their activation.28 To determine the selectivity of several novel derivatives (Table 1) and TG8–69 for EP2 over other Gas-coupled prostanoid receptors, we created C6-glioma cell lines overexpressing DP1, EP4 and IP receptors and used them for counter screening.22 The novel compounds shown in Table 1 showed low selectivity to either DP1 or IP receptors, except compound 3e (TG8–184) which showed > 200-fold selectivity against DP1 and IP receptors. Three other compounds 3c (TG8–130), 3g (TG8–239) and 3h (TG8–186) displayed > 200-fold selectivity against DP1 receptor, but their selectivity against IP receptor was < 100-fold (Table 1). However, compound TG8–69 showed >300-fold selectivity against DP1 and EP4, and >1000-fold selectivity against IP receptors (Fig 3B), so it should be useful for in vitro and in vivo proof of concept studies. To learn the selectivity against EP1 and EP3 receptors, we tested TG8–69 against radiolabeled H3-PGE2 binding to EP1 and EP3 receptors at Cerep laboratories (CRO). Interestingly TG8–69 did not show any significant inhibition of H3-PGE2 binding to EP1 and EP3 at 3 μM, but it inhibited 90% H3-PGE2 binding to EP2 receptor at 1 μM concentration (see SI Fig 3). Additional dose-response studies are needed to establish the fold selectivity to EP2 against EP1 and EP3 receptors.
To determine ADME properties, we first tested several modestly selective novel compounds 3c, 3e, 3h, 3i (Table 1) for stability in mouse liver microsomes, but, these compounds showed < 10 minutes of half-life. These compounds were not tested in human liver microsomes for stability. However interestingly compound TG8–69 showed > 60 minutes half-life in mouse liver microsomes and subsequently in human liver microsomes, when incubated at 1 μM concentration (Fig 4A). We then tested TG8–69 against a panel of seven cytochrome-450 (CYP450) enzymes in binding assays. The assays were conducted as reported according to the methods reported in24, and the details are provided in SI. Interestingly when tested at 10 μM concentration, TG8–69 showed < 30% inhibition in six out of seven CYP450 tested, but CYP2C8 was inhibited about 70%. Additional studies are needed to determine the IC50 against this and other CYP450 enzymes. CYP2C8 is an epoxygenase enzyme, associated mainly with metabolism of long chain fatty acids, and relatively less involved (in comparison with CYP3A4 and CYP2D6) in the metabolism of xenobiotics. Nonetheless, additional studies using a known CYP2C8 metabolizing substrate are needed to confirm whether TG8–69 is a strong inhibitor that would influence a drug-drug interaction. Furthermore, TG8–69 did not inhibit the binding of dofetilide to potassium channel hERG at 10 μM concentration (Fig 4B). These data indicate that TG8–69 may not have a potential cardiotoxicity liability.
Figure 4.

TG8–69 displayed high stability in liver microsomes and low off-target inhibition of most CYP450 enzymes and potassium channel hERG. (A) TG8–69 displayed >60 minutes half-life in pooled liver microsomal fractions. 1 μM compound was incubated in 0.5 mg/mL liver microsomes for the indicated time and the TG8–69 levels were measured by LC-MS/MS. (B). Inhibition of ligand binding to various CYP450 enzymes and fiERG. TG8–69 (10 μM) showed < 30 % inhibition of six out of seven CYP450s tested, and no inhibition of hERG.
The high potency and selectivity, and good in vitro ADME characteristics, prompted us to explore in vivo pharmacokinetics for TG8–69. As shown in Table 2 and Figure 5, when C57BL/6 male mice were dosed at 5 mg/kg intravenously (i.v.), or 10 mg/kg by oral gavage (p.o.), TG8–69 exhibited a terminal half-life 6.7 h and 10.5 h respectively. Using AUCinf, the calculated oral bioavailability for TG8–69 is 14.5%. To evaluate brain-to-plasma ratio (B/P ratio) for TG8–69, we conducted a separate study with oral dosing (10 mg/kg) and determined plasma and brain tissue concentrations at 0.5, 2 and 6 hrs. The data indicated that B/P ratio is 0.01–0.02 (see Table 2) suggesting it is largely peripherally restricted and should be useful for investigating chronic peripheral disease models.
Table 2:
Pharmacokinetic parameters of TG8–69 in plasma following a single intravenous (i.v.) (dose: 5 mg/kg) and oral (p.o.) (dose: 10 mg/kg) administration to male C57BL/6 mice.
| Compd. | Route | Tmax | aCmax (nM) | AUClast (hr*nM) | AUCinf (hr*nM) | T1/2 (hr) | CL (mL/min/kg) | Vss (L/kg) | %Fb | B/P ratioe |
|---|---|---|---|---|---|---|---|---|---|---|
| TG8-69 | i.v.c | - | 41000 | 14900 | 15000 | 6.7 | 16.0 | 1.11 | - | nd |
| p.o.d | 0.25 | 1360 | 3900 | 4335 | 10.5 | - | - | 14.5 | 0.01–0.02 |
back extrapolated conc. for i.v. group.
AUCinf is used for calculating bioavailability.
n = 3 animals were used at each of time point between 0.08 h-24 h (9 time points).
n = 5 animals were used at each time point between 0.25 h–24 h (9 time points).
Brain-to-plasma is determined by a separate oral dosing experiment by looking at concentrations in the brain tissue and plasma at 0.5, 2, 6 h time points.Originally, the plasma concentrations were determined in ng/mL concentrations, but we converted them to nM concentrations and presented in Table 2 and Fig 5. Similarly, the concentrations in the brain tissue were determined in ng/g, which were used to calculate brain to plasma ratio.
Figure 5:

Plasma pharmacokinetics of TG8–69. TG8–69 was dissolved in N-methyl-2-pyrrolidone (NMP) (5%), solutol-HS-15 (5%) and saline (90%) and injected to mice via tail-vein intravenous (i.v.) or oral gavage (p.o.) routes. The blood was harvested and the compound extracted and analyzed by LCMS/MS.
To determine whether we can formulate this compound in an aqueous solution for in vivo use, we tested its solubility in PBS containing 1% DMSO at pH 7.4 using a nephelometry assay.26 In this assay, when visible light is passed through a solution, part of the incident radiant energy will be scattered. The measurement of the intensity of the scattered light as a function of the concentration of the dispersed phase is the basis of the nephelometric assay. The nephelometric assay can be employed to determine either the point at which a solute begins to precipitate out of a true solution to form a suspension or the concentration at which a suspension when diluted further becomes a solution. The scattered light will remain at a constant intensity until precipitation occurs, at which point it will increase sharply as shown in SI Figure 4. In this assay, TG8–69 showed solubility of 172 ± 15 μg/mL (500 ± 40 μM) indicating it is highly aqueous soluble (SI Fig 4).
The applications of an EP2 antagonist will be found in several chronic inflammatory diseases such as rheumatoid arthritis. EP2 receptor activation exacerbates symptoms of experimental inflammatory bowel disease (colitis) by increasing IL-23 expression and reducing IL-12, together causing T-cells to differentiate to Th17 effectors.29 Moreover, deleterious roles of EP2 are reported in other peripheral events such as tumor angiogenesis and preterm delivery.30–35 Therefore, a well characterized preclinical candidate will enable proof-of-concept tests in these and similar indications. Having developed a lead candidate EP2 antagonist TG8–69, we next investigated anti-inflammatory properties in vitro. A routine isolation of primary macrophages from peritoneal region, or microglia from mouse brain proved to be low throughput because the primary cells behave variably depending on the animal. Thus, we created a BV2 cell line, which was previously derived from mouse microglia, overexpressing human EP2 receptors (Rojas et al., in preparation). Upon activation of this cell line with 100 ng/mL lipopolysaccharide (LPS), mRNA levels of several proinflammatory genes were induced including COX-2, IL-6 and IL-1β. An EP2 specific agonist 0N0-AE1-259-01 at 30 nM further exacerbated the induction of these inflammatory genes. Gratifyingly, TG8–69 (0.3 μM and 1.0 μM) blunted the upregulation of these inflammatory genes in a concentration dependent manner (Fig 6). EP2 mRNA expression was not affected by either the agonist or the antagonist. In a control experiment, incubation of BV2-EP2 cell line with TG8–69 (1 uM) in the presence or absence of LPS did not show a significant effect on induction of inflammatory mediators and EP2 (Fig 7). These data support the use of this EP2 antagonist as anti-inflammatory agent in chronic disease models.
Figure 6.

Mean fold change in mRNA expression of cytokines in BV2-hEP2 cells upon TG8–69, Ono and LPS treatment. 200,000 cells/well were grown overnight and treated with vehicle or TG8–69 for 1 hr, then vehicle or ONO-AE1-259-01 (abbreviated to Ono in the Figure 6 and 7) for 1 hr, then vehicle or LPS for 2 hrs. Analyte mRNAs were measured by qRT-PCR. For statistical analysis, AACT values were used as they were normally distributed compared to fold changes. ANOVA-with Holm-Sidak multiple comparisons test for post-hoc analysis was used. P values were considered significant at *<0.05. Experimental repeats n = 7 for COX-2, IL-1β and IL-6; n = 5 for hEP2.
Figure 7:

Mean fold change in mRNA expression of cytokines in BV2-hEP2 cells upon TG8–69 and LPS treatment. Cells were grown overnight and treated with vehicle or TG8–69 (1 μM) for 1 hr and vehicle or LPS (100 ng/ml) for 2 hrs. Analyte mRNAs were measured by qRT-PCR. For statistical analysis, ΔΔCT values were used as they were normally distributed compared to fold changes. Student t-test was used to compare among the LPS treated and untreated groups. P values were found to be non-significant (>0.05) for all the comparisons. n = 3 experimental repeats.
In summary, we have developed a novel, selective, aqueous-soluble peripherally restricted EP2 antagonist with suitable pharmaceutical properties and showed that this compound dampens pro-inflammatory gene expression in an in vitro cell culture model indicating this compound will be useful to explore in vivo models where EP2 is suspected to play a deleterious role.
Supplementary Material
ACKNOWLEDGMENT
Ono Pharmaceutical company co, Osaka, Japan, for providing the ONO-AE1-259-01.
Funding Sources
This work was supported by NIH/NIA grant U01 AG052460 (T.G.), ADDF grant 20131001 (T.G.), R01 NS097776 (R.D.) and R21 NS101167 (T.G.).
Footnotes
Contains experimental details of radioligand [H3-PGE2] binding, liver microsomal stability, CYP450 inhibition, human-ERG potassium channel inhibition assays and in vivo pharmacokinetics studies.
Also contains synthetic procedure for novel compounds, NMR and Mass spectral characteristics for novel compounds, and additional pharmacological characterization data of TG8–69 and PF-04419848, selectivity and aqueous-solubility data of TG8–69. A table containing PCR primers (SI Fig 2) is also included. This material is available at http:/pubs.acs.org.
REFERENCES
- 1.Crofford LJ; Wilder RL; Ristimaki AP; Sano H; Remmers EF; Epps HR; Hla T Cyclooxygenase-1 and −2 expression in rheumatoid synovial tissues. Effects of interleukin-1 beta, phorbol ester, and corticosteroids. J Clin Invest 1994, 93, 1095–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westman M; Korotkova M; af Klint E; Stark A; Audoly LP; Klareskog L; Ulfgren AK; Jakobsson PJ Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum 2004, 50, 1774–80. [DOI] [PubMed] [Google Scholar]
- 3.Myers LK; Kang AH; Postlethwaite AE; Rosloniec EF; Morham SG; Shlopov BV; Goorha S; Ballou LR The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum 2000, 43, 2687–93. [DOI] [PubMed] [Google Scholar]
- 4.Trebino CE; Stock JL; Gibbons CP; Naiman BM; Wachtmann TS; Umland JP; Pandher K; Lapointe JM; Saha S; Roach ML; Carter D; Thomas NA; Durtschi BA; McNeish JD; Hambor JE; Jakobsson PJ; Carty TJ; Perez JR; Audoly LP Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A 2003, 100, 9044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodie MJ; Hensby CN; Parke A; Gordon D Is prostacyclin in the major pro-inflammatory prostanoid in joint fluid? Life Sci 1980, 27, 603–8. [DOI] [PubMed] [Google Scholar]
- 6.Sheibanie AF; Khayrullina T; Safadi FF; Ganea D Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum 2007, 56, 2608–19. [DOI] [PubMed] [Google Scholar]
- 7.Ganesh T Prostanoid receptor EP2 as a therapeutic target. J Med Chem 2014, 57, 4454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grosser T; Yu Y; FitzGerald GA Emotion Recollected in Tranquility: Lessons Learned from the COX-2 Saga. Annual Review of Medicine 2010, 61, 17–33. [DOI] [PubMed] [Google Scholar]
- 9.Arehart E; Stitham J; Asselbergs FW; Douville K; MacKenzie T; Fetalvero KM; Gleim S; Kasza Z; Rao Y; Martel L; Segel S; Robb J; Kaplan A; Simons M; Powell RJ; Moore JH; Rimm EB; Martin KA; Hwa J Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: potential implications for cyclooxygenase-2 inhibition. Circ Res 2008, 102, 986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egan KM; Lawson JA; Fries S; Koller B; Rader DJ; Smyth EM; Fitzgerald GA COX-2-derived prostacyclin confers atheroprotection on female mice. Science 2004, 306, 1954–7. [DOI] [PubMed] [Google Scholar]
- 11.Quan Y; Jiang J; Dingledine R EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem 2013, 288, 9293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rojas A; Ganesh T; Lelutiu N; Gueorguieva P; Dingledine R Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology 2015, 93, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rojas A; Ganesh T; Manji Z; O’Neill T; Dingledine R Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology 2016, 110, 419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woodling NS; Wang Q; Priyam PG; Larkin P; Shi J; Johansson JU; Zagol-Ikapitte I; Boutaud O; Andreasson KI Suppression of Alzheimer-associated inflammation by microglial prostaglandin-E2 EP4 receptor signaling. JNeurosci 2014, 34, 5882–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCoy JM; Wicks JR; Audoly LP The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest 2002, 110, 651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Attur M; Al-Mussawir HE; Patel J; Kitay A; Dave M; Palmer G; Pillinger MH; Abramson SB Prostaglandin E2 exerts catabolic effects in osteoarthritis cartilage: evidence for signaling via the EP4 receptor. J Immunol 2008, 181, 5082–8. [DOI] [PubMed] [Google Scholar]
- 17.Rausch-Derra LC; Huebner M; Rhodes L Evaluation of the safety of long-term, daily oral administration of grapiprant, a novel drug for treatment of osteoarthritic pain and inflammation, in healthy dogs. Am J Vet Res 2015, 76, 853–9. [DOI] [PubMed] [Google Scholar]
- 18.Kirkby Shaw K; Rausch-Derra LC; Rhodes L Grapiprant: an EP4 prostaglandin receptor antagonist and novel therapy for pain and inflammation. Vet Med Sci 2016, 2, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.af Forselles KJ; Root J; Clarke T; Davey D; Aughton K; Dack K; Pullen N In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP(2) receptor antagonist. Br J Pharmacol 2011, 164, 1847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox BM; Beck HP; Roveto PM; Kayser F; Cheng Q; Dou H; Williamson T; Treanor J; Liu H; Jin L; Xu G; Ma J; Wang S; Olson SH A selective prostaglandin E2 receptor subtype 2 (EP2) antagonist increases the macrophage-mediated clearance of amyloid-beta plaques. J Med Chem 2015, 58, 5256–73. [DOI] [PubMed] [Google Scholar]
- 21.Ganesh T; Jiang J; Dingledine R Development of second generation EP2 antagonists with high selectivity. Eur J Med Chem 2014, 82, 521–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesh T; Jiang J; Yang MS; Dingledine R Lead optimization studies of cinnamic amide EP2 antagonists. J Med Chem 2014, 57, 4173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J; Ganesh T; Du Y; Quan Y; Serrano G; Qui M; Speigel I; Rojas A; Lelutiu N; Dingledine R Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proc Natl Acad Sci U S A 2012, 109, 3149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller VP; Stresser DM; Blanchard AP; Turner S; Crespi CL Fluorometric high-throughput screening for inhibitors of cytochrome P450. Ann N Y Acad Sci 2000, 919, 26–32. [DOI] [PubMed] [Google Scholar]
- 25.Huang XP; Mangano T; Hufeisen S; Setola V; Roth BL Identification of Human Ether a-go-go Related Gene Modulators by Three Screening Platforms in an Academic Drug-Discovery Setting. Assay and Drug Development Technologies 2010, 8, 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bevan CD; Lloyd RS A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal Chem 2000, 72, 1781–7. [DOI] [PubMed] [Google Scholar]
- 27.Jiang J; Ganesh T; Du Y; Thepchatri P; Rojas A; Lewis I; Kurtkaya S; Li L; Qui M; Serrano G; Shaw R; Sun A; Dingledine R Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc Natl Acad Sci U S A 2010, 107, 2307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirata T; Narumiya S Prostanoid Receptors. Chemical Reviews 2011, 111, 6209–6230. [DOI] [PubMed] [Google Scholar]
- 29.Sheibanie AF; Yen JH; Khayrullina T; Emig F; Zhang M; Tuma R; Ganea D The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23-->IL-17 axis. J Immunol 2007, 178, 8138–47. [DOI] [PubMed] [Google Scholar]
- 30.Donnini S; Finetti F; Solito R; Terzuoli E; Sacchetti A; Morbidelli L; Patrignani P; Ziche M EP2 prostanoid receptor promotes squamous cell carcinoma growth through epidermal growth factor receptor transactivation and iNOS and ERK1/2 pathways. FASEB J 2007, 21, 2418–30. [DOI] [PubMed] [Google Scholar]
- 31.Gross G; Imamura T; Vogt SK; Wozniak DF; Nelson DM; Sadovsky Y; Muglia LJ Inhibition of cyclooxygenase-2 prevents inflammation-mediated preterm labor in the mouse. Am J Physiol Regul Integr Comp Physiol 2000, 278, R1415–23. [DOI] [PubMed] [Google Scholar]
- 32.Kabashima K; Nagamachi M; Honda T; Nishigori C; Miyachi Y; Tokura Y; Narumiya S Prostaglandin E2 is required for ultraviolet B-induced skin inflammation via EP2 and EP4 receptors. Lab Invest 2007, 87, 49–55. [DOI] [PubMed] [Google Scholar]
- 33.Kishore AH; Owens D; Word RA Prostaglandin E2 regulates its own inactivating enzyme, 15-PGDH, by EP2 receptor-mediated cervical cell-specific mechanisms. J Clin EndocrinolMetab 2014, 99, 1006–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sung YM; He G; Hwang DH; Fischer SM Overexpression of the prostaglandin E2 receptor EP2 results in enhanced skin tumor development. Oncogene 2006, 25, 5507–16. [DOI] [PubMed] [Google Scholar]
- 35.Kishore AH; Liang H; Kanchwala M; Xing C; Ganesh T; Akgul Y; Posner B; Ready JM; Markowitz SD; Word RA Prostaglandin dehydrogenase is a target for successful induction of cervical ripening. Proc Natl Acad Sci U S A 2017, 114, E6427–E6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.