

Abstract
Contemporary immunotherapies, e.g. those that target the CTLA-4 and PD-1/PD-L1 axis, act on T cells to reinstate their antitumor activity. An alternative, and possibly more powerful approach is to target and reprogram the innate immune system within the tumor microenvironment. To this end, blockade of CD47 has been demonstrated as an attractive approach. Blockade of CD47 inhibits antiphagocytic signals therefore inducing macrophage phagocytosis of cancer cells. CD47 blockade also primes antitumor T-cell responses by either activating antigen presenting cells or inhibiting interactions between CD47 on cancer cells and the matricellular protein thrombospondin-1 (TSP1) on T cells. Here, we identified a combination immunotherapy using cowpea mosaic virus (CPMV) in situ vaccination and CD47-blocking antibodies. The CPMV in situ vaccine synergizes with CD47 blockade, because CPMV in situ vaccination activates the innate immune system, leading to recruitment and activation of phagocytes. Therefore, the combination therapy targets monocytes and boosts their ability of cancer cell phagocytosis, in turn priming the adaptive immune system leading to a potent antitumor immune response. This work presents a novel strategy to promote macrophage activity to kill tumor cells, and hold promise to enhance T cells targeted immunotherapies by inducing both innate and adaptive arms of immune system.
Keywords: plant virus, cancer immunotherapy, cytotoxicity, ovarian cancer, breast cancer, immunotherapy
Graphical Abstract
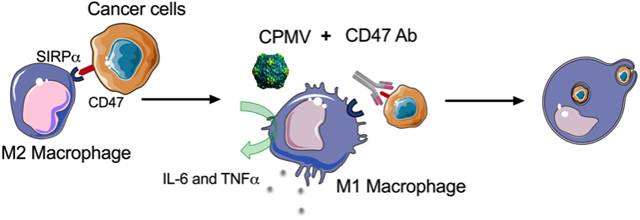
Overexpression of CD47 on cancer cells inhibits phagocytic activity through engagement of its counter receptor SIRPα on macrophages. Cowpea mosaic virus (CPMV) in situ vaccination and CD47-blocking antibodies combination therapy induces synergistic macrophage-mediated cytotoxicity against 4T1 breast cancer cells and ID8-Defb29/Vegf-A ovarian cancer cells. In vivo experiments show anticipated tumor inhibition of combination treatment in 4T1 tumor model.
Immunotherapies manipulate the immune system of host-self to eliminate tumor cells. Checkpoint immunotherapies focus on targeting inhibitory signals on T cells allowing T cells to retain their cytotoxic ability to attack cancer cells.[1] However, mounting evidence suggests that immunostimulatory agents able to recruit and stimulate both antigen-presenting cells (APCs) and T cells at the tumor site would be essential to synergize with checkpoint inhibitors therapies to further boost the antitumor effect. CD47, widely expressed on a variety of tumor cells, acts as a “don’t eat me” signal that binds the inhibitory receptor signal regulatory protein alpha (SIRPα) on phagocytic cells.[2] Human CD47-blocking monoclonal antibodies treatment in immunodeficient mice bearing human tumors enhances therapeutic antitumor response by promoting phagocytosis of antibody-bound tumor cells.[3, 4] CD47 also functions as a signaling receptor of matricellular protein thrombospondin-1 (TSP1) expressed on T cells. Binding of TSP1 to CD47 causes a profound inhibition of T cells activation.[5] Therefore, CD47 blockade holds potential to directly enhance tumor immunesurveillance by T cells. Furthermore, recent studies using syngeneic tumor models show anti-CD47 monotherapy primes antitumor CD8+ T cells responses and regulates regression of established tumor lesions through tumor infiltrating dendritic cells (DCs) or macrophages activation.[6] Together, those results suggest anti-CD47 treatment may act as a novel immune checkpoint blockade therapy to target both innate and adaptive immune systems.
While targeting the CD47 axis is an attractive strategy, most immunotherapies are not powerful enough when administered as solo-therapy. Therefore, there is need to develop combination therapies that synergize and prolong antitumor immunity through activation of multiple different mechanisms. We hypothesized that a particularly powerful combination would be with 1) an immunotherapy that modulates the local tumor microenvironment to recruit immune cells, i.e. monocytes and T cells, into the tumor bed, and 2) the CD47 checkpoint therapy to enhance activity of recruited immune cells. Toward this goal we combined a plant virus in situ vaccination nanotechnology with CD47 blockade.
Plant virus nanoparticles (VNPs) have been widely investigated as promising immunotherapeutic adjuvants and nanoparticle vaccines.[7] More recently we demonstrated that the immunostimulatory properties of VNPs can be harnessed also in the context of tumors: when applied as in situ vaccine, the VNPs formed by the cowpea mosaic virus (CPMV) reverse the immunosuppressive tumor microenvironment and promote the antigen-presenting ability of innate immune cells, therefore restarting the cancer immunity cycle.[8–10] Preclinical data in several mouse models indicate potent efficacy; we also demonstrated the translational potential by successfully treating canine patients with spontaneous melanoma using a combination of CPMV in situ vaccination and radiation therapy.[11]
The therapeutic effect of CPMV is associated with the infiltration and activation of macrophages, tumor infiltrated neutrophils, and Natural killer (NK) cells.[12] Data indicate that CPMV remodels the tumor microenvironment leading to the infiltration and activation of the innate immune system – hence the combination with CD47 blockade acting on both the innate and adaptive immune system is expected to enhance therapy success. We tested this hypothesis in two mouse models using the ID8-Defb29/Vegf-A ovarian cancer model and 4T1 breast cancer model.
Various human tumors cells, including glioblastoma, myeloma, breast cancer, and ovarian clear cell carcinoma, overexpress CD47 on their surface to avoid removal by macrophages and the interruption of CD47-SIRP α interactions has been shown efficacious in immunodeficient mice bearing human tumors.[3, 13] However, the expression levels of CD47 on murine tumor cells have not been systemically summarized. Therefore, we firstly investigated the expression levels of CD47 on tumor tissues of both tumor models that were used in this study, a breast (4T1) and ovarian cancer (ID8-Defb29/Vegf-A) model. All animal experiments were carried out in accordance with Case Western Reserve University’s Institutional Animal Care and Use Committee (IACUC). Detailed experimental procedures are provided in the Supporting Information. In brief, we inoculated 4T1 cells intradermally (i.d.) and ID8-Defb29/Vegf-A cells intraperitoneally (i.p.) tumor cells in BALB/c or C57BL/6 mice, respectively, and on day 30 (for 4T1 tumor) and day 35 (for ID8-Defb29/Vegf-A tumor), mice were sacrificed for tumor tissue collection. The expression levels of CD47 on tumor cells were visualized using immunofluorescence staining and confocal microscopy (Figure 1). In addition, we confirmed CD47 expression levels on both tumor cells lines using flow cytometry. A recent study shows that CD47 is expressed on primary mouse macrophages (RAW 264.7 and U937 cell lines);[14] therefore RAW 264.7 cells were used as a positive control. The results indicated that CD47 was highly expressed in both tumor cell lines (Figure S1, Supporting Information).
Figure 1.
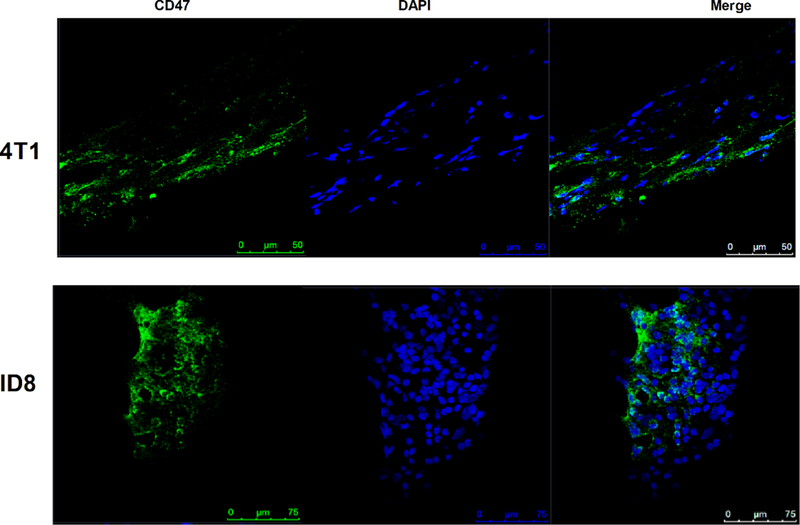
Confirmation of CD47 expression on 4T1 and ID8-Defb29/Vegf-A tumor tissues. Representative immunofluorescence of CD47 expression levels in 4T1 tumors (upper); ID8-Defb29/Vegf-A tumors (bottom). Blue, DAPI; green, CD47 Ab. Tumor tissues are collected on day 30 (4T1) and 35 (ID8-Defb29/Vegf-A) post inoculation from non-treated mice. Scale bars are indicated in figures.
It is known that CD47 blockade turns off “don’t eat me” signals on cancer cells,[2] therefore enhancing the phagocytic activity of innate cells against cancer cells. CD47 is widely expressed on tumor cells and we confirmed its expression on ID8-Defb29/Vegf-A and 4T1 tumors (Figure 1). Next, we tested the effect of CPMV and CPMV+CD47 blockade on the cytotoxic potential of macrophage against 4T1 or ID8-Defb29/Vegf-A cancer cells. Luciferase-labeled 4T1 or ID8-Defb29/Vegf-A tumor cells were stimulated with either anti-CD47 antibody (CD47 Ab, Clone MIAP410), CPMV, or the combination and then co-cultured with murine macrophage (RAW 264.7) at different ratios (Figure 2). The number of viable tumor cells was determined after 20 h by measuring their bioluminescence signals. The methods are detailed in the Supporting Information.
Figure 2.
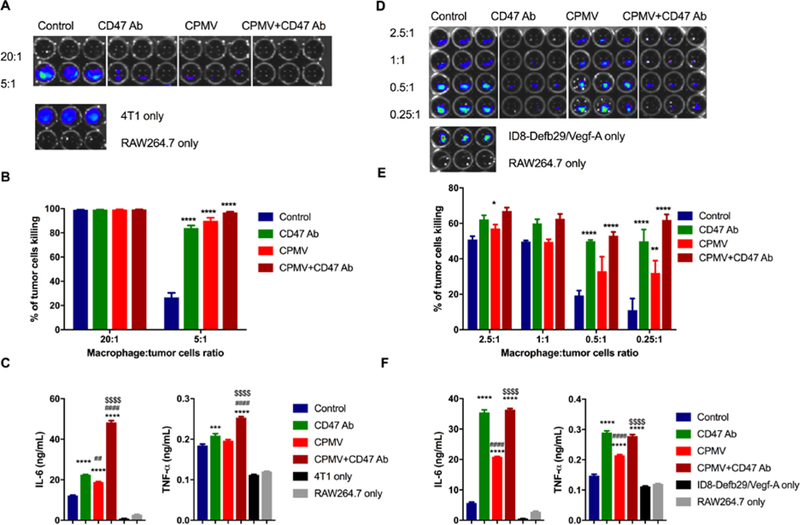
Tumor cytotoxicity improved by CD47 antibody blockade and CPMV. Murine macrophage (RAW 264.7) were co-cultured with mammary fat pad cells (4T1-Luc, A) and ovarian tumor cells (ID8-Defb29/Vegf-A-Luc, D) with different ratio, from 20:1 to 0.25 :1. 10 μg CPMV, anti-CD47 Ab or combination were added as treatment for 20 h. Bioluminescence intensity (BLI) was measured to quantify the percentage of live/dead 4T1 cells (B) and ID8-Defb29/Vegf-A cells (E) by phagocytosis. Data are means ± SEM. Statistical significance was calculated by two-way ANOVA with Tukey test. *vs. control. *p<0.05, **p<0.001, ****p<0.0001. At ratio 2.5:1 (4T1: RAW 264.7, C; ID8-Defb29/Vegf-A: RAW 264.7, F), the supernatant of each well was collected and released cytokines (IL-6 and TNF-α) were measured using ELISA. Data are means ± SEM. Statistical significance was calculated by one-way ANOVA with Tukey test. *vs. control; #vs. CD47 antibody; $vs. CPMV. ***p<0.0005, ****p<0.0001.
At high macrophage-to-tumor cell ratios, i.e. 20:1 RAW 264.7:4T1, cytotoxic activity of the macrophages was observed whether or not CPMV and/or CD47 blockade was added. This demonstrated the therapeutic potential of the innate immune system. However, without therapies added, the therapeutic effect of the macrophages was only observed at high macrophage-to-cancer cell ratios. At lower ratios, without additional stimuli, macrophages alone were not able to control cancer cell growth, i.e. at a RAW 264.7:4T1 ratio of 5:1, no cytotoxicity against tumor cells was observed. The addition of CD47 blockade, CPMV stimulation, and the combination therapy, however, demonstrated significant cytotoxicity enhancement of macrophages against the tumor cells (p<0.0001, Figure 2A, B). While there were no statistically significant differences between the treatment arms, the trend indicated more efficient tumor killing when CD47 blockade and CPMV are combined: CD47 Ab or CPMV alone resulted in 81.6% or 88.6% tumor cell killing; the combination therapy achieved tumor cell kill with an efficiency of 96.4% (Figure 2B, Figure S2, Supporting Information). Statistically significant differences could be observed between control and combination therapy. However, the expression of luciferase from tumor cells was not sensitive enough to identify the difference between control and single CD47 Ab or CPMV treated groups.
The trends were matched using the ovarian tumor model (Figure 2D, E). Again, at higher macrophage-to-tumor cell ratios, i.e. 2.5:1 RAW 264.7:ID8-Defb29/Vegf-A, macrophages alone demonstrated their ability to inhibit tumor cell growth. At lower macrophage-to-tumor cell ratios blockade or stimulation was needed to induce effective tumor cell killing. For example, at a cell ratio of RAW 264.7:ID8-Defb29/Vegf-A of 0.25:1, macrophages alone were not sufficient to control cancer cell growth. The addition of CPMV increased the cytotoxic potential of macrophages and tumor cell killing with an efficacy of 32.0 % (p<0.001); CD47 blockade alone enhanced cancer cell killing to 49.9% (p<0.0001); the CPMV+CD47 blockade combination further enhanced the cytotoxicity to 62.0% (p<0.0001).
Differences in macrophage behavior may result from differing activation stimuli. When macrophages are exposed to Th1 cytokines, they are subjected to classical (type 1, M1) activation with anti-tumor properties. Alternative activation by Th2 cytokine induces M2 type macrophages with pro-tumor properties.[15] The populations of tumor-associated macrophages are distinct in different human or animals tumor models, however, an increase in the overall M1/M2 ratios generally correlates with an improved prognosis.[16] M1 macrophages secrete inflammatory cytokines such as tumor necrosis factor (TNF), IL-1, IL-6, IL-8, and IL-12.[17] To identify whether CD47 Ab and CPMV and the combination could enhance the classical activation of macrophages, we measured the released inflammatory cytokines (IL-6 and TNF-α) in the supernatant of 2.5:1 (tumor cell: macrophage) co-cultured samples with different stimuli (Figure 2C, 2F). For the 4T1 tumor model (Figure 2C), treatment with CPMV significantly increased the secretion of IL-6 but not TNF-α; CD47 Ab treatment resulted in a statistically significant difference for both IL-6 and TNF-α; the combination further enhanced the secretion and manifested a synergistic effect in macrophage activation. In the ID8-Defb29/Vegf-A (Figure 2F) tumor model, we observed a similar trend: any treatment significantly enhanced IL-6 and TNF-α secretion from macrophages compared to the control group (p<0.0001); differences were noted comparing CD47 vs. CPMV and CPMV vs. combination (p<0.0001), but no significant difference could be observed when comparing CD47 to the combination.
Together, the results indicate that both CD47 Ab and CPMV treatment promote tumor cell phagocytosis by macrophages. In addition, the combination of CD47 Ab and CPMV has synergistic potential to induce tumor cell death through macrophage activation compared to single treatments in both tumor cell models. It is interesting to observe a significant enhancement of macrophage cytotoxicity and activation by CPMV stimulation. We further stimulated RAW 264.7 cells with CPMV in vitro and found that CPMV stimulation increased the levels of surface co-expression molecules CD86 and major histocompatibility complex class II (MHC II) on RAW 264.7 cells, which was associated with enhancement in antigen presentation and proliferation of T cells (Figure S3, Supporting Information). It may suggest that the activated macrophage phenotype (CD86+MHCII+) caused by CPMV correlates with higher cytotoxic potential and M1-type activation.[18]
Next, we evaluated the therapeutic potential of the proposed CPMV+CD47 blockade in the 4T1 breast tumor model using BALB/c mice and ID8-Defb29/Vegf-A ovarian tumor model using C57BL/6 mice. In brief, 4T1 mammary carcinoma tumors were intradermally (i.d.) implanted on the right flank of female BALB/c mice. Mice were in situ treated with Phosphate-Buffered Saline (PBS), 70 μg of CPMV, 100 μg CD47 Ab, or combination on day 10 and day 17 post-tumor challenge. The tumors of PBS-treated mice grew progressively (Figure 3A), while all treatment groups led to reduced tumor burden, with most significant delay in tumor growth being achieved with the combination therapy. On day 32, the mean tumor volume of PBS-treated group was 1041 mm3, while the mean tumor volumes of solo CD47 blockade, solo CPMV and the combination were 716 mm3 and 371 mm3, and 274 mm3; therefore these tumors measured 1.4 times (p<0.0005), 2.8 times (p<0.0001), and 3.8 times (p<0.0001) smaller than tumors from PBS-treated animals, respectively. CD47 Ab monotherapy resulted in a modest delay in tumor growth but this delay in tumor growth did not translate to a statistically significant survival advantage (Figure 3B, median survival: CD47 Ab treated mice vs. PBS-treated mice, 40 days vs. 33 days). In contrast, CPMV in situ vaccination and CPMV+CD47 blockade combination more significantly delayed tumor development and therefore significantly prolonged median survival compared with the control mice (median survival: CPMV treated mice vs. PBS-treated mice, 44 days vs. 33 days, p<0.01; combination treated mice vs. PBS-treated mice, 48 days vs. 33 days, p<0.01).
Figure 3.
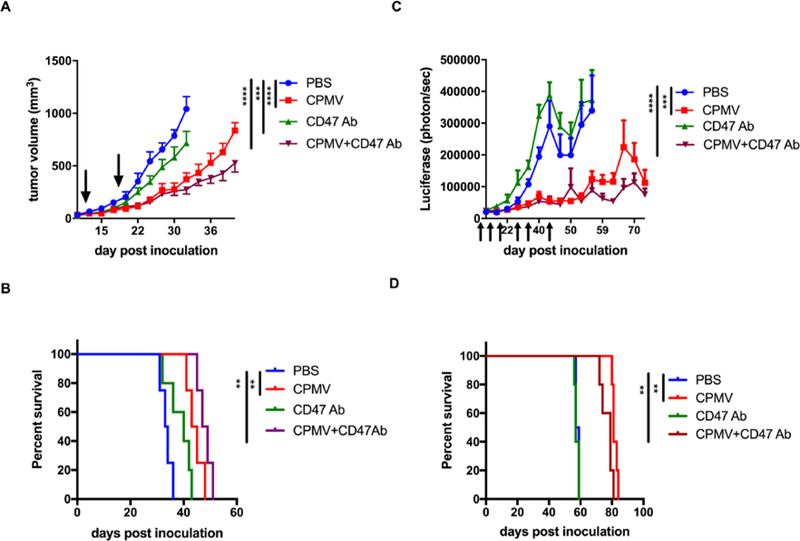
Potentiation of CD47 antibody and CPMV for treatment of 4T1 and ID8-Defb29/Vegf-A tumors. A-B. BALB/c mice were implanted intradermally with 4T1 mammary carcinoma cells (1.25× 105) on the right flank. Mice were treated with PBS, 70 μg of CPMV, 100 μg CD47 Ab, or combination on day 10 and day 17 post-tumor challenge (indicated by black arrow). A. Tumor growth curves shown as relative tumor volume. Growth curves were stopped when the first animal of the corresponding group was sacrificed (tumor volume ≥ 1000 mm3). Statistical significance (on day 32) was calculated by two-way ANOVA with Tukey test. ***p<0.0005, ****p<0.0001. B. Survival rates of treated and control mice. Statistical significance of survival was calculated by Log-rank (Mantel-Cox) test. **p<0.01. C-D. ID8-Defb29/Vegf-A tumor-challenged C57BL/6 mice were treated with 30 μg of CPMV, 100 μg CD47 Ab, or combination weekly beginning on day 7 post-tumor challenge (indicated by black arrow). C. Tumor growth was followed by measuring the luciferase expression in peritoneal cavity. Statistical significance (on day 56) was calculated by two-way ANOVA with Tukey test. ***p<0.0005, ****p<0.0001. Growth curves were stopped when the first animal of the corresponding group was sacrificed (weight ≥ 33 grams). D. Survival rates of treated and control mice. Data are means ± SEM (n=4–5). Statistical significance of survival was calculated by Log-rank (Mantel-Cox) test. **p<0.01.
The observations were not matched in the ovarian tumor model: while CPMV demonstrated anti-tumor efficacy, CD47 blockade did not appear to be effective. C57BL/6 mice were inoculated with ID8-Defb29/Vegf-A ovarian tumor cells intraperitoneally (i.p.) on day 0. Treatment started at day 7 post tumor inoculation: 30 μg of CPMV, 100 μg CD47 Ab, or combination thereof was administered weekly via i.p. injection. Tumor growth was monitored by measuring the luciferase expression in peritoneal cavity (Figure 3C). Mice treated with CD47 Ab had no discernable effect on tumor growth or survival compared with the PBS-treated group (Figure 3C, D). In contrast, solo CPMV treatment was effective; as expected because we had previously demonstrated efficacy of CPMV in this tumor model.[19] Due to the lack of efficacy of the CD47 Ab, there was also no enhancement in treatment efficacy comparing solo CPMV and CPMV+CD47 blockade. On day 56 mean luciferase signal intensity of solo CPMV and combination treatment were 123400 photon/sec (p<0.0005) and 88520 photon/sec (p<0.0001), respectively, which were significantly lower than that of PBS-treated group (339360 photon/sec). Solo CPMV therapy significantly prolonged the median survival compared to PBS-treated group (median survival: CPMV treated mice vs. PBS-treated mice, 81 days vs. 58 days, p<0.01). Again, no additional survival benefit was obtained with combination CD47 Ab and CPMV therapy compared to solo CPMV (Figure 3D). We performed dosing studies and found that blockade of CD47 with a higher dose (300 μg) resulted in a reduction of tumor luciferase expression in peritoneal cavity (Figure S4A, Supporting Information), however, no difference in survival efficacy compared with PBS-treated or lower doses treated groups (Figure S4B, Supporting Information). Furthermore, a more severe malignant ascites and weight increase (Figure S4C, Supporting Information) were overserved in the higher dose CD47 Ab treated mice.
While efficacy of CD47 Ab and its combination with CPMV was demonstrated in the 4T1 model, efficacy was not observed in the ID8-Defb29/Vegf-A model. Nevertheless, we confirmed CD47 expression in the ID8-Defb29/Vegf-A model and also in vitro studies indicated that CD47 blockade had the potential to turn macrophage activity against ID8-Defb29/Vegf-A cells inducing cell killing, which was enhanced in the presence of CPMV. However, data suggest that turning off the ‘eat me not signals’ through blockade of the CD47 axis is not sufficient in the ID8-Defb29/Vegf-A model to slow down tumor progression. Differences in the tumor model, the tumor microenvironment, and different anatomical physiology may explain this discrepancy.
ID8-Defb29/Vegf-A is a highly aggressive ovarian tumor cell line which is engineered to overexpress mouse vascular endothelial growth factor-A (Vegf-A164) and β-defensin-29 (Defb29). It has been demonstrated that tumor vasculogenesis can be mediated by immature DCs through the cooperation of β-defensins and Vegf-A. It is the main reason that intraperitoneal ID8-Defb29/Vegf-A tumors cause rapid ascites accumulation and result in significantly shorter survival than the parent ID8 or ID8-Vegf cell lines.[20] The role of CD47 as a “don’t eat me” signal has been well established. However, CD47 engagement also inhibits inflammatory cytokine production and maintains DCs in an immature state.[21] A hypothesis why CD47 blockade does not show any survival benefit in ID8-Defb29/Vegf-A tumor model is that blocking CD47 may recruit more immature DCs which specifically promotes tumor vasculogenesis. Therefore, it may compromise the benefit of phagocytosis enhancement caused by CD47 blockade therapy.
CD47, as a universal marker in normal healthy tissue, is involved in different physiological processes, such as programmed cell removal, regulation of cardiovascular homeostasis.[22] Hence, blocking CD47 in an indiscriminate manner may result in decreased antibody bioavailability because of antigen sink and on-target toxicities.[23] The ascites fluids of the ID8-Defb29/Vegf-A tumor-bearing mice contain a high concentration of red blood cells and leukocytes. Therefore, the inefficacy of blockade with anti-CD47 monoclonal antibodies is likely due to the large antigen sink of CD47 ubiquitously expressed on those healthy cells in ascites of this malignant ovarian tumor model. In addition, the anti-CD47 monoclonal antibody clone we used (MIAP410) may also results in different efficacies in different tumor models.[3]
Our results indicate that CD47 blockade (at the dosages and administration schedules employed) is insufficient to induce durable antitumor effects, especially long-lasting adaptive immune response, against either the 4T1 breast cancer or ID8-Defb29/Vegf-A ovarian cancer model. While therapeutic efficacy was demonstrated in the 4T1 model, the combination strategy with added VNP in situ vaccination was proven more effective. Plant VNPs, as a novel nano-biomaterials platform, show great potential as immunotherapies and adjuvants to boost the immune system in the context of tumors.[9, 12, 24] From a translational point of view, the plant viral system is attractive for several reasons: compared to mammalian viral vectors and oncolytic viral therapies the plant VNP approach is more bio-safe because plant viruses are non-infectious toward mammals. Furthermore, biopharming allows the large-scale manufacture of VNPs or virus-like particles (VLPs) in plants or by heterologous expression.[25]
In conclusion, in this study we show CD47 blockade alone has the capacity to enhance macrophage phagocytosis to kill tumor cells in vitro, however, is insufficient to induce systemic antitumor effects in 4T1- and ID8-Defb29/Vegf-A-bearing immunocompetent hosts; no efficacy was observed in the ovarian tumor model. When combined with in situ CPMV vaccination, synergistic activity in the 4T1 breast tumor model was demonstrated. This work presents a novel strategy to promote macrophage activity to kill tumor cells, and hold promise to also enhance T cells targeted immunotherapies by inducing both innate and adaptive arms of immune system in the future.
Supplementary Material
Acknowledgements
This work was funded in part by a grant from the NIH, R01CA224605 to NFS and a gift to Case Western Reserve University by Michael R. Shaughnessy. We thank Prof. Steve Fiering (Dartmouth College, NH) for intellectual discussions. We thank Dr. Sourabh Shukla for assisting in vitro stimulation experiment conduction.
Footnotes
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Chao. Wang, Department of NanoEngineering, University of California, San Diego, La Jolla CA 92093, United States. Department of Biomedical Engineering, Case Western Reserve University School of Medicine, Cleveland, OH 44106, United States.
Nicole. F. Steinmetz, Department of Radiology, Moores Cancer Center, Department of Bioengineering, University of California, San Diego, La Jolla CA 92093, United States. nsteinmetz@ucsd.edu
Reference
- [1].a) Disis ML, Patel MR, Pant S, Infante JR, Lockhart AC, Kelly K, Beck JT, Gordon MS, Weiss GJ, Ejadi S, Taylor MH, von Heydebreck A, Chin KM, Cuillerot JM, Gulley JL, J. Clin. Oncol. 2015, 33; [Google Scholar]; Varga A, Piha-Paul SA, Ott PA, Mehnert JM, Berton-Rigaud D, Johnson EA, Cheng JD, Yuan S, Rubin EH, Matei DE, Journal of Clinical Oncology 2015, 33. [Google Scholar]
- [2].Weiskopf K, Eur. J. Cancer 2017, 76, 100. [DOI] [PubMed] [Google Scholar]
- [3].Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, Lovelace P, Scheeren FA, Chao MP, Weiskopf K, Tang C, Volkmer AK, Naik TJ, Storm TA, Mosley AR, Edris B, Schmid SM, Sun CK, Chua MS, Murillo O, Rajendran P, Cha AC, Chin RK, Kim D, Adorno M, Raveh T, Tseng D, Jaiswal S, Enger PO, Steinberg GK, Li G, So SK, Majeti R, Harsh GR, van de Rijn M, Teng NNH, Sunwoo JB, Alizadeh AA, Clarke MF, Weissman IL, Porc. Natl. Acad. Sci. U. S. A. 2012, 109, 6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao F, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R, Cell 2010, 142, 699; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weiskopf K, Ring AM, Ho CCM, Volkmer JP, Levin AM, Volkmer AK, Ozkan E, Fernhoff NB, van de Rijn M, Weissman IL, Garcia KC, Science 2013, 341, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Soto-Pantoja DR, Terabe M, Ghosh A, Ridnour LA, DeGraff WG, Wink DA, Berzofsky JA, Roberts DD, Cancer Res. 2014, 74, 6771; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kaur S, Kuznetsova SA, Pendrak ML, Sipes JM, Romeo MJ, Li ZQ, Zhang LJ, Roberts DD, J. Biol. Chem. 2011, 286, 14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu YX, Xu MM, Nat. Med. 2015, 21, 1209; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, Weissman IL, Porc. Natl. Acad. Sci. U. S. A. 2013, 110, 11103; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sockolosky JT, Dougan M, Ingram JR, Ho CCM, Kauke MJ, Almo SC, Ploegh HL, Garcia KC, Porc. Natl. Acad. Sci. U. S. A. 2016, 113, E2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Brennan FR, Gilleland LB, Staczek J, Bendig MM, Hamilton WDO, Gilleland HE, Microbiology 1999, 145, 2061; [DOI] [PubMed] [Google Scholar]; b) Lomonossoff GP, Hamilton WDO, in Plant Biotechnology: New Products and Applications (Eds.: Hammond J, McGarvey P, Yusibov V), Springer, Berlin, Heidelberg, 2000, pp. 177–189; [Google Scholar]; c) Sainsbury F, Liu L, Lomonossoff GP, in Recombinant Proteins From Plants: Methods and Protocols (Eds.: Faye L, Gomord V), Humana Press, Totowa, NJ: 2009, pp. 25–39. [Google Scholar]
- [8].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S, Nat. Nanotechnol. 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lebel M-È, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A, Nano Lett. 2016, 16, 1826. [DOI] [PubMed] [Google Scholar]
- [10].Jobsri J, Allen A, Rajagopal D, Shipton M, Kanyuka K, Lomonossoff GP, Ottensmeier C, Diebold SS, Stevenson FK, Savelyeva N, Plos One 2015, 10, e0118096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hoopes PJ, Wagner RJ, Duval K, Kang K, Gladstone DJ, Moodie KL, Crary-Burney M, Ariaspulido H, Veliz FA, Steinmetz NF, Fiering SN, Mol. Pharmaceutics 2018, 15, 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Murray AA, Wang C, Fiering S, Steinmetz NF, Mol. Pharmaceutics 2018, 15, 3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL, Cell 2009, 138, 271–285; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Poels LG, Peters D, van Megen Y, Vooijs GP, Verheyen RNM, Willemen A, van Niekerk CC, Jap PHK, Mungyer G, Kenemans P, JNCI: J. Natl. Cancer Inst. 1986, 76, 781; [PubMed] [Google Scholar]; c) Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, Gill H, Presti J, Chang HY, van de Rijn M, Shortliffe L, Weissman IL, Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14016; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jannie M. Rendtlew Danielsen, Lene M. Knudsen, Inger M. Dahl, Lodahl M, Rasmussen T, Br. J. Haematol. 2007, 138, 756; [DOI] [PubMed] [Google Scholar]; e) Feliz-Mosquea YR, Christensen AA, Wilson AS, Westwood B, Varagic J, Meléndez GC, Schwartz AL, Chen Q-R, Griner L. Mathews, Guha R, Thomas CJ, Ferrer M, Merino MJ, Cook KL, Roberts DD, Soto-Pantoja DR, Breast Cancer Res. Treat. 2018, 172, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Smith AA, Wilson AS, Soto-Pantoja DR, Can. Res. 2017, 77, 5661. [Google Scholar]
- [15].Balkwill F, Charles KA, Mantovani A, Cancer Cell 2005, 7, 211; [DOI] [PubMed] [Google Scholar]; Biswas SK, Mantovani A, Nat. Immunol. 2010, 11, 889. [DOI] [PubMed] [Google Scholar]
- [16].a) Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM, Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2796; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, Di W, J. Ovarian Res. 2014, 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Duque G. Arango, Descoteaux A, Front. Immunol. 2014, 5, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, Ring AM, Connolly AJ, Weissman IL, Nature 2017, 545, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Czapar AE, B BD. Tiu, Veliz FA, Pokorski JK, Steinmetz NF, Adv. Sci. 2018, 5, 1700991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Conejo-Garcia JR, Benencia F, Coureges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na HN, Zhang L, Wagner DS, Katsaros D, Caroll R, Coukos G, Nat. Med. 2004, 10, 950; [DOI] [PubMed] [Google Scholar]; b) Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, Benencia F, Stan RV, Keler T, Sarobe P, Sentman CL, Conejo-Garcia JR, Cancer Res. 2008, 68, 7684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Braun D, Galibert L, Nakajima T, Saito H, Quang VV, Rubio M, Sarfati M, J. Immunol. 2006, 177, 8550; [DOI] [PubMed] [Google Scholar]; b) Demeure CE, Tanaka H, Mateo V, Rubio M, Delespesse G, Sarfati M, J. Immunol. 2000, 164, 2193. [DOI] [PubMed] [Google Scholar]
- [22].a) Barclay AN, van den Berg TK, Annu. Rev. Immunol. 2014, 32, 25; [DOI] [PubMed] [Google Scholar]; b) Marika S, Genevieve F, S. Marianne Raymond and Santos, Curr. Drug Targets 2008, 9, 842. [DOI] [PubMed] [Google Scholar]
- [23].a) Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, Willingham S, Howard M, Prohaska S, Volkmer J, Chao M, Weissman IL, Majeti R, Plos One 2015, 10, e0137345; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, van Rooijen N Jr., Weissman IL, Cell 2009, 138, 286; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, Cons L, Calloud S, Majocchi S, Nelson R, Rousseau F, Ferlin W, Kosco-Vilbois M, Fischer N, Masternak K, Mol. Ther. 2017, 25, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Patel R, Czapar AE, Fiering S, Oleinick NL, Steinmetz NF, ACS Omega 2018, 3, 3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) D’Aoust M-A, Couture MMJ, Charland N, Trépanier S, Landry N, Ors F, Vézina L-P, Plant Biotechnol. J. 2010, 8, 607; [DOI] [PubMed] [Google Scholar]; b) Tschofen M, Knopp D, Hood E, Stöger E, Annu. Rev. Anal. Chem. 2016, 9, 271. [DOI] [PubMed] [Google Scholar]
- [26].Wen AM, Le N, Zhou X, Steinmetz NF, Popkin DL, ACS Biomaterials Science & Engineering 2015, 1, 1050–1054; Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ, Mol Pharm 2013, 10, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wallat JD, Czapar AE, Wang C, Wen AM, Wek KS, Yu X, Steinmetz NF, Pokorski JK, Biomacromolecules 2017, 18, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hashimoto K, Man S, Xu P, Cruz-Munoz W, Tang T, Kumar R, Kerbel RS, Molecular Cancer Therapeutics 2010, 9, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM, Cancer Cell 2009, 16, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.