

Abstract
This chemoenzymatic protocol describes a strategy for the preparation of 4′-thioribose nicotinamide adenine dinucleotide (S-NAD+), including chemical synthesis of nicotinamide 4′-riboside (S-NR), recombinant expression and purification of two NAD+ biosynthesis enzymes nicotinamide riboside kinase (NRK) and nicotinamide mononucleotide adenylyltransferase (NMNAT), and enzymatic synthesis of S-NAD+. The first part describes the procedures for introduction of nicotinamide onto 4’-thioribose and subsequent deprotections to generate S-NR as the key intermediate for enzymatically synthesizing S-NAD+. In the second part, experimental methods are detailed for the production of recombinant human NRK1 and NMNAT1 to catalyze conversion of S-NR to S-NAD+. The last part presents the enzymatic approach for the generation of S-NAD+ from S-NR precursor.
Keywords: chemoenzymatic synthesis, nicotinamide riboside, 4′-thioribose, NAD+, NRK, NMNAT
INTRODUCTION
This unit provides a chemoenzymatic methodology for the synthesis of 4′-thioribose nicotinamide adenine dinucleotide (S-NAD+), a novel stable mimic of nicotinamide adenine dinucleotide (NAD+) (Figure 1A) (Dai et al., 2018). Inspired by nicotinamide riboside kinase (NRK)- and nicotinamide mononucleotide adenylyltransferase (NMNAT)-mediated biosynthesis of NAD+ from its NR precursor (Figure 1B) (Berger, Lau, Dahlmann, & Ziegler, 2005; Sorci et al., 2007; Tempel et al., 2007), we envisioned that S-NAD+ could be enzymatically synthesized in high efficiency and yields from nicotinamide 4′-thioriboside (S-NR) by exploiting recombinant NRK and NMNAT enzymes. The first set of methods presented in this unit (Basic Protocol 1) outlines the chemical synthesis of S-NR. In this part, introduction of nicotinamide onto 4′-thioribose is a key step for successfully making S-NR nucleoside. Next, experimental procedures for expression and purification of human NRK1 and NMNAT1 are reported in Basic Protocol 2. On the basis of this protocol, resulting NRK1 and NMNAT1 enzymes could be applied to subsequent enzymatic synthesis of S-NAD+. In Basic Protocol 3, the approach is described in detail for NRK1- and NMNAT1-catalyzed preparation of S-NAD+ from S-NR molecule. By following these procedures, S-NAD+ could be efficiently generated in high yields from S-NR through one-pot, two-step enzymatic synthesis.
Figure 1.

Enzymatic synthesis of S-NAD+ from S-NR. (A) Chemical structures of NAD+, S-NAD+ and S-NR. (B) NRK- and NMNAT-catalyzed formation of NAD+ from NR. ATP: adenosine triphosphate; ADP: adenosine diphosphate; PPi: pyrophosphate.
NOTE: All glassware should be dried, and all reactions should be performed under anhydrous conditions.
CAUTION: All chemical reactions must be run in a suitable fume hood with efficient ventilation. Proper safety glasses, gloves, and lab coats should be worn at all times.
Compound characterization.
Chemical characterization data are provided for all compounds. 1H NMR spectra were recorded on an Oxford AM-400 spectrometer for solution in CDCl3, CD3OD or D2O. Coupling constants J are shown in Hz. 13C NMR spectra were recorded on an Oxford AM-400 spectrophotometer (100 MHz) with a complete proton decoupling spectrophotometer. Flash column chromatography was performed using 230–400 mesh silica gel (Sigma-Aldrich, St. Louis, MO). For thin-layer chromatography (TLC), silica gel plates coated with fluorescent indicator F254 (Sigma-Aldrich) were used. High-performance liquid chromatography (HPLC) was performed using a Waters 2487 series equipped with a C18 Kinetex column (5 µm, 100 Å, 150×10.0 mm, Phenomenex Inc, Torrance, CA, USA). All other reagents were purchased from readily available commercial sources and used without further purification.
BASIC PROTOCOL 1: PREPARATION OF NICOTINAMIDE 4′-THIORIBOSIDE (S-NR)
The methods for chemical synthesis of nicotinamide riboside (NR) were previously reported (Dowden, Brown, Moreau, Galione, & Potter, 2005; Dowden et al., 2004; Franchetti et al., 2004; Tanimori, Ohta, & Kirihata, 2002) and applied for making NR derivatives (Carter-O’Connell, Jin, Morgan, David, & Cohen, 2014; Cen & Sauve, 2010). In these reported synthetic methods, NR and its analogues were typically generated through introduction of the nicotinamide onto protected riboses and subsequent deprotections. However, the chemical synthesis of S-NR had not been reported until 2018 (Dai et al., 2018). In this protocol, the methodology is presented for chemical synthesis of S-NR through introduction of nicotinamide onto 4′-thioribose (Figure 2). The protected 4′-thioribose 1 was prepared first according to a reported procedure (Jeong et al., 2006), followed by sequential treatments with HBr (33% (wt) in acetic acid) in toluene at 0°C and nicotinamide in CH3CN at room temperature to afford 2 in a 58% yield (Figure 2). The removal of acetonide protecting group was then carried out using a mixture of TFA/H2O (9/1) at 0°C to generate 3 with a final yield of 76%. Lastly, deprotection of 3 using ammonia in methanol resulted in S-NR nucleoside for a 64% yield.
Figure 2.

Chemical synthesis of S-NR.
Materials
((3aS,4R,6aR)-6-acetoxy-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)methyl benzoate (1)(Jeong et al., 2006)
Dry argon (Ar)
Anhydrous toluene (Acros Organics)
Hydrogen bromide, HBr, pure, 33 wt% solution in glacial acetic acid (Acros Organics)
Nicotinamide (Acros Organics)
Anhydrous acetonitrile (CH3CN, Acros Organics)
Trifluoroacetic Acid (TFA, Oakwood Chemical)
Ammonia (NH3), ca. 7N solution in methanol (Acros Organics)
Methanol (MeOH, HPLC grade, Fisher Chemical)
Anhydrous ethyl ether (Et2O, HPLC grade, Fisher Chemical)
Petroleum ether (Certified ACS, Fisher Chemical)
Ethyl acetate (HPLC grade, Fisher Chemical)
Methylene chloride (HPLC grade, Fisher Chemical)
Deuterated methanol (CD3OD) for NMR characterization, >99.5% pure (Oakwood Chemical)
Deuterium oxide (D2O) for NMR characterization, 99.9% pure (Oakwood Chemical)
Silica gel (60 Å, 70–230 mesh, 63–200 μm, Sigma-Aldrich)
Silica gel (60 Å, 230–400 mesh, 40–63 μm, Sigma-Aldrich)
Deionized water
50- and 100-mL round-bottom flasks
Magnetic stir bars
Magnetic stirring plates
Rotary evaporator equipped with vacuum pump
Oil vacuum pump
Glass flash chromatography columns
Analytical TLC plates (aluminum-backed, precoated with silica gel 60 matrix, fluorescent indicator F254; Sigma-Aldrich)
UV light source
Glass funnels
Filter papers
Preparation of 1-((3aR,4R,6R,6aS)-6-((benzoyloxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-3-carbamoylpyridin-1-ium bromide (2)
-
1
Place 106 mg (0.3 mmol) of ((3aS,4R,6aR)-6-acetoxy-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)methyl benzoate (1) in a 100-mL round-bottom flask containing a magnetic stir bar, and replace air in the flask with dry argon.
-
2
Add 10 mL of anhydrous toluene.
-
3
Place the flask in an ice bath and cool the solution of 1 in toluene to 0°C.
-
4
While stirring, add 110 mg (0.45 mmol) of HBr (33% (wt) in acetic acid) at 0°C and continue stirring for 5 hours under an argon atmosphere.
-
5
After 1 is consumed, evaporate the solvent under reduced pressure using a rotary evaporator to offer a brown residue.
-
6
Azeotrope the residue with anhydrous toluene (20 mL) for 3 times to remove remaining acetic acid and dry it in vacuo to provide a brominated crude product.
-
7
Add 44 mg (0.36 mmol) of nicotinamide and a magnetic stir bar to the flask containing the brominated crude product and replace air in the flask with dry argon.
-
8
Add 10 mL of anhydrous acetonitrile and start to stir under argon atmosphere at ambient temperature for 24 hours.
-
9
Evaporate the reaction mixture to dryness using a rotary evaporator under reduced pressure while keeping temperature of the water bath below 35°C.
-
10
Dissolve the crude product in a minimum amount of CH2Cl2 and carefully place the solution on top of a glass flash chromatography column packed with silica gel in petroleum ether. Elute with pure ethyl acetate and a gradient solution of CH2Cl2/MeOH (99:1 to 93:7 v/v).
-
11
Examine the fractions by TLC using UV light, combine those fractions containing products and evaporate to dryness using a rotary evaporator under reduced pressure while keeping temperature of the water bath below 35°C to afford 86 mg of 1-((3aR,4R,6R,6aS)-6-((benzoyloxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-3-carbamoylpyridin-1-ium bromide (2) in a 58% yield as a colorless solid.
-
12
Characterize the compound by 1H NMR and 13C NMR.
1H NMR (400 MHz, CD3OD): δ 1.45 (s, 3H, CH3), 1.67 (s, 3H, CH3), 4.31 (t, 1H, J = 4.4 Hz, CH), 4.57 (dd, 1H, J = 12.0, 4.4 Hz, CH2), 4.75 (dd, 1H, J = 12.0, 4.4 Hz, CH2), 5.22 (dd, 1H, J = 4.8, 0.8 Hz, CH), 5.46 (d, 1H, J = 4.8 Hz, CH), 6.51 (s, 1H, CH), 7.36 (t, 2H, J = 8.0 Hz, ArH), 7.55–7.59 (m, 1H, ArH), 7.65–7.67 (m, 2H, ArH), 8.03 (t, 1H, J = 6.8 Hz, ArH), 8.70 (d, 1H, J = 8.0 Hz, ArH), 9.44 (d, 1H, J = 6.8 Hz, ArH), 9.70 (s, 1H, ArH).
13C NMR (100 MHz, CD3OD): δ 23.8, 26.0, 54.4, 66.4, 86.2, 86.6, 92.6, 112.9, 127.9, 128.4, 128.8, 129.3, 133.2, 133.8, 142.7, 144.1, 144.4, 163.0, 165.8.
Preparation of 1-((2R,3R,4S,5R)-5-((benzoyloxy)methyl)-3,4-dihydroxytetrahydrothiophen-2-yl)-3-carbamoylpyridin-1-ium bromide (3)
-
13
Prepare a 9:1 (v/v) mixture of TFA and water (18/2 mL) and cool the mixture to 0°C using an ice bath.
-
14
Place 50 mg (0.1 mmol) of 1-((3aR,4R,6R,6aS)-6-((benzoyloxy)methyl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)-3-carbamoylpyridin-1-ium bromide (2) in a 100-mL round-bottom flask containing a magnetic stir bar, and replace air in the flask with dry argon.
-
15
Add 15 mL of the prepared 0°C 9/1 (v/v) mixture of TFA/H2O.
-
16
Place the flask in an ice bath and continue stirring at 0°C until the reaction complete while monitoring by TLC.
-
17
Evaporate the reaction mixture to dryness using a rotary evaporator under reduced pressure while keeping temperature of the water bath below 35°C.
-
18
Add 0.3 mL of MeOH to dissolve the crude product.
-
19
Add 20 mL of anhydrous ethyl ether to precipitate the desired product.
-
20
Collect the precipitation by filtration.
-
21
Repeat procedures 18, 19 and 20 sequentially for two more times to afford 35 mg of compound 3 in a 76% yield as a colorless solid.
-
22
Characterize the compound by 1H NMR and 13C NMR.
1-((2R,3R,4S,5R)-5-((benzoyloxy)methyl)-3,4-dihydroxytetrahydrothiophen-2-yl)-3-carbamoylpyridin-1-ium bromide (3). 1H NMR (400 MHz, CD3OD): δ 3.92 (td, 1H, J = 6.8, 2.8 Hz, CH), 4.43 (t, 1H, J = 3.2 Hz, CH), 4.52 (dd, 1H, J = 6.8, 3.2 Hz, CH), 4.74 (d, 2H, J = 6.8 Hz, CH2), 6.23 (d, 1H, J = 6.8 Hz, CH), 7.51 (t, 2H, J = 8.0 Hz, ArH), 7.63–7.67 (m, 1H, ArH), 8.05–8.07 (m, 2H, ArH), 8.21 (dd, 1H, J = 8.0, 6.4 Hz, ArH), 9.03 (d, 1H, J = 8.0 Hz, ArH), 9.46 (d, 1H, J = 6.4 Hz, ArH), 9.68 (s, 1H, ArH).
13C NMR (100 MHz, CD3OD): δ 51.5, 65.1, 73.9, 80.4, 81.4, 128.0, 128.4, 129.2, 129.4, 133.3, 134.5, 143.6, 144.9, 145.1, 163.4, 166.2.
Preparation of 3-carbamoyl-1-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrothiophen-2-yl)pyridin-1-ium bromide (S-NR)
-
23
Place 23 mg (0.05 mmol) of 1-((2R,3R,4S,5R)-5-((benzoyloxy)methyl)-3,4-dihydroxytetrahydrothiophen-2-yl)-3-carbamoylpyridin-1-ium bromide (3) in a 50-mL round-bottom flask containing a magnetic stir bar and replace air in the flask with dry argon.
-
24
Add 5 mL of ammonia (7 N solution in methanol).
-
25
Place the flask in an ice bath and stir at 0°C for 48 hours.
-
26
Evaporate the reaction mixture to dryness using a rotary evaporator under reduced pressure while keeping temperature of the water bath below 35°C to give a residue.
-
27
Add 0.2 mL of MeOH to dissolve the crude product.
-
28
Add 20 mL of anhydrous ethyl ether to precipitate the desired product.
-
29
Collect the precipitation by filtration.
-
30
Repeat procedures 27, 28 and 29 sequentially for two more times to afford 11 mg of compound S-NR in a 64% yield as a colorless solid.
-
31
Characterize the compound by 1H NMR, 13C NMR and high-resolution mass spectrometry (HRMS).
3-carbamoyl-1-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrothiophen-2-yl)pyridin-1-ium (S-NR). 1H NMR (400 MHz, D2O): δ 3.67–3.70 (m, 1H, CH), 3.92–4.01 (m, 2H, CH2), 4.38 (t, 1H, J = 3.6 Hz, CH), 4.62 (dd, 1H, J = 5.6, 3.6 Hz, CH), 6.19 (d, 1H, J = 5.6 Hz, CH), 8.26 (dd, 1H, J = 8.0, 6.4 Hz, ArH), 8.97 (dt, 1H, J = 8.0, 1.2 Hz, ArH), 9.45 (dt, 1H, J = 6.4, 1.2 Hz, ArH), 9.78 (t, 1H, J = 1.2 Hz, ArH).
13C NMR (100 MHz, D2O): δ 53.9, 61.5, 73.8, 80.6, 80.9, 128.4, 134.1, 143.2, 145.4, 145.5, 165.6.
HRMS (ESI) for C11H15N2O4S+ (M)+ Calcd.: 271.0763 Da; Obs: 271.0753 Da.
BASIC PROTOCOL 2: PREPARATION OF HUMAN NRK1 AND NMNAT1
Human NRK1 and NMNAT1 were expressed and purified from Escherichia coli based on previous reports (Khan, Xiang, & Tong, 2007; Raffaelli et al., 2002). A His6-tag was placed at the C-termini of the open reading frames (ORFs) of human NRK1 (UniProt Entry: Q9NWW6) and NMNAT1 (UniProt Entry: Q9HAN9). The resulting gene fragments were then ligated into pET-28a (+) expression vector.
It should be noted that to construct the bacterial expression vectors of human NRK1 and NMNAT1 and express both enzymes in E. coli, different cloning reagents (i.e. DNA restriction enzymes), procedures (i.e. transformation methods), expression vectors, and compatible E. coli strains can be used, depending on resource availability.
Prior to the expression in 1 liter of bacterial culture, E. coli harboring the constructed expression vectors of NRK1 or NMNAT1 were inoculated into 5 mL of Lysogeny broth (LB) media supplemented with 50 µg/mL kanamycin and cultured overnight at 37°C in a shaker incubator with a speed of 250 rpm. The overnight culture was transferred into sterile 1 liter of LB with 50 µg/mL kanamycin for expansion in a shaker incubator at 37°C until optical density at 600 nm (OD600nm) reached 0.6, followed by additions of isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 0.5 mM for inducing protein expression at 18°C for overnight. E. coli cells were then harvested by centrifugation and lysed for protein purification through affinity chromatography.
Materials
Synthetic DNA fragments encoding human NRK1 and NMNAT1 ORFs (Integrated DNA Technologies (IDT))
Primers for amplifying NRK1 and NMNAT1 with C-terminal His6-tags and restriction enzyme sites (IDT)
pET-28a (+) expression vector (EMD Millipore)
DH10B electrocompetent cells (Thermo Fisher Scientific)
BL21 (DE3) electrocompetent cells (New England BioLabs)
Agarose (Thermo Fisher Scientific, MA)
Ethidium bromide (1% solution) (Thermo Fisher Scientific)
Zymoclean Gel DNA Recovery Kit (Zymo Research)
ZR Plasmid Miniprep (Zymo Research)
AccuPrime Pfx DNA Polymerase (Thermo Fisher Scientific)
DNA restriction enzymes NcoI and XhoI (New England BioLabs)
T4 DNA ligase (New England BioLabs)
Electroporation cuvettes 1 mm width (Thermo Fisher Scientific)
Kanamycin sulfate (Thermo Fisher Scientific)
LB Broth, Miller (Thermo Fisher Scientific)
Imidazole (Thermo Fisher Scientific)
Sodium chloride (Thermo Fisher Scientific)
Tris hydrochloride (Thermo Fisher Scientific)
IPTG (VWR International)
Deionized water
Equilibrium buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 20 mM imidazole)
Wash buffer (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 30 mM imidazole)
Elution buffer (20 mM Tris-HCl, pH 8.0, 200mM NaCl, 400 mM imidazole)
Storage buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1 mM DTT, 10% glycerol)
Disposable plastic cuvettes for measurements of OD600nm (VWR International)
Whatman cellulose acetate membrane filters 0.45 μm (GE Healthcare)
Ni-NTA agarose resin (Thermo Fisher Scientific)
Amicon Ultra Centrifugal Filters 10 kDa (EMD Millipore)
14-mL sterile round-bottom culture tubes with caps
1-L centrifuge bottles
Nalgene Oak Ridge high-speed polypropylene copolymer centrifuge tubes
2-L sterile shaker flasks
Petri dishes, 100 mm X 15 mm
Molecular cloning of human NRK1 and NMNAT1 into bacterial expression vectors
-
1
Amplify ORFs of human NRK1 and NMNAT1 with C-terminal His6-tags and NcoI and XhoI restriction enzyme sites from synthesized DNA fragments using designed primers via polymerase chain reaction (PCR) reactions.
-
2
Resolve the amplified DNA fragments through DNA gel electrophoresis (0.2 μg/mL ethidium bromide in DNA agarose gels), followed by purification using DNA gel recovery kits.
-
3
Digest pET-28a (+) vector and the purified DNA fragment inserts with NcoI and XhoI enzymes for 3 hours at 37°C, followed by DNA gel electrophoresis and purification of the digested vector backbone and gene inserts using DNA gel recovery kits.
-
4
Incubate the purified gene inserts with pET-28a vector backbone at a molar ratio of 7:1 in the presence of T4 DNA ligase using the supplied reaction buffer for 1 hour at room temperature. Using the purified pET-28a vector backbone alone as a negative control.
-
5
Transform 50 µL DH10B electrocompetent cells with 1 µL of ligation products through electroporation, followed by rescue with 300 µL pre-wared LB medium and incubation at 37°C in a shaker incubator (250 rpm) for 30 minutes.
-
6
Spread the transformants onto LB agar plates with 50 µg/mL kanamycin and incubate overnight at 37°C. Inoculate colonies appeared after 18-hour incubation into 5 mL of LB media with 50 µg/mL kanamycin for overnight growth at 37°C. Extract plasmids using ZR Plasmid Miniprep and confirm the expression constructs through DNA sequencing.
Expression of human NRK1 and NMNAT1 in E. coli
-
7
Transform BL21(DE3) electrocompetent cells with sequence-verified pET-28a expression vectors of human NRK1 or NMNAT1. Spread the transformants onto LB agar plates with 50 µg/mL kanamycin and incubate overnight at 37°C. Inoculate colonies into 5 mL LB media with 50 µg/mL of kanamycin and incubate overnight at 37°C in a shaker incubator (250 rpm).
-
8
Dilute overnight culture into 1 liter of sterile LB media with 50 µg/mL of kanamycin and incubate in a shaker incubator at 37°C with a speed of 250 rpm.
-
9
At OD600nm of 0.6, add IPTG to a final concentration of 0.5 mM and incubate at 18°C in a shaker incubator at 250 rpm for overnight expression.
Purification of human NRK1 and NMNAT1
-
10
Harvest bacterial cells after overnight protein expression by centrifugation at 4,000 g for 30 minutes.
-
11
Resuspend cell pellets in equilibrium buffer, followed by lysis using a French Press at 25,000 psi by passing through the cell resuspension for three cycles.
-
12
Spin the cell lysates at 14,000 g for 1 hour, collect supernatants, and pass through 0.45 μm filter membranes.
-
13
Load the filtered supernatants onto a gravity-flow column with 1 mL Ni-NTA agarose resin, followed by washing the columns with 15 column volume (CV) of equilibrium buffer and 15 CV of wash buffer.
-
14
Elute the bound human NRK1 and NMNAT1 with 15 CV of elution buffer, followed by dialysis against storage buffer overnight at 4°C. Then, change the dialysis buffer and incubate for another 6 hours at 4°C.
-
15
Concentrate the purified human NRK1 and NMNAT1 using Amicon centrifugal concentrators (10 kDa molecular weight cut-off) and determine the protein concentrations based on UV absorbance at 280 nm. The calculated molecular extinction coefficient values are 1.537 for NRK1-His6 and 1.592 for NMNAT1-His6.
-
16
Analyze the purified NRK1 and NMNAT1 by SDS-PAGE gel stained with Coomassie blue (Figure 3). The yields are 1.265 mg per liter for human NRK1 and 3.264 mg per liter for human NMNAT1.
Figure 3.
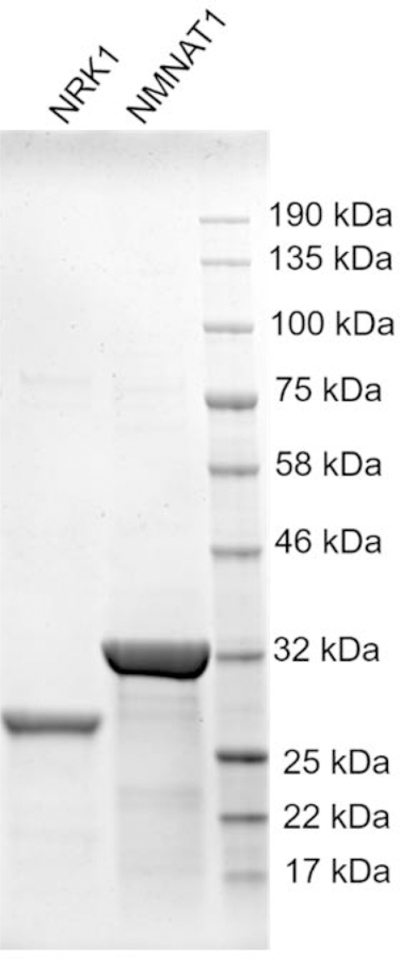
SDS-PAGE gel of purified human NRK1 and NMNAT1 enzymes stained with Coomassie blue. Protein loadings were normalized to 3 µg per lane.
BASIC PROTOCOL 3: PREPARATION OF NICOTINAMIDE 4’-THIORIBOSE NAD+ (S-NAD+)
NAD+ could be biosynthesized from its NR precursor through two sequential reactions catalyzed by NRK and NMNAT. Considering the high levels of similarity in geometry and electrostatics between NAD+ and S-NAD+, the chemically synthesized S-NR could also be utilized by NRK1 and NMNAT1 for efficient formation of S-NAD+. This protocol presents a facile and efficient approach for the generation of S-NAD+ through one-pot, two-step enzymatic reactions using recombinant human NRK1 and NMNAT1 (Figure 4).
Figure 4.

Enzymatic synthesis of S-NAD+.
Materials
Adenosine 5′-triphosphate (ATP) disodium salt hydrate (Sigma-Aldrich)
Tris base (Tris, Bioland Scientific LLC)
Sodium chloride (NaCl, Fisher Chemical)
Magnesium chloride (MgCl2, Fisher Chemical)
Dithiothreitol (DTT, VWR International)
Formic acid (Acros Organics)
Methanol (MeOH, HPLC grade, Fisher Chemical)
Deionized water
Deuterium oxide (D2O) for NMR characterization, 99.9% pure (Oakwood Chemical)
100-mL round-bottom flasks
Rotary evaporator equipped with vacuum pump
1.5 mL centrifuge tubes (VWR International)
15 mL centrifuge tubes (VWR International)
Semipreparative HPLC (Waters 2695 separations module; Waters 2487 dual λ absorbance detector; Kinetex C18 150 × 10.0 mm reversed-phase column)
Fast-freeze flasks
Lyophilizer
Preparation of nicotinamide 4′-thioribose NAD+ (S-NAD+)
Prepare a 100 mL of reaction buffer which consists of 50 mM Tris, pH 7.5, 100 mM NaCl, 12 mM MgCl2 and 1 mM DTT.
Place 0.5 mg (1.4 μmol) of S-NR (final concentration 2 mM) in a 1.5 mL centrifuge tube.
Add 693 μL of the reaction buffer.
Add 3.5 μL of purified human NRK1 and 3.5 μL of human NMNAT1 (1 mM) to final concentrations of 5 μM for both.
Add 2.3 mg (4.2 μmol) of ATP (final concentration 6 mM) to the reaction mixture.
Incubate the reaction mixtures at room temperature for 4 hours.
Centrifuge the reactions and transfer the supernatants into two 400 μL HPLC vials for HPLC analysis.
Analyze the reactions by HPLC based on UV absorbance at 260 nm using a semipreparative C18 Kinetex column (5 µm, 100 Å, 150×10.0 mm, from Phenomenex Inc, Torrance, CA) and the following method: mobile phase A: 0.1% formic acid (aq); mobile phase B: 0.1% formic acid in methanol; flow rate: 2.0 mL/min; 0–2 min: 0–4% B, 2–4 min: 4–10% B; 4–6 min: 10–20% B; 6–9 min: 20% B; 9–12 min: 20–50% B; and 12–14 min: 50–0% B (Figure 5).
Collect the fractions containing S-NAD+.
Combine and evaporate the fractions to dryness using a rotary evaporator under reduced pressure while keeping temperature of the water bath below 35°C to yield the desired product.
Add 1 mL of water to dissolve the product and lyophilize the aqueous solution to offer 0.68 mg of S-NAD+ as a colorless powder in a 70% yield.
-
Characterize the compound by 1H NMR, 13C NMR and HRMS.
S-NAD+. 1H NMR (400 MHz, D2O): δ 3.76–3.79 (m, 1H, CH), 4.22–4.40 (m, 5H, 2CH2+CH), 4.48 (t, 1H, J = 3.2 Hz, CH), 4.51–4.54 (m, 1H, CH), 4.68 (dd, 1H, J = 6.4, 3.6 Hz, CH), 4.75 (t, 1H, J = 5.2 Hz, ArH), 6.13–6.15 (m, 2H, 2CH), 8.26 (dd, 1H, J = 8.0, 6.4 Hz, ArH), 8.41 (s, 1H, ArH), 8.62 (s, 1H, ArH), 8.92 (d, 1H, J = 8.0 Hz, ArH), 9.55 (d, 1H, J = 6.4 Hz, ArH), 9.60 (s, 1H, ArH).
13C NMR (100 MHz, D2O): δ 52.6 (d, J = 8.0 Hz), 65.0–65.1 (m), 65.9–66.1 (m), 70.2, 74.5, 74.9, 81.0, 81.2, 84.0 (d, J = 8.9 Hz), 87.7, 128.7, 133.9, 142.2, 142.8, 145.2, 145.4, 145.8, 150.2, 165.4.
HRMS (ESI) for C21H27N7O13P2SNa+ (M+Na)+: Calcd.: 702.0766 Da; Obs: 702.0760 Da.
Figure 5.

HPLC analysis of enzymatic synthesis of S-NAD+ by purified NRK1 and NMNAT1 as measured by UV absorbance at 260 nm. Adapted with permission from Chem. Sci., 2018, 9, 8337–8342 Published by The Royal Society of Chemistry.
COMMENTARY
Background Information
As an essential cofactor, NAD+ participates in numerous biological processes (Imai & Guarente, 2014; Lin, 2007; K. W. Ryu, Kim, & Kraus, 2015; Verdin, 2015). Recent studies indicate that NAD+-dependent enzymes are extensively involved in pathophysiological conditions (Blacher et al., 2015; Chatterjee et al., 2018; Morandi et al., 2015; D. Ryu et al., 2016; Scarpa, Fabrizio, & Di Girolamo, 2013; Tarragó et al., 2018). Therefore, elucidation of their mechanisms of action is a key to the development of better therapies for many human diseases. Toward this end, x-ray crystallography for reactive enzyme complexes provides direct information about the catalytic reactions at the molecular levels (Hassa, Haenni, Elser, & Hottiger, 2006). Due to the short lives for reactive complexes with bound NAD+, stable NAD+ analogues are often used for this purpose. However, current NAD+ analogues are suboptimal in mimicking NAD+ due to either the lack of nicotinamide leaving group for redox reactions or the changed ribosyl geometry resulted from methylene substitution (Jiang et al., 2009; Langelier, Zandarashvili, Aguiar, Black, & Pascal, 2018; Shrimp et al., 2014; Szczepankiewicz et al., 2012; Zatorski, Watanabe, Carr, Goldstein, & Pankiewicz, 1996). To create a stable analogue closely mimicking NAD+ in both geometry and electrostatics, the endocyclic oxygen was replaced by sulfur atom to afford the S-NAD+ molecule, which features a UV absorption spectrum nearly identical to that NAD+ (Figure 6). The sulfur substitution is expected to improve stability of N-glycosidic bond while minimizing changes in ribosyl geometry, resulting in a novel stable NAD+ mimic.
Figure 6.

UV absorption spectra of NAD+ and S-NAD+. 0.3 mM NAD+ (A) or S-NAD+ (B) was used to measure the UV absorption spectra (190–400 nm) by a NanoDrop 2000C spectrophotometer. Adapted with permission from Chem. Sci., 2018, 9, 8337–8342 Published by The Royal Society of Chemistry.
Despite established procedures for preparing 4′-thionucleosides (Jeong et al., 2006), a method to generate S-NR had not been reported until 2018 (Dai et al., 2018). Thus, introduction of nicotinamide to 4′-thioribose 1 to form the intermediate 2 is a key step for generation of S-NAD+ (Figure 4).
Previous studies report a series of chemically synthesized NAD+ analogues for investigating NAD+-dependent ADP-ribosylation (Buntz et al., 2016; Carter-O’Connell et al., 2014; Carter-O’Connell et al., 2016; Gibson et al., 2016; Jiang, Kim, Frizzell, Kraus, & Lin, 2010; Wallrodt, Buntz, Wang, Zumbusch, & Marx, 2016; Wang, Rösner, Grzywa, & Marx, 2014). These synthetic methodologies for preparing NAD+ analogues have proven challenging because of the synthetic complexity, low efficiency for the last difficult pyrophosphate coupling step, and tedious and time-consuming product purification. To synthesize S-NAD+ in a more efficient manner, we exploit NRK and NMNAT enzymes involved in the biosynthesis of NAD+. Our enzymatic conversion of S-NR to S-NAD+ demonstrates a facile and efficient approach for the generation of S-NAD+ (Figure 4).
Critical Parameters and Troubleshooting
The successful introduction of nicotinamide onto 4′-thioribose to generate 2 in Basic Protocol 1 is critical for the preparation of S-NR. By testing conditions to replace the OAc group of 1 with nicotinamide, it was found that 2 could be successfully generated in a good yield by sequentially treating 1 with HBr (33% (wt) in acetic acid) in toluene at 0°C for 4 hours and nicotinamide in CH3CN at room temperature for 18 hours. In these two-step reactions, the equivalence of HBr (33% (wt) in acetic acid) to 1 (1.5) and the reaction temperature (0°C) for the first step are crucial to achieve a good yield of 2. Increasing the equivalence of HBr (33% (wt) in acetic acid) or the reaction temperature would reduce the yield of 2.
The concentrations of S-NR, NRK1, NMNAT1 and ATP used for the enzymatic synthesis of S-NAD+ in Basic Protocol 3 allow scale-up of the reaction in high efficiency.
The chemoenzymatic procedures presented in this unit are intended for experienced users with common laboratory techniques in organic chemistry and biochemistry. Product characterization requires knowledge of NMR spectrum and mass spectroscopy. Careful attention to details of the experiments is required. Laboratory safety and appropriate personal protection are of primary concern when handling hazardous materials.
Understanding Results
The method applied in Basic Protocol 1 to introduce nicotinamide onto 4′-thioribose for substituting the OAc group can be utilized to prepare S-NR derivatives with modifications at different positions of 4′-thioribose.
The demonstrated high efficiency in enzymatic synthesis of S-NAD+ suggests that NRK and NMNAT enzymes may also allow facile and efficient synthesis of novel NAD+ analogues.
Time Considerations
It takes five days to prepare and purify S-NR. Bacterial expression and purification of human NRK1 and NMNAT1 require four days. The enzymatic synthesis, purification, and characterization of S-NAD+ could be finished within just three days, including the lyophilization.
REAGENTS AND SOLUTIONS
Non-sterile or sterile deionized water is used in all recipes and protocol steps.
ACKNOWLEDGEMENT
This work was supported by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, Sharon L. Cockrell Cancer Research Fund, The V Foundation for Cancer Research V Scholar Grant V2016–021 (to Y.Z.), and University of Southern California Research Center for Liver Diseases Pilot Grant P30DK048522 (to Y.Z.).
LITERATURE CITED
- Berger F, Lau C, Dahlmann M, & Ziegler M (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. Journal of Biological Chemistry, 280(43), 36334–36341. [DOI] [PubMed] [Google Scholar]
- Blacher E, Dadali T, Bespalko A, Haupenthal VJ, Grimm MO, Hartmann T, & Levy A (2015). Alzheimer’s disease pathology is attenuated in a CD 38-deficient mouse model. Annals of neurology, 78(1), 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buntz A, Wallrodt S, Gwosch E, Schmalz M, Beneke S, Ferrando-May E, Zumbusch A (2016). Real-Time Cellular Imaging of Protein Poly (ADP-ribos) ylation. Angewandte Chemie International Edition, 55(37), 11256–11260. [DOI] [PubMed] [Google Scholar]
- Carter-O’Connell I, Jin H, Morgan RK, David LL, & Cohen MS (2014). Engineering the substrate specificity of ADP-ribosyltransferases for identifying direct protein targets. Journal of the American Chemical Society, 136(14), 5201–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O’Connell I, Jin H, Morgan RK, Zaja R, David LL, Ahel I, & Cohen MS (2016). Identifying family-member-specific targets of mono-ARTDs by using a chemical genetics approach. Cell reports, 14(3), 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen Y, & Sauve AA (2010). Transition state of ADP-ribosylation of acetyllysine catalyzed by Archaeoglobus fulgidus Sir2 determined by kinetic isotope effects and computational approaches. Journal of the American Chemical Society, 132(35), 12286–12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, & Kang I (2018). CD38-NAD+ axis regulates immunotherapeutic anti-tumor T cell response. Cell metabolism, 27(1), 85–100. e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Zhang X-N, Nasertorabi F, Cheng Q, Pei H, Louie SG, & Zhang Y (2018). Facile chemoenzymatic synthesis of a novel stable mimic of NAD+. Chemical science, 9(44), 8337–8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowden J, Brown RS, Moreau C, Galione A, & Potter BV (2005). Chemical synthesis of the novel Ca2+ messenger NAADP. Nucleosides, Nucleotides and Nucleic Acids, 24(5–7), 513–518. [DOI] [PubMed] [Google Scholar]
- Dowden J, Moreau C, Brown RS, Berridge G, Galione A, & Potter BV (2004). Chemical synthesis of the second messenger nicotinic acid adenine dinucleotide phosphate by total synthesis of nicotinamide adenine dinucleotide phosphate. Angewandte Chemie International Edition, 43(35), 4637–4640. [DOI] [PubMed] [Google Scholar]
- Franchetti P, Pasqualini M, Petrelli R, Ricciutelli M, Vita P, & Cappellacci L (2004). Stereoselective synthesis of nicotinamide β-riboside and nucleoside analogs. Bioorganic & medicinal chemistry letters, 14(18), 4655–4658. [DOI] [PubMed] [Google Scholar]
- Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, & Kraus WL (2016). Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science, 353(6294), 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, & Hottiger MO (2006). Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiology and Molecular Biology Reviews, 70(3), 789–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S. i., & Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends in cell biology, 24(8), 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, & Moon HR (2006). Structure−Activity Relationships of 2-Chloro-N6-substituted-4’-thioadenosine-5’-uronamides as Highly Potent and Selective Agonists at the Human A3 Adenosine Receptor. Journal of Medicinal Chemistry, 49(1), 273–281. 10.1021/jm050595e [DOI] [PubMed] [Google Scholar]
- Jiang H, Congleton J, Liu Q, Merchant P, Malavasi F, Lee HC, & Lin H (2009). Mechanism-based small molecule probes for labeling CD38 on live cells. Journal of the American Chemical Society, 131(5), 1658–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Kim JH, Frizzell KM, Kraus WL, & Lin H (2010). Clickable NAD analogues for labeling substrate proteins of poly (ADP-ribose) polymerases. Journal of the American Chemical Society, 132(27), 9363–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan JA, Xiang S, & Tong L (2007). Crystal structure of human nicotinamide riboside kinase. Structure, 15(8), 1005–1013. [DOI] [PubMed] [Google Scholar]
- Langelier M-F, Zandarashvili L, Aguiar PM, Black BE, & Pascal JM (2018). NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nature communications, 9(1), 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H (2007). Nicotinamide adenine dinucleotide: beyond a redox coenzyme. Organic & biomolecular chemistry, 5(16), 2541–2554. [DOI] [PubMed] [Google Scholar]
- Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, & Pistoia V (2015). CD56brightCD16− NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. The Journal of Immunology, 1500591. [DOI] [PubMed]
- Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, & Magni G (2002). Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochemical and biophysical research communications, 297(4), 835–840. [DOI] [PubMed] [Google Scholar]
- Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mázala DA, Mouchiroud L, & Knowels GM (2016). NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Science translational medicine, 8(361), 361ra139–361ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Kim D-S, & Kraus WL (2015). New facets in the regulation of gene expression by ADP-ribosylation and poly (ADP-ribose) polymerases. Chemical reviews, 115(6), 2453–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa ES, Fabrizio G, & Di Girolamo M (2013). A role of intracellular mono-ADP-ribosylation in cancer biology. The FEBS journal, 280(15), 3551–3562. [DOI] [PubMed] [Google Scholar]
- Shrimp JH, Hu J, Dong M, Wang BS, MacDonald R, Jiang H, & Lin H (2014). Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. Journal of the American Chemical Society, 136(15), 5656–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, & Magni G (2007). Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry, 46(16), 4912–4922. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz BG, Dai H, Koppetsch KJ, Qian D, Jiang F, Mao C, & Perni RB (2012). Synthesis of carba-NAD and the structures of its ternary complexes with SIRT3 and SIRT5. The Journal of organic chemistry, 77(17), 7319–7329. [DOI] [PubMed] [Google Scholar]
- Tanimori S, Ohta T, & Kirihata M (2002). An efficient chemical synthesis of nicotinamide riboside (NAR) and analogues. Bioorganic & medicinal chemistry letters, 12(8), 1135–1137. [DOI] [PubMed] [Google Scholar]
- Tarragó MG, Chini CC, Kanamori KS, Warner GM, Caride A, de Oliveira GC, & Huang R (2018). A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell metabolism, 27(5), 1081–1095. e1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel W, Rabeh WM, Bogan KL, Belenky P, Wojcik M, Seidle HF, & Park H-W (2007). Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS biology, 5(10), e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E (2015). NAD+ in aging, metabolism, and neurodegeneration. Science, 350(6265), 1208–1213. [DOI] [PubMed] [Google Scholar]
- Wallrodt S, Buntz A, Wang Y, Zumbusch A, & Marx A (2016). Bioorthogonally Functionalized NAD+ Analogues for In-Cell Visualization of Poly (ADP-Ribose) Formation. Angewandte Chemie International Edition, 55(27), 7660–7664. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rösner D, Grzywa M, & Marx A (2014). Chain-Terminating and Clickable NAD+ Analogues for Labeling the Target Proteins of ADP-Ribosyltransferases. Angewandte Chemie International Edition, 53(31), 8159–8162. [DOI] [PubMed] [Google Scholar]
- Zatorski A, Watanabe KA, Carr SF, Goldstein BM, & Pankiewicz KW (1996). Chemical synthesis of benzamide adenine dinucleotide: inhibition of inosine monophosphate dehydrogenase (types I and II). Journal of Medicinal Chemistry, 39(12), 2422–2426. [DOI] [PubMed] [Google Scholar]