

Abstract
Fatty acid binding proteins (FABPs) are essential for energy production and long-chain polyunsaturated fatty acid-related signaling in the brain and other tissues. Of various FABPs, heart-type fatty acid binding protein (H-FABP, FABP3) is highly expressed in neurons of mature brain and plays a role in arachidonic acid incorporation into brain and heart cells. However, the precise function of H-FABP in brain remains unclear. We previously demonstrated that H-FABP is associated with the dopamine D2 receptor long isoform (D2LR) in vitro. Here, we confirm that H-FABP binds to dopamine D2 receptor (D2R) in brain extracts and colocalizes immunohistochemically with D2R in the dorsal striatum. We show that H-FABP is highly expressed in acetylcholinergic interneurons and terminals of glutamatergic neurons in the dorsal striatum of mouse brain but absent in dopamine neuron terminals and spines in the same region. H-FABP knock-out (KO) mice showed lower responsiveness to methamphetamine-induced sensitization and enhanced haloperidol-induced catalepsy compared with wild-type mice, indicative of D2R dysfunction. Consistent with the latter, aberrant increased acetylcholine (ACh) release and depolarization-induced glutamate (Glu) release were observed in the dorsal striatum of H-FABP KO mice. Furthermore, phosphorylation of CaMKII (Ca2+/calmodulin-dependent protein kinase II) and ERK (extracellular signal-regulated kinase) was significantly increased in the dorsal striatum. We confirmed elevated ERK phosphorylation following quinpirole-mediated D2R stimulation in H-FABP-overexpressing SHSY-5Y human neuroblastoma cells. Together, H-FABP is highly expressed in ACh interneurons and glutamatergic terminals, thereby regulating dopamine D2R function in the striatum.
Introduction
Perturbed metabolism of long-chain polyunsaturated fatty acids (LCPUFAs) has been reported in human neurodegenerative and psychiatric diseases, such Alzheimer's disease and schizophrenia. Schizophrenic patients exhibit significantly lower levels of arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) in red blood cells (Peet et al., 1995; Arvindakshan et al., 2003a,b). Compared with nonschizophrenic individuals, reduced levels of LCPUFAs, particularly AA and DHA, were observed in never-medicated patients but those reductions were much less significant in patients treated with antipsychotic drugs (Arvindakshan et al., 2003b). Several clinical studies indicate that oral administration of EPA can improve emotional and cognitive function in schizophrenic patients (Mellor et al., 1995; Arvindakshan et al., 2003a,b). Likewise, AA (Kotani et al., 2006) and DHA (Horrocks and Yeo, 1999) supplementation can improve cognitive dysfunction seen in human disorders such as Alzheimer's disease.
Since LCPUFAs are insoluble in an aqueous cellular environment, fatty acid binding proteins (FABPs) are essential to function as cellular shuttles to transport LCPUFAs to appropriate intracellular compartments (Coe and Bernlohr, 1998). Small 14–15 kDa cytoplasmic FABPs belong to a family consisting of at least 13 different widely distributed proteins (Banaszak et al., 1994; Veerkamp and Maatman, 1995; Glatz and van der Vusse, 1996). Among various FABPs, brain (B-), epidermal (E-), and heart (H-) type FABPs are expressed in brain (Owada et al., 1996). B-FABP knockdown by small interfering RNA impairs cell proliferation and promotes neuronal differentiation in cortical neuroepithelial cells (Arai et al., 2005). B-FABP knock-out (KO) mice show abnormalities in emotional behavior (Owada et al., 2006), decreased neurogenesis in the dentate gyrus, and impaired prepulse inhibition (Watanabe et al., 2007). H-FABP KO mice also exhibit a 24% reduction in incorporation of [14C] AA (20:4n-6) into brain cells and a reduced proportion of total n-6 fatty acids in major phospholipid classes (Murphy et al., 2005), suggesting that H-FABP is critical for AA uptake and metabolism in neurons.
Using a yeast two-hybrid screen of a mouse brain cDNA library combined with coimmunoprecipitation assays, we previously demonstrated that the 29-aa insert region in the third cytoplasmic loop of the dopamine (DA) D2 receptor long isoform (D2LR) interacts with H-FABP (Takeuchi and Fukunaga, 2003a). Furthermore, overexpressed and endogenous H-FABP colocalized with D2LR but not with the dopamine D2 receptor short isoform (D2SR) intracellularly in NG108-15 cells (Takeuchi and Fukunaga, 2003a). Therefore, we asked what was the function of H-FABP/D2LR binding in brain dopaminoceptive neurons in the brain.
Here, to address this issue, we investigated the precise localization of H-FABP in the mouse CNS and analyzed dopamine-related behaviors using H-FABP KO mice. We found that H-FABP KO mice exhibit dopamine D2 receptor (D2R) dysfunction in the striatum, where we observed aberrant cholinergic and glutamatergic neurotransmission. Moreover, D2R-stimulated phosphorylation of ERK (extracellular signal-regulated kinase) was markedly enhanced in H-FABP-overexpressing human neuroblastoma SHSY-5Y cells. Our findings demonstrate that H-FABP regulates functions of the dopamine D2R in the brain, through neuronal D2LR/H-FABP interaction.
Materials and Methods
Animals.
Generation of homozygous H-FABP KO mice (on a C57BL/6 genetic background) has been described by Binas et al. (1999). H-FABP KO mice exhibit normal phenotypes with regard to fertility, sex ratio, and weight gain (Binas et al., 1999). Adult 12-week-old mice were used in all experiments. Mice were housed under climate-controlled conditions with a 12 h light/dark cycle and provided standard food and water ad libitum. The experiments were approved by Institutional Animal Care and Use Committee at Tohoku University (Permission No. 21-Pharm-Animal-1).
Immunoprecipitation and immunoblotting analysis.
Immunoprecipitation and immunoblotting analysis was performed as previously described (Shioda et al., 2007). Mouse brains were immediately removed and perfused in ice-cold buffer for 3 min (0.32 m sucrose, 20 mm Tris-HCl, pH 7.4). Dorsal striatum (DS) tissues were dissected and homogenized in 200 μl of buffer containing 50 mm Tris-HCl, pH 7.5, 0.5% Triton X-100, 0.5 m NaCl, 4 mm EDTA, 4 mm EGTA, 1 mm Na3VO4, 50 mm NaF, 1 mm DTT, 2 μg/ml pepstatin A, 1 μg/ml leupeptin, 100 nm calyculin A). Dorsal striatum position was identified by Paxinos and Franklin (2001). Antibodies used included: rabbit polyclonal antibodies against CaMKII (calcium/calmodulin-dependent protein kinase II) (1:5000, Fukunaga et al., 1988); phospho-CaMKII (α-Thr286/β-Thr287) (1:5000, Fukunaga et al., 1988); D2R (1:2500, Narushima et al., 2006); D2LR (1:1000, Millipore); tyrosine hydroxylase (TH) (Millipore); ERK (1:1000, Cell Signaling Technology); phospho-ERK (1:1000, Cell Signaling Technology); VGLUT1 [vesicular glutamate (Glu) transporter 1] (1:500, WAKO); mouse monoclonal antibodies against H-FABP (1:50, Hycult Biotechnology); β-tubulin (1:10,000, Sigma); guinea pig polyclonal antibodies against dopamine D1 receptor (D1R) (1:2500, Narushima et al., 2006); goat polyclonal antibodies against choline acetyltransferase (ChAT) (1:1000, Millipore); and vesicular acetylcholine transporter (VAChT) (1:1000, Millipore). Images were scanned and analyzed semiquantitatively using NIH image (Research Services Branch of the National Institute of Mental Health). Background density within the same image was subtracted from optical densities of corresponding immunoreactive bands.
Immunohistochemistry and cell counting.
Immunohistochemical studies were performed as previously described (Shioda et al., 2007). Briefly, after fixation, 50 μm thick coronal sections were cut using a vibrating microtome (Dosaka EM). Sections were incubated as follows: 30 min in PBS, pH 7.4, containing 0.01% Triton X-100; 1 h in PBS containing 3% bovine serum albumin (BSA) (blocking solution); and overnight with various primary antibodies in blocking solution. Antibodies included mouse monoclonal antibodies against H-FABP (1:50, Hycult Biotechnology); rabbit polyclonal antibodies against D2R (1:2500, Narushima et al., 2006); TH (1:1000, Millipore); spinophilin (1:1000, Millipore); guinea pig polyclonal antibodies against VGLUT1 (1:1000, Millipore); and goat polyclonal antibodies against VAChT (1:1000, Millipore) and ChAT (1:1000, Millipore). Double staining with antibodies against ChAT and D2R was performed using biotinylated donkey anti-goat IgG (1:500; Jackson ImmunoResearch) with a mixture of streptavidin-HRP (1:500, NEN Life Science Products) and Alexa 594 anti-rabbit IgG (1:500, Invitrogen). Double staining with antibodies against H-FABP and various proteins was performed using biotinylated donkey anti-mouse IgG (1:500; Jackson ImmunoResearch) with a mixture of streptavidin-HRP (1:500, NEN Life Science Products) and Alexa 594 anti-rabbit, guinea pig or goat IgG (1:500, Invitrogen). Finally, sections were stained with tetramethylrhodamine tyramide for 10 min using a TSA-Direct kit (NEN Life Science Products). To count phospho-ERK-positive cells in dorsal striatum, coronal brain sections 50 μm thick were incubated 1 h in blocking solution and then overnight with rabbit polyclonal anti-phospho-ERK antibody (1:500, Millipore). After thorough washing, sections were incubated for 3 h with Alexa 448-labeled anti-rabbit IgG (1:500, Invitrogen). After several PBS washes, sections were incubated with the nuclear stain propidium iodide (PI, Invitrogen, 1:200 in PBS) for 10 min to count ERK-activated cells. Phospho-ERK and PI double-positive cells were counted in four areas (350 × 350 μm/area) per section of the dorsal striatum region (four sections per mouse, four mice per condition). All steps were performed at room temperature, and all primary and secondary antibodies were diluted in the same BSA solution as above. Sections were mounted in Vectashield (Vector Laboratories) and examined with a confocal laser scanning light microscope (Leica TCS SP2). The positions of the dorsal striatum, prefrontal cortex (PFC), substantia nigra pars compacta (SNc), and ventral tegmental area were identified by Paxinos and Franklin (2001).
Receptor binding assay.
D2R ligand binding assays using striatal membrane fractions were performed as described previously (Baik et al., 1995). Briefly, striatal tissues were homogenized with a Polytron in ice-cold buffer containing 50 mm Tris-HCl, pH 7.5. Homogenates were centrifuged at 45,000 × g for 10 min at 4°C. Pellets were resuspended in the same solution and then recentrifuged to obtain striatal membrane fractions. Ligand-binding assays were performed with 30 μg of striatal membrane fractions using [3H]-spiperone (specific activity 15 Ci/mmol; PerkinElmer) at concentrations of 0.01–0.6 nm; nonspecific binding was determined in the presence of 1 μm (+)-butaclamol (Sigma). Experiments were repeated four times in each group.
Behavioral tests.
Behavioral tests were conducted in an isolated room under dim light between 8:30 A.M. and 8:30 P.M. To measure locomotor activity during a 24 h period, mice housed individually in standard plastic cages were positioned in an automated open-field activity monitor using digital counters with an infrared sensor (DAS system, Neuroscience). To measure locomotor activity of methamphetamine (METH)-administered animals, wild-type and H-FABP KO mice were administered saline only on the first day (day 0) and then injected with METH (Dainihon Pharmaceutical; 0.25 or 0.5 mg/kg, i.p.) once a day for 7 consecutive days (days 1–7), followed by a 6 d withdrawal interval and a challenge injection of METH on day 14. Locomotor activity was measured every 5 min for 120 min using digital counters equipped with an infrared sensor (DAS system, Neuroscience). Wild-type and H-FABP KO mice were habituated to the test environment for 60 min before measurement of locomotor activity.
Catalepsy was evaluated as described by Boulay et al. (2000). Time spent in a cataleptic position was monitored by positioning the mouse so that both front paws rested on a 0.3 cm diameter steel rod (covered with nonslippery tape) 3.5 cm above a bench surface. The time during which each mouse maintained this position was recorded up to a maximum of 2 min. For some studies, haloperidol (Sigma) dissolved in 5% DMSO with distilled water was administered i.p. in a volume of 10 ml/kg. SCH23390 (Sigma) was dissolved in saline and administered i.p. in a volume of 10 ml/kg. Doses are expressed as weights of the free base. For haloperidol or SCH23390 administration, mice were injected four times (first vehicle alone and then three doses of drug). Catalepsy was measured 1 h after haloperidol or SCH23390 administration in each test session. Two consecutive test sessions were separated by 2 h. Experiments with an acetylcholine (ACh) receptor antagonist were performed as described (Moo-Puc et al., 2003). Atropine (Sigma, 4 mg/kg, i.p.) was injected 30 min before haloperidol (1 mg/kg, i.p.) administration.
Microdialysis of freely moving mice.
Microdialysis studies were performed as described (Han et al., 2008). A guide cannula (AG-4; Eicom) was lowered into the dorsal striatum 2 mm above the location of the probe tip under anesthesia (1.5% halothane, Takeda Chemical Industries). Mouse coordinates relative to the bregma were as follows (in mm): anterior = 0, lateral to the midline = +2 and down from the dura surface = −2, according to Paxinos and Franklin (2001). Twenty-four hours after recovery, microdialysis probes (A-I-4-02, 2-mm-long dialysis membrane, outer diameter 0.22 mm; Eicom) were inserted. Microdialysis was performed using a fully automated online system in HTEC-500ACA (Eicom) and HTEC-500GAA (Eicom) for ACh and Glu analyses using freely moving mice. The microdialysis probe was perfused with Ringer's solution (1.3 mm CaCl2, 3 mm KCl, 146 mm NaCl, and 1 mm MgSO4) at a 2.0 μl/min flow rate using a microsyringe pump (ESP-64; Eicom) over the entire period. Haloperidol-induced ACh and Glu release was measured under the catalepsy protocol described above. KCl-induced ACh and Glu concentrations were measured and averaged at the acute phase with higher KCl content (1.3 mm CaCl2, 146 mm KCl, 3 mm NaCl, and 1 mm MgSO4) at a flow rate of 2.0 μl/min using a microsyringe pump (ESP-64; Eicom) during a 30 min period. Dialysates were collected every 19.5 min in the sample loop of an auto-injector (EAS-20; Eicom) and then analyzed using an HPLC-Electrochemical detector system (Eicom).
Cell culture and transfection.
Human neuroblastoma SHSY-5Y cells (a kind gift from Dr. T. Uehara, Hokkaido University, Sapporo, Japan) and rodent neuroblastoma × glioma hybrid NG108-15 cells were grown in DMEM-containing additions as described (Takeuchi and Fukunaga, 2003a). Cells were plated in 35 mm dishes, cultured in standard medium for 24 h, and then transfected using Lipofectamine 2000 (Invitrogen) reagent at a volume (μl) to DNA (μg) ratio of 1:1 in 1.5 ml of serum-free medium for 6 h. The culture medium was changed to the standard medium, and cells were cultured an additional 48 h. D2LR cDNA was obtained as described (Takeuchi and Fukunaga, 2003a). Sense and antisense H-FABP cDNA was kindly provided by Professor Y. Owada (Yamaguchi University, Ube, Japan).
Statistical evaluation.
All values were expressed as means ± SEM. Comparison between two experimental groups (wild-type versus H-FABP KO mice) was made using the unpaired Student's t test for experiments of immunoblotting analysis, immunohistochemical analysis and catalepsy analysis using atropine. Analyses of locomotion activity, catalepsy duration, microdialysis and ERK phosphorylation with different doses of quinpirole in SHSY-5Y cells were subjected to two-way ANOVA, followed by one-way ANOVA for each group and Dunnett's tests. Between and within factors were as follows; locomotion activity (genotype × day and METH dose × day), catalepsy (genotype × dose), haloperidol-treated microdialysis (genotype × dose), KCl-treated microdialysis (genotype × time) and ERK phosphorylation in SHSY-5Y cells (the type of transfected plasmid × quinpirole concentrations). p < 0.05 was considered significant.
Results
H-FABP forms a complex and colocalizes with D2R in the dorsal striatum
In a previous study, we searched for proteins binding to a 29-aa sequence in the third cytoplasmic loop of D2LR using a yeast two-hybrid system and identified H-FABP as an interactor (Takeuchi and Fukunaga, 2003a). To confirm that interaction in vivo, we performed immunoprecipitation of brain extracts using an D2R antibody followed by immunoblotting with anti-H-FABP antibody and observed an immunoreactive H-FABP band in extracts from wild-type mice (Fig. 1 A, lane 1) but not in extracts made from H-HABP KO mice (Fig. 1 A, lane 2). Conversely, after H-FABP immunoprecipitation from brain extracts, immunocomplexes were immunoblotted with an anti-D2R antibody (Fig. 1 B). As expected, an H-FABP/D2R complex was observed in wild-type mice (Fig. 1 B, lane 1), but the D2R band was absent in H-FABP KO mice (Fig. 1 B, lane 2). Immunoreactive bands of molecular weights corresponding to H-FABP and D2R were detected in cell extracts from dorsal striatum and prefrontal cortex and from NG108-15 cells cotransfected with H-FABP and D2LR expression vectors (Fig. 1 A,B, lanes 3, 4, and 5). No significant differences were observed in protein expression of dopamine D1R, D2R, or D2LR in H-FABP KO versus wild-type mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To define the property of D2R binding in H-FABP KO mice, we performed a binding assay of [3H]-spiperone using striatal membrane fractions. The results showed that the maximum binding capacities (B max = 488.1 ± 16.9 fmol/mg protein) and affinities (K d = 185.7 ± 14.3 pm) of D2R in H-FABP KO mice were not significantly different from those seen in wild-type mice (B max = 458.5 ± 24.1 fmol/mg protein, K d = 161.9 ± 3.1 pm) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Furthermore, no significant differences in immunohistochemical localization of D2R in striatal cholinergic neurons were observed between wild-type and H-FABP KO mice (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
Figure 1.

H-FABP forms a complex and colocalizes with D2R in the dorsal striatum. A, B, Coimmunoprecipitation of H-FABP and D2R in brain extracts. Extracts were obtained from DS of wild-type (L1; lane 1) and H-FABP KO (L2; lane 2) mice. Extracts were immunoprecipitated (IP) with anti-D2R (A) or anti-H-FABP (B) antibody, and immunoprecipitates were then immunoblotted (IB) with anti-H-FABP (A) or with anti-D2R (B) antibody. As control H-FABP and D2R immunoreactive bands, cell extracts (Input) are shown from DS (L3; lane 3), PFC (L4; lane 4) or NG108-15 cells cotransfected with H-FABP and D2LR (L5; lane 5) constructs. C, Squares in schematic drawing are after Paxinos and Franklin (2001) and indicate dorsal striatum brain regions. D, Confocal images showing colocalization of H-FABP (green) and D2R (red) in the dorsal striatum. H-FABP colocalized with D2R in large aspiny cells likely to be cholinergic neurons. Scale bars, 20 μm. WT, wild-type mice; KO, H-FABP KO mice.
To confirm H-FABP and D2R colocalization in vivo, double-staining with H-FABP and D2R was performed in the dorsal striatum region (Fig. 1 C) of wild-type mice. H-FABP and D2R immunoreactivity colocalized in specific neurons of the dorsal striatum. As shown in Figure 1 D, H-FABP/D2R-double positivity was observed in the soma and neurites of large aspiny neurons, which were likely cholinergic (Fig. 1 D). Colocalization of H-FABP and D2R immunoreactivity was also detected in the PFC and SNc (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Unique localization of H-FABP in the dorsal striatum
The striatal microcircuit is composed of medium-sized spiny neurons that receive excitatory corticostriatal glutamatergic innervation (Dubé et al., 1988), dopaminergic nigrostriatal fibers (Pickel et al., 1981) and of cholinergic interneuron terminals (Holt et al., 1997). To determine the precise localization of H-FABP in the dorsal striatum, we performed double-staining for H-FABP and markers of each cell type, using antibody to VGLUT1 to label glutamatergic terminals of cortical and subcortical origins (Fujiyama et al., 2001), and antibodies to VAChT and TH to mark cholinergic interneurons and dopaminergic terminals, respectively. Strong H-FABP immunoreactivity was detected in all VAChT-positive cholinergic neurons (Fig. 2 A). In bouton-like structures showing H-FABP immunoreactivity, almost all boutons were VGLUT1 positive (Fig. 2 B). In contrast, colocalization of H-FABP with TH (Fig. 2 C) or the postsynaptic marker spinophilin (Fig. 2 D) was rarely observed in dorsal striatum. These results suggest that H-FABP protein is present in cholinergic interneurons and glutamatergic terminals in the dorsal striatum, whereas H-FABP is not present in dopaminergic terminals or in postsynaptic densities in dendritic spines of medium-sized spiny neurons in the dorsal striatum.
Figure 2.
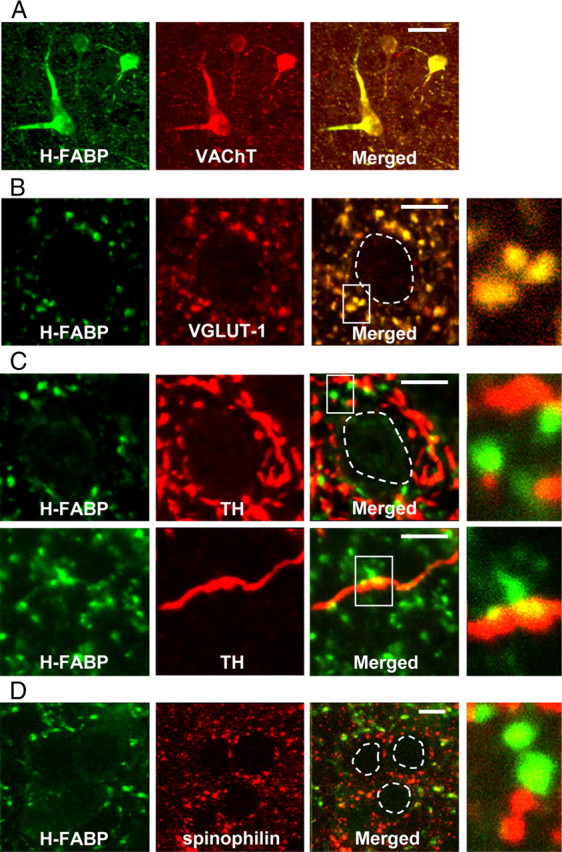
H-FABP localization in the dorsal striatum. Confocal images showing colocalization of H-FABP (green) and markers of three classical neurotransmitters (acetylcholine, glutamate, and dopamine) or spinophilin (red) in the dorsal striatum. A, Immunoreactivities of H-FABP and VAChT almost completely merge. B, Most H-FABP-containing boutons show VGLUT1 immunoreactivity. C, D, Most H-FABP-positive structures do not show immunoreactivity for either TH or spinophilin. At right in B–D are high-magnification images. Areas circled with dashed lines are cell soma. Scale bars: A, 30 μm; B–D, 10 μm.
H-FABP KO mice show low responsiveness to METH-induced sensitization and enhanced haloperidol-induced catalepsy
The basal ganglia, including the dorsal striatum, function in sensitization to the drugs METH and cocaine and control motor coordination such as the sequence of movements (Cools, 1980; Aosaki et al., 1994). To investigate potential changes in basal ganglia-dependent functions in H-FABP KO mice, we first examined METH-induced behavioral sensitization. Since there was no difference in spontaneous daytime locomotor activity observed between wild-type and H-FABP KO mice (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), behavioral experiments were conducted during that period. Interestingly, H-FABP KO mice showed significantly reduced drug sensitization against repeated METH administration (Fig. 3 A,B). H-FABP KO mice showed significantly reduced locomotor activity after repeated administration of either 0.25 or 0.5 mg/kg METH compared with wild-type mice (Fig. 3 A) [n = 5 each, genotype × day interaction (F (8,80) = 24.5, p = 0.0017)] (Fig. 3 B) [n = 6 each, genotype × day interaction (F (8,98) = 32.7, p < 0.001); METH dose × day interaction in wild-type (F (8,89) = 15.9, p < 0.01), in H-FABP KO (F (8,89) = 0.85, p = 0.56)].
Figure 3.

Behavioral responses to METH, haloperidol, and SCH23390 in H-FABP KO mice. A, B, Locomotor activity of H-FABP KO mice was significantly reduced following repeated administration of 0.25 mg/kg METH (A) and 0.5 mg/kg METH (B) compared with wild-type mice. Each bar of points represents the mean ± SEM. ***p < 0.001, versus saline-treated (day 0) wild-type mice; ## p < 0.01 and ### p < 0.001, versus saline-treated (day 0) H-FABP KO mice. C, H-FABP KO mice exhibited strong haloperidol-induced catalepsy compared with wild-type mice. Each bar of points represents the mean ± SEM. **p < 0.01, versus vehicle-treated (Veh.) wild-type mice; # p < 0.05, ## p < 0.01, versus vehicle-treated (Veh.) H-FABP KO mice. § p < 0.05, §§ p < 0.01 in H-FABP KO versus wild-type mice at the same dosage. D, SCH23390-induced catalepsy did not differ significantly between wild-type and H-FABP KO mice. Each bar of points represents the mean ± SEM. *p < 0.05 and **p < 0.01 versus vehicle-treated (Veh.) wild-type mice; # p < 0.05 and ## p < 0.01 versus vehicle-treated (Veh.) H-FABP KO mice. WT, wild-type mice; KO, H-FABP KO mice.
To determine whether H-FABP functions in the response to typical antipsychotic drugs acting via D2R, we examined haloperidol-induced catalepsy behavior. Haloperidol-induced catalepsy was markedly enhanced in H-FABP KO compared with wild-type mice, particularly at low haloperidol doses (Fig. 3 C) [n = 6 each, genotype × dose interaction (F (3,47) = 8.16, p = 0.0171)]. To determine whether H-FABP KO mice exhibit altered D1R function, we examined catalepsy induced by SCH23390, a D1 antagonist. SCH23390-induced catalepsy was not significantly different between wild-type and H-FABP KO mice (Fig. 3 D) [n = 6 each, genotype × dose interaction (F (3,43) = 0.28, p = 0.61)], indicating no significant change in D1R function in H-FABP KO mice. Together, H-FABP KO mice show dysfunction only of D2LR with no change in D1R function with regard to catalepsy activity.
H-FABP KO mice exhibit enhanced ACh release associated with increased haloperidol-induced catalepsy
Since ACh and Glu release in striatum is reportedly critical for D2LR-stimulated catalepsy (Usiello et al., 2000), we hypothesized that ACh and/or Glu release is enhanced in H-FABP KO mice. To investigate this question, we used microdialysis to analyze changes in release of both neurotransmitters in the dorsal striatum of freely moving animals. Although basal levels of ACh efflux between wild-type and H-FABP KO mice were similar, haloperidol administration-induced ACh release was markedly enhanced at different doses in the dorsal striatum of H-FABP KO mice compared with wild-type animals (Fig. 4 A). Enhanced ACh release observed at 0.25 and 0.5 mg/kg haloperidol was closely correlated with haloperidol-induced cataleptic responses, particularly at 0.5 mg/kg, as shown in Figure 3 C (Fig. 4 A) [n = 4 each, genotype × dose interaction (F (3,31) = 10.96, p = 0.005)]. In contrast, no significant elevation in haloperidol-induced Glu release in the dorsal striatum was observed either in wild-type or H-FABP KO mice (Fig. 4 B) (n = 4 each). To confirm whether haloperidol-induced catalepsy is related to ACh release, we measured the haloperidol-induced cataleptic response following pretreatment with the muscarinic ACh receptor antagonist atropine (administered 30 min before haloperidol injection). As expected, the duration of haloperidol-induced catalepsy was significantly decreased by atropine pretreatment both in wild-type and H-FABP KO mice [wild-type: haloperidol, 120 s, atropine plus haloperidol, 30.4 ± 9.0 s, t = 9.96, degrees of freedom (df) = 8, p < 0.01, n = 5 each; H-FABP KO: haloperidol, 102 ± 18 s, atropine plus haloperidol, 46.2 ± 13.1 s, t = 2.51, df = 8, p = 0.0182, n = 5 each].
Figure 4.
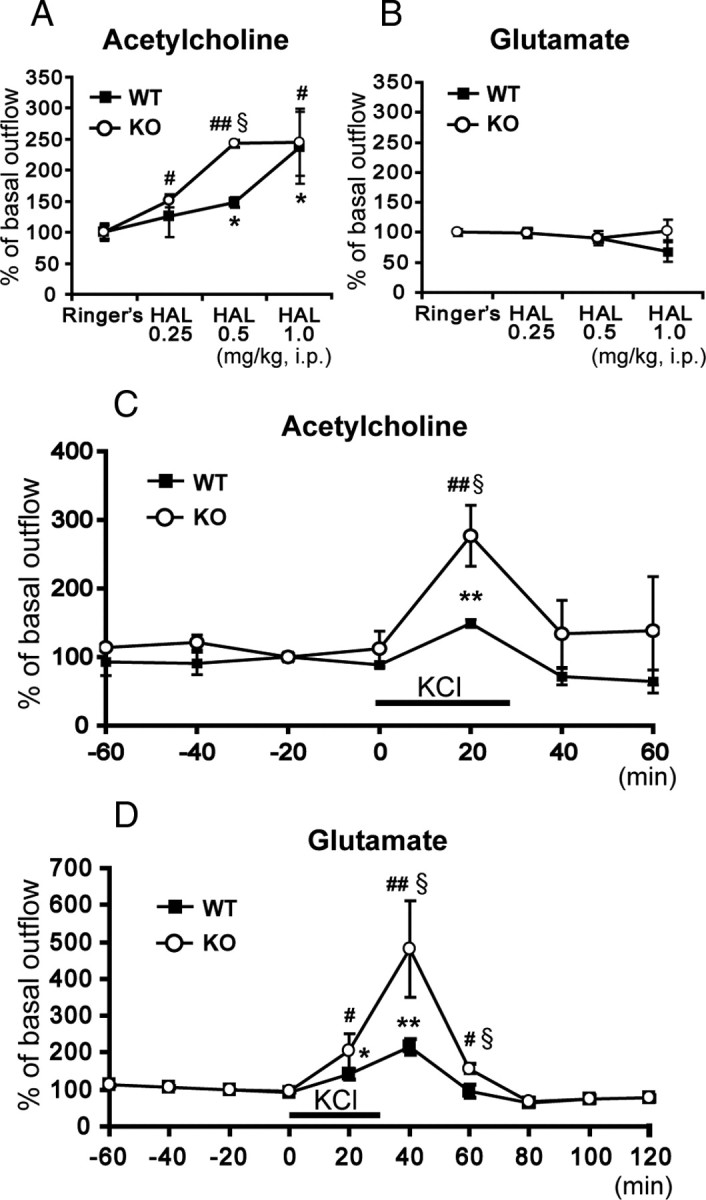
Extracellular ACh and Glu concentrations in the dorsal striatum seen in freely moving animals. A, Extracellular ACh changes following intraperitoneal haloperidol (HAL) administration. H-FABP KO mice showed markedly increased haloperidol-induced ACh release compared with wild-type mice at a dosage of 0.5 mg/kg. § p < 0.05, in H-FABP KO mice versus wild-type mice at the same dose. *p < 0.05, versus basal levels in wild-type mice; # p < 0.05 and ## p < 0.01, versus basal levels in H-FABP KO mice. B, Extracellular Glu changes following intraperitoneal haloperidol administration. C, KCl depolarization-induced ACh release in the dorsal striatum. Each bar of points represents the mean ± SEM. § p < 0.05, H-FABP KO mice versus wild-type mice at the same dosage. **p < 0.01, versus basal levels in wild-type mice; ## p < 0.01, versus basal levels in H-FABP KO mice. D, KCl depolarization-induced Glu release in the dorsal striatum. Each bar of points represents the mean ± SEM. § p < 0.05, in H-FABP KO versus wild-type mice at the same dosage. *p < 0.05 and **p < 0.01, versus basal levels in wild-type mice; # p < 0.05 and ## p < 0.01, versus basal levels in H-FABP KO mice. WT, wild-type mice; KO, H-FABP KO mice.
We further determined whether H-FABP deficiency alters depolarization-induced ACh and Glu release in the dorsal striatum. A microdialysis probe was perfused with a high KCl solution to depolarize neurons in the dorsal striatum. After monitoring basal ACh and Glu release using normal Ringer's buffer, high KCl-containing buffer was introduced into the microdialysis probe for 30 min. High KCl-induced ACh and Glu release was markedly enhanced in H-FABP KO compared with wild-type mice (Fig. 4 C,D) [ACh, n = 4 each, genotype × time interaction (F (6,55) = 3.16, p = 0.011); Glu, n = 4 each, genotype × time interaction (F (9,79) = 8.22, p < 0.001)]. We also measured VGLUT1, VChAT, ChAT and TH protein expression in the dorsal striatum and observed no significant differences in expression levels of these neurotransmitter markers between wild-type and H-FABP KO mice (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These results suggest that increased ACh release observed in the striatum predominantly accounts for the onset of haloperidol-induced catalepsy.
Aberrant activation of CaMKII and ERK in the dorsal striatum of H-FABP KO mice
Since microdialysis studies revealed increased depolarization-induced Glu release in the dorsal striatum of H-FABP KO mice and since NMDA receptor functions in drug sensitization (Karler et al., 1989; Stewart and Druhan, 1993), we hypothesized that CaMKII autophosphorylation and ERK phosphorylation through the NMDA receptor were upregulated in H-FABP KO mice. As expected, CaMKII autophosphorylation of both the α and β subunits markedly increased in KO compared with wild-type mice (α subunit, 372.6 ± 37.9% of wild-type mice, t = 6.71, df = 10, p < 0.01; β subunit, 378.0 ± 80.0% of wild-type mice, t = 3.12, df = 10, p = 0.018, n = 6 each) without changes in protein levels of either subunit. Moreover, phospho- (p-) ERK levels were also significantly increased in H-FABP KO mice (Fig. 5 B) (505.9 ± 35.5% of wild-type mice, t = 8.76, df = 10, p < 0.01, n = 6 each). To confirm upregulation of ERK phosphorylation in striatal cells, we counted dorsal striatum neurons double-positive for pERK and the nuclear stain PI. Consistent with the immunoblot results, pERK/PI double-positive cells were remarkably increased in the dorsal striatum of H-FABP KO mice relative to wild-type mice (Fig. 5 C) (42.5 ± 1.9 cells in wild-type mice versus 84.5 ± 4.7 cells/360 × 360 μm in H-FABP KO mice, n = 4 each, t = 8.24, df = 6, p < 0.01).
Figure 5.
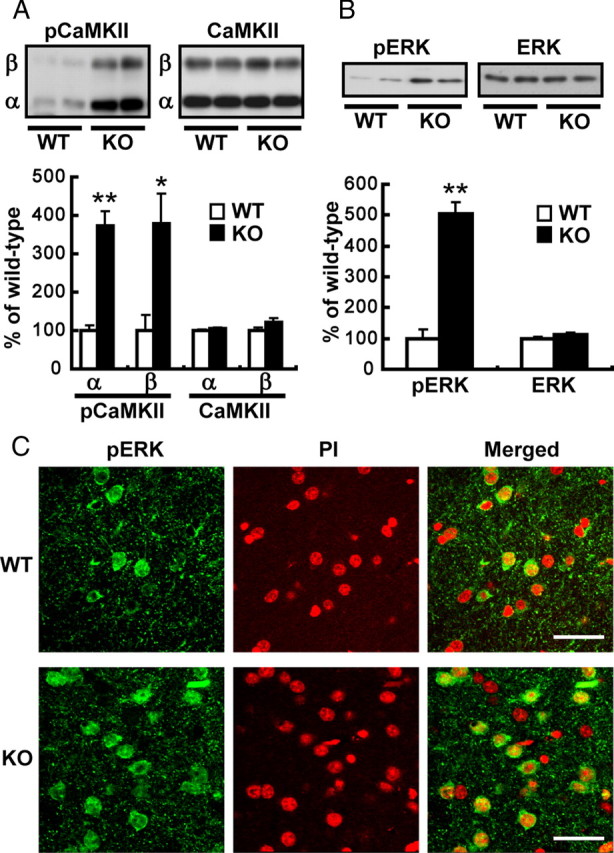
CaMKII and ERK phosphorylation is significantly increased in H-FABP KO mice. A, B, Top, representative immunoblots probed with various antibodies are shown. Bottom, Quantitative analyses by densitometry are shown. Each bar represents the mean ± SEM. *p < 0.05 and **p < 0.01 versus wild-type mice. C, Confocal microscopy images of double staining in dorsal striatum for phospho- (p)ERK (green) and PI (red), as well as merged images. Scale bar, 40 μm. WT, Wild-type mice; KO, H-FABP KO mice.
H-FABP overexpression increases sensitivity of D2R to quinpirole in human neuroblastoma cells
We previously documented that D2R stimulation with quinpirole increases ERK activity and concomitant transcriptional activity of genes encoding factors such as NF-kappaB in NG108-15 cells transfected with D2LR (Takeuchi and Fukunaga, 2003b, 2004). Here we measured ERK phosphorylation after D2R stimulation with quinpirole in human neuroblastoma SHSY-5Y cells, with or without H-FABP overexpression. The schematic diagram in Figure 6 A shows H-FABP sense and antisense (control) pcDNA3 constructs used in these studies. As expected, quinpirole stimulation clearly activated ERK, and ERK phosphorylation was significantly potentiated in H-FABP sense-transfected compared with antisense-transfected cells (Fig. 6 B) (antisense with quinpirole, 236.3 ± 7.4%; sense with quinpirole, 324.3 ± 21.2% of antisense with distilled water, t = 3.92, df = 10, p < 0.01, n = 6 each). ERK protein levels remained unchanged after transfection with any construct, and with or without quinpirole stimulation. We also examined ERK activation in response to different concentrations (3 or 30 μm) of quinpirole. In H-FABP-overexpressing cells, ERK phosphorylation was significantly increased at either dose compared with H-FABP antisense-transfected cells (supplemental Fig. 6, available at www.jneurosci.org as supplemental material) (F (3,31) = 3.22, p < 0.05, n = 4 each). Thus, interaction of H-FABP with D2LR likely upregulates D2LR signaling, as evidenced by ERK activation.
Figure 6.

Effect of quinpirole on H-FABP-overexpressing SHSY-5Y human neuroblastoma cells. A, Schematic diagrams of H-FABP sense (top) and antisense (bottom) orientation plasmids in pcDNA3. H-FABP cDNA is the open reading frame of Mus musculus fatty acid binding protein 3 (NM_010174). B, Representative immunoblots of cell extract proteins from treated (Quin.) and control (D.W.) cells probed with anti-pERK, anti-D2LR, H-FABP and anti-ERK (conventional) antibodies. C, Quantitative analysis of ERK activity was performed by measuring phosphorylated ERK immunoreactivity. After treatment of cells with 10 μm quinpirole or distilled water (D.W.) for 10 min in standard medium, cell extracts (20 μg) were prepared and subjected to immunoblot analysis. Each bar represents the mean ± SEM. **p < 0.01 versus distilled water-treated group; ## p < 0.01 versus antisense with quinpirole-treated group.
Discussion
In this study we used H-FABP KO mice to confirm that H-FABP binds to D2R and regulates D2R function in dopamine-related locomotor activity and motor coordination. Indeed, H-FABP and D2R proteins are colocalized in cholinergic interneurons in the dorsal striatum and in terminals of glutamatergic neurons innervated from the cortex to the dorsal striatum. A pharmacological approach also demonstrated a decrease in drug-induced locomotor sensitization (drug sensitization) with METH treatment and enhanced haloperidol-induced catalepsy in H-FABP KO mice compared with wild-type mice. The most critical observation we made in H-FABP KO mice was that enhanced haloperidol-induced catalepsy is closely associated with increased ACh release in the dorsal striatum and marked increases in KCl-induced ACh and Glu release, suggesting that H-FABP regulates dopamine-regulated ACh and Glu release through D2R in the striatum (Fig. 7).
Figure 7.

Schematic representation of altered neurotransmission in the dorsal striatum following H-FABP deletion. Left, The striatal microcircuit is composed of medium spiny neurons (MSNs) that receive input from excitatory corticostriatal glutamatergic projections, dopaminergic nigrostriatal fibers, and cholinergic terminals. D2R is present at the postsynapse (spine) of MSN and in each terminal. However, H-FABP is present only in glutamatergic and cholinergic terminals. H-FABP binds to the third cytoplasmic loop of D2R. Right, Following H-FABP deletion, D2R inhibitory effects are disrupted, resulting in increased Glu and ACh release from respective glutamatergic and cholinergic terminals and CaMKII and ERK activation at MSN postsynapses.
Striatal cholinergic interneurons highly express D2R, whose activation reduces ACh release (DeBoer et al., 1996, Yan et al., 1997; Pisani et al., 2000). In glutamatergic terminals, electron microscopy (Fisher et al., 1994; Sesack et al., 1994) and electrophysiology (Bamford et al., 2004) studies indicate that D2R stimulation inhibits Glu release from corticostriatal terminals. Animals with genetic D2R ablation exhibit greatly reduced cocaine-induced behavioral sensitization (Welter et al., 2007) with concomitant facilitation of Glu release at corticostriatal synapses (Cepeda et al., 2001). Thus, we hypothesized that H-FABP plays important roles in modulating D2R function, particularly that of D2LR, in striatum-related behaviors. Loss of H-FABP may reduce D2LR function. However, in D2LR gene-ablated mice, a reduction in haloperidol-induced catalepsy was observed (Usiello et al., 2000, Wang et al., 2000). Thus loss of D2LR in the whole brain may impair cataleptic behavior, whereas reduced D2LR function through loss of H-FABP in discrete populations of cholinergic neurons and glutamatergic terminals is associated with enhanced haloperidol-induced catalepsy. D2LR is also expressed in postsynaptic regions of medium spiny neurons in the dorsal striatum (Khan et al., 1998), in which H-FABP is not expressed. Furthermore, H-FABP KO mice exhibit no change in total D2R protein levels compared with wild-type mice, suggesting that H-FABP does not alter D2LR stability or turnover in the striatum. Likewise, expression levels of other D2R binding proteins, such as spinophilin (Smith et al., 1999), calmodulin (Bofill-Cardona et al., 2000), and the NMDA receptor 2B subunit (Liu et al., 2006), remain unchanged in H-FABP KO mice, suggesting that H-FABP does not function in interactions between D2LR and these signaling molecules. A calmodulin-binding domain is located in the third cytoplasmic loop of D2R (Bofill-Cardona et al., 2000, Liu et al., 2007). The interaction with calmodulin attenuates D2R signaling by blocking D2R-mediated G-protein activation in a calcium-dependent manner (Bofill-Cardona et al., 2000) and inversely enhances D2R signaling (Liu et al., 2007). Thus, we speculate that H-FABP regulates ERK activity through D2LR-mediated G-protein activation directly through interaction of the D2R third cytoplasmic loop or indirectly through competition with other proteins such as calmodulin and spinophilin interacting with the third cytoplasmic loop (Kabbani and Levenson, 2007). Further extensive studies are required to resolve mechanisms underlying D2LR activation by H-FABP.
Importantly, we provide direct evidence that haloperidol-induced catalepsy is associated with increased ACh release but not with Glu release in the striatum. Haloperidol can directly stimulate striatal ACh release by disinhibition of D2R-induced ACh release (Guyenet et al., 1975) and consequently increasing ACh release (Scatton, 1982). Haloperidol administration activates ERK and induces c-fos expression in mouse striatopallidal neurons (Bertran-Gonzalez et al., 2008). Thus, haloperidol may block D2R-mediated inhibition of cholinergic neurons, leading to increased ACh release and in turn facilitating haloperidol-induced catalepsy. Likewise, blockade of D2R located on presynaptic glutamatergic fibers increases Glu release, thereby eliciting haloperidol-induced catalepsy (Maura et al., 1988). Moreover, Glu stimulates ACh release via NMDA receptors on cholinergic interneurons (Scatton and Lehmann, 1982; Ikarashi et al., 1998). Therefore, we hypothesized initially that increased Glu release seen following haloperidol administration enhanced ACh release, which in turn caused catalepsy. However, we show here that haloperidol administration does not affect presynaptic glutamatergic terminals; therefore, the glutamatergic pathway is likely not involved in haloperidol-induced catalepsy, at least in the dorsal striatum. In pharmacological studies, MK-801, an NMDA receptor antagonist, has been shown to reduce haloperidol-induced catalepsy in rats (Dragunow et al., 1990; de Souza and Meredith, 1999). Notably, in this study depolarization-induced Glu release was markedly enhanced in H-FABP KO mice. Although haloperidol-induced glutamate release was unchanged in a microdialysis assay, the basal glutamate tone from the cortex to the striatum is likely elevated in H-FABP KO mice. Thus, glutamatergic input may be enhanced in H-FABP KO mice. This enhancement coincides with markedly elevated striatal CaMKII and ERK phosphorylation that we observed in H-FABP KO mice.
Intraperitoneal administration of haloperidol increased striatal DA levels in mice (Usiello et al., 2000). DA suppresses and ACh enhances D2R-expressing striatopallidal neuronal activity (Day et al., 2008). Therefore, cholinergic action opposes the effects of DA on the D2R in striatopallidal neurons. In H-FABP KO mice, there were no significant differences in D2R and TH protein levels compared with wild-type mice in the substantia nigra, which is the origin of dopaminergic neurons (our unpublished data). Thus, increased haloperidol sensitivity seen in H-FABP KO mice may not be due to deficits in dopaminergic neurons. In future studies, we plan to measure DA release in the prefrontal cortex, in which H-FABP and D2R are colocalized (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Interestingly, strong H-FABP immunoreactivity was detected in the substantia nigra. H-FABP and TH double-positive cells likely to be neurons were observed in the SNc (supplemental Figs. 3, 7, available at www.jneurosci.org as supplemental material). Recently, it was reported that high H-FABP levels are observed in sera from Parkinson's disease patients (Wada-Isoe et al., 2008), suggesting that H-FABP could be a useful biomarker to detect cell damage in these patients.
H-FABP has a high affinity for AA (Hanhoff et al., 2002), and H-FABP KO mice show 24% decreases in AA incorporation into brain cells (Murphy et al., 2005). Our preliminary studies show that H-FABP KO mice exhibit increased fear/anxiety and cognitive function deficits (Motohashi et al., in preparation). Stimulation of D2R increases AA production in Chinese hamster ovary (CHO) cells (Kanterman et al., 1991; Piomelli et al., 1991) and in primary striatal neuron cultures (Schinelli et al., 1994). In the present study, ERK activity was significantly increased after treatment of H-FABP-overexpressing SHSY-5Y cells with a D2 agonist, indicating that AA could be a beneficial molecule to promote D2R function. Future studies should address whether reduced AA levels in the brain in part mediate D2R dysfunction in H-FABP KO mice. A crucial role for dopamine D2R has been demonstrated based on the types of therapeutics used to treat neuropsychiatric disorders, such as schizophrenia, and addictive, stress or movement disorders (Noble, 2003). It is also important to determine whether AA administration rescues D2R dysregulation in these disorders through H-FABP.
In conclusion, we have demonstrated that H-FABP KO mice exhibit D2R dysfunction and show aberrant activation of cholinergic and glutamatergic neurotransmission in the dorsal striatum. H-FABP KO mice also showed abnormal striatal-related behaviors, such as reduced METH-induced behavioral sensitization and enhanced haloperidol-induced catalepsy. Both immunohistochemical and immunoprecipitation studies also strongly suggest that H-FABP is essential to regulate D2R function.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (20659008 to K.F.) and the Smoking Research Foundation (to K.F.).
References
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. 1994;14:3969–3984. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Funatsu N, Numayama-Tsuruta K, Nomura T, Nakamura S, Osumi N. Role of Fabp7, a downstream gene of Pax6, in the maintenance of neuroepithelial cells during early embryonic development of the rat cortex. J Neurosci. 2005;25:9752–9761. doi: 10.1523/JNEUROSCI.2512-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvindakshan M, Ghate M, Ranjekar PK, Evans DR, Mahadik SP. Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr Res. 2003a;62:195–204. doi: 10.1016/s0920-9964(02)00284-0. [DOI] [PubMed] [Google Scholar]
- Arvindakshan M, Sitasawad S, Debsikdar V, Ghate M, Evans D, Horrobin DF, Bennett C, Ranjekar PK, Mahadik SP. Essential polyunsaturated fatty acid and lipid peroxide levels in never-medicated and medicated schizophrenia patients. Biol Psychiatry. 2003b;53:56–64. doi: 10.1016/s0006-3223(02)01443-9. [DOI] [PubMed] [Google Scholar]
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Banaszak L, Winter N, Xu Z, Bernlohr DA, Cowan S, Jones TA. Lipid-binding proteins: a family of fatty acid and retinoid transport proteins [review] Adv Protein Chem. 1994;45:89–151. doi: 10.1016/s0065-3233(08)60639-7. [DOI] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binas B, Danneberg H, McWhir J, Mullins L, Clark AJ. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999;13:805–812. doi: 10.1096/fasebj.13.8.805. [DOI] [PubMed] [Google Scholar]
- Bofill-Cardona E, Kudlacek O, Yang Q, Ahorn H, Freissmuth M, Nanoff C. Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J Biol Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- Boulay D, Depoortere R, Oblin A, Sanger DJ, Schoemaker H, Perrault G. Haloperidol-induced catalepsy is absent in dopamine D(2), but maintained in dopamine D(3) receptor knock-out mice. Eur J Pharmacol. 2000;391:63–73. doi: 10.1016/s0014-2999(99)00916-4. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Altemus KL, Flores-Hernández J, Calvert CR, Jokel ES, Grandy DK, Low MJ, Rubinstein M, Ariano MA, Levine MS. Facilitated glutamatergic transmission in the striatum of D2 dopamine receptor-deficient mice. J Neurophysiol. 2001;85:659–670. doi: 10.1152/jn.2001.85.2.659. [DOI] [PubMed] [Google Scholar]
- Coe NR, Bernlohr DA. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim Biophys Acta. 1998;1391:287–306. doi: 10.1016/s0005-2760(97)00205-1. [DOI] [PubMed] [Google Scholar]
- Cools AR. Role of the neostriatal dopaminergic activity in sequencing and selecting behavioural strategies: facilitation of processes involved in selecting the best strategy in a stressful situation. Behav Brain Res. 1980;1:361–378. doi: 10.1016/0166-4328(80)90035-2. [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer P, Heeringa MJ, Abercrombie ED. Spontaneous release of acetylcholine in striatum is preferentially regulated by inhibitory dopamine D2 receptors. Eur J Pharmacol. 1996;317:257–262. doi: 10.1016/s0014-2999(96)00761-3. [DOI] [PubMed] [Google Scholar]
- de Souza IE, Meredith GE. NMDA receptor blockade attenuates the haloperidol induction of Fos protein in the dorsal but not the ventral striatum. Synapse. 1999;32:243–253. doi: 10.1002/(SICI)1098-2396(19990615)32:4<243::AID-SYN1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Robertson GS, Faull RL, Robertson HA, Jansen K. D2 dopamine receptor antagonists induce fos and related proteins in rat striatal neurons. Neuroscience. 1990;37:287–294. doi: 10.1016/0306-4522(90)90399-o. [DOI] [PubMed] [Google Scholar]
- Dubé L, Smith AD, Bolam JP. Identification of synaptic terminals of thalamic or cortical origin in contact with distinct medium-size spiny neurons in the rat neostriatum. J Comp Neurol. 1988;267:455–471. doi: 10.1002/cne.902670402. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Levine MS, Sibley DR, Ariano MA. D2 dopamine receptor protein location: Golgi impregnation-gold toned and ultrastructural analysis of the rat neostriatum. J Neurosci Res. 1994;38:551–564. doi: 10.1002/jnr.490380508. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Furuta T, Kaneko T. Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol. 2001;435:379–387. doi: 10.1002/cne.1037. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Goto S, Miyamoto E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J Neurochem. 1988;51:1070–1078. doi: 10.1111/j.1471-4159.1988.tb03070.x. [DOI] [PubMed] [Google Scholar]
- Glatz JF, van der Vusse GJ. Cellular fatty acid-binding proteins: their function and physiological significance [review] Prog Lipid Res. 1996;35:243–282. doi: 10.1016/s0163-7827(96)00006-9. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Javory AF, Beaujouan JC, Rossier BJ, Glowinski J. Effects of dopaminergic receptor agonists and antagonists on the activity of the neo-striatal cholinergic system. Brain Res. 1975;84:227–244. doi: 10.1016/0006-8993(75)90978-6. [DOI] [PubMed] [Google Scholar]
- Han F, Shioda N, Moriguchi S, Qin ZH, Fukunaga K. Downregulation of glutamate transporters is associated with elevation in extracellular glutamate concentration following rat microsphere embolism. Neurosci Lett. 2008;430:275–280. doi: 10.1016/j.neulet.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Hanhoff T, Lücke C, Spener F. Insights into binding of fatty acids by fatty acid binding proteins. Mol Cell Biochem. 2002;239:45–54. [PubMed] [Google Scholar]
- Holt DJ, Graybiel AM, Saper CB. Neurochemical architecture of the human striatum. J Comp Neurol. 1997;384:1–25. doi: 10.1002/(sici)1096-9861(19970721)384:1<1::aid-cne1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Horrocks LA, Yeo YK. Health benefits of docosahexaenoic acid (DHA) Pharmacol Res. 1999;40:211–225. doi: 10.1006/phrs.1999.0495. [DOI] [PubMed] [Google Scholar]
- Ikarashi Y, Yuzurihara M, Takahashi A, Ishimaru H, Shiobara T, Maruyama Y. Direct regulation of acetylcholine release by N-methyl-d-aspartic acid receptors in rat striatum. Brain Res. 1998;795:215–220. doi: 10.1016/s0006-8993(98)00293-5. [DOI] [PubMed] [Google Scholar]
- Kabbani N, Levenson R. A proteomic approach to receptor signaling: molecular mechanisms and therapeutic implications derived from discovery of the dopamine D2 receptor signalplex [review] Eur J Pharmacol. 2007;572:83–93. doi: 10.1016/j.ejphar.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Kanterman RY, Mahan LC, Briley EM, Monsma FJ, Jr, Sibley DR, Axelrod J, Felder CC. Transfected D2 dopamine receptors mediate the potentiation of arachidonic acid release in Chinese hamster ovary cells. Mol Pharmacol. 1991;39:364–369. [PubMed] [Google Scholar]
- Karler R, Calder LD, Chaudhry IA, Turkanis SA. Blockade of “reverse tolerance” to cocaine and amphetamine by MK-801. Life Sci. 1989;45:599–606. doi: 10.1016/0024-3205(89)90045-3. [DOI] [PubMed] [Google Scholar]
- Khan ZU, Mrzljak L, Gutierrez A, de la Calle A, Goldman-Rakic PS. Prominence of the dopamine D2 short isoform in dopaminergic pathways. Proc Natl Acad Sci U S A. 1998;95:7731–7736. doi: 10.1073/pnas.95.13.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani S, Sakaguchi E, Warashina S, Matsukawa N, Ishikura Y, Kiso Y, Sakakibara M, Yoshimoto T, Guo J, Yamashima T. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci Res. 2006;56:159–164. doi: 10.1016/j.neures.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Liu XY, Chu XP, Mao LM, Wang M, Lan HX, Li MH, Zhang GC, Parelkar NK, Fibuch EE, Haines M, Neve KA, Liu F, Xiong ZG, Wang JQ. Modulation of D2R-NR2B interactions in response to cocaine. Neuron. 2006;52:897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Liu Y, Buck DC, Macey TA, Lan H, Neve KA. Evidence that calmodulin binding to the dopamine D2 receptor enhances receptor signaling. J Recept Signal Transduct Res. 2007;27:47–65. doi: 10.1080/10799890601094152. [DOI] [PubMed] [Google Scholar]
- Maura G, Giardi A, Raiteri M. Release-regulating D-2 dopamine receptors are located on striatal glutamatergic nerve terminals. J Pharmacol Exp Ther. 1988;247:680–684. [PubMed] [Google Scholar]
- Mellor JE, Laugharne JD, Peet M. Schizophrenic symptoms and dietary intake of n-3 fatty acids. Schizophr Res. 1995;18:85–86. doi: 10.1016/0920-9964(95)00084-4. [DOI] [PubMed] [Google Scholar]
- Moo-Puc RE, Góngora-Alfaro JL, Alvarez-Cervera FJ, Pineda JC, Arankowsky-Sandoval G, Heredia-López F. Caffeine and muscarinic antagonists act in synergy to inhibit haloperidol-induced catalepsy. Neuropharmacology. 2003;45:493–503. doi: 10.1016/s0028-3908(03)00202-8. [DOI] [PubMed] [Google Scholar]
- Murphy EJ, Owada Y, Kitanaka N, Kondo H, Glatz JF. Brain arachidonic acid incorporation is decreased in heart fatty acid binding protein gene-ablated mice. Biochemistry. 2005;44:6350–6360. doi: 10.1021/bi047292r. [DOI] [PubMed] [Google Scholar]
- Narushima M, Uchigashima M, Hashimoto K, Watanabe M, Kano M. Depolarization-induced suppression of inhibition mediated by endocannabinoids at synapses from fast-spiking interneurons to medium spiny neurons in the striatum. Eur J Neurosci. 2006;24:2246–2252. doi: 10.1111/j.1460-9568.2006.05119.x. [DOI] [PubMed] [Google Scholar]
- Noble EP. D2 dopamine receptor gene in psychiatric and neurologic disorders and its phenotypes. Am J Med Genet B Neuropsychiatr Genet. 2003;116B:103–125. doi: 10.1002/ajmg.b.10005. [DOI] [PubMed] [Google Scholar]
- Owada Y, Yoshimoto T, Kondo H. Spatio-temporally differential expression of genes for three members of fatty acid-binding proteins in developing and mature rat brains. J Chem Neuroanat. 1996;12:113–122. doi: 10.1016/s0891-0618(96)00192-5. [DOI] [PubMed] [Google Scholar]
- Owada Y, Abdelwahab SA, Kitanaka N, Sakagami H, Takano H, Sugitani Y, Sugawara M, Kawashima H, Kiso Y, Mobarakeh JI, Yanai K, Kaneko K, Sasaki H, Kato H, Saino-Saito S, Matsumoto N, Akaike N, Noda T, Kondo H. Altered emotional behavioral responses in mice lacking brain-type fatty acid-binding protein gene. Eur J Neurosci. 2006;24:175–187. doi: 10.1111/j.1460-9568.2006.04855.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Peet M, Laugharne J, Rangarajan N, Horrobin D, Reynolds G. Depleted red cell membrane essential fatty acids in drug-treated schizophrenic patients. J Psychiatr Res. 1995;29:227–232. doi: 10.1016/0022-3956(95)00001-l. [DOI] [PubMed] [Google Scholar]
- Pickel VM, Beckley SC, Joh TH, Reis DJ. Ultrastructural immunocytochemical localization of tyrosine hydroxylase in the neostriatum. Brain Res. 1981;225:373–385. doi: 10.1016/0006-8993(81)90843-x. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Pilon C, Giros B, Sokoloff P, Martres MP, Schwartz JC. Dopamine activation of the arachidonic acid cascade as a basis for D1/D2 receptor synergism. Nature. 1991;353:164–167. doi: 10.1038/353164a0. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Centonze D, Calabresi P, Bernardi G. Activation of D2-like dopamine receptors reduces synaptic inputs to striatal cholinergic interneurons. J Neurosci. 2000;20:RC69. doi: 10.1523/JNEUROSCI.20-07-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scatton B. Further evidence for the involvement of D2, but not D1 dopamine receptors in dopaminergic control of striatal cholinergic transmission. Life Sci. 1982;31:2883–2890. doi: 10.1016/0024-3205(82)90679-8. [DOI] [PubMed] [Google Scholar]
- Scatton B, Lehmann J. N-methyl-d-aspartate-type receptors mediate striatal 3H-acetylcholine release evoked by excitatory amino acids. Nature. 1982;297:422–424. doi: 10.1038/297422a0. [DOI] [PubMed] [Google Scholar]
- Schinelli S, Paolillo M, Corona GL. Opposing actions of D1- and D2-dopamine receptors on arachidonic acid release and cyclic AMP production in striatal neurons. J Neurochem. 1994;62:944–949. doi: 10.1046/j.1471-4159.1994.62030944.x. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14:88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda N, Han F, Moriguchi S, Fukunaga K. Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J Neurochem. 2007;102:1506–1517. doi: 10.1111/j.1471-4159.2007.04600.x. [DOI] [PubMed] [Google Scholar]
- Smith FD, Oxford GS, Milgram SL. Association of the D2 dopamine receptor third cytoplasmic loop with spinophilin, a protein phosphatase-1-interacting protein. J Biol Chem. 1999;274:19894–19900. doi: 10.1074/jbc.274.28.19894. [DOI] [PubMed] [Google Scholar]
- Stewart J, Druhan JP. Development of both conditioning and sensitization of the behavioral activating effects of amphetamine is blocked by the non-competitive NMDA receptor antagonist, MK-801. Psychopharmacology (Berl) 1993;110:125–132. doi: 10.1007/BF02246961. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Fukunaga K. Differential subcellular localization of two dopamine D2 receptor isoforms in transfected NG108-15 cells. J Neurochem. 2003a;85:1064–1074. doi: 10.1046/j.1471-4159.2003.01763.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Fukunaga K. Differential regulation of NF-kappaB, SRE and CRE by dopamine D1 and D2 receptors in transfected NG108-15 cells. J Neurochem. 2003b;85:729–739. doi: 10.1046/j.1471-4159.2003.01711.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Fukunaga K. Dopamine D2 receptor activates extracellular signal-regulated kinase through the specific region in the third cytoplasmic loop. J Neurochem. 2004;89:1498–1507. doi: 10.1111/j.1471-4159.2004.02446.x. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rougé-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Veerkamp JH, Maatman RG. Cytoplasmic fatty acid-binding proteins: their structure and genes [review] Prog Lipid Res. 1995;34:17–52. doi: 10.1016/0163-7827(94)00005-7. [DOI] [PubMed] [Google Scholar]
- Wada-Isoe K, Imamura K, Kitamaya M, Kowa H, Nakashima K. Serum heart-fatty acid binding protein levels in patients with Lewy body disease. J Neurol Sci. 2008;266:20–24. doi: 10.1016/j.jns.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xu R, Sasaoka T, Tonegawa S, Kung MP, Sankoorikal EB. Dopamine D2 long receptor-deficient mice display alterations in striatum-dependent functions. J Neurosci. 2000;20:8305–8314. doi: 10.1523/JNEUROSCI.20-22-08305.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Toyota T, Owada Y, Hayashi T, Iwayama Y, Matsumata M, Ishitsuka Y, Nakaya A, Maekawa M, Ohnishi T, Arai R, Sakurai K, Yamada K, Kondo H, Hashimoto K, Osumi N, Yoshikawa T. Fabp7 maps to a quantitative trait locus for a schizophrenia endophenotype. PLoS Biol. 2007;5:e297. doi: 10.1371/journal.pbio.0050297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter M, Vallone D, Samad TA, Meziane H, Usiello A, Borrelli E. Absence of dopamine D2 receptors unmasks an inhibitory control over the brain circuitries activated by cocaine. Proc Natl Acad Sci U S A. 2007;104:6840–6845. doi: 10.1073/pnas.0610790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein kinase C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]