

Abstract
Zinc transporter-3 (ZnT3) protein controls synaptic vesicular Zn2+ levels, which is predicted to regulate normal cognitive function. Surprisingly, previous studies found that 6- to 10-week-old ZnT3 knock-out (KO) mice did not show impairment in the Morris water maze. We hypothesized that older ZnT3 KO animals would display a cognitive phenotype. Here, we report that ZnT3 KO mice exhibit age-dependent deficits in learning and memory that are manifest at 6 months but not at 3 months of age. These deficits are associated with significant alterations in key hippocampal proteins involved in learning and memory, as assessed by Western blot. These include decreased levels of the presynaptic protein SNAP25 (−46%; p < 0.01); the postsynaptic protein PSD95 (−37%; p < 0.01); the glutamate receptors AMPAR (−34%; p < 0.01), NMDAR2a (−64%; p < 0.001), and NMDAR2b (−49%; p < 0.05); the surrogate marker of neurogenesis doublecortin (−31%; p < 0.001); and elements of the BDNF pathway, pro-BDNF (−30%; p < 0.05) and TrkB (−22%; p < 0.01). In addition, there is a concomitant decrease in neuronal spine density (−6%; p < 0.05). We also found that cortical ZnT3 levels fall with age in wild-type mice (−50%; p < 0.01) in healthy older humans (ages, 48–91 years; r 2 = 0.47; p = 0.00019) and particularly in Alzheimer's disease (AD) (−36%; p < 0.0001). Thus, age-dependent loss of transsynaptic Zn2+ movement leads to cognitive loss, and since extracellular β-amyloid is aggregated by and traps this pool of Zn2+, the genetic ablation of ZnT3 may represent a phenocopy for the synaptic and memory deficits of AD.
Introduction
The zinc transporter-3 (ZnT3) gene (Slc30a3) was originally cloned and characterized in 1996 (Palmiter et al., 1996), and the ZnT3 protein has subsequently been shown to be essential for loading Zn2+ into synaptic vesicles (Cole et al., 1999; Linkous et al., 2008). ZnT3 is primarily localized to glutamatergic synapses (Palmiter et al., 1996) present in regions of the brain, such as the hippocampus and neocortex, that mediate higher cognitive functions. ZnT3 is likely to represent the major, possibly sole, synaptic vesicular Zn2+ transporter and therefore may govern the downstream effects of synaptic Zn2+ on a variety of signaling pathways. Zn2+, which rises to high micromolar concentrations at the synapse during activity (Frederickson et al., 2006), is believed to play a key role in learning and memory via its function as a neuronal messenger and a modulator of synaptic transmission and plasticity (Smart et al., 2004; Paoletti et al., 2009). Specifically, Zn2+ may achieve this through targeted interactions with numerous proteins, including ZnR (GPR39) (Besser et al., 2009), TrkB (Huang et al., 2008), NMDAR2b (Paoletti et al., 2009), and p75(NTR) (Lee et al., 2008). Thus, ZnT3 has been predicted to regulate cognition.
It was therefore surprising when characterization of ZnT3 knock-out (KO) mice failed to express a cognitive phenotype. These mice exhibit a 20% reduction in total zinc levels and a specific absence of histochemically reactive Zn2+ in synaptic vesicles (Cole et al., 1999), yet behavioral characterization did not detect any impairments in spatial learning, memory, or sensorimotor functions (Cole et al., 2001). This suggested that vesicular Zn2+ (and hence, ZnT3) is either not essential for cognitive function or that there are sufficient compensatory mechanisms in place to overcome any deficits incurred by the ablation of ZnT3. However, these studies were conducted in young animals (6–10 weeks of age), and it is known that ZnT3 mediates age-related Alzheimer's disease (AD)-like β-amyloid (Aβ) neuropathology in transgenic mice (Lee et al., 2002; Stoltenberg et al., 2007). Therefore, we hypothesized that ZnT3 KO animals may express a cognitive phenotype that only emerges with aging.
Materials and Methods
Animal tissues.
ZnT3 KO and wild-type (WT) animals were housed in the animal facility at The Mental Health Research Institute. All animal experimentation was approved by the Howard Florey Animal Ethics Committee and conformed to the Code of Practice established by the National Health and Medical Research Council of Australia for the Care and Use of Animals for Scientific Purposes, Seventh Edition (2004).
Animals were culled at 3 and 6 months of age. The group sizes and gender split are as reported in the main text. Before culling, animals were tested for spatial memory and visual ability in the Morris water maze. On completion of behavioral studies, animals were anesthetized (pentobarbital, intraperitoneal injection) and transcardially perfused with ice-cold PBS. Brains were removed and hemisected; right hemispheres were further microdissected and frozen on dry ice, whereas left hemispheres were snap-frozen in ice-cold isopentane for histological analysis or fixed for Golgi staining. A separate cohort of animal tissues were used for the assessment of age-related changes in ZnT3 protein. These animals were normal (B6C3) mice killed at 1.9 and 8.7 months of age.
Human tissues.
A cohort of human brain tissue was obtained from the National Neural Tissue Resource Centre, National Neuroscience Facility (Victoria, Australia). All the human cases used were classified as healthy controls based on histopathological examination and clinical data.
Behavioral studies.
We used the Morris water maze to assess spatial learning and memory, as previously reported (Adlard et al., 2008). Briefly, after nonspatial pretraining, animals were subjected to 6 consecutive days of place discrimination training, with four trials per day (random quadrant entry; maximum 90 s per trial), followed by a probe trial (one 90 s free swim without the escape platform present) 24 h later to assess retention of the task. After the probe trial, animals were subject to a visible platform task, in which the escape platform was raised above the water line and the time taken to reach the platform assessed on two consecutive trials done in separate quadrants of the pool. Data were analyzed using the EthoVision automated tracking system. There were no changes in swimming speed between groups (all age-matched) that would account for the behavioral differences.
We also assessed motor function using an accelerating rotarod, measuring the time on the rod, the total distance traveled, and the associated maximal speed reached. Three consecutive trials were performed (maximum time of 5 min; speed increases every 8 s). A separate cohort of 6-month-old ZnT3 KO and WT animals (n = 8/group) were used for these experiments.
Metal analyses.
Lyophilized hippocampal homogenates were digested in concentrated high purity nitric acid (Aristar; BDH) overnight at room temperature, and then at 90°C for 20 min. Samples were diluted with 1% nitric acid, and measurements were made using a Varian UltraMass inductively coupled plasma mass spectroscopy (ICPMS) instrument under operating conditions suitable for routine multielement analysis, as previously described (Maynard et al., 2006). The instrument was calibrated using blank, 10, 50, and 100 ppb of a certified multielement ICPMS standard solution (ICP-MS-CAl2-1; AccuStandard) for Mn, Fe, Cu, and Zn in 1% nitric acid. Results were normalized to tissue wet weight before analysis.
Biochemical analyses.
Western blot was used for quantification. Individual hippocampi were weighed and homogenized in 15 vol of ice-cold PBS containing protease inhibitors (Complete Protease Inhibitor Cocktail Tablets; Roche Applied Science) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 1 and 2; Sigma-Aldrich). Samples were sonicated (Branson Sonifier 450) on ice at a 40% duty cycle. Protein concentration was estimated using the Pierce BCA protein assay. Homogenates were stored at −80°C until required. Samples were then prepared for PAGE by the addition of 4× NuPAGE LDS Sample Buffer (Invitrogen) and 10× NuPAGE Sample Reducing Agent (Invitrogen) (to a final 1× concentration). Samples were heated to 70°C for 10 min, loaded onto NuPage Novex 4–12% Bis-Tris gels (Invitrogen), and run at 130 V for 90 min. Gels were transferred to nitrocellulose using the iBlot Gel Transfer Device (Invitrogen) set to program 3. Membranes were heated for 5 min in PBS, blocked in Tris-buffered saline and Tween-20 (TBST) containing 5% skim milk powder, and then incubated with primary antibody overnight at 4°C. Blots were rinsed in TBST and incubated in secondary antibody (1 h; room temperature), followed by additional rinsing, development with ECL reagent, and imaging using a Fujifilm Luminescent Image Analyser LAS-3000 and Image Reader LAS-3000 software package (Fujifilm).
For the assessment of age-related changes in ZnT3 protein levels in mice, the only difference to the above protocol was that a whole hemisphere (without cerebellum) was homogenized for subsequent biochemical assessment. Similarly, ZnT3 protein levels in human cases were assessed in the same way but assessed in Brodmann's area 8/9.
For all Western blot analyses, data were normalized to GAPDH (glyceraldehyde 3-phosphate dehydrogenase) as a loading control.
Statistical analysis.
Data were analyzed using JMP, version 5.0.1a (SAS), and R, version 2.9.0 (2009-04-17) (R Development Core Team, 2009). For statistical comparisons of water maze data, repeated-measures ANOVA were conducted, followed by independent one-way ANOVAs for each interday comparison between groups (as was also done for other two-sample comparisons in this study).
Results
ZnT3 KO animals display an age-dependent cognitive phenotype
We assessed 3- and 6-month-old ZnT3 KO mice (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) in the Morris water maze (Morris, 1984), a behavioral task that has been used to quantify memory defects in AD mouse models (Adlard et al., 2008). Three-month-old ZnT3 KO mice were not impaired in learning (Fig. 1 a) or recall (Fig. 1 b) performance on this task compared with WT controls, consistent with previous findings in 6- to 10-week-old ZnT3 KO mice (Cole et al., 2001). In fact, the ZnT3 KO animals actually performed significantly better on the first 3 trial days, suggesting that they learned the task faster than the WTs. However, there was no difference in the maximum performance on this task or in the recall of the task. This enhanced performance in the KO animals may be reflected by some of the biochemical differences observed in the hippocampus of these animals (see below).
Figure 1.

Morris water maze assessment of ZnT3 KO mice. a, Learning trials for 3-month-old WT (C57×129Sv background strain for the ZnT3 KOs; 6 males, 6 females) and ZnT3 KO mice (4 males, 6 females). ZnT3 KOs performed better than WTs on the task, driven by differences on trial days 1–3, but maximal performance was not different between the two genotypes. b, Probe trial for 3-month-old mice, showing no significant difference between genotypes. c, Learning trials for 6-month-old WT (8 males, 4 females) and ZnT3 KO mice (8 males, 8 females). ZnT3 KO mice performed significantly worse than age-matched WT overall (p < 0.0001), with the performance of the ZnT3 KO mice separating from WT on the last 4 trial days (days 3–6, respectively; p = 0.0003; p = 0.001; p = 0.0002; p = 0.003, compared with WT). d, Probe trial for 6-month-old WT and ZnT3 KO animals, showing that ZnT3 KO mice are significantly impaired in their retention of the task, compared with WT animals (p < 0.0001). e, Quadrant analysis, showing that 6-month-old WT animals have a significant preference for the correct northwest quadrant and a diminished preference for both the southeast and southwest quadrants, whereas the age-matched ZnT3 KO animals show equal preference for all four quadrants, indicating a lack of learning/memory. The asterisks (*) represent means that are different from age-matched WT means. *p < 0.05; **p < 0.01; ***p < 0.001.
In contrast, 6-month-old ZnT3 KO animals exhibited a marked impairment in learning (p < 0.0001) (Fig. 1 c) and memory (p < 0.0001) (Fig. 1 d,e), compared with age-matched WT controls. In addition, 6-month-old ZnT3 KO animals were significantly impaired in learning this task compared with 3-month-old ZnT3 KOs (p = 0.0005), whereas the learning curves for the WT animals were not significantly different between age groups (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
The swim speed comparisons and the quadrant analysis data suggest that there are no deficits in neuromuscular strength or exploratory locomotion in the KO mice. To explore the possibility that ZnT3 KO mice develop other age-related changes that cause poor water maze performance, we examined both visual ability and motor coordination. There were no differences between 6-month-old WT and KO mice in ability to find a visible platform (WT, 19.3 ± 4 s; KO, 21.0 ± 5.3; p = 0.8). Also, there were no differences in motor function detected on the accelerating rotarod, for total time on the rod (WT, 0.38 ± 0.08 s; KO, 0.32 ± 0.13 s; p = 0.68), average speed obtained (WT, 7.88 ± 0.57; KO, 6.63 ± 0.98; p = 0.29), or total distance traveled (WT, 500.0 ± 115.8 cm; KO, 341.0 ± 174.9 cm; p = 0.46).
Aged ZnT3 KO animals have biochemical and anatomical alterations in the brain associated with the cognitive phenotype
We explored possible neurochemical changes underlying the cognitive phenotype. There was a marked 23% decline (p < 0.0001) in hippocampal zinc between 3 and 6 months of age in the normal mice (Fig. 2 a). Compared with age-matched WT mice, ZnT3 KO animals had significantly decreased hippocampal zinc levels at both 3 (−39%; p < 0.0001) and 6 (−32%; p < 0.0001) months of age, consistent with the loss of the synaptic vesicular Zn2+ pool (Palmiter et al., 1996; Cole et al., 1999; Lee et al., 2002; Linkous et al., 2008). However, there was also a 15% decline (p < 0.0001) in ZnT3 KO hippocampal zinc from 3 to 6 months of age (Fig. 2 a), indicating that the age-dependent loss of hippocampal zinc is not exclusively from the vesicular pool. We suspected that the decline in hippocampal zinc with age, exaggerated in the ZnT3 KO animals, might adversely impact signaling cascades involved in learning and memory. Therefore, we assayed synaptic and plasticity-related proteins that may be affected by synaptic Zn2+ (Table 1).
Figure 2.

Hippocampal metal analysis of ZnT3 KO mice. a, Hippocampal zinc levels were assayed using inductively coupled plasma mass spectrometry in 3- and 6-month-old mice, showing that aging in both the WT and ZnT3 KO mice causes a significant decline. There is a significant additional zinc deficit in the ZnT3 KO animals at both ages. b, Zn levels were normalized to the Cu levels for each individual sample, showing that there is a significant drop in the Zn/Cu ratio with aging in WT and ZnT3 KO animals. The asterisks (*) represent means that are different from age-matched WT means, and delta (Δ) represents a difference between 3- and 6-month-old ZnT3 KO mice. ***, ΔΔΔ p < 0.001.
Table 1.
Levels of brain neuronal and synaptic proteins in ZnT3 KO mice compared with normal background strain
| ZnT3 KO |
||
|---|---|---|
| 3 months | 6 months | |
| Presynaptic | ||
| SNAP-25 | 76.5 ± 9.5 | 53.7 ± 3.3** |
| Synaptotagmin I | 79.6 ± 6 | 123 ± 12.5 |
| Synaptophysin | 94.6 ± 2.4 | 142.4 ± 18* |
| Postsynaptic | ||
| Spinophilin | 90 ± 11.7 | 114 ± 7.9 |
| PSD-95 | 87.8 ± 4.9 | 62.7 ± 5.7** |
| Cell support | ||
| TrkB | 105.4 ± 18 | 78 ± 4.8** |
| Pro-BDNF | 54 ± 5*** | 70.8 ± 2.8* |
| BDNF | 99 ± 8 | 86.7 ± 4.8 |
| DCX | ND | 69 ± 4.5*** |
| Glutamate receptors | ||
| AMPAR | 120 ± 8* | 66 ± 6.5** |
| NMDAR1 | 115 ± 19.7 | 83.2 ± 13 |
| NMDAR2a | 20.5 ± 4.5** | 36.3 ± 6*** |
| NMDAR2b | ND | 50.9 ± 9.8* |
Values are mean (±SEM) Western blot densities normalized to 100% of age-matched WT means. For antibody details, see supplemental Table S1 (available at www.jneurosci.org as supplemental material). DCX, Doublecortin; ND, not detected.
The asterisks represent means that are different from age-matched WT means.
*p < 0.05;
**p < 0.01;
***p < 0.001.
Although some changes were evident at 3 months of age, the most pronounced biochemical differences between ZnT3 KO and WT were observed at 6 months of age, where presynaptic SNAP25 (−46%; p = 0.008) and postsynaptic PSD95 (−37%; p = 0.002) were markedly decreased. We also found elevated presynaptic synaptophysin (+42%; p = 0.03), although no change in synaptotagmin, suggesting possible compensation for deficits in synaptic exocytosis. PSD95 tethers ionotropic glutamate receptors, NMDA and AMPA, which are involved in learning and memory. We found marked reductions in AMPAR (−34%; p = 0.003), NMDAR2a (−64%; p = 0.001), and NMDAR2b (−49%; p = 0.02) and unchanged levels of NMDAR1, supporting glutamatergic dysfunction as contributing to cognitive deficits observed at this age.
In addition, there was a small (∼6%; p = 0.04) decrease in total dendritic spines per neuron in 6-month-old ZnT3 KO animals (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Although there was no discernible change in the spine marker, spinophilin, there were other biochemical deficiencies consistent with decreased trophic support: decreased pro-BDNF (−30%; p = 0.02), TrkB (−22%; p = 0.002), and the surrogate marker of neurogenesis, doublecortin (−31%; p < 0.0001) (Table 1). Extracellular Zn2+ increases the secretion and conversion of pro-BDNF/BDNF (Hwang et al., 2005) and, in the absence of neurotrophic ligands, can transactivate TrkB by postsynaptic entry through NMDA receptors (Huang et al., 2008). Thus, our findings are consistent with ZnT3-associated presynaptic Zn2+ being used to activate TrkB, an essential component of hippocampal long-term potentiation.
Some neurochemical changes were also observed in cognitively unimpaired 3-month-old ZnT3 KO hippocampi (Table 1), including deficits in pro-BDNF (−46%; p < 0.0001) and NMDAR2a (−79.5%; p = 0.002). There was also an elevation in AMPAR (+20%; p = 0.05), which may represent a compensatory mechanism that accounts for their lack of a cognitive phenotype at this age.
ZnT3 levels decrease with age in both murine and human cortical tissue
To assess the potential relevance of ZnT3 to age-related cognitive disorders, we measured changes in brain ZnT3 levels with aging. Normal mice (B6C3) exhibited a ≈50% decrease in ZnT3 (p = 0.0026) between 1.9 and 8.7 months of age (Fig. 3 a). This decrease was recapitulated in humans, in which there was a significant decline (p = 0.0002) in cortical ZnT3 levels from age 48 to 91 in healthy people (Fig. 3 b). ZnT3 levels decreased even further in AD cortex (Fig. 3 c). Therefore, synaptic Zn2+ release may be decreased in aging and more so in AD, which, our animal data indicate, could contribute to cognitive loss. So ZnT3 knock-out mice may model an important aspect of the synaptic pathology of AD.
Figure 3.
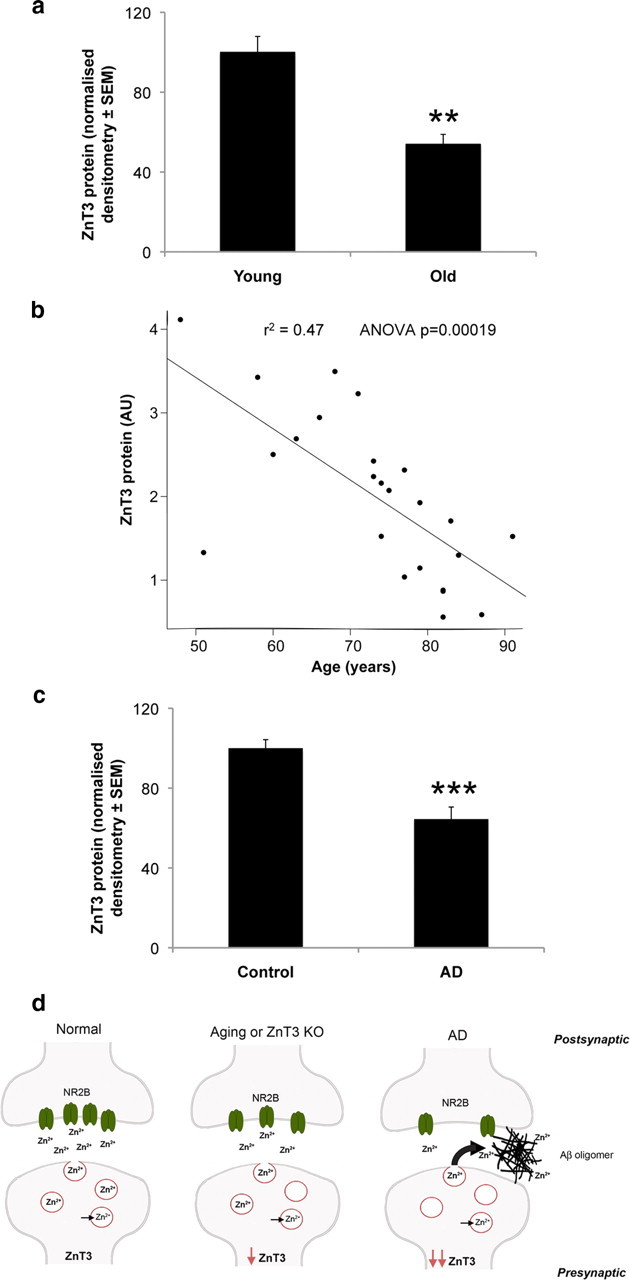
Decline of brain ZnT3 levels in murine and human aging. a, Cerebral ZnT3 levels over age assayed by Western blot in normal mice (B6C3 strain; n = 8 per group; 50% male; average ages: young, 1.9 months; old, 8.7 months), demonstrating a significant decline in ZnT3 with age (ANOVA, p = 0.0026). b, ZnT3 levels in BA8/9 of normal humans (n = 16 males, 8 females; age range, 48–91 years; average age ± SD, 73 ± 11 years). The linear regression line is shown (r 2 = 0.47; p = 0.00019), demonstrating a significant age-related decline in ZnT3. c, ZnT3 levels in BA8/9 are further decreased (−36%; ANOVA, p < 0.0001) in AD (n = 4 males, 5 females; 81 ± 9 years) compared with the healthy controls (n = 8 males, 6 females; 73 ± 13 years). d, ZnT3-associated decline in synaptic zinc in age and Alzheimer's disease. This diagrammatic model shows a zinc-containing glutamatergic synapse in health, aging, and disease. In health, ZnT3 loads Zn2+ into vesicles, whereupon it is transported and released at the synapse. After release, Zn2+ can interact with postsynaptic targets such as NMDA receptors (NR2B subunit shown here). With aging, or after the genetic ablation of ZnT3, the loss of ZnT3 inhibits Zn2+ delivery and leads to a decline in NR2B and other postsynaptic targets involved in learning and memory (see text). In AD, there is a profound impairment of synaptic Zn2+ reaching its postsynaptic targets because there is both an accentuated drop in ZnT3 levels as well as entrapment of extracellular Zn2+ by Aβ oligomers. Loss of transsynaptic Zn2+ signaling leads to decreased expression of select postsynaptic targets such as NR2B and cognitive decline. The asterisks (*) represent means that are different from age-matched WT means. **p < 0.01; ***p < 0.001.
Discussion
This is the first report of a cognitive phenotype resulting from the ablation of ZnT3 and demonstrates that vesicular Zn2+ is a requirement for normal memory function in adulthood. From these findings, we propose that Zn2+ is needed to sustain synaptic health during aging through modulation of metabotropic and other postsynaptic targets. In the mouse brain, ZnT3 expression and the presence of synaptic vesicular Zn2+ commence in late embryonic development and increase until weaning (Valente and Auladell, 2002). Here, we find that with aging hippocampal zinc and ZnT3 decline for uncertain reasons, possibly related to the need for energy to sustain zinc levels in the brain (Melov et al., 2007). We hypothesize that, in the absence of ZnT3, this drop in hippocampal zinc becomes critical earlier (perhaps reaching a biologically relevant minimal zinc threshold), causing an exaggerated cognitive aging phenotype. This would be consistent with the decrease in NR2B and TrkB that we observed since the levels of these proteins are modulated by zinc (Kim et al., 2002; Chowanadisai et al., 2005; Huang et al., 2008). Our current findings are also consistent with reports of cognitive impairment caused by nutritional zinc deficiency in both humans and animals (Keller et al., 2001; Stoecker et al., 2009; Tahmasebi Boroujeni et al., 2009).
The Zn2+ released by ZnT3 is also known to mediate parenchymal and cerebrovascular amyloid formation in Tg2576 amyloid precursor protein (APP) transgenic (Tg) mice (Lee et al., 2002; Friedlich et al., 2004; Stoltenberg et al., 2007). Furthermore, recent data have shown that the attraction of Aβ oligomers to Zn2+ emanating from the glutamatergic synapse occludes the NR2B NMDA receptor (Deshpande et al., 2009). Although Tg2576 mice display cognitive impairments across various behavioral tasks (Chapman et al., 1999; Westerman et al., 2002), the previous reports on the ablation of ZnT3 relieving amyloid pathology did not test cognition (Lee et al., 2002; Friedlich et al., 2004). Our findings show that genetic ablation of ZnT3 may actually generate a phenocopy for the synaptic and memory deficits present in the cognitively impaired APP Tg mouse model for AD. This notion would be paradoxical if β-amyloid accumulation were the sole cause of neurotoxicity in this model. Instead, we propose a mechanism where β-amyloid pathology could cause cognitive impairment by trapping synaptic zinc rather than through direct toxicity. In AD and APP Tg mice, Zn2+ accumulates in extracellular parenchymal amyloid (Lovell et al., 1998; Lee et al., 1999). We hypothesize that the trapping of Zn2+ by amyloid might generate neurophysiological sequelae similar to ZnT3 ablation (Fig. 3 d). Consistent with this notion, we found that ZnT3 levels fall in aging and fall further in AD (Fig. 3 a–c). Therefore, the transsynaptic movement of Zn2+ may be severely compromised in AD both by the lack of ZnT3 expression and by being sequestered in amyloid. Thus, sufficient Zn2+ fails to reach its postsynaptic targets, further exacerbating cognitive decline (Fig. 3 d). This conclusion is also consistent with those of Deshpande et al. (2009), who postulated that the sequestration of zinc in oligomeric Aβ–Zn complexes may lead to a reduction in zinc availability at the synapse, resulting in a loss of zinc modulatory activity and leading to cognitive deficits in AD.
Our findings support therapeutic strategies for age-related cognitive decline that sustain intracellular zinc in the cortex by facilitating its reuptake from the extracellular compartment. This proposed mechanism may explain the rapid recovery of cognitive performance in APP transgenic mice treated with zinc ionophores, CQ (clioquinol) and PBT2 (Adlard et al., 2008), and may underlie their potential therapeutic benefits in AD (Ritchie et al., 2003; Lannfelt et al., 2008) and age-related cognitive decline.
Note added in proof.
Our results of decreased ZnT3 in AD are consistent with a decrease in ZnT3 mRNA recently reported (Beyer et al., 2009).
Footnotes
This work was supported by funds from the Australian Research Council, the National Health and Medical Research Council, Operational Infrastructure Support from the Victorian State Government, and the Alzheimer's Association. We thank Dr. Noel Faux for statistical assistance, Irene Volitakis for ICPMS analysis, Fairlie Hinton and Dr. Catriona McLean for the provision of neuropathologically characterized human brain samples (through the National Neural Tissue Resource Centre), and Andrew Tsatsanis, Bruce Etherton, and Sarah Evans for assistance with behavioral studies.
References
- Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT, Hershfinkel M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci. 2009;29:2890–2901. doi: 10.1523/JNEUROSCI.5093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer N, Coulson DT, Heggarty S, Ravid R, Irvine GB, Hellemans J, Johnston JA. ZnT3 mRNA levels are reduced in Alzheimer's disease post-mortem brain. Mol Neurodegener. 2009;4:53. doi: 10.1186/1750-1326-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Chowanadisai W, Kelleher SL, Lönnerdal B. Maternal zinc deficiency reduces NMDA receptor expression in neonatal rat brain, which persists into early adulthood. J Neurochem. 2005;94:510–519. doi: 10.1111/j.1471-4159.2005.03246.x. [DOI] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A. 1999;96:1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TB, Martyanova A, Palmiter RD. Removing zinc from synaptic vesicles does not impair spatial learning, memory, or sensorimotor functions in the mouse. Brain Res. 2001;891:253–265. doi: 10.1016/s0006-8993(00)03220-0. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J. A role for synaptic zinc in activity-dependent Aβ oligomer formation and accumulation at excitatory synapses. J Neurosci. 2009;29:4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, 3rd, Balaji RV, Rengarajan B, Masalha R, Frederickson CJ, Zeng Y, Lopez EV, Koh JY, Chorin U, Besser L, Hershfinkel M, Li Y, Thompson RB, Krezel A. Synaptic release of zinc from brain slices: factors governing release, imaging, and accurate calculation of concentration. J Neurosci Methods. 2006;154:19–29. doi: 10.1016/j.jneumeth.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Friedlich AL, Lee JY, van Groen T, Cherny RA, Volitakis I, Cole TB, Palmiter RD, Koh JY, Bush AI. Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer's disease. J Neurosci. 2004;24:3453–3459. doi: 10.1523/JNEUROSCI.0297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57:546–558. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Hwang JJ, Park MH, Choi SY, Koh JY. Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J Biol Chem. 2005;280:11995–12001. doi: 10.1074/jbc.M403172200. [DOI] [PubMed] [Google Scholar]
- Keller KA, Grider A, Coffield JA. Age-dependent influence of dietary zinc restriction on short-term memory in male rats. Physiol Behav. 2001;72:339–348. doi: 10.1016/s0031-9384(00)00421-2. [DOI] [PubMed] [Google Scholar]
- Kim TY, Hwang JJ, Yun SH, Jung MW, Koh JY. Augmentation by zinc of NMDA receptor-mediated synaptic responses in CA1 of rat hippocampal slices: mediation by Src family tyrosine kinases. Synapse. 2002;46:49–56. doi: 10.1002/syn.10118. [DOI] [PubMed] [Google Scholar]
- Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- Lee JY, Mook-Jung I, Koh JY. Histochemically reactive zinc in plaques of the Swedish mutant β-amyloid precursor protein transgenic mice. J Neurosci. 1999;19(RC10):1–5. doi: 10.1523/JNEUROSCI.19-11-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim YJ, Kim TY, Koh JY, Kim YH. Essential role for zinc-triggered p75NTR activation in preconditioning neuroprotection. J Neurosci. 2008;28:10919–10927. doi: 10.1523/JNEUROSCI.3421-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous DH, Flinn JM, Koh JY, Lanzirotti A, Bertsch PM, Jones BF, Giblin LJ, Frederickson CJ. Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J Histochem Cytochem. 2008;56:3–6. doi: 10.1369/jhc.6A7035.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Maynard CJ, Cappai R, Volitakis I, Cherny RA, Masters CL, Li QX, Bush AI. Gender and genetic background effects on brain metal levels in APP transgenic and normal mice: implications for Alzheimer beta-amyloid pathology. J Inorg Biochem. 2006;100:952–962. doi: 10.1016/j.jinorgbio.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li QX, Laughton K, Hubbard A, Cherny RA, Gibson B, Bush AI. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One. 2007;2:e536. doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci U S A. 1996;93:14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2009. [Google Scholar]
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- Smart TG, Hosie AM, Miller PS. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- Stoecker BJ, Abebe Y, Hubbs-Tait L, Kennedy TS, Gibson RS, Arbide I, Teshome A, Westcott J, Krebs NF, Hambidge KM. Zinc status and cognitive function of pregnant women in southern Ethiopia. Eur J Clin Nutr. 2009;63:916–918. doi: 10.1038/ejcn.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltenberg M, Bush AI, Bach G, Smidt K, Larsen A, Rungby J, Lund S, Doering P, Danscher G. Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience. 2007;150:357–369. doi: 10.1016/j.neuroscience.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Tahmasebi Boroujeni S, Naghdi N, Shahbazi M, Farrokhi A, Bagherzadeh F, Kazemnejad A, Javadian M. The effect of severe zinc deficiency and zinc supplement on spatial learning and memory. Biol Trace Elem Res. 2009;130:48–61. doi: 10.1007/s12011-008-8312-7. [DOI] [PubMed] [Google Scholar]
- Valente T, Auladell C. Developmental expression of ZnT3 in mouse brain: correlation between the vesicular zinc transporter protein and chelatable vesicular zinc (CVZ) cells. Glial and neuronal CVZ cells interact. Mol Cell Neurosci. 2002;21:189–204. doi: 10.1006/mcne.2002.1159. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]