

Abstract
Tauopathy comprises hyperphosphorylation of the microtubule-associated protein tau, causing intracellular aggregation and accumulation as neurofibrillary tangles and neuropil treads. Some primary tauopathies are linked to mutations in the MAPT gene coding for protein tau, but most are sporadic with unknown causes. Also, in Alzheimer's disease, the most frequent secondary tauopathy, neither the cause nor the pathological mechanisms and repercussions are understood. Transgenic mice expressing mutant Tau-P301L suffer cognitive and motor defects and die prematurely from unknown causes. Here, in situ electrophysiology in symptomatic Tau-P301L mice (7–8 months of age) revealed reduced postinspiratory discharges of laryngeal motor outputs that control laryngeal constrictor muscles. Under high chemical drive (hypercapnia), postinspiratory discharge was nearly abolished, whereas laryngeal inspiratory discharge was increased disproportionally. The latter may suggest a shift of postinspiratory laryngeal constrictor activity into inspiration. In vivo double-chamber plethysmography of Tau-P301L mice showed significantly reduced respiratory airflow but significantly increased chest movements during baseline breathing, but particularly in hypercapnia, confirming a significant increase in inspiratory resistive load. Histological analysis demonstrated hyperphosphorylated tau in brainstem nuclei, directly or indirectly involved in upper airway motor control (i.e., the Kölliker–Fuse, periaqueductal gray, and intermediate reticular nuclei). In contrast, young Tau-P301L mice did not show breathing disorders or brainstem tauopathy. Consequently, in aging Tau-P301L mice, progressive upper airway dysfunction is linked to progressive tauopathy in identified neural circuits. Because patients with tauopathy suffer from upper airway dysfunction, the Tau-P301L mice can serve as an experimental model to study disease-specific synaptic dysfunction in well defined functional neural circuits.
Introduction
Neurodegenerative diseases, with Alzheimer's disease (AD) as a prime example, are inherently associated with tauopathy, pathologically demonstrated postmortem as neurofibrillary tangles (NFTs) and neuropil treads. In AD, the mainstream hypothesis holds amyloid peptides, cleaved from the amyloid precursor protein, as the primary pathological cause. Neither the connection to phosphorylation of protein tau nor the contributions of tauopathy to the clinical symptoms are understood in molecular and pathological detail (Terwel et al., 2002; Janus, 2008; Takashima, 2008; Denk and Wade-Martins, 2009). Because amyloid pathology is not observed as a separate entity, the associated tauopathy in AD should contribute to the neurodegeneration and clinical problems. In this context, we developed an experimental model for tauopathy (i.e., Tau-P301L transgenic mice) (Terwel et al., 2005, 2008) that we have studied here specifically for autonomic functions that become compromised by the developing tauopathy with aging.
Protein tau functions in the assembly and stabilization of microtubuli in neurons, a process regulated mainly by phosphorylation in adult brain (Andorfer and Davies, 2000; Buée et al., 2000; Goedert and Jakes, 2005). In frontotemporal dementia with parkinsonism, mutations in the tau gene on chromosome 17 are closely associated with disease and with accumulation of hyperphosphorylated tau as NFTs, diagnostic for all tauopathies, including AD (Goedert and Jakes, 2005). In patients and transgenic mice, tauopathy correlates well with memory impairment (Tatebayashi et al., 2002). Conversely, hyperphosphorylation of tau at later stages was often associated with premature death in transgenic animals (Lewis et al., 2001; Oddo et al., 2003; Götz et al., 2004; Terwel et al., 2005, 2008).
Tau-P301L transgenic mice progressively develop severe neuronal tauopathy in midbrain and brainstem, and neurological symptoms start around age 6–7 months (Terwel et al., 2005). The mice become progressively lethargic and motor impaired, lose weight, and die of unknown causes, most at age 9–10 months (Terwel et al., 2005, 2008).
Because of these well known motor defects of Tau-P301L mice (Terwel et al., 2005, 2008), we investigated the impact of the brainstem tauopathy on breathing as a vital motor function at 3 and 7–8 months of age (i.e., before and at symptomatic ages, respectively). Analysis of the respiratory motor pattern using in situ approaches (Paton, 1996; Stettner et al., 2007) and in vivo plethysmography revealed increased inspiratory resistive load in Tau-P301L mice potentially because of laryngeal constrictor activation during inspiration. The observed dysregulation of upper airway resistance during the breathing cycle correlates with the expression of hyperphosphorylated protein tau in brainstem upper airway premotor populations [i.e., Kölliker–Fuse (KF) and intermediate reticular nuclei]. At 3 months of age, Tau-P301L mice neither showed alteration in the breathing pattern nor tauopathy in vital brainstem circuits. The identified tau-laden neuron clusters in various midbrain and brainstem areas are anatomically linked directly to the observed upper airway dysfunction. They further suggest disturbances in other autonomic functions, ranging from the regulation of the sleep–wake cycle to the mediation of defensive behavior, all to be explored in detail.
Materials and Methods
The experimental procedures were performed in accordance with European and national guidelines for care and use of laboratory animals and were approved by the local ethical committees.
Animals.
Unless indicated otherwise, male Tau-P301L transgenic mice in the FVB/N genetic background (3 and 7–8 months of age) were studied and compared with age- and sex-matched FVB/N nontransgenic mice (Terwel et al., 2005, 2008). In these transgenic mice, the longest human tau isoform bearing the P301L mutation (tau-4R/2N-P301L) is expressed under control of the mouse thy1 gene promoter aiming for neuron-specific expression starting in the third postnatal week (Terwel et al., 2005).
Transgenic mice used were homozygous for the Tau-P301L transgene and were genotyped by PCR with forward primer specific for the mouse thy1 gene promoter and reversed primer specific for human tau cDNA (Terwel et al., 2005).
In vivo single- and double-chamber plethysmography.
The breathing pattern of 64 mice has been studied in vivo thanks to two different noninvasive plethysmographic approaches. As reported previously (Viemari et al., 2005; Zanella et al., 2008), breathing was recorded from 40 unrestrained, conscious mice [n = 16 wild type (WT) and n = 12 Tau-P301L, 7–8 months of age; mean weight, 29.5 ± 0.9 g; n = 6 WT and n = 6 Tau-P301L, 3 months of age; mean weight, 28.1 ± 0.7 g] using a standard whole-body flow plethysmograph (EMKA Technologies) in which a constant flow pump connected to the animal chamber ensures proper and continuous inflow of fresh air (600 ml/min) or of a hypercapnic gas mixture (CO2, 4%). The animal and reference chambers (volume, 200 ml) were maintained at 25 ± 0.5°C (temperature sensor Checktemp 1; Hanna Instruments). The spirogram, reflecting the strength of the mouse respiratory movement, was obtained by recording the pressure difference between the two chambers. The signal was amplified, filtered (DC, 50 Hz), fed to an analog-to-digital converter (sampling frequency, 1 kHz), and stored on a PC disk via the Spike 2 interface and software (Cambridge Electronic Design). To avoid stress, the mice were habituated to the plethysmograph by putting them in the chamber 2 d before the recording during 2 h. The next day, only those recording periods were analyzed during which the animals were quiet (i.e., without major limb, body, and head movements). Routinely, breathing of four mice was simultaneously recorded in the four-chamber plethysmograph, allowing comparison between WT and Tau-P301L mice placed in identical conditions. The mouse temperature was monitored before and after recording sessions (Infrared thermosensor S 01500; Technimed). Temperature did not change during the recording session, and no significant differences were observed in WT versus Tau-P301L mice. The mean respiratory frequency (fR) [expressed in cycle per minute (c min−1)], durations of inspiratory (TI) and expiratory (TE) periods (expressed in milliseconds), and spirogram amplitude (SA) were measured during 10–20 min baseline and during 5 min hypercapnic challenges. Because plethysmographic recordings do not provide absolute information about actual tidal volumes, SA values were expressed in arbitrary units (a.u.). In addition, the apnea index (AI) [i.e., the number of long-lasting apneas (>2 respiratory cycles) per minute] and the irregularity score (IS) of successive respiratory cycles (>200 cycles) were calculated as reported previously (Zanella et al., 2008) to check whether the respiratory rhythmogenic mechanisms were altered in Tau-P301L mice.
A double-chamber plethysmograph was developed to separately record the respiratory chest movements and respiratory airflow in 24 other conscious mice (n = 6 WT, n = 6 Tau-P301L, 7–8 months of age; n = 6 WT, n = 6 Tau-P301L, 3 months of age). The double-chamber plethysmograph was built from two 50 ml syringes, with a rubber collar separating the body chamber (50 ml) from the head chamber (20 ml). Air tightness between body and head chambers was verified by imposing small pressure changes in one chamber and checking the lack of pressure changes in the other. The head chamber was connected to a constant flow pump to ensure proper and continuous flow of either fresh air (200 ml/min) or hypercapnic gas mixture (CO2, 4% in air). Body and head chambers were connected to respective reference chambers, all maintained at 25 ± 0.5°C, and pressure changes were recorded by pressure gauges (EMKA Technologies) to measure spirogram amplitude from the chest and the airflow (chest SA and airflow SA). In contrast to single-chamber analysis, double-chamber recordings are more stressful, and the mice were habituated to the double-chamber plethysmograph before actual recordings were made, according the same protocol described above.
In situ working heart–brainstem preparation.
Experiments were performed using the arterially perfused working heart–brainstem preparation (Paton, 1996). Mice (n = 4 WT; n = 6 Tau-P301L; 8 months of age) were anesthetized deeply in a saturated atmosphere of isoflurane (1-chloro-2,2,2-trifluoroethyl-difluoromethylether) (Abbott). Once the animal failed to respond to noxious pinch to the tail or a hindpaw, it was transected below the diaphragm, transferred into ice-cooled (5°C) artificial CSF (aCSF) gassed with carbogen (95% O2 and 5% CO2), decerebrated at the precollicular level, and cerebellectomized. The lungs were removed. The left phrenic nerve was separated and cut at the level of the diaphragm. The descending aorta was isolated from the ventral surface of the spinal column. The right vagal nerve was also dissected and prepared for recording.
These initial procedures took ∼15 min. The preparation was then transferred to a recording chamber. The descending aorta was cannulated and perfused using a peristaltic pump (Watson-Marlow) with carbogen-gassed aCSF at 31°C containing Ficoll (1.25%; Sigma-Aldrich) to maintain colloid-osmotic pressure. The perfusate contained the following (in mm): 125 NaCl, 3 KCl, 1.25 KH2PO4, 2.5 CaCl2, 1.25 MgSO4, 25 NaHCO3, 10 d-glucose, and 1.25% Ficoll. The osmolarity of the perfusate was 300 ± 10 mosmol L−1, and the pH was 7.35 ± 0.05 by gassing with carbogen. The perfusate was filtered and passed through bubble traps to remove gas bubbles. The perfusate leaking from the preparation was collected and recirculated after reoxygenation. Cardiac activity returned within seconds, and rhythmic contractions of respiratory muscles within a few minutes after onset of reperfusion. Respiratory-related movements were abolished by vecuronium bromide (0.3 μg ml−1; Inresa).
After paralysis, phrenic nerve activity (PNA) was primarily used to fine-tune the preparations by adjusting the flow rate (10–22 ml min−1) and perfusion pressure (40–70 mmHg). For measurement of aorta perfusion pressure, a double lumen catheter was used and connected to a pressure transducer. During the tuning, the flow rates were individually adjusted to fulfill the following criteria: reappearance of a clearly identifiable respiratory pattern, rhythmic respiratory motor discharges of at least 40 bursts/min, and maintenance of the respiratory pattern for at least 30 min before starting any experiments or data analysis. To test the respiratory response to hypercapnia, the perfusate was gassed with carbogen containing 12% CO2 for 1 min.
Nerve recordings.
Respiratory motor nerve activities were recorded simultaneously from the phrenic and central vagus nerve in all preparations. The suction electrodes were connected to differential amplifiers (Neurolog 100). Nerve activity was amplified and filtered (8 Hz to 3 kHz; Neurolog modules 104 and 125). All data were digitized using a MacLab 8s interface and stored on a computer using Chart software (version 5.2; ADInstruments). Respiratory motor nerve activity was integrated (time constant, 100 ms). The electrocardiogram was recorded simultaneously with the PNA but not analyzed in present study.
Data analysis.
Using the PNA and central vagal nerve activity (cVNA) recordings, we analyzed the duration of the following respiratory parameters: total respiratory cycle length (Ttot), time of inspiration (Ti), postinspiration (Tπ), and late expiration (Te2). In contrast to cat in vivo investigations, PNA recording does not display a clearly identifiable postinspiratory activity in mice. Therefore, we classified the entire expiratory interval as Te (including Tπ and Te2). Tπ and Te2 were identified and measured from the cVNA. The inspiratory cVNA coincided with the PNA. The duration of the expiratory cVNA was defined as Tπ and the phase with absent cVNA in the late expiration as Te2. To quantify changes in the discharge strength inspiratory and postinspiratory cVNA (I-VNA and PI-VNA, respectively), we measured the surface area of the integrated signals. Since the cVNA showed prominent changes in its discharge pattern, a more detailed analysis was performed on the timing of the peak amplitude of the integrated cVNA. Peak cVNA amplitude was demonstrated in numerous publications to coincide with the activation of laryngeal adductor muscles (for review, see Dutschmann and Paton, 2002a). We determined the inspiratory off-switch from the abrupt cessation of PNA and used it as reference point (T = 0) to comparatively analyze the occurrence of VNA peak amplitude in inspiration or early expiration. A negative latency of cVNA peak thereby reflects its occurrence in expiration, and a positive latency, its occurrence during inspiration (see Fig. 3).
Figure 3.
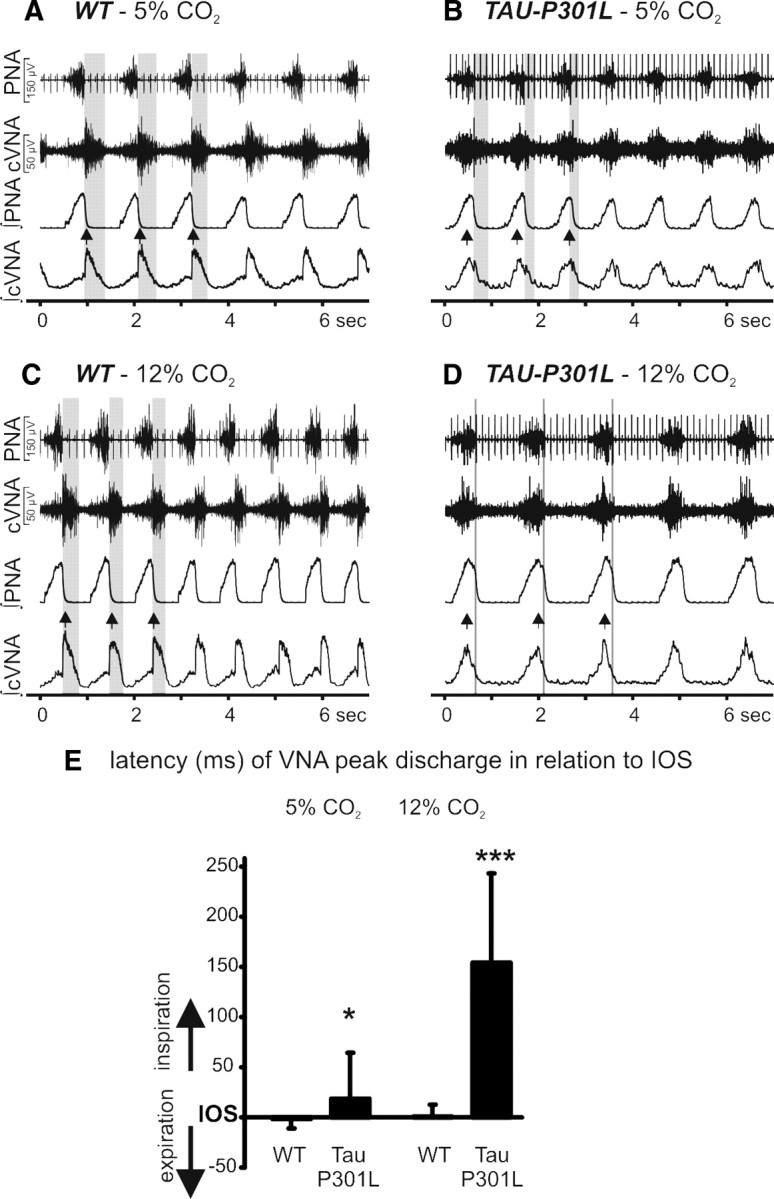
In situ recordings of the respiratory motor pattern from WT and Tau-P301L mice. A–D, Recordings of inspiratory phrenic and inspiratory/postinspiratory vagal nerve activity (PNA; cVNA) of WT (A) and Tau-P301L mice (B) under control condition, 5% CO2 in perfusate and hypercapnic conditions 12% CO2 in perfusate (C, D). The gray-shaded areas highlight the reduced postinspiratory activity in Tau-P301L mice. Postinspiratory activity was virtually absent, whereas inspiratory-related cVNA discharge was disproportionally increased. For additional details and statistical analysis, see text. The double arrowheads in the integrated signals (A–D) illustrate cVNA peak discharge in coincidence with inspiratory PNA. Group data for the occurrence of cVNA peak discharge during the respiratory cycle are illustrated in E (*p < 0.05, ***p < 0.001). IOS, Inspiratory off-switch.
Finally, we focused the present study on respiratory dysfunctions in Tau-P301L mice; thus, cardiovascular parameters such as ECG or perfusion pressure were not analyzed.
Statistics.
Data analysis was performed with the Statistica 7.1 software package (StatSoft). All results are presented as mean ± SEM. Respiratory parameters of WT and Tau-P301L preparations were compared using the two-sided unpaired (differences between WT and Tau-P301L) or paired (differences between normocapnia and hypercapnia) Student t test in the case of normally distributed data and using the Mann–Whitney U test in the case of non-normally distributed data. A value of p < 0.05 was considered to be significant.
Mouse brain immunohistochemistry.
Mice were anesthetized (Nembutal; 120 mg/kg, i.p.) for transcardiac perfusion with ice-cold saline (2 ml/min, 2 min). Brain and spinal cord were removed, fixed overnight in 4% paraformaldehyde in PBS at 4°C, and stored in 0.1% sodium azide in PBS at 4°C until sectioning. Coronal vibratome sections of 40 μm were cut from brain, brainstem, and spinal cord. Immunohistochemistry was performed essentially as described previously (Terwel et al., 2005, 2008). After rinsing in PBS, sections were treated for 15 min with 1.5% H2O2 in 50% methanol/PBS to eliminate endogenous peroxidase activity. Nonspecific binding sites were blocked by incubation in 10% fetal calf serum, 0.1% Triton X-100 in PBS (blocking buffer). The sections were incubated with primary monoclonal antibodies AT8 (mouse anti-AT8, 0.2 μg/ml) (Terwel et al., 2005) or PG5 (mouse anti-PG5, 1/5000) (Andorfer and Davies, 2000) in blocking buffer at 4°C overnight. After rinsing in blocking buffer, sections were incubated for 1 h with the secondary goat anti-mouse IgG antiserum (1:500 in blocking buffer) (Dako). Sections were rinsed in PBS and incubated in 50 mm Tris·HCl, pH 7.6, for 5 min, before enzymatic staining with 3,3′-diaminobenzidine (0.5 mg/ml), 0.3% H2O2 in 50 mm Tris·HCl, pH 7.6. Sections were counterstained with hematoxylin, before dehydration by passage through a graded series of ethanol solutions. After delipidation in xylol, the sections were mounted for microscopic analysis.
We analyzed continuous series of between 90 and 100 sections (20 μm; WT, n = 3; Tau-P301L, n = 7) from the caudal medulla oblongata to rostral end of the superior colliculus in the midbrain. In addition, sections of cervical and thoracic spinal cord segments containing respiratory motor neurons were analyzed (Tau-P301L, n = 2; WT, n = 1).
The expression of phospho-epitopes of protein tau, defined by monoclonal antibodies AT8 and PG5, was analyzed in brainstem and midbrain (n = 7; n = 5, respectively) series of adjacent sections from Tau-P301L mice at symptomatic age (8 months) and compared with young mice (3 months of age). The tauopathy pattern in brainstem and midbrain nuclei is expressed on a relative score (i.e., Ø, no expression; and scores + to +++ refer to 1–5, 5–20, and 20–50 positive neurons per 104 μm2). It is important to note that brainstem and midbrain from young Tau-P301L mice (3 months of age) and from WT mice at ages 3 and 8 months did not show any accumulation of hyperphosphorylated Tau into tau aggregates, let alone NFTs. Faint staining for AT8 but not PG5 could be observed in some midbrain and brainstem areas (e.g., reticular formation). Consequently, statistical analysis of NFT expression in Tau-P301L versus WT mice or during development is per se highly significant and therefore not reported.
Results
Abnormal breathing pattern of 7- to 8-month-old Tau-P301L mice (in vivo approach)
The premature death of Tau-P301L mice at age 9–10 months is preceded by generalized motor deficiency starting around age 7–8 months. Here, we analyzed Tau-P301L mice for breathing disorders as potential cause of the premature death in this transgenic mouse line, extensively characterized previously for forebrain tauopathy and cognition (Terwel et al., 2005, 2008; Boekhoorn et al., 2006).
First, we used a standard whole-body plethysmograph to record breathing in unrestrained, conscious WT and Tau-P301L mice (Fig. 1A) under normocapnic conditions (air breathing) and during moderate hypercapnic challenges (5 min; 4% CO2 in air). Analyzing breathing parameters under normocapnia during periods when mice were quiet, not moving revealed no significant differences between young WT and Tau-P301L mice (3 months of age) but significant abnormalities in 7- to 8-month-old Tau-P301L mice (Fig. 1).
Figure 1.

Whole-body plethysmographic recordings in unrestrained, conscious WT and Tau-P301L mice. A, Schematic representation of the experimental setup used to record breathing in unrestrained, conscious mouse placed in a 200 ml recording chamber (to be compared with the experimental setup illustrated in Fig. 2A, in which the mouse is restrained). B, C, Individual traces show spirograms (inspiration upward) recorded from 8-month-old WT (B) and Tau-P301L (C) mice placed under control (B1, C1) (air; room air, 0.03% CO2) and hypercapnic (B2, C2) (CO2; 4% CO2 in air, 5 min) breathing conditions. SA values are expressed in arbitrary units (vertical calibration bars: 5 a.u. for B1 and C1; 10 a.u. for B2 and C2); note the increased SA under hypercapnic conditions in B2 and C2. D, The bars in the histograms show the mean SA (and SEM) from 6 WT and 6 Tau-P301L mice at 3 months of age (D1) and from 16 WT and 12 Tau-P301L mice at 7–8 months of age (D2). Note that the mean SA did not significantly differ in young WT versus Tau-P301L mice placed under identical breathing conditions (in D1; either air or CO2), whereas the mean SA was significantly larger in Tau-P301L versus WT old mice under air (D2; *p < 0.01) and CO2 (D2; **p < 0.05) breathing conditions. In all groups, hypercapnia significantly increased mean SA when compared with air.
In young WT (n = 6) and Tau-P301L (n = 6) mice under normocapnia, the mean respiratory frequency (fR = 174 ± 9 vs 179 ± 10 c min−1; NS) and mean durations of inspiratory (TI = 144 ± 20 vs 130 ± 10 ms; NS) and expiratory (TE = 207 ± 10 vs 207 ± 10 ms; NS) periods did not significantly differ. In addition, the spirogram amplitudes were not statistically different (SA = 6.6 ± 0.9 vs 6.6 ± 0.5 a.u.; NS) (Fig. 1D1), as well as the mean apnea index (AI = 0.1 ± 0.1 vs 0.1 ± 0.2; NS) and the mean irregularity score (IS = 10.3 ± 1.5 vs 6.7 ± 0.8; NS). When subjected to moderate hypercapnia, similar breathing changes were observed in WT and Tau-P301L mice: both types of mice significantly increased fR and SA to reach similar mean values (fR = 254 ± 10 vs 246 ± 8 c min−1; NS; SA = 18.5 ± 0.9 vs 17.4 ± 0.9 a.u.; NS) (Fig. 1D1).
In 7- to 8-month-old WT (n = 16) and Tau-P301L (n = 12) mice under normocapnia, no differences were observed in the mean fR (150 ± 4 vs 155 ± 4 c min−1; NS), TI (148 ± 5 vs 139 ± 9 ms; NS), TE (258 ± 10 vs 251 ± 8 ms; NS), AI (0.1 ± 0.1 and 0.1 ± 0.1; NS), and IS (8.6 ± 1.1 and 10.6 ± 1.1; NS). However, the mean SA was significantly higher in Tau-P301L than WT mice (9.5 ± 0.9 vs 5.6 ± 0.3 a.u.; p < 0.005) (Fig. 1B1,C1,D2). When subjected to moderate hypercapnia, old WT (n = 10) and Tau-P301L (n = 12) mice increased their fR by ∼50% to reach similar mean fR values (240 ± 10 vs 252 ± 4 c min−1; NS), but Tau-P301L mice had a significantly shorter TI than WT (91 ± 2 vs 105 ± 5 ms, respectively; p < 0.05) and retained a significantly larger SA than WT (30.7 ± 3.6 vs 20.9 ± 1.7 a.u.; p < 0.05) (Fig. 1B2,C2,D2).
We developed a double-plethysmographic approach to simultaneously record the chest respiratory movements and the resulting airflow in conscious old mice at 7–8 months of age (Fig. 2A) because SA gained from simple plethysmographic recordings does not provide absolute information about actual tidal volumes. Consequently, the increased SA observed in old Tau-P301L mice could be related to an increase of respiratory movements potentially caused by increase of upper airway resistance. Indeed, compared with old WT mice (n = 6), old Tau-P301L mice (n = 6) showed significantly weaker airflow SA (123 ± 12 vs 84 ± 11 a.u.; p < 0.05) but significantly larger chest SA (42 ± 5 vs 99 ± 5 a.u.; p < 0.001) (Fig. 2B–D). Therefore, the ratio of airflow/chest SA was more than threefold larger in WT than Tau-P301L mice (3.1 ± 0.4 vs 0.9 ± 0.1, respectively; p < 0.01) (Fig. 2D3). Neither fR (252 ± 13 vs 247 ± 7 c min−1; NS) nor TI (123 ± 6 vs 112 ± 2 ms; NS) nor TE (115 ± 7 vs 132 ± 7 ms; NS) differed significantly between WT versus Tau-P301L mice. In both strains, the baseline fR measured with double plethysmography was significantly higher than that previously measured with simple plethysmography. The increase of fR probably reflected the immobilization stress caused by the restrained double-chamber plethysmography. The fR appeared elevated close to the physiological maximum, because hypercapnia did not further increase the fR in both genotypes. However, hypercapnia differently affected the chest and airflow SA of old WT and Tau-P301L mice (Fig. 2D1,D2). In old WT mice, hypercapnia increased both airflow and chest SA by ∼50%, and the ratio airflow/chest SA under hypercapnia (3.2 ± 0.5) was not statistically different from the ratio under normocapnia (Fig. 2D3). In old Tau-P301L mice, hypercapnia had opposite effects on airflow and chest SA, further depressing the airflow SA by 27% (61 ± 10 a.u.; p < 0.001) and further increasing the chest SA by 31% (130 ± 9 a.u.; p < 0.001). Therefore, the ratio airflow/chest SA of old Tau-P301L mice, which was already weak under normocapnia, was further reduced under hypercapnia (0.5 ± 0.1 a.u.; p < 0.01) (Fig. 2D3). The observed mismatch between airflow and chest SA in old Tau-P301L during both normocapnia and hypercapnia provides strong evidence for an increased work load of inspiratory pump muscles (e.g., diaphragm) against a potentially constricted upper airway, resulting in a significantly reduced airflow.
Figure 2.

Double-chamber plethysmographic recordings in conscious WT and Tau-P301L mice. A, Schematic representation of the experimental setup used for simultaneous recordings of chest and airflow spirograms in conscious but restrained mouse placed in a 50 ml recording chamber (to be compared with the experimental setup illustrated in Fig. 1A, in which the mouse is not restrained). B, C, Individual traces show chest and airflow spirograms (inspiration upward) recorded from 8-month-old WT (B) and Tau-P301L (C) mice placed under control (B1, C1) (air; room air, 0.03% CO2) and hypercapnic (B2, C2) (CO2; 4% CO2 in air, 5 min) breathing conditions. SA values are expressed in arbitrary units (vertical calibration bars, 50 a.u.). Note that hypercapnia increased both chest SA and airflow SA in the WT mouse (B), whereas hypercapnia increased chest SA but reduced airflow SA in the Tau-P301L mouse (C). D, The bars in the histograms show the mean (and SEM) values of chest SA (D1), airflow SA (D2), and ratio airflow versus chest SA (D3) obtained from double-plethysmographic recordings in 7- to 8-month-old WT (n = 6) and Tau-P301L (n = 6) mice placed under control (air, white bars) and hypercapnic (CO2, black bars) breathing conditions. The asterisk (*) indicated a significant difference between WT and Tau-P301L mice placed under identical breathing conditions (either air or CO2) (*p < 0.05; **p < 0.01; ***p < 0.001). The dagger (†) indicated a significant difference in control versus hypercapnic conditions between either paired WT or paired Tau-P301L mice (†p < 0.05; ††p < 0.01). Note chest SA was significantly larger in Tau-P301L than WT mice under both breathing conditions (D1), whereas airflow SA was significantly smaller in Tau-P301L than WT mice under both breathing conditions (D2). D3 shows that the ratio airflow/chest SA was significantly lower in Tau-P301L than WT mice under air and was further lowered by hypercapnia.
In young WT (n = 6) and Tau-P301L (n = 6) mice, double-chamber plethysmographic approach revealed identical ratio airflow/chest SA during normocapnia (1.1 ± 0.2 vs 1.1 ± 0.1 a.u.; NS) and during hypercapnia (1.1 ± 0.1 vs 1.1 ± 0.1 a.u.; NS). However, under the experimental condition of restrained double plethysmography, the mean fR was found significantly higher in Tau-P301L than WT mice (256 ± 11 vs 204 ± 8 c min−1, respectively; p < 0.05), a result possibly suggesting a greater sensitivity to immobilization stress in Tau-P301L than WT mice.
Pathophysiological changes of respiratory-related upper airway activity in 7- to 8-month-old Tau-P301L mice (in situ approach)
We analyzed the three-phase motor pattern (inspiration, I; postinspiration, PI; late expiration, l-E) of breathing in WT (n = 4) and Tau-P301L mice (n = 6) to reveal significant differences in the respiratory motor pattern determined from PNA and cVNA (Fig. 3). Experimental baseline condition (5% CO2) revealed a significant shortening of the expiratory interval (TE, postinspiration plus late expiration) because of a significant reduction of postinspiratory phase duration (TPI, 383 ± 177 vs 141 ± 39 ms; p < 0.05) and integrated PI-cVNA in Tau-P301L mice (33.5 ± 16.5 vs 3.5 ± 1.5 μV * 100 ms; p < 0.05). No significant differences in inspiratory duration (TI, 357 ± 93 vs 479 ± 87 ms; NS) and integrated I-VNA (16.7 ± 8.0 vs 8.4 ± 2.6 μV * 100 ms; NS), late expiratory phase duration (359 ± 77 vs 485 ± 145 ms; NS) were observed between WT and Tau-P301L mice. The phrenic burst frequency was also the same in Tau-P301L and WT mice (54.6 ± 11 vs 54.6 ± 18 c min−1; NS). The significantly reduced postinspiratory motor discharge in Tau-P301L mice was further depressed when excitatory drive and arousal in the ponto-medullary respiratory network was enhanced with hypercapnic challenge evoked by gassing the perfusate with 12% CO2 (Fig. 3C,D). In WT mice, an enhanced integrated I-VNA (16.7 ± 8.0 to 29.1 ± 18.5 μV * 100 ms) was observed, whereas PI-VNA remained constant compared with baseline (33.5 ± 16.5 vs 28.4 ± 14.4 μV * 100 ms). In contrast, in mutant Tau-P301L mice, a massive increase in integrated I-VNA (8.4 ± 2.6 to 40.0 ± 11.3 μV * 100 ms; p < 0.05) compared with baseline was observed, whereas integrated PI-VNA was almost abolished (3.5 ± 1.5 vs 0.6 ± 0.15 μV * 100 ms; p < 0.05).
The peak discharge of cVNA occurs physiologically in the transition from inspiration to expiration and is tightly linked to the activation of laryngeal adductor muscles, which breaks the expiratory airflow and counteracts elastic lung recoil after lung inflation (for review, see Dutschmann and Paton, 2002a). To further classify the changes in cVNA, we analyzed the occurrence of peak cVNA in relation to the inspiratory off-switch determined from the abrupt cessation of PNA (Fig. 3E). The cVNA peak discharge in WT always occurred with the termination of the inspiratory PNA discharge in normocapnia (5% CO2) and hypercapnia (12% CO2), thus matching the physiological role of expiratory airflow breaking. In contrast, in Tau-P301L mice, cVNA peak discharge occurred during the late inspiratory phase in normocapnia and was shifted deeper into the inspiratory phase during hypercapnia (Fig. 3E). The shift of the peak discharge of cVNA in Tau-P301L mice suggested paradoxical laryngeal constrictor activation during inspiration causing pathological inspiratory flow obstruction (Dutschmann and Paton 2002a,b). Therefore, in situ preparations of 7- to 8-month-old Tau-P301L mice revealed a paradoxical discharge pattern in laryngeal motor nerves, which could explain the upper airway dysfunction and breathing abnormalities suggested by the plethysmographic approaches.
Another difference between WT and Tau-P301L mice was observed in the response to hypercapnia. WT mice showed a physiological response with a subtle increase in PNA burst frequency from 55.1 ± 3.5 to 61.8 ± 7.4 c min−1 (NS) and significant increase in PNA peak discharge from 124 ± 19 to 184 ± 34 μV (p < 0.05). In contrast, Tau-P301L mice responded with a significant decrease in PNA burst frequency from 56.1 ± 4.8 to 37.9 ± 4.8 c min−1 (p < 0.05) (Fig. 3) accompanied by an increase in PNA peak discharge from 75 ± 19 to 139 ± 34 μV (p < 0.05).
Expression pattern of phosphorylated Tau in brainstem
Tau-P301L mice develop a clinical phenotype around age 7–8 months involving a progressive motor deficiency, most easily identified by hindlimb and later forelimb clasping. The mice die prematurely around age 9–10 months and none survive for >12 months of age (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) (Terwel et al., 2005, 2008). The lethality correlates with significant brainstem tauopathy, exemplified by the number of MC1-positive neurons (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). MC1 is a monoclonal antibody that defines a conformational form of Tau that is typical for tauopathy, including AD (Weaver et al., 2000). Although the initial analysis suggested that the premature death of Tau-P301L mice relates to brainstem tauopathy, neither the precise location of Tau aggregates in the brainstem nor their physiological consequences have been investigated previously.
In the present study, we performed detailed histochemical analysis of the expression of protein Tau phospho-epitopes AT8 and PG5, widely accepted as pretangle and pathological epitopes, respectively (Figs. 4–7). Accumulated hyperphosphorylated Tau and tau aggregates were revealed in very distinct patterns in the brainstem and midbrain substructures of mutant Tau-P301L mice at age 7–8 months (summarized in Table 1, Fig. 4). Immunohistochemistry revealed typically stained somata in specific brain nuclei, whereas in some brain nuclei also labeling of putative nerve terminals was detected (for details, see below). Staining for epitope AT8 was more pronounced in all analyzed brain areas compared with PG5 and MC1 epitopes, which fits the definition of the AT8 epitope as “pretangle” (i.e., on the borderline between physiology and pathology) (Terwel et al., 2005, 2008, and references therein). Tauopathy was completely absent in the CNS of nontransgenic mice at any age (data not shown) and in brain of young Tau-P301L mice, analyzed here and previously (Terwel et al., 2005, 2008; Boekhoorn et al., 2006).
Figure 4.
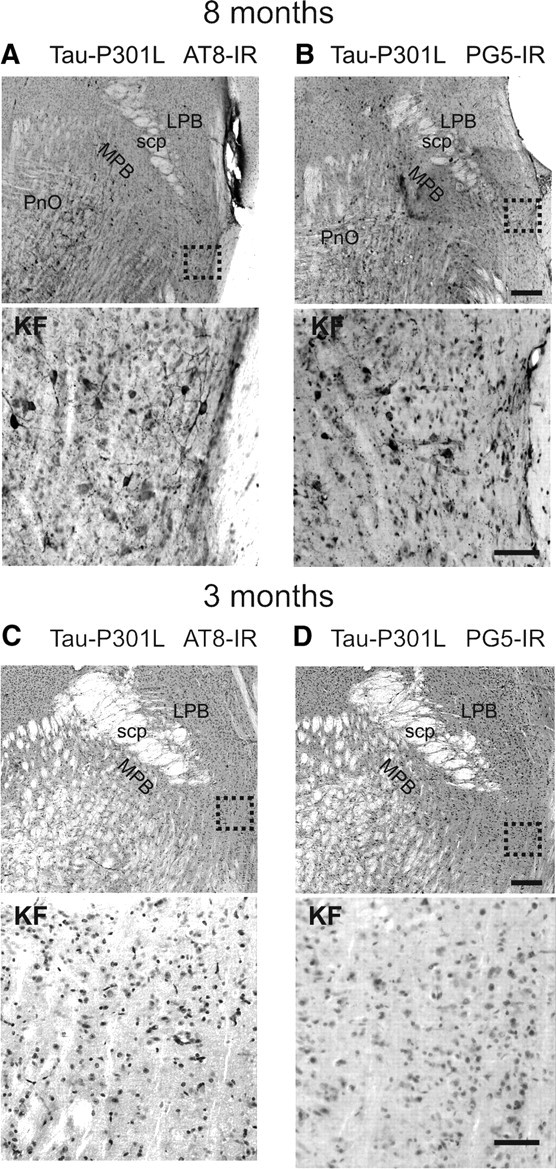
Tauopathy in the Kölliker–Fuse nucleus. The photomicrographs in A–D illustrate staining for phospho-epitopes AT8 and PG5 in the KF nucleus at 8 and 3 months of age. Scale bars: low magnification, 300 μm; high magnification, 50 μm. Abbreviations: LPB, Lateral parabrachial nuclei; MPB, medial parabrachial nucleus; PnO, pontine reticular nucleus, oral part; scp, superior cerebellar peduncle.
Figure 5.

Relative intensity of tauopathy in brainstem and midbrain nuclei by immunohistochemistry with phospho-tau epitopes AT8 and PG5. Schematic rendering of the location of tauopathy [i.e., neurons containing NFTs within the caudal midbrain and ponto-medullary brainstem]. A–I, Sections from the caudal medulla oblongata to the caudal midbrain. The gray-shaded areas with black diamonds (NFTs) and points (AT8- and PG-5-IR staining of terminal-like structures) symbolize dense staining. 7, Facial nerve; 7N, facial nucleus; XII, hypoglossal nucleus; A5, A5 noradrenaline cells; Aq, aqueduct; C1, C1 adrenaline cells; Cu, cuneate nucleus; Dll, dorsal nucleus of the lateral lemniscus; PDTg, posterodorsal tegmental nucleus; DR, dorsal raphe nucleus; Gi, gigantocellular reticular nucleus; IC, inferior colliculus; IRt, intermediate reticular nucleus; KF, Kölliker–Fuse nucleus; LC, locus ceruleus; LPB, lateral parabrachial nuclei, LRt, spinal trigeminal nucleus, caudal part; LSO, lateral superior olive; Mo5, motor trigeminal nucleus; MPB, medial parabrachial nucleus; PAG, periaqueductal gray; PnC, pontine reticular nucleus, caudal part; PnO, pontine reticular nucleus, oral part; Pr5, principal trigeminal nucleus; RMg, raphe magnus nucleus; ROb, raphe obscurus nucleus; RPa, raphe pallidus nucleus; RVLM, rostral ventrolateral medulla oblongata; scp, superior cerebellar peduncle; SolC, nucleus of the solitary tract, commissural part; SolM, nucleus of the solitary tract, medial part; SolVL, nucleus of the solitary tract, ventrolateral part; Sp5C, spinal trigeminal nucleus, caudal part; Sp5I, spinal trigeminal nucleus, interpolar part; Sp5O, spinal trigeminal nucleus, oral part; LVe, lateral vestibular nucleus.
Figure 6.
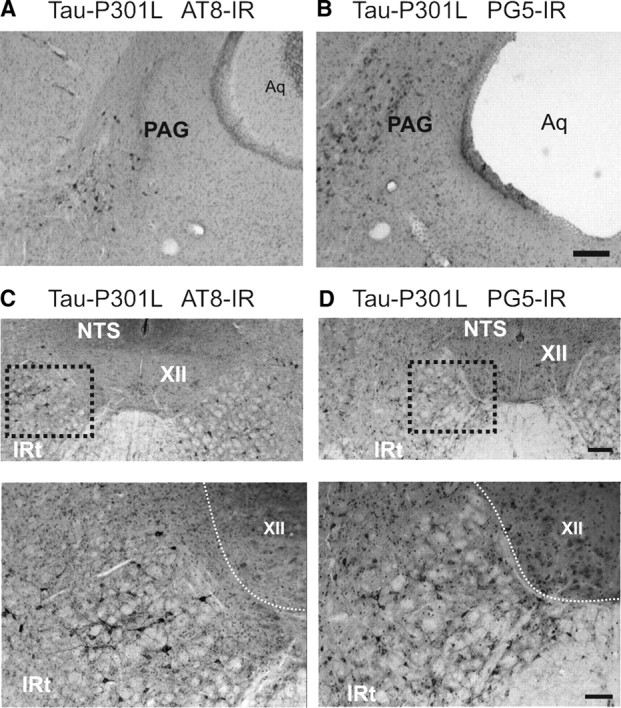
Tauopathy in lateral periaqueductal gray and intermediate reticular nucleus. The photomicrographs in A and B illustrate staining for phospho-epitopes AT8 and PG5 in the lateral periaqueductal gray (PAG) of Tau-P301L mouse. The photomicrographs in C and D illustrate staining in a Tau-P301L mouse for phospho-epitopes AT8 and PG5 in the intermediate reticular nucleus (IRt), which is located ventrolateral to the nucleus of the solitary tract (NTS) and lateral to hypoglossal nucleus (XII). Scale bars: low magnification, 300 μm; high magnification, 50 μm. Aq, Aqueduct.
Figure 7.
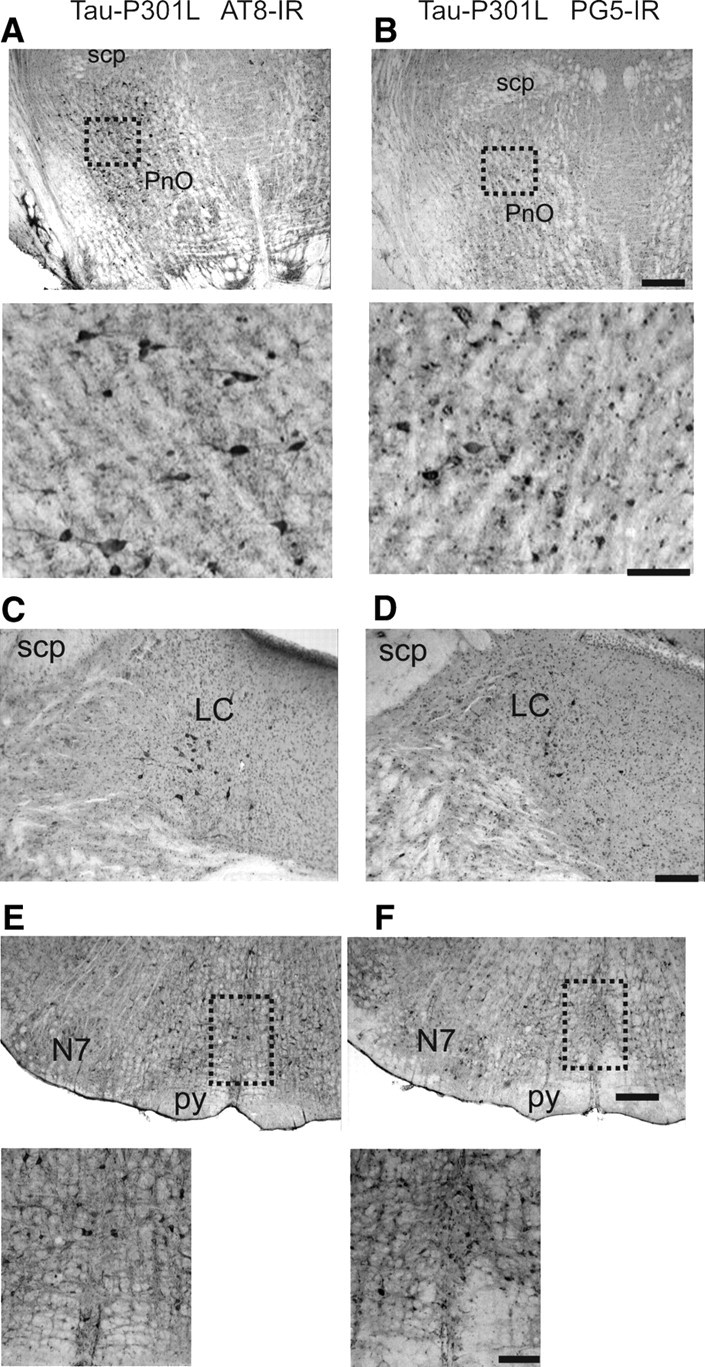
Tauopathy in pontine reticular nucleus, locus ceruleus, and raphe obscurus. A and B show AT8 and PG5 immunoreactivity (IR) in the dorsal aspect of the oral part of pontine reticular nucleus (PnO). Scale bars: low magnification, 300 μm; high magnification, 100 μm. The photomicrographs in C and D illustrate staining for phospho-epitopes AT8 and PG5 in the locus ceruleus (LC) of Tau-P301L mouse. E and F show the same immunoreactivity (IR) in the raphe obscurus nucleus (ROb), mediolateral to the facial nucleus (7N). Scale bars: low magnification, 300 μm; high magnification, 100 μm. scp, Superior cerebellar peduncle; py, pyramidal tract.
Table 1.
Summary of the relative density of tauopathy, based on phospho-tau epitopes AT8 and PG5, within neurons of brainstem and midbrain nuclei
| Brain nuclei | Wild typeAT8/PG5 | Tau-P301L |
|
|---|---|---|---|
| AT8 | PG5 | ||
| Nuclei of the medullary and pontinereticular formation | |||
| LRt | Ø | + | ++ |
| CVLM | Ø | Ø | Ø |
| Gi | Ø | +++ | |
| RVLM | Ø | Ø | Ø |
| C1 | Ø | Ø/+ | Ø |
| Pre-BötC | Ø | Ø | Ø |
| BötC | Ø | Ø/+ | Ø/+ |
| pFRG/RTN | Ø | Ø | Ø |
| A5 | Ø | + | + |
| PnC | Ø | +++ | +++ |
| ITR | Ø | + | Ø/+ |
| KF | Ø | ++ | ++ |
| LPB | Ø | Ø/+ | Ø/+ |
| MPB | Ø | Ø/+ | Ø/+ |
| PnO | Ø | +++ | +++ |
| IRt | Ø | +++ | +++ |
| NRA | Ø | Ø/+ | Ø/+ |
| Nuclei of the arousal system | |||
| RPa | Ø | + | + |
| ROb | Ø | +++ | +++ |
| RMg | Ø | ++ | ++ |
| Dorsal raphe | Ø | Ø | Ø |
| LC | Ø | ++ | + |
| Cranial motor nuclei | |||
| Amb | Ø | Ø/+ | Ø/+ |
| Mo5 | Ø | Ø/+ | Ø/+ |
| 7N | Ø | Ø/+ | + |
| 10N | Ø | Ø/+ | Ø |
| 12N | Ø | + | + |
| Cerebellum | |||
| Deep | Ø | +++ | ++ |
| Other cerebellar nuclei | Ø | Ø | Ø |
| Midbrain nuclei | |||
| PAG | Ø | +++ | +++ |
| Visceral and somatic sensory relay nuclei | |||
| Sp5C | Ø | Ø | Ø |
| Sp5O | Ø | Ø | Ø |
| Sp5I | Ø | Ø/+ | |
| SolVL | Ø | + (only terminals) | + |
| SolM | Ø | + | Ø/+ |
| SolC | Ø | Ø/+ | Ø/+ |
| Cu | Ø | ++ | + |
| IO | Ø | Ø/+ | Ø/+ |
| LSO | Ø | Ø/+ | Ø |
| IC | Ø | Ø | Ø |
| NC | Ø | Ø | Ø |
| Ve | Ø | ++ | + |
| Cervical and thoracic spinal cord | |||
| Dorsal horn | Ø | Ø | Ø |
| Ventral horn | Ø | +++/++ | ++ |
+++, Very dense; ++, dense; +, moderate; Ø, no tauopathy. For details, see Materials and Methods. Abbreviations (Paxinos and Franklin, 2001) in alphabetical order are as follows: 7N, facial nucleus; 10N, dorsal motor nucleus of vagus; 12N, hypoglossal nucleus; A5, A5 noradrenaline cells; Amb, ambiguus nucleus; BötC, Bötzinger complex; C1, C1 adrenaline cells; Cu, cuneate nucleus; CVLM, caudal ventrolateral medulla oblongata; Deep, lateral and interposed cerebellar nucleus; Gi, gigantocellular reticular nucleus; IC, inferior colliculus; IO, inferior olive; IRt, intermediate reticular nucleus; ITR, intertrigeminal region; KF, Kölliker–Fuse nucleus; LC, locus ceruleus; LPB, lateral parabrachial nuclei; LRt, spinal trigeminal nucleus, caudal part; LSO, lateral superior olive; Mo5, motor trigeminal nucleus; MPB, medial parabrachial nucleus; NC, cochlear nucleus; NRA, nucleus retroambiguus; PAG, periaqueductal gray; pFRG/RTN, parafacial respiratory group/retro trapezoid nucleus; PnC, pontine reticular nucleus, caudal part; PnO, pontine reticular nucleus, oral part; pre-BötC, pre-Bötzinger complex; RMg, raphe magnus nucleus; Rob, raphe obscurus nucleus; RPa, raphe pallidus nucleus; RVLM, rostral ventrolateral medulla oblongata; SolC, nucleus of the solitary tract, commissural part; SolM, nucleus of the solitary tract, medial part; SolVL, nucleus of the solitary tract, ventrolateral part; Sp5C, spinal trigeminal nucleus, caudal part; Sp5I, spinal trigeminal nucleus, interpolar part; Sp5O, spinal trigeminal nucleus, oral part; Ve, vestibular nuclei.
Tauopathy in the pontineKölliker–Fuse nucleus
Closely correlating with the upper airway phenotype of Tau-P301L mice was the accumulation of hyperphosphorylated tau and tau aggregates in the KF of Tau-P301L mice at age 7–8 months (Fig. 4A,B). The KF nucleus is known to specifically gate postinspiratory laryngeal adductor activity in the respiratory motor pattern of rodents (Dutschmann and Herbert, 2006). In addition, at age 3 months, no indications for tauopathy were observed in the KF of Tau-P301L mice (Fig. 4C,D). Because of the complete absence of tauopathy in WT mice, quantification of tauopathy-positive neurons was performed only on the intermediate part of the KF in Tau-P301L mice at age 8 months. For the analysis, the presence of the external and internal lateral subnuclei of the lateral parabrachial complex was used as landmark. In Tau-P301L mice (8 months of age; n = 5), we found an average of 24 ± 5 KF neurons per 104 μm2 that expressed Tau phospho-epitope AT8, whereas the density of neuronal expression of the PG5 epitope was lower (20 ± 7 per 104 μm2).
Tauopathy in midbrain and ponto-medullary nuclei in old Tau-P301L mice
Both phospho-epitopes on protein tau defined by Mab AT8 and PG5 showed a strongly overlapping expression pattern in the midbrain, ponto-medullary brainstem (Fig. 5). In contrast, these and other Tau phospho-epitopes were completely absent in CNS of WT mice (Terwel et al., 2005, 2008). The intensity of the tauopathy in Tau-P301L mice was scored on a relative scale, based on the counted number of neurons expressing the indicated tau phospho-epitopes (for details, see Materials and Methods).
Midbrain
Tauopathy was most prominently present in the midbrain intermediate and caudal periaqueductal gray (PAG). Dense clusters of neurons containing tau aggregates were observed within the dorsal, dorsolateral, lateral, and ventrolateral columns of the PAG (Figs. 5H,I, 6A,B).
Ponto-medullary reticular formation
Tauopathy was dense in the oral and caudal parts of pontine reticular nucleus (PnO; PnC). Whereas expression in the PnO was more dorsal (Figs. 5H,I, 7A,B), staining in the PnC was predominantly in the ventral aspects involving reticular giant neurons (Fig. 5F). Heavily attained were the posterodorsal tegmental nucleus (PDTg) (Fig. 5F), the Kölliker–Fuse nuclei (KF) (Figs. 4, 5G,H) of the parabrachial complex (PB), whereas staining in the lateral or medial nuclei (LBP; MPB) was rare (Table 1). Some cells were labeled in the intertrigeminal region (ITR) and at the level of the A5 noradrenergic cell group (Fig. 5F). At the medullary level, AT8 and PG5 stained neurons predominantly in the mediolateral reticular formation, involving strongly the gigantocellular reticular nucleus (Gi) (Fig. 5C,D). Another particular dense cluster of tauopathy was observed in the dorsal intermediate reticular nucleus (IRt) (Figs. 5A,B, 6C,D). Finally, some somata were labeled in the lateral reticular nucleus, whereas moderate-to-dense labeling was evident in putative nerve terminals in this nucleus (Fig. 5A). Tauopathy was rare in the “classical” cardiorespiratory autonomic centers of the medulla oblongata [i.e., the caudal and rostral ventro-lateral medulla oblongata (CVLM; RVLM) including the parafacial respiratory group/retro trapezoid nucleus (pFRG/RTN), pre-Bötzinger (pre-BötC), Bötzinger complex (BötC), or nucleus retroambiguus (NRA) tauopathy was sparse] (Fig. 5B–D, Table 1).
Ponto-medullary arousal system
Dense tauopathy-laden neurons were evident in key nuclei of the ponto-medullary reticular arousal system. In the pons, a dense cluster of AT8- and PG5-positive neurons was located in the locus ceruleus (Figs. 5F, 7C,D), whereas the dorsal raphe nuclei were devoid of staining. In the medullary raphe nuclei, AT8- and PG5-labeled cells were most dominant in the raphe obscurus (Rob) (Figs. 5C–E, 7E,F) and raphe magnus (RMg), whereas in the raphe pallidus (RPa) fewer neurons were stained.
Cranial motor nuclei
In cranial motor nuclei such as the nucleus ambiguus (Amb), trigeminal motor nucleus (Mo5), facial vagal and hypoglossal motor nuclei (7N, 10N, 12N), tauopathy was generally sparse, although moderate labeling of putative nerve terminals was observed in the Mo5, 7N, and 12N (Fig. 5, Table 1). In only one Tau-P301L animal, dense staining for PG5 was observed in the 7N and 12N nuclei.
Visceral and somatic relay nuclei
In the caudal, interpolar, and oral part of the spinal trigeminal nucleus (Sp5C, Sp5I, Sp5O), tauopathy was absent, only occasionally evident in Sp5l (Fig. 5B,C). In the commissural, medial, and ventrolateral parts of the nucleus of the solitary tract (SolC, SolM, SolVL), moderate staining was observed in the SolM, whereas labeled terminal-like structures were observed in the SolVL (Fig. 5A,B). Medullary and pontine nuclei of the auditory system such as the cochlear nucleus (NC), lateral superior olive (LSO), and inferior colliculus (IC) were devoid of tauopathy. In contrast, tauopathy was dense in the vestibular and cuneate nuclei, whereas it was absent in the inferior olive (Table 1).
Cerebellum and spinal cord
The cerebellum of Tau-P301L mice was completely devoid of tauopathy, as described previously (Terwel et al., 2005, 2008). A dense cluster of tau-laden neurons was identified in deep cerebellar nuclei such as the lateral and interposed cerebellar nucleus (Table 1). At the level of the cervical and thoracic spinal cord, dense tauopathy, defined by AT8 or PG5 staining, was observed in the ventral horn (Table 1). It is important to note that the spinal cord at these levels contains crucial populations of respiratory motor neurons innervating the diaphragm and intercostal muscles. In contrast, the sensory relays in the dorsal horn showed no obvious tauopathy (Table 1).
Discussion
Postmortem analysis of AD patients reveals tauopathy, not only in limbic regions but also in midbrain and hindbrain regions (Parvizi et al., 1998; Rüb et al., 2001, 2002; Thai, 2002). The generation of transgenic mice that express mutant protein tau associated with frontotemporal dementia allows detailed investigation into the pathophysiological changes in brain functions in mouse models for tauopathy (Lewis et al., 2001; Oddo et al., 2003; Götz et al., 2004; Terwel et al., 2005, 2008). Here, we analyzed the transgenic mouse strain with neuron-specific, postnatal expression at near-physiological levels of mutant human Tau-P301L (Terwel et al., 2005, 2008; Boekhoorn et al., 2006). In old Tau-P301L mice, our study reveals a consistent pattern of expression Tau protein phospho-epitopes AT8 and PG5 in defined nuclei of midbrain pons and brainstem that are closely linked to the observed airway dysfunction and breathing deficits in vivo and in situ. In the many clinical types of primary and secondary tauopathies, vast regional differences are observed in the CNS structures that are affected. However, neither analyses of human patient nor animal models has so far yielded satisfactory explanations for the vast differential susceptibility of brain circuits for tau pathology.
Tauopathy in neural circuits associated with upper airway control
The breathing phenotype we detected in Tau-P301L mice strongly points toward a dysfunction in the laryngeal valve that dynamically controls upper airway resistance and respiratory airflow across the respiratory cycle and during various oropharyngeal behaviors. The data obtained from in situ preparations showed reduced postinspiratory activity recorded from the vagal nerve, which drives laryngeal adductor (constrictor) muscles under physiological conditions. The laryngeal adductors are important to break expiratory airflow, counteract elastic lung recoil after lung inflation, and regulate expiratory airflow, and are crucially involved in vocal–respiratory coupling (Dutschmann and Paton, 2002a; Dutschmann et al., 2004, 2008). The potential shift of postinspiratory laryngeal activity into inspiration and the reduced airflow accompanying increased chest movements fully support a paradoxical increase of airway resistance during inhalation in old Tau-P301L mice. The potential laryngeal dysfunction in old Tau-301L mice can be well correlated with tauopathy in specific brain circuits. The KF was reported to gate the postinspiratory motor output to laryngeal adductor (constrictor) muscles in the upper airways (Dutschmann and Herbert, 2006) and has also important functions in the mediation of inspiratory–expiratory phase switch, in the control of respiratory phase durations, and in vocal–respiratory coupling (Chamberlin and Saper, 1994; Dick et al., 1994; Lara et al., 2002; Alheid et al., 2004; Dutschmann et al., 2004; Dutschmann and Herbert, 2006; Ezure and Tanaka, 2006; Smotherman et al., 2006). Tauopathy is particularly observed in KF neurons and correlates well with the observed breathing phenotype in Tau-P301L mice, but the effects of tauopathy on synaptic function of KF neurons remain unknown. In addition, it has to be considered that the respiratory motor pattern in situ is generated in absence of relevant afferent inputs from the periphery. Under in vivo conditions, strength and duration of postinspiratory activity may be modulated by upper brain structures and by sensory feedback arising from pulmonary stretch receptors (Kubin et al., 2006). Interestingly, the terminal field of stretch receptor afferents within the ventrolateral nucleus of the solitary tract (NTS) revealed tauopathy in terminal-like structures. Thus, the observation of decreased postinspiratory activity in situ and airflow reduction in vivo is linked to tauopathy within NTS/KF axis, which has an essential role in the regulation of postinspiratory motor activity (for review, see Dutschmann et al., 2008).
Importantly, autopsy of brains from patients with AD revealed strong tauopathy in the medial parabrachial and KF nuclei (Figs. 2, 3) (Rüb et al., 2001) (note that the anatomical position of the KF overlaps with subpeduncular pigmented nucleus in this study). However, tauopathy in the NTS of AD patients is not reported yet. Our mouse data may connect the described brainstem tauopathy of AD patients to upper airway disorder, which is a common feature in various forms of dementias. For instance, progressive aphasia in frontotemporal dementia associated with downstream motor speech deficits (Hillert, 1999; Tolnay and Probst, 2002). In addition, postinspiratory activity is essential for laryngeal closure during swallowing to prevent aspiration of food into the airways (Gestreau et al., 2005). Indeed, dysphagia and aspiration pneumonia belong to the most serious complications, and eventual cause of death, in AD patients (Kalia, 2003).
Nevertheless, tauopathy in old Tau-P301L mice was widespread in midbrain, pons, and brainstem, and was also observed in respiratory motor neurons in cervical and thoracic spinal cord. Therefore, various other nuclei may also contribute to the upper airway phenotype observed in Tau-P301L mice. Of main interest here are the PAG, which strongly modulates breathing during behavioral tasks associated with emotion and particularly during vocalization (Davis et al., 1996; Jürgens, 2002; Subramanian et al., 2008), and the intermediate reticular nucleus in the medulla oblongata. The latter brain region contains a large population of hypoglossal premotor neurons involved in the control of tongue movements during oropharyngeal behaviors such as swallowing and vocalization (Gestreau et al., 2005). Thus, overlapping and closely interacting neural circuits for upper airway control are stressed by the tauopathy in old Tau-P301L mice.
In old Tau-P301L mice, in vivo and in situ studies did not show altered respiratory rhythmogenesis, and immunohistochemistry revealed that primary medullary respiratory centers such as the entire ventral respiratory column including the primary rhythm generating areas (e.g., parafacial respiratory group and pre-Bötzinger complex) were mostly devoid of tauopathy. We propose two explanations for these observations. First, dementia is not lethal per se and thus unaltered rhythmogenic circuits would make sense. Second, it was reported that Thy1 promoter used for the generation of Tau-P301L mice has limitations in targeting neurons in the ventral medulla (Winter et al., 2007), and therefore tauopathy did not develop in the ventral respiratory group for technical reasons.
Enhanced upper airway dysfunction during hypercapnia
Tauopathy was detected in brainstem areas such as the serotonergic medullary raphe nuclei, the noradrenergic locus ceruleus, and the A5 region, which principally belong to the arousal system (for discussion, see below), but have also major implications in central chemosensitivity and the state-dependent modulation of breathing (Bou-Flores et al., 2000; Nattie, 2001; Richter et al., 2003; Ballantyne et al., 2004; Hilaire et al., 2004; Richerson, 2004; Viemari et al., 2005; Zanella et al., 2008; Besnard et al., 2009). The tauopathy in these nuclei may contribute to the enhanced upper airway phenotype during exposure to hypercapnia in situ and in vivo. Our data provide evidence that activation of the respiratory network and associated arousal/chemosensitive systems during hypercapnia contribute to evoke paradoxical activation of laryngeal adductor muscles during the inspiratory phase, leading to inspiratory airflow limitation. This phenomenon was reported to occur specifically after blockade of brainstem glycinergic neurotransmission (Dutschmann and Paton, 2002a,b) and during severe hypoxia (Dutschmann and Paton, 2005). However, the verification of potentially reduced glycinergic inhibition within the ponto-medullary brainstem of Tau-P301L mice requires future experimental analysis.
Finally, the described upper airway dysfunction with reduced respiratory airflow versus enlarged chest muscular work could trigger spells of systemic hypoxia. In turn, hypoxia is considered to contribute (Zhang et al., 2008) or even trigger AD (Peers et al., 2007). Therefore, the onset of breathing disorders most likely aggravates neurodegenerative processes in the brains of Tau-P301L mice, causing spread of tauopathy to forebrain structures, as previously reported (Terwel et al., 2005, 2008).
Summary
Aged Tau-P301L mice show defined patterns of tauopathy in vital brain circuits in midbrain, pons, brainstem, and spinal cord. The present study identified breathing deficits in older Tau-P301L mice, which strongly connect to tauopathy in defined neural circuits controlling upper airway function and to reported upper airway dysfunction in dementia patients. The observed tauopathy in other brain regions of Tau-P301L mice also correlates well with other dysfunctions of the autonomic nervous system described in dementia patients, which begs for additional experimental analysis.
Footnotes
This work was supported by the Bernstein Center for Computational Neurosciences, Göttingen, Grant 01GQ0432 (M.D.) and by Fonds Wetenschappelijk Onderzoek–Vlaanderen, Katholieke Universiteit (KU) Leuven Research Fund, and KU Leuven R&D. We are most grateful to Peter Davies (Litwin-Zucker Center for Research on Alzheimer's Disease, Feinstein Institute for Medical Research, Manhasset NY) and Eugeen Van Mechelen (Innogenetics, Gent, Belgium) for generously providing us with antibodies.
References
- Alheid GF, Milsom WK, McCrimmon DR. Pontine influences on breathing: an overview. Respir Physiol Neurobiol. 2004;143:105–114. doi: 10.1016/j.resp.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Andorfer CA, Davies P. PKA phosphorylations on tau: developmental studies in the mouse. Dev Neurosci. 2000;22:303–309. doi: 10.1159/000017454. [DOI] [PubMed] [Google Scholar]
- Ballantyne D, Andrzejewski M, Mückenhoff K, Scheid P. Rhythms, synchrony and electrical coupling in the locus coeruleus. Respir Physiol Neurobiol. 2004;143:199–214. doi: 10.1016/j.resp.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Besnard S, Denise P, Cappelin B, Dutschmann M, Gestreau C. Stimulation of the rat medullary raphe nuclei induces differential responses in respiratory muscle activity. Respir Physiol Neurobiol. 2009;165:208–214. doi: 10.1016/j.resp.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, Ramakers GJ, de Vos K, Krugers H, Tomiyama T, Mori H, Joels M, van Leuven F, Lucassen PJ. Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci. 2006;26:3514–3523. doi: 10.1523/JNEUROSCI.5425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou-Flores C, Lajard AM, Monteau R, De Maeyer E, Seif I, Lanoir J, Hilaire G. Abnormal phrenic motoneuron activity and morphology in neonatal monoamine oxidase A-deficient transgenic mice: possible role of a serotonin excess. J Neurosci. 2000;20:4646–4656. doi: 10.1523/JNEUROSCI.20-12-04646.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Chamberlin NL, Saper CB. Topographic organization of respiratory responses to glutamate microstimulation of the parabrachial nucleus in the rat. J Neurosci. 1994;14:6500–6510. doi: 10.1523/JNEUROSCI.14-11-06500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PJ, Zhang SP, Winkworth A, Bandler R. Neural control of vocalization: respiratory and emotional influences. J Voice. 1996;10:23–38. doi: 10.1016/s0892-1997(96)80016-6. [DOI] [PubMed] [Google Scholar]
- Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging. 2009;30:1–13. doi: 10.1016/j.neurobiolaging.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick TE, Bellingham MC, Richter DW. Pontine respiratory neurons in anesthetized cats. Brain Res. 1994;636:259–269. doi: 10.1016/0006-8993(94)91025-1. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Herbert H. The Kölliker-Fuse nucleus gates the post-inspiratory phase of the respiratory cycle to control inspiratory off-switch and upper airway resistance in rat. Eur J Neurosci. 2006;24:1071–1084. doi: 10.1111/j.1460-9568.2006.04981.x. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Paton JF. Inhibitory synaptic mechanisms regulating upper airway patency. Respir Physiol Neurobiol. 2002a;131:57–63. doi: 10.1016/s1569-9048(02)00037-x. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Paton JF. Glycinergic inhibition is essential for co-ordinating cranial and spinal respiratory motor outputs in the neonatal rat. J Physiol. 2002b;543:643–653. doi: 10.1113/jphysiol.2001.013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutschmann M, Paton JF. Dynamic changes in glottal resistance during exposure to severe hypoxia in neonatal rats in situ. Pediatr Res. 2005;58:193–198. doi: 10.1203/01.PDR.0000169968.07488.AD. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Mörschel M, Kron M, Herbert H. Development of adaptive behaviour of the respiratory network: implications for the pontine Kolliker-Fuse nucleus. Respir Physiol Neurobiol. 2004;143:155–165. doi: 10.1016/j.resp.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Mörschel M, Reuter J, Zhang W, Gestreau C, Stettner GM, Kron M. Postnatal emergence of synaptic plasticity associated with dynamic adaptation of the respiratory motor pattern. Respir Physiol Neurobiol. 2008;164:72–79. doi: 10.1016/j.resp.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Ezure K, Tanaka I. Distribution and medullary projection of respiratory neurons in the dorsolateral pons of the rat. Neuroscience. 2006;141:1011–1123. doi: 10.1016/j.neuroscience.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Gestreau C, Dutschmann M, Obled S, Bianchi AL. Activation of XII motoneurons and premotor neurons during various oropharyngeal behaviors. Respir Physiol Neurobiol. 2005;147:159–176. doi: 10.1016/j.resp.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F. Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry. 2004;9:664–683. doi: 10.1038/sj.mp.4001508. [DOI] [PubMed] [Google Scholar]
- Hilaire G, Viemari JC, Coulon P, Simonneau M, Bévengut M. Modulation of the respiratory rhythm generator by the pontine noradrenergic A5 and A6 groups in rodents. Respir Physiol Neurobiol. 2004;143:187–197. doi: 10.1016/j.resp.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Hillert D. On processing lexical meanings in aphasia and Alzheimer's disease: some (re)considerations. Brain Lang. 1999;69:95–118. doi: 10.1006/brln.1999.2053. [DOI] [PubMed] [Google Scholar]
- Janus C. Conditionally inducible tau mice—designing a better mouse model of neurodegenerative diseases. Genes Brain Behav. 2008;7(Suppl 1):12–27. doi: 10.1111/j.1601-183X.2007.00375.x. [DOI] [PubMed] [Google Scholar]
- Jürgens U. Neural pathways underlying vocal control. Neurosci Biobehav Rev. 2002;26:235–258. doi: 10.1016/s0149-7634(01)00068-9. [DOI] [PubMed] [Google Scholar]
- Kalia M. Dysphagia and aspiration pneumonia in patients with Alzheimer's disease. Metabolism. 2003;52(Suppl 2):36–38. doi: 10.1016/s0026-0495(03)00300-7. [DOI] [PubMed] [Google Scholar]
- Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol. 2006;101:618–627. doi: 10.1152/japplphysiol.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara JP, Dawid-Milner MS, López MV, Montes C, Spyer KM, González-Barón S. Laryngeal effects of stimulation of rostral and ventral pons in the anaesthetized rat. Brain Res. 2002;934:97–106. doi: 10.1016/s0006-8993(02)02364-8. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Nattie EE. Central chemosensitivity, sleep, and wakefulness. Respir Physiol. 2001;129:257–268. doi: 10.1016/s0034-5687(01)00295-x. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Van Hoesen GW, Damasio A. Severe pathological changes of parabrachial nucleus in Alzheimer's disease. Neuroreport. 1998;9:4151–4154. doi: 10.1097/00001756-199812210-00028. [DOI] [PubMed] [Google Scholar]
- Paton JFR. A working heart-brainstem preparation of the mouse. J Neurosci Methods. 1996;65:63–68. doi: 10.1016/0165-0270(95)00147-6. [DOI] [PubMed] [Google Scholar]
- Peers C, Pearson HA, Boyle JP. Hypoxia and Alzheimer's disease. Essays Biochem. 2007;43:153–164. doi: 10.1042/BSE0430153. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci. 2004;5:449–461. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- Richter DW, Manzke T, Wilken B, Ponimaskin E. Serotonin receptors: guardians of stable breathing. Trends Mol Med. 2003;9:542–548. doi: 10.1016/j.molmed.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Rüb U, Del Tredici K, Schultz C, Thal DR, Braak E, Braak H. The autonomic higher order processing nuclei of the lower brain stem are among the early targets of the Alzheimer's disease-related cytoskeletal pathology. Acta Neuropathol. 2001;101:555–564. doi: 10.1007/s004010000320. [DOI] [PubMed] [Google Scholar]
- Rüb U, Del Tredici K, Schultz C, de Vos RA, Jansen Steur EN, Arai K, Braak H. Progressive supranuclear palsy: neuronal and glial cytoskeletal pathology in the higher order processing autonomic nuclei of the lower brainstem. Neuropathol Appl Neurobiol. 2002;28:12–22. doi: 10.1046/j.0305-1846.2001.00374.x. [DOI] [PubMed] [Google Scholar]
- Smotherman M, Kobayasi K, Ma J, Zhang S, Metzner W. A mechanism for vocal-respiratory coupling in the mammalian parabrachial nucleus. J Neurosci. 2006;26:4860–4869. doi: 10.1523/JNEUROSCI.4607-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettner GM, Huppke P, Brendel C, Richter DW, Gärtner J, Dutschmann M. Breathing dysfunctions associated with impaired control of post-inspiratory activity in Mecp2−/y knockout mice. J Physiol. 2007;579:863–876. doi: 10.1113/jphysiol.2006.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian HH, Balnave RJ, Holstege G. The midbrain periaqueductal gray control of respiration. J Neurosci. 2008;28:12274–12283. doi: 10.1523/JNEUROSCI.4168-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima A. Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J Alzheimers Dis. 2008;14:371–375. doi: 10.3233/jad-2008-14403. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci U S A. 2002;99:13896–13901. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. Neuromolecular Med. 2002;2:151–165. doi: 10.1385/NMM:2:2:151. [DOI] [PubMed] [Google Scholar]
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L mice underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F. Mutant APP and GSK-3β aggravate tauopathy similarly in cortex and hippocampus of tau bigenic mice. Am J Pathol. 2008;172:786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai DR. Excitatory amino acid transporter EAAT-2 in tangle-bearing neurons in Alzheimer's disease. Brain Pathol. 2002;12:405–411. doi: 10.1111/j.1750-3639.2002.tb00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolnay M, Probst A. Frontotemporal lobar degeneration—tau as a pied piper? Neurogenetics. 2002;4:63–75. doi: 10.1007/s10048-002-0140-x. [DOI] [PubMed] [Google Scholar]
- Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Peña F, Zanella S, Bévengut M, Barthelemy-Requin M, Herzing LB, Moncla A, Mancini J, Ramirez JM, Villard L, Hilaire G. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–727. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Winter SM, Hirrlinger J, Kirchhoff F, Hülsmann S. Transgenic expression of fluorescent proteins in respiratory neurons. Respir Physiol Neurobiol. 2007;159:108–114. doi: 10.1016/j.resp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Zanella S, Watrin F, Mebarek S, Marly F, Roussel M, Gire C, Diene G, Tauber M, Muscatelli F, Hilaire G. Necdin plays a role in the serotonergic modulation of the mouse respiratory network: implication for Prader–Willi syndrome. J Neurosci. 2008;28:1745–1755. doi: 10.1523/JNEUROSCI.4334-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Dong Y, Zhang G, Moir RD, Xia W, Yue Y, Tian M, Culley DJ, Crosby G, Tanzi RE, Xie Z. The inhalation anesthetic desflurane induces caspase activation and increases amyloid beta-protein levels under hypoxic conditions. J Biol Chem. 2008;283:11866–11875. doi: 10.1074/jbc.M800199200. [DOI] [PMC free article] [PubMed] [Google Scholar]