

Abstract
Nav1.7 sodium channels can amplify weak stimuli in neurons and act as threshold channels for firing action potentials. Neurotrophic factors and pro-nociceptive cytokines that are released during development and under pathological conditions activate mitogen-activated protein kinases (MAPKs). Previous studies have shown that MAPKs can transduce developmental or pathological signals by regulating transcription factors that initiate a gene expression response, a long-term effect, and directly modulate neuronal ion channels including sodium channels, thus acutely regulating dorsal root ganglion (DRG) neuron excitability. For example, neurotrophic growth factor activates (phosphorylates) ERK1/2 MAPK (pERK1/2) in DRG neurons, an effect that has been implicated in injury-induced hyperalgesia. However, the acute effects of pERK1/2 on sodium channels are not known. We have shown previously that activated p38 MAPK (pp38) directly phosphorylates Nav1.6 and Nav1.8 sodium channels and regulates their current densities without altering their gating properties. We now report that acute inhibition of pERK1/2 regulates resting membrane potential and firing properties of DRG neurons. We also show that pERK1 phosphorylates specific residues within L1 of Nav1.7, inhibition of pERK1/2 causes a depolarizing shift of activation and fast inactivation of Nav1.7 without altering current density, and mutation of these L1 phosphoacceptor sites abrogates the effect of pERK1/2 on this channel. Together, these data are consistent with direct phosphorylation and modulation of Nav1.7 by pERK1/2, which unlike the modulation of Nav1.6 and Nav1.8 by pp38, regulates gating properties of this channel but not its current density and contributes to the effects of MAPKs on DRG neuron excitability.
Introduction
Nav1.7 is a threshold voltage-gated sodium channel (Rush et al., 2007) that plays a significant role in inherited pain disorders (Dib-Hajj et al., 2007). Mitogen-activated protein kinases (MAPKs) are transducers of cell signaling that have been implicated in neuronal responses to developmental cues and pathologies including pain (Ji and Strichartz, 2004; Ji et al., 2009). Recently, we have shown that Nav1.7 and Nav1.8 sodium channels and activated (phosphorylated) MAPKs (pERK1/2 and pp38), accumulate within blind-ending axons in painful human neuromas (Black et al., 2008), in agreement with previous findings of pERK1/2 in proximal axons after axonal injury (Sheu et al., 2000; Agthong et al., 2006). These data suggest possible modulation of these channels by MAPKs and is consistent with a pp38-dependent increase in current density of tetrodotoxin-resistant sodium currents contributing to DRG neuron hyperexcitability after acute treatment with tumor necrosis factor-α or interleukin-1β (Jin and Gereau, 2006; Binshtok et al., 2008). We have shown previously that pp38 directly phosphorylates Nav1.6 (Wittmack et al., 2005) and Nav1.8 (Hudmon et al., 2008) sodium channels and regulates their current densities. However, direct effects of pERK1/2 on voltage-gated sodium channels and DRG neuron firing are not well understood.
Activation of ERK1/2 by trophic factors in DRG neurons (Averill et al., 2001; Delcroix et al., 2003; Woodall et al., 2008) has been implicated in hyperalgesia (Ji et al., 1999; Dai et al., 2002; Obata et al., 2004). Acute nerve growth factor (NGF) application depolarizes PC12 membrane potential in a sodium-dependent manner (Shimazu et al., 2005), suggesting involvement of sodium conductances without de novo protein synthesis. Although Nav1.2 and Nav1.7 sodium channels are expressed in PC12 cells (Toledo-Aral et al., 1995), Nav1.2 is not detectable at significant levels in adult DRG neurons (Waxman et al., 1994; Black et al., 1996), pointing to Nav1.7 as a potential target of pERK1/2 in these neurons. However, molecular mechanisms underlying pERK1/2 putative effects on sodium channels are not known.
pERK1/2 has been shown to phosphorylate Cav2.2 channels (Martin et al., 2006) and regulate calcium currents (ICa2+) in DRG neurons (Fitzgerald and Dolphin, 1997; Fitzgerald, 2000; Woodall et al., 2008). NGF- and glial cell line-derived neurotrophic factor (GDNF)-induced acute activation of pERK1/2 in DRG neurons appears to regulate ICa2+ density (NGF and GDNF) and gating properties (NGF alone) in a developmental-specific manner, which suggests a complex pERK1/2 regulation of calcium channels in these neurons. pERK1/2 also phosphorylates Kv4.2 (Adams et al., 2000) and reduces the A-type potassium current in hippocampal neurons (Yuan et al., 2002). We report in this study that pERK1/2 regulates resting membrane potential and firing properties of adult DRG neurons. We also identify pERK1/2 phosphorylation sites within Nav1.7 L1 and show that acute inhibition of pERK1/2 induces a depolarizing shift of activation and fast inactivation of Nav1.7, without altering current density, and that this effect is abrogated by mutating pERK1/2 phosphoacceptor sites within L1 of the channel. Thus, unlike the pp38 effect on Nav1.6 and Nav1.8 current densities, pERK1/2 appears to specifically modulate gating properties of Nav1.7, an effect that may contribute to the role of this channel in DRG neuron excitability.
Materials and Methods
Reagents.
The rabbit polyclonal antibody against activated p44/42 MAPK (pERK1/2; Thr202/Tyr204) was purchased from Cell Signaling Technology. The mouse monoclonal antibody against total (nonphosphorylated and phosphorylated) ERK1/2 (TERK1/2) was purchased from Zymed, and the rabbit polyclonal antibody against TERK1/2 was purchased from Sigma-Aldrich. Rabbit polyclonal Nav1.7-specific antibody (Y083; 1:250; generated from the rat amino acid sequence 514–532) has been described previously (Black et al., 2008). The monoclonal antibody against PGP9.5 was purchased from Ultraclone. The secondary antibodies for Western blot analysis, anti-rabbit IgG and anti-mouse IgG, were purchased from Dako. The active ERK1 kinase was purchased from Millipore Corporation. The active MAP kinase kinase (MEK) inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] and its inactive analog U0124 [1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene] were purchased from Calbiochem. The [γ-32P] ATP was purchased from PerkinElmer Life and Analytical Sciences.
DRG culture.
The protocol for the care and sacrifice of rats used in the study was approved by the Veterans Administration Connecticut Healthcare system institutional animal care and use committee. Adult Sprague Dawley rats (1–2 months of age) were deeply anesthetized with ketamine/xylazine (80 and 10 mg/kg, i.p., respectively) and decapitated, L4 and L5 DRGs were quickly removed and desheathed, and DRG neurons were isolated using a protocol (Dib-Hajj et al., 2009b) that was adapted from Rizzo et al. (1994). Briefly, dissected ganglia were placed in ice-cold oxygenated complete saline solution (CSS) that contained the following (in mm): 137 NaCl, 5.3 KCl, 1 MgCl2, 25 sorbitol, 3 CaCl2, and 10 HEPES, pH 7.2. DRGs were then transferred to an oxygenated, 37°C CSS containing 1.5 mg/ml Collagenase A (Roche Applied Science) and 0.6 mm EDTA and incubated with gentle agitation at 37°C for 20 min. This solution was then exchanged with an oxygenated, 37°C CSS containing 1.5 mg/ml Collagenase D (Roche Applied Science), 0.6 mm EDTA, and 30 U/ml papain (Worthington Biochemical) and incubated with gentle agitation at 37°C for 20 min. The solution was then aspirated, and the ganglia were triturated in DRG media (DMEM/F-12 at 1:1) with 100 U/ml penicillin, 0.1 mg/ml streptomycin (Invitrogen), and 10% fetal calf serum (Hyclone), which contained 1.5 mg/ml bovine serum albumin (Sigma-Aldrich) and 1.5 mg/ml trypsin inhibitor (Roche Applied Science). Cells were then plated on poly-d-lysine/laminin-coated glass coverslips, flooded with DRG media after 30 min, and incubated at 37°C in a humidified 95% air–5% CO2 incubator.
Immunohistochemistry.
Rats were deeply anesthetized with ketamine/xylazine (80 and 10 mg/kg, i.p., respectively) and perfused with 4% paraformaldehyde in 0.14 m Sorensen's phosphate buffer, pH 7.4. L4 and L5 DRGs were harvested, rinsed in PBS, and cryoprotected overnight in 30% sucrose in PBS. Cryosections were incubated with rabbit polyclonal antibodies against TERK1/2 (Sigma-Aldrich) and pERK1/2 (1:1000; Cell Signaling Technology) and monoclonal antibody against PGP9.5 (1:1000; Ultraclone). Secondary antibodies were goat anti-rabbit cyanine 3 and goat anti-mouse-Alexa Fluor 488 (1:1000). For DRG cultures and DRG tissue, coverslips were incubated sequentially in the following: (1) blocking solution (PBS with 5% normal goat serum, 1% bovine serum albumin, 0.1% Triton X-100, and 0.02% sodium azide) for 30 min, (2) primary antibodies, (3) PBS wash, (4) goat anti-rabbit IgG Alexa Fluor 546 and goat anti-mouse IgG Alexa Fluor 488, and (5) PBS wash. Sections were examined with a Zeiss LSM510META confocal microscope or Nikon E800 light microscope. Images were collected with LSM 510 or Meta-Vue (Molecular Devices) software and were arranged with Adobe Photoshop (Adobe Systems).
Immunostaining and image analysis for DRG culture.
At designated times after treatments, DRG cultures were fixed with 4% paraformaldehyde, followed by ice-cold methanol. For examination of pERK1/2 and TERK1/2 and Nav1.7 expression, DRG cultures were blocked in 10% normal donkey serum/0.3% Triton X-100 and incubated with primary antibodies [mouse monoclonal TERK1/2 (1:500; Zymed), rabbit polyclonal pERK1/2 (1:500; Cell Signaling Technology), or rabbit polyclonal Nav1.7 (rabbit Y083; 1:250)]. The cells were then washed with PBS and incubated in secondary antibodies (donkey anti-mouse, Alexa Fluor 488; donkey anti-rabbit, Alexa Fluor 555). After washing, coverslips were mounted with anti-fade Gelmount (BioMeda). Images were taken using a Zeiss LSM 510 confocal microscope. The intensity of pERK1/2 signal in small DRG neurons (diameter of ∼20 μm) was quantitated using Zeiss LSM 510 image program (histogram analysis function). Briefly, an indicator line was drawn across the full diameter of the cell of interest to identify cell size and cover region for analysis. Mean pixel intensity (from 0 to 255) across the cell was calculated and presented in a histogram table. For analysis of increased numbers of DRG neurons with pERK1/2 activation after treatment with neurotrophic factors, we measured the intensity of the fluorescence in the green channel (TERK1/2) and in the red channel (pERK1/2). We considered cells positive for either TERK1/2 (green) or pERK1/2 (red), a fluorescence intensity that was ≥2.5× the average intensity of cells deemed negative by visual inspection [fluorescence intensity, ≤50 arbitrary units (AU)]. The fraction of green fluorescent cells (≥150 AU green) that manifested red fluorescence (≥150 AU red) represents small/medium DRG neurons with pERK1/2. For analysis of increased pERK1/2 intensity after treatment with neurotrophic factors, an average of red fluorescence intensity of 20 randomly selected small DRG neurons were measured for each condition.
Plasmids and HEK 293 cell transfections.
The plasmid carrying the TTX-resistant (TTX-R) version of human Nav1.7 cDNA (hNav1.7R) was described previously (Herzog et al., 2003). Human embryonic kidney cells (HEK 293) were transfected with channel constructs and β1 and β2 subunits (Lossin et al., 2002) using Lipofectamine 2000 (Invitrogen). Transfected HEK 293 cells were grown under standard culture conditions (5% CO2, 37°C) in 50% DMEM/50% F-12 supplemented with 10% fetal bovine serum.
For fusion protein production, DNA inserts encoding the N and C termini and the loops joining domains I and II (L1), domains II and III (L2), and domains III and IV (L3) were amplified by PCR using the Nav1.7 plasmid as a template and subcloned into the BamHI–EcoRI (for the N and C termini and L3) and the BamHI–SmaI (L2) sites of pGEX (GE Healthcare), whereas L1 was subcloned into the Nde1–Sap1 sites of pTXB1 plasmid (New England Biolabs). The plasmids are designated pGEX–Nav1.7–N (amino acids 1–121), pTXB1–Nav1.7–L1 (amino acids 405–718), pGEX–Nav1.7–L2 (amino acids 959–1175), pGEX–Nav1.7–L3 (amino acids 1446–1499), and pGEX–Nav1.7–C (amino acids 1749–1977). The C-terminal fusion of Intein to Nav1.7–L1 sequence (pTXB1–Nav1.7–L1 construct) resulted in a more stable fusion protein in bacteria compared with N-terminus fusion to glutathione S-transferase (GST) (data not shown). Amino acid substitutions were introduced using QuikChange XL site-directed mutagenesis reagents (Stratagene). The identity of all constructs was confirmed by sequencing the inserts (Howard Hughes Medical Institute/Keck Biotechnology Resource Laboratory at Yale University).
Recombinant protein expression and purification.
BL21 Rosetta competent cells (Novagen) were transformed with constructs encoding the GST-tag or Intein-tag fusion proteins and grown in the presence of ampicillin (100 μg/ml) and Chloramphenicol (34 μg/ml). Large-scale growths were inoculated using an overnight starter culture (1:100) and grown to an OD595 of 0.6 at 37°C in 2XYT media. Cell cultures were chilled on ice to reach 20°C, 0.5 mm isopropyl-d-thiogalactoside was added to the culture to induce recombinant protein synthesis, and the cells were then incubated with shaking for 4 h at 27°C for the GST fusion proteins and overnight at 12°C for the Intein fusion proteins. Bacterial pellets were collected by centrifugation at 6000 × g for 10 min at 4°C, washed in ice-cold PBS, and frozen at −80°C. Frozen bacterial pellets were resuspended in 20 ml of ice-cold lysis buffer [10 mm Tris, pH 8, 0.1 mm EDTA, 300 mm NaCl, 0.5% Triton X-100 supplemented with protease inhibitor mixture minus EDTA (Roche)]. Lysed cells were sonicated and incubated at 4°C for 30 min and then centrifuged at 12,000 × g for 20 min at 4°C. Supernatants with soluble GST fusion proteins were applied to glutathione-Sepharose beads (Invitrogen) for 2 h at 4°C. Supernatants with Intein fusion proteins were applied to Chitin Beads (New England Biolabs) overnight at 4°C. The columns were washed with 10 bed column volume of lysis buffer. GST fusion proteins were eluted with 20 mm glutathione. For Intein fusion protein purification, Chitin beads were incubated for 168 h in cleavage buffer (100 mm Tris, 300 mm NaCl, 2 m urea, 100 mm DTT, and 0.1% Tween 20), and proteins were eluted in the same buffer. Intein fusion proteins were desalted using PD10 columns (GE Healthcare) and concentrated using Amicon Ultra 4 concentrators (Millipore Corporation). Purified proteins were quantified using Bradford assay (BSA as standard), and equal proteins levels were visually inspected using Coomassie blue staining.
pERK1/2 kinase assay.
Kinase reactions were performed in 100 μl when proteins were bound to the beads or 50 μl when soluble proteins were used and contained 20 mm 3-(N-morpholino)-propanesulfonic acid (MOPS), 25 mm β-glycerophosphate, 5 mm EGTA, 75 mm MgCl2, 0.25 mm ATP, and 10 μCi of [γ-32P]ATP. The reaction mixture was warmed to 30°C for 2 min before the addition of 250 ng of pERK1. The reaction was allowed to proceed for an additional 10 min. The kinase reactions were terminated with 5 vol (1 ml) of PBS (0.14 m NaCl, 0.0027 m KCl, and 0.10 m phosphate buffer, pH 7.4) containing 20 mm EDTA. When proteins on beads were used as substrates, beads were collected by centrifugation and washed three times in PBS plus EDTA, and proteins were released from the beads using 2× lithium dodecyl sulfate–PAGE sample buffer (Invitrogen) and loaded on gels. For the gel analysis, 20 μl of samples was loaded and run on NUPAGE Bis–Tris 10% SDS-PAGE gel (Invitrogen). Gels were stained with Coomassie blue, destained, and placed in water for 24 h before drying. The dried gel was exposed to a Kodak x-ray film (Invitrogen) to visualize the phosphorylated bands by autoradiography, individual bands were then cut, and incorporated [32P]ATP was measured by Cerenkov count in a scintillation counter.
Western blot and autoradiography.
DRG or HEK 293 cells were grown under standard conditions and were untreated or treated with U0126 (ERK1/2 phosphorylation inhibitor), U0124 (inactive analog), NGF, or GDNF. Cells were then washed twice with PBS and resuspended in a MOPS-based buffer system as a sample buffer. Proteins were separated on NUPAGE Bis–Tris 4–12% gels (Invitrogen) and were processed on gels using MES as the running buffer (Invitrogen). Proteins were transferred to nitrocellulose membranes for 1 h at 30 V using the XCell II system (Invitrogen) and blocked overnight in TBST/10% nonfat milk buffer. The membrane was washed in TBST (Tris-buffered saline plus 0.1% Tween 20) four times for 5 min each after primary and secondary antibody incubations. Chemiluminescent detection was performed using Lightening Plus (PerkinElmer Life and Analytical Sciences) and detected on Biomax MR Kodak film. Gels for autoradiography were first stained with Coomassie blue, and the intensity of protein bands was measured to determine whether comparable levels of fusion proteins were used in the kinase assays.
Electrophysiological recordings.
Whole-cell current-clamp recordings from small-diameter (20–27 μm) DRG neurons were obtained at room temperature between 16 and 30 h after plating, using the MultiClamp 700B amplifier and the Digidata 1440A interface. Micropipettes with resistances from 1 to 2 MΩ were pulled from a capillary glass (PG10165-4; WPI) using the P97 puller (Sutter Instruments) and polished on a microforge. Pipette tips were wrapped with Parafilm to diminish pipette capacitance. The internal solution contained the following (in mm): 140 KCl, 1 EGTA, 10 NaCl, 2 Mg-ATP, and 10 HEPES, pH 7.3 (adjusted to 310 mOsm/L with sucrose). The external solution contained the following (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 dextrose, and 10 HEPES, pH 7.3 (adjusted to 315 mOsm/L with sucrose). Pipette potentials were adjusted to zero before seal formation, and liquid junction potentials were not corrected. Whole-cell configuration was obtained by adjusting pipette capacitance neutralization and bridge balance automatically in current-clamp mode. Action potentials were filtered at 10 kHz and acquired at 100 kHz, using Clampex 10.2 software (Molecular Devices). Cells with stable resting membrane potentials more negative than −40 mV were used for data analysis.
Wild-type (WT) hNav1.7R channels (Herzog et al., 2003) were cotransfected together with the human β1 and β2 subunits (Lossin et al., 2002) into HEK 293 cells using Lipofectamine 2000, and currents were recorded 24–36 h later. Whole-cell voltage-clamp recordings were obtained at room temperature (21–23°C) with Axopatch 200B amplifiers (Molecular Devices) using the following solutions: internal (in mm), 140 CsF, 1 EGTA, 10 NaCl, and 10 HEPES, pH 7.3 (adjusted to 310 mOsm/L with sucrose); external (in mm), 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 glucose, and 20 HEPES, pH 7.3 (adjusted to 320 mOsm/L with sucrose). Micropipettes (0.6–0.9 MΩ) were pulled from capillary glass (PG10165-4; WPI) with a Flaming Brown P80 micropipette puller (Sutter Instruments). The pipette potential was zeroed before seal formation; voltages were not corrected for liquid junction potential, and voltage errors were minimized using 90% series resistance compensation. Recordings were started 4–5 min after establishing whole-cell configuration to allow currents to stabilize. Currents were elicited from a holding potential of −120 mV, filtered at 5 kHz, and acquired at 50 kHz using pClamp 8.2 software. Only cells with series resistance <2 MΩ were studied.
Electrophysiological protocols and data analysis.
A stock solution of the active MEK inhibitor U0126 (Calbiochem) and the inactive analog U0124 (Calbiochem) were prepared in DMSO and added to the culture media and the recording chamber to produce a final concentration of 10 μm in 0.1% DMSO. To inactivate ERK1/2, transfected HEK293 cells were pretreated with 10 μm U0126 for 20 min; the inactive analog U0124 was used in control experiments. The media was then replaced with the bath solution in the presence of 10 μm U0126 or its inactive analog U0124, and electrophysiological recordings were performed.
Statistics.
Except when otherwise indicated, Student's unpaired t test was used with the criterion for statistical significance set at 0.05. For multigroup statistical analysis, the F statistic associated with ANOVA was used to test the null hypothesis of no differences between experimental conditions, and a post hoc Tukey's test was conducted to examine significance at the level of 0.05. Descriptive data are presented as mean ± SEM. The ANOVA and post hoc analyses and Student's t test were conducted using Origin7.5 (Microcal).
Results
ERK1/2 MAP kinases are coexpressed with Nav1.7 sodium channel in small DRG neurons
To investigate the possible modulation of Nav1.7 by ERK1/2, we first examined the expression of ERK1/2 in DRG neurons. Immunostaining of rat DRG sections (Fig. 1A) with anti-TERK1/2 antibody demonstrated that TERK1/2 (red) was expressed in small- and medium-sized PGP9.5-positve (neuronal marker) DRG neurons (green). TERK1/2 immunostaining was generally not observed in large DRG neurons. We also examined the co-localization of Nav1.7 and TERK1/2 in DRG neurons in culture. Figure 1B shows that Nav1.7-specific immunolabeling (red) colocalizes with immunolabeling of TERK1/2 in adult rat small- and medium-sized DRG neurons. Thus, we hypothesized that pERK1/2 may modulate Nav1.7 sodium channels and influence neuronal firing in small DRG neurons.
Figure 1.

ERK1/2 is widely expressed in DRG neurons and colocalized with Nav1.7 sodium channels. A, Immunofluorescence labeling of adult rat DRG sections probed with anti-TERK1/2 and anti-PGP9.5 antibodies. Merged image (yellow) shows that TERK1/2 (red) and PGP9.5 (green) immunofluorescent signals colocalize within small- and medium-diameter DRG neurons. Large-diameter DRG neurons generally do not express TERK1/2. Scale bar, 50 μm. B, Immunostaining of TERK1/2 (green) and Nav1.7 (red) in DRG neurons in culture. Merged image (yellow) shows that TERK1/2 and Nav1.7 colocalize in small- and medium-diameter DRG neurons. Scale bar, 50 μm.
NGF and GDNF activate ERK1/2 in cultured DRG neurons
We investigated the effect of acute treatment of DRG neurons with neurotrophic factors on ERK1/2 activation. Adult rat DRG neurons maintained in culture overnight were treated with neurotrophic factors, NGF and GDNF, and ERK1/2 activation was measured by anti-pERK1/2-specific immunostaining. Figure 2 shows DRG neurons in culture for 24 h that have been acutely treated with NGF (50 ng/ml) or GDNF (50 ng/ml) for 30 min. ERK1/2 activation was assessed by immunostaining (Fig. 2A) using a rabbit polyclonal anti-pERK1/2-specific antibody. The number of DRG neurons with a signal intensity of pERK1/2 above a threshold (2.5× intensity of background signal under control conditions) increased from 2% in control to 19% after GDNF treatment and 34% after NGF treatment. Quantitation of the immunostaining signal (Fig. 2B) shows an increase of pERK1/2 in small DRG neurons (≥20 μm) 30 min after treatment with NGF (95.8 ± 16 AU) and GDNF (77.3 ± 13 AU) compared with control (33.5 ± 2 AU). Thus, both the number of cells with pERK1/2 and the level of pERK1/2-immunoreactive signal are increased after acute treatment with the neurotrophic factors. Neurotrophic factor activation of ERK1/2 in DRG cultures was also confirmed by Western blot analysis (Fig. 2C). The apparently greater increase in pERK1/2-immunoreactive signal on the Western blot (Fig. 2C) compared with that determined by measuring this signal by immunocytochemistry (Fig. 2B) is likely attributable to the contribution of pERK1/2 signal from Schwann and supporting cells in the culture to the signal on the Western blot.
Figure 2.
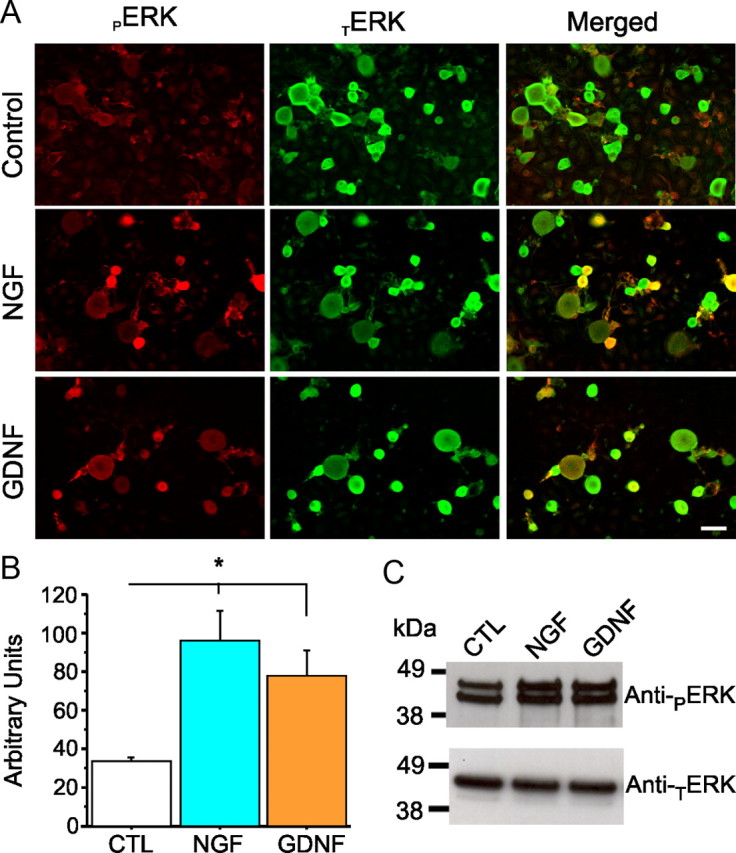
ERK1/2 are widely expressed in DRG neurons in culture containing NGF or GDNF. A, Immunostaining of ERK1/2 in adult rat DRG neurons in culture (24 h). In the morning, DRG neurons were treated for 30 min with NGF (50 μg/ml) or GDNF (50 μg/ml), and levels of TERK1/2 and pERK1/2 were examined using anti-TERK1/2 (green) and anti-pERK1/2 (red). Scale bar, 50 μm. TERK1/2 signal is present in DRG neurons in culture, and a 20 min treatment with NGF or GDNF increases the signal of pERK1/2 (merged image). B, Histogram showing pERK1/2 expression level for each culture condition. Treatment with NGF or GDNF elicits a significant increase of pERK1/2 compared with the control condition (CTL). Data are mean ± SEM. *p < 0.05, Student's t test. C, Western blot using anti-pERK1/2 antibody to assess activation of ERK1/2 in DRG neurons in culture after 30 min treatment with NGF (50 μg/ml) or GDNF (50 μg/ml). Basal levels of pERK1/2 are detected in these cultures (which contain Schwann cells and fibroblasts in addition to neurons), and levels of pERK1/2 are increased acutely by the presence of NGF or GDNF in the culture media.
pERK1/2 inhibition decrease excitability in DRG neurons in culture
We investigated the effect of pERK1/2 on action potential firing properties of DRG neurons. DRG cells were treated overnight with NGF (50 ng/ml) and GDNF (50 ng/ml) to maintain ion channel conductances that permit studying firing of action potentials in the current-clamp mode (Dib-Hajj et al., 2005, 2009; Rush et al., 2006; Cummins et al., 2009). The protocol for acute isolation and short-term culture of DRG neurons has been shown to activate p38 MAPK, whereas allowing the cultures to incubate overnight reduces pp38 to background levels (Hudmon et al., 2008). Additionally, ERK1/2 is constitutively activated under normal DRG culture conditions (Fig. 2, control panel). Therefore, ERK1/2 activation was acutely inhibited by treatment with the MEK inhibitor U0126, and firing properties of treated DRG neurons were investigated using current-clamp recordings.
On the morning of the recording sessions, DRG cultures were acutely (20 min) treated with U0126 (10 μm) or with its inactive analog U0124 (10 μm). Quantitation of the pERK1/2 immunostaining signal in Figure 3A shows an approximately fivefold decrease in the pERK1/2 signal intensity within small DRG neurons after treatment with U0126 (U0124, 127.2 ± 15.0 AU; U0126, 26.4 ± 3.0 AU; p < 0.01); immunolabeling of pERK1/2 (red) is strongly decreased after 20 min incubation with the active inhibitor U0126, whereas TERK1/2 signal (green) does not change. Western blot analysis (Fig. 3B) shows total loss of pERK1/2 signal in sister cultures but no effect on TERK1/2 signal (signal from neuron and non-neuronal cells) after 20 min treatment with U0126, confirming the immunostaining results.
Figure 3.

Activation of ERK1/2 in DRG neurons is inhibited by acute treatment with U0126. A, Immunostaining of ERK1/2 in adult rat DRG neurons in culture (24 h) containing NGF (50 ng/ml) and GDNF (50 ng/ml). ERK1/2 activation inhibitor U0126 (10 μm) or its inactive analog U0124 (10 μm) were added to the media on the morning of the experiment for 20 min before cells are fixed and immunolabeled for TERK1/2 (green) and pERK1/2 (red). Scale bar, 50 μm. B, Western blot using anti-pERK1/2 (top) and anti-TERK1/2 (bottom) to assess effect of U0126 treatment on DRG neurons in culture (24 h) in the presence of NGF (50 ng/ml) and GDNF (50 ng/ml). U0126 (10 μm) or the inactive analog U0124 (10 μm) were added to the cultures for 20 min before cell lysis. Treatment with U0126 abolishes ERK1/2 activation in DRG cultures.
To investigate whether the inhibition of pERK1/2 influences DRG neuron excitability, whole-cell current-clamp recordings were performed in small (20–27 μm diameter) DRG neurons after incubation for 20 min with U0124 (inactive) or U0126 (active) ERK1/2 activation inhibitor. Cell capacitance and input resistance of DRG neurons treated with U0124 (24.1 ± 2.4 pF, 768.5 ± 138.9 MΩ, n = 20) were not significantly different (p > 0.05) from those treated with U0126 (24.0 ± 1.6 pF, 581.0 ± 72.6 MΩ, n = 20). However, the resting membrane potential of DRG neurons treated with U0126 (−69.3 ± 1.8 mV, p < 0.005) was significantly more hyperpolarized compared with that of neurons treated with U0124 (−61.4 ± 1.1 mV).
Although it is expected that pERK1/2 can act on multiple ion conductances, modulation of Nav1.7 would be predicted to affect threshold for single action potentials and firing frequency because this channel has been reported to act as a threshold channel in DRG neurons (Dib-Hajj et al., 2007, 2009a; Rush et al., 2007). When DRG neurons were stimulated by 0.5 ms step current injection from their resting membrane potentials, current threshold of single action potential in the presence of U0126 (3.2 ± 0.3 nA) was significantly higher (p < 0.01) than in the presence of U0124 (2.1 ± 0.3 nA). The peak amplitudes (50.6 ± 1.0 mV for U0124; 46.7 ± 2.2 mV for U0126) and voltage threshold (−33.6 ± 1.3 mV for U0124; −31.1 ± 1.8 mV for U0126) for action potentials in cells treated with the active and inactive ERK1/2 inhibitor were not significantly different (p > 0.05).
Because Nav1.7 is known to produce a current in response to a ramp stimulus, thus amplifying weak stimuli (Cummins et al., 1998; Rush et al., 2007), we compared the firing frequency in DRG neurons treated with U0124 or U0126 by applying 1 s ramp current injection from 0.5 to 2 nA, in 0.5 nA increments (0.5–2 nA/s). Figure 4A shows responses of representative DRG neurons to 1.0 and 1.5 nA ramp stimuli in the presence of the inactive inhibitor U0124 (left column) or the active inhibitor U0126 and shows that cells treated with U0126 fire reduced number of action potentials compared with cells treated with U0124. For all DRG neurons in this set of recordings, the number of action potentials (Fig. 4B) was significantly (p < 0.01) lower for DRG neurons treated with U0126 at every stimulus level (0.9 ± 0.8 at 0.5 nA; 1.9 ± 0.8 at 1 nA; 2.6 ± 0.8 at 1.5 nA; 2.6 ± 0.7 at 2 nA) compared with U0124 (11.8 ± 3.4 at 0.5 nA; 13.6 ± 3.1 at 1 nA; 13.1 ± 2.6 at 1.5 nA; 12.0 ± 2.5 at 2 nA). The reported reduction of voltage-gated calcium currents attributable to inhibition of pERK1/2 (Fitzgerald and Dolphin, 1997; Fitzgerald, 2000) may contribute to the reduced firing frequency in DRG neurons treated with U0126. To ascertain the effect of pERK1/2 on sodium conductances, we measured the time-to-first action potential peak, which is less likely to be affected by ion channels such as calcium channels (Renganathan et al., 2001). The time-to-first peak is increased in DRG neurons treated with U0126 compared with U0124 (Fig. 4C). Thus, inhibition of pERK1/2 by U0126 reduces excitability in DRG neurons in response to ramp stimuli, consistent with a possible effect on Nav1.7, and suggests that Nav1.7 may be a substrate for modulation by pERK1/2.
Figure 4.

Inhibition of pERK1/2 reduces excitability of DRG neurons. Whole-cell current-clamp recordings were performed in DRG neurons pretreated for 20 min with U0124 or U0126. Action potentials were elicited by 1 s ramp current injection ranging from 0.5 to 2 nA in 0.5 nA increments from resting membrane potential. A, Top row shows representative trains of action potentials in response to a 1 nA (top traces) and 1.5 nA (bottom traces) ramp stimuli. DRG neurons treated with U0126 show reduced numbers of action potentials and longer time-to-peak for the first action potential spike compared with neurons treated with the inactive analog U0124. B, The mean number of action potentials (defined as action potentials that overshoot 0 mV) for the population of DRG neurons studied was significantly reduced for neurons pretreated with U0126 (filled symbols; n = 20) compared with U0124 (open symbols; n = 20). C, For the population of DRG neurons studied, the mean time-to-peak for the first action potential (AP) spike was significantly (*p < 0.05) longer in neurons treated with U0126 (filled symbols; n = 20) compared with U0124 (open symbols; n = 20). Data are expressed as means ± SEM.
ERK1/2 inhibition alters gating of Nav1.7 sodium channel
To investigate whether pERK1/2 specifically modulates Nav1.7 currents, we inhibited ERK1/2 activation acutely in HEK 293 cells transfected with Nav1.7 channels. We show that, similar to DRG neurons, HEK 293 cells grown under normal culture conditions show constitutively activated ERK1/2 [Fig. 5A, control (CTL) lane]. Figure 5A shows that 20 min treatment with U0126 (active inhibitor) causes a strong reduction of ERK1/2 activation compared with treatment with the control, inactive analog U0124. Thus, we replicated these conditions in HEK 293 cells transfected with Nav1.7 channels to study the effect of pERK1/2 inhibition on Nav1.7 current properties using whole-cell voltage-clamp recordings.
Figure 5.

pERK1/2 inhibitor U0126 shifts activation and fast inactivation of Nav1.7. A, Western blot using anti-pERK1/2 antibody (top panel) and anti-TERK1/2 antibody (bottom panel) showing the effect of treatment with U0124 (10 μm) or U0126 (10 μm) on ERK1/2 activation in HEK 293 cells transiently transfected with hNav1.7 together with β1 and β2 subunits. At 24–36 h after transfection, HEK 293 cells were incubated with 10 μm of the drug for 20 min at 37°C. Cells were then washed with ice-cold PBS, lysed, and resuspended in 2× sample buffer. Untreated [control (CTL)] or U0124-treated cells show pERK1/2 signal, whereas treatment with U0126 abolishes the pERK1/2 signal. B, Representative sodium current traces were recorded from HEK 293 cells transiently expressing hNav1.7 channels together with hβ1 and hβ2. Cells were treated with either U0124 or U0126 for 20 min before recording. Peak hNav1.7 current amplitude was comparable in cells treated with U0124 (n = 14) or U0126 (n = 13). C, For activation (right curves), whole-cell Na+ currents were elicited by 50 ms test pulses to potentials between −80 and +40 mV in steps of 5 mV from a holding potential of −120 mV. For steady-state fast inactivation (left curves), currents were elicited with test pulses to −10 mV after 500 ms conditioning pulses. The V1/2 for activation and steady-state fast inactivation were measured from curves that were fitted by Boltzmann distribution equations. For activation, V1/2,Act = −27.5 ± 1.8 mV and k = 7.0 ± 0.4 mV for U0124 (■; n = 14); V1/2,Act = −20.9 ± 1.8 mV and k = 7.8 ± 0.4 mV for U0126 (●; n = 13). For steady-state fast inactivation, V1/2,Fast = −85.8 ± 1.4 mV and k = 6.4 ± 0.1 mV for U0124; V1/2,Fast = −81.0 ± 1.7 mV and k = 7.2 ± 0.3 mV for U0126. Both activation and steady-state fast inactivation curves for the cells pretreated with U0126 were significantly different from those for U0124 (p < 0.05).
HEK 293 cells were cotransfected with hNav1.7, hβ1 and hβ2 constructs, treated on the day of recording with either 10 μm U0124 or 10 μm U0126 for 20 min in culture media, and then transferred to the recording bath solution that included the same compounds. Data were collected 4–5 min after establishing whole-cell configuration to ensure stable currents, and the coverslip was replaced with another coverslip after 40 min of recordings. Figure 5B shows representative families of hNav1.7 currents recorded in the presence of U0124 (left) or U0126 (right) in the bath solution. Although the average current amplitudes were not changed (422 ± 50 pA/pF for U0124, n = 14; 407 ± 89 pA/pF for U0126, n = 13), the voltage dependence for both activation and steady-state fast inactivation of hNav1.7 channels in the presence of U0126 were significantly shifted in the depolarized direction (p < 0.05) compared with those in the presence of U0124 (Fig. 5C). The I–V data were converted to conductances and were fitted by Boltzmann distribution equations. The voltage-dependent activation curves in the presence of U0126 (V1/2 = −20.9 ± 1.8 mV, k = 7.8 ± 0.4 mV) were significantly depolarized by ∼7 mV (p < 0.05) compared with curves obtained in the presence of U0124 (V1/2 = −27.5 ± 1.8 mV, k = 7.0 ± 0.4 mV), without a significant change in the slope factor. The voltage dependence of steady-state fast inactivation in the presence of U0126 (V1/2 = −81.0 ± 1.7 mV, k = 7.2 ± 0.3 mV) was also significantly depolarized by ∼5 mV (p < 0.05) and the slope was shallower compared with curves obtained with U0124 (V1/2 = −85.8 ± 1.4 mV, k = 6.4 ± 0.1 mV). We also tested the effect of pERK1/2 on slow inactivation of hNav1.7. Slow inactivation was elicited by 30 s prepulses at various voltages ranging from −130 to 10 mV, in 10 mV increments, followed by a brief hyperpolarizing pulse to −120 mV for 100 ms to recover from fast-inactivation, followed by a test pulse to 0 mV to determine available channels. The voltage dependence of steady-state slow inactivation in the presence of U0126 (V1/2 = −66.7 ± 2.2 mV, k = 12.2 ± 0.2 mV, n = 12) was not significantly different from that in the presence of U0124 (V1/2 = −69.8 ± 2.2 mV, k = 13.0 ± 0.4 mV, n = 12).
The first intracellular loop of the sodium channel Nav1.7 is phosphorylated by pERK1
The voltage-clamp recordings suggest that pERK1/2 can modulate voltage-dependent properties of the Nav1.7 channel, which, given the short preincubation time, suggests that this action occurs through fast posttranslational effects on the channel. Nevertheless, these data do not demonstrate a direct effect on the channel by phosphorylation of specific channel residues. Sequence analysis of Nav1.7, however, shows the presence of several S/TP dipeptide motifs that may act as phosphoacceptor sites for pERK1/2: N terminus (three sites); L1 (four sites); and C terminus (three sites).
To determine whether any of the major intracellular fragments of Nav1.7 are substrates for pERK1/2, fusion proteins of these regions were produced in Escherichia coli, purified on affinity columns, and tested by in vitro kinase assays. The N, L2, L3, and C intracellular regions were produced as GST fusion proteins. The L1 intracellular region was produced as an Intein-tag fusion protein. All channel fragments were tested to guard against the presence of non-canonical phosphorylation sites in these fragments. Equal samples of purified proteins (Fig. 6B, top, Coomassie blue-stained strips) were incubated with pERK1 in the presence of [γ-32P]ATP to determine whether they are phosphorylation substrates for this enzyme. The autoradiogram (ARG) in Figure 6B shows that L1 is the only substrate for pERK1. Quantitation of the phosphorylation signal by Cerenkov counts (Fig. 6C) shows significantly greater 32P incorporation into L1 (2103 ± 600 cpm) compared with the other fragment: N terminal (443 ± 134 cpm; note, however, lack of a signal on the ARG), L2 (82 ± 33 cpm), L3 (47 ± 8 cpm), C terminal (44 ± 3 cpm), or control GST (34 ± 5 cpm) and intein (30 ± 7 cpm). Statistical analysis using ANOVA and post hoc Tukey's test show that L1 is a good substrate for pERK1 compared with the other intracellular polypeptides (p < 0.01, Tukey's test, n = 3).
Figure 6.
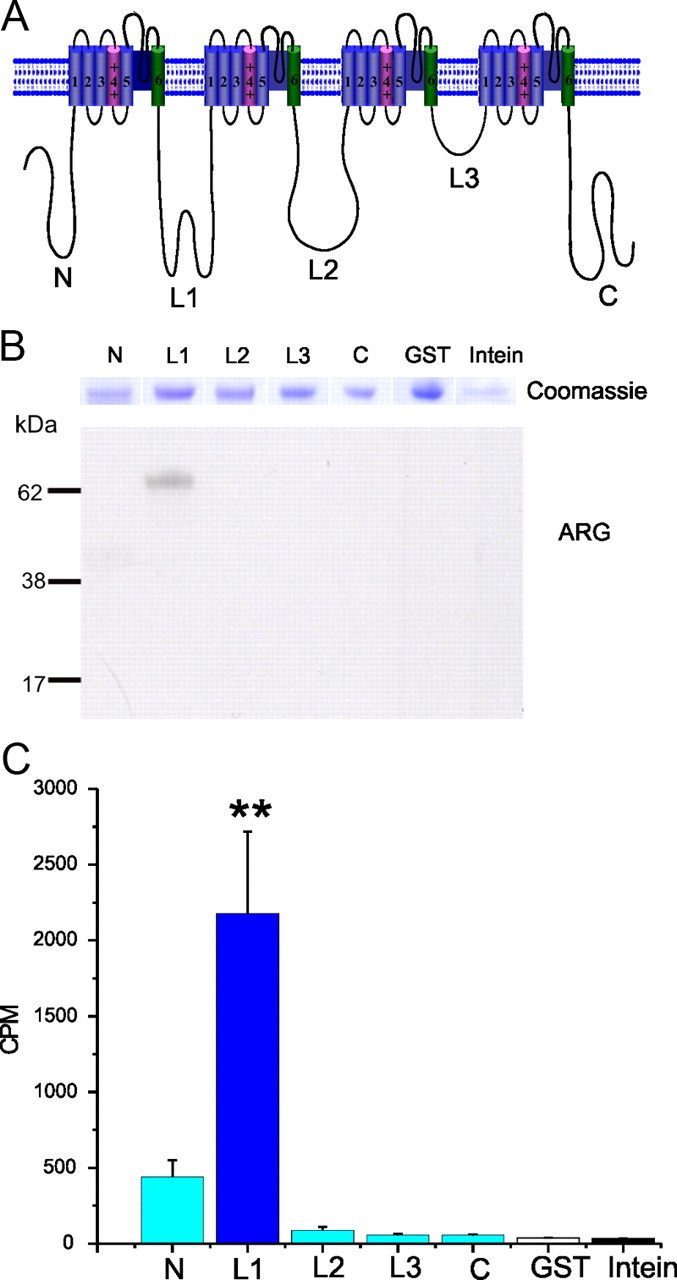
The first intracellular loop of the sodium channel Nav1.7 is phosphorylated by pERK1. A, Schematic of a sodium channel polypeptide showing the organization in four domains (DI–DIV) joined by three intracellular loops (L1–L3) and the intracellular N and C termini. The N, L2, L3, and C intracellular regions were produced as GST fusion proteins. The L1 intracellular region was produced as an Intein fusion protein. B, L1 is the only substrate for pERK1. Comparable amounts of fusion proteins, as determined by Coomassie blue staining (top panel), were used in the kinase assay. ARG shows that only L1 is phosphorylated by pERK1 in this assay. C, Histogram showing the Cerenkov counts of incorporated [32P]ATP in the different fusion proteins, after the kinase assay. **p < 0.01. Data are expressed as mean ± SEM; n = 3.
Nav1.7 L1 is phosphorylated at multiple sites
We evaluated the contribution of the four putative phosphoacceptor sites within L1 (Fig. 7A): threonine 531 (T531), serine 535 (S535), S608, and S712. We mutated each of these putative phosphorylation sites individually to the nonphosphorylatable amino acid alanine (Fig. 7B) and tested the ability of pERK1 to phosphorylate the mutant L1. The Coomassie blue staining profile shows that equal amounts of WT L1 and its mutant derivatives are used in these kinase assays. Figure 7C shows that the mutants T531A, S535A, and S608A are less efficient pERK1 substrates compared with WT. To better understand the contribution of each residue and compare the results from multiple experiments, we expressed the results as a percentage of the WT. The histogram in Figure 7D shows reduced phosphorylation for all mutants except S712A: 54 ± 6% (T531A), 71.5 ± 8% (S535A), 68 ± 3% (S608A), 108 ± 4% (S712A), and 5 ± 2 for Intein (the fusion tag for L1). These results show that phosphorylation of T531, S535, and S608 contribute to the total phosphorylation states of L1 of Nav1.7 by in vitro kinase assay.
Figure 7.
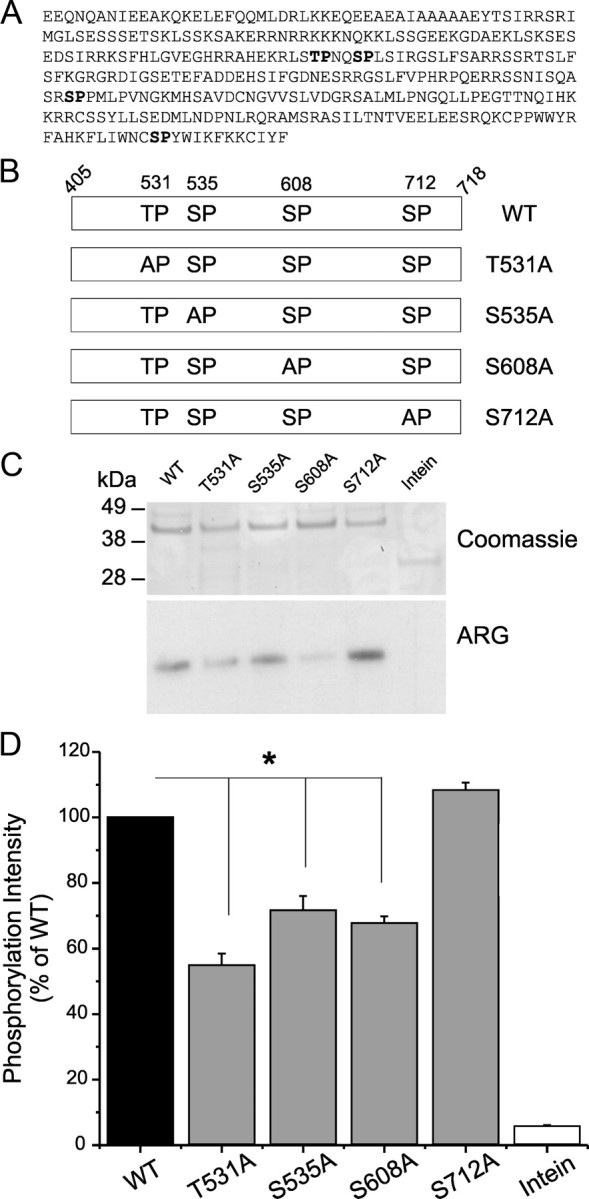
Identification of the pERK1 phosphorylation sites in L1 of Nav1.7. A, Four putative SP/TP phosphoacceptor sites (bold type) are identified within Nav1.7/L1. B, Schematic showing the replacement of S/T residues by the nonphosphorylatable alanine (T531A, S535A, S608A, S712A) to determine the contribution of each of these sites to pERK1/2 phosphorylation of Nav1.7/L1. C, Comparable amounts of WT and mutant fusion proteins, as determined by Coomassie blue staining (top), were used in the kinase assay. ARG shows that T531A, S535A, and S608A reduce [32P]ATP incorporation compared with WT, indicating that they contribute to the total phosphorylation of L1 (bottom). D, The [32P]ATP incorporation into WT L1 was corrected by the intensity of Coomassie blue staining of the band (top) and was set as 100% to measure the relative effect of individual replacement of the putative phosphorylation sites in L1. Histogram of the corrected [32P]ATP incorporation (Cerenkov count/Coomassie blue intensity) shows significant decrease for T531A, S535A, and S608A, but not S712A, mutant proteins compared with WT. *p < 0.05. Data are expressed as mean ± SEM; n = 4.
Because individual phosphorylation sites within the full-length channel may have an impact on channel properties that is not reflected by the extent of their contribution to the phosphorylation signal in an in vitro kinase assay using a fragment of the channel as a fusion protein substrate, we substituted each of the four phosphorylation sites individually with alanine and tested the effect on the gating properties of the mutant Nav1.7 channel. Voltage-clamp recordings showed that no single mutation produced a significant change on the effect of pERK1/2 inhibition on Nav1.7 voltage dependence of activation and fast inactivation in transfected HEK 293 cells (data not shown), consistent with the view that there is no one dominant site, or a preferred order of phosphorylation is required, among these three sites. Therefore, we reasoned that phosphorylation of multiple sites is required for pERK1/2 to alter Nav1.7 gating properties.
Triple-site substitution is required to abrogate phosphorylation of Nav1.7 L1 in vitro
Based on the data from single-site substitutions in L1 (Fig. 6), we generated several different mutant combinations (Fig. 8A): T531A/S608A (TASA), S535A/S608A (SASA), T531A/S535A/S608A (3MUT1), and T531A/S608A/S712A (3MUT2). Equal amounts of WT L1 and mutant derivatives were used in the in vitro kinase assay (Fig. 8B, Coomassie panel). The ARG panel in Figure 8B shows that the combination of two substitutions (TASA and SASA) significantly reduces the phosphorylation signal, whereas the triple substitutions reduce the signal to background levels. Compared with WT, the percentages of phosphorylation for mutant derivatives are as follows: 27 ± 3% (TASA), 25 ± 5% (SASA), 2 ± 0.5% (3MUT1), 2 ± 0.4% (3MUT2), and 1 ± 0.2% (Intein fusion tag). Interestingly, these results show that, despite the absence of a decrease of phosphorylation for S712A individually, its inclusion in a triple mutant further reduced the phosphorylation signal (compare phosphorylation of TASA with 3MUT2). These data suggest that phosphorylation of individual sites may enhance the phosphorylation of other sites, although there does not appear to be a dominant order for phosphorylation of individual sites.
Figure 8.
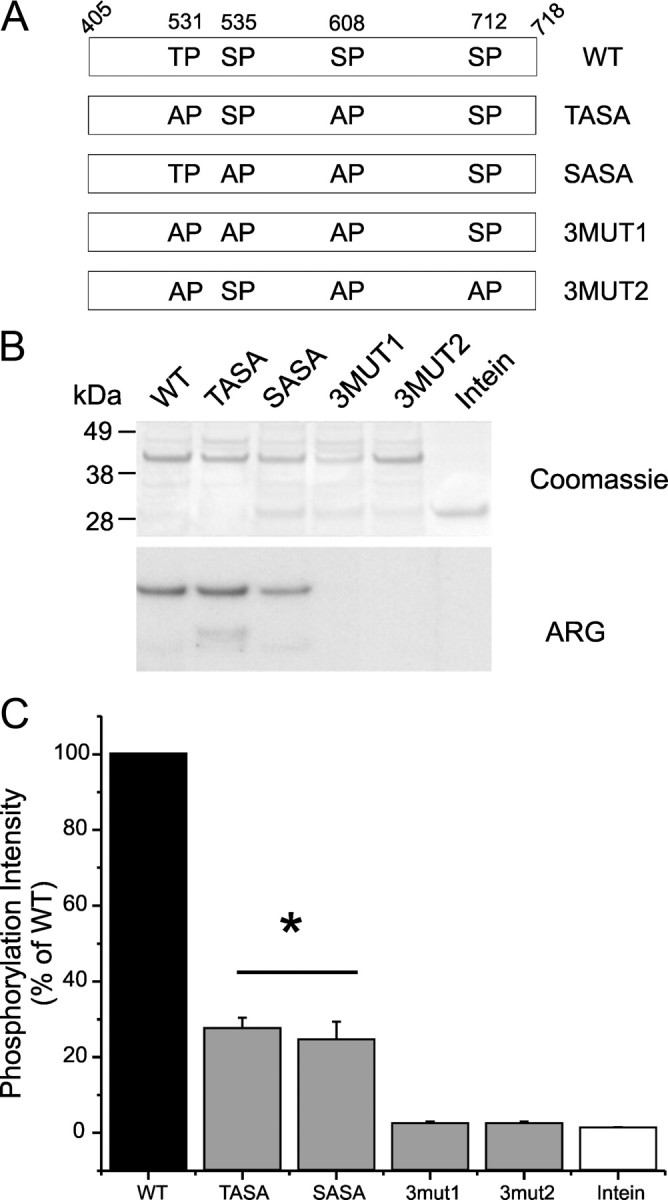
Triple substitutions are required to abolish phosphorylation of L1. A, Schematic diagram illustrating several double and triple replacements of phosphorylation residues by alanine [WT, TASA (T531S535/AA), SASA (S535S608/AA), 3MUT1 (T531S535S608/AAA), and 3MUT2 (T531S608S712/AAA)] that were tested as substrates for pERK1. B, Comparable amounts of WT and mutant fusion proteins, as determined by Coomassie blue staining (top), were used in the kinase assay. ARG shows that TASA and SASA are substrates for pERK1, whereas both triple mutants 3MUT1 and 3MUT2 are no longer phosphorylated by pERK1, indicating that they contribute to the total phosphorylation of L1 (bottom). C, The [32P]ATP incorporation into WT L1 was corrected by the intensity of Coomassie blue staining of the band (top) and was set as 100% to measure the relative efficiency of several double- and triple-mutant derivatives as pERK1 substrates. Histogram of the corrected [32P]ATP incorporation (Cerenkov count/Coomassie blue intensity) shows significant decrease for TASA and SASA mutant proteins, whereas 3MUT1 and 3MUT2 triple-mutant proteins reach only background levels (same as the control Intein tag), indicating that S712 may contribute to phosphorylation of L1. *p < 0.05. Data are expressed as mean ± SEM; n = 3.
Multiple phosphorylation sites are required for modulation of Nav1.7 by pERK1/2
We tested one of the double mutations (T531A/S608A), which showed loss of 63% of phosphorylation signal compared with WT (Fig. 8C), for the effect on pERK1/2 modulation of the voltage dependence of activation and steady-state fast inactivation of Nav1.7. Although the shift of steady-state fast inactivation of the double-mutant channel by U0126 disappeared (V1/2 = −84.8 ± 0.8 mV for U0124; V1/2 = −83.6 ± 0.9 mV for U0126), the shift of activation was reduced but not eliminated (V1/2 = −24.9 ± 1.2 mV for U0124; V1/2 = −21.5 ± 0.7 mV for U0126), indicating that other phosphorylation sites contribute to pERK1/2 modulation of Nav1.7. Therefore, we tested the effect of the two triple mutants (3MUT1 and 3MUT2) on pERK1/2 modulation of Nav1.7; these triple mutations reduced phosphorylation by pERK1 in vitro to background levels (Fig. 8C). The data (Table 1) show that V1/2 for activation and fast inactivation are shifted in a depolarized direction in the triple mutant channels compared with WT channels. To determine whether pERK1/2 can alter gating properties of these triple-mutant channels, we also examined the effects of inhibiting pERK1/2 and observed that treatment with U0126 has no significant effect on the V1/2 of activation and fast inactivation of 3MUT1 and 3MUT2 channels.
Table 1.
Activation and fast inactivation properties of WT and mutant hNav1.7 channels
| U0124 |
U0126 |
|||
|---|---|---|---|---|
| V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | |
| WT | ||||
| Activation | −27.5 ± 1.8 | 7.0 ± 0.4 | −20.9 ± 1.8* | 7.8 ± 0.4 |
| Inactivation | −85.8 ± 1.4 | 6.4 ± 0.1 | −81.0 ± 1.7* | 7.2 ± 0.3* |
| T531A, S608A | ||||
| Activation | −24.9 ± 1.2 | 7.6 ± 0.2 | −21.5 ± 0.7* | 8.1 ± 0.3 |
| Inactivation | −84.8 ± 0.8 | 6.0 ± 0.2 | −83.6 ± 0.9 | 7.1 ± 0.2* |
| T531A, S535A, S608A | ||||
| Activation | −23.1 ± 1.6 | 8.0 ± 0.3 | −21.9 ± 1.6 | 7.8 ± 0.3 |
| Inactivation | −82.4 ± 1.2 | 6.3 ± 0.1 | −83.0 ± 1.5 | 6.7 ± 0.2 |
| T531A, S608A, S712A | ||||
| Activation | −22.3 ± 0.8 | 7.9 ± 0.2 | −22.3 ± 0.9 | 7.8 ± 0.1 |
| Inactivation | −82.1 ± 0.9 | 6.5 ± 0.1 | −83.1 ± 1.3 | 6.7 ± 0.1 |
*p < 0.05 versus U0124.
Discussion
Human studies have shown that Nav1.7 is central to pain signaling, with gain-of-function mutations causing painful syndromes and loss-of-function mutations causing congenital insensitivity to pain (Cox et al., 2006; Fertleman et al., 2006; Dib-Hajj et al., 2007). Additionally, animal studies have shown that inflammation increases Nav1.7 levels that are paralleled by an increase in the TTX-sensitive current density in DRG neurons (Black et al., 2004) and have demonstrated that ERK1/2 are activated in DRG neurons in response to neurotrophic factors and proinflammatory cytokines (Obata and Noguchi, 2004). We report in this study that inhibition of ERK1/2 alters firing properties of DRG neurons and show that Nav1.7 sodium channels are modulated in a manner that is predicted to contribute to pERK1/2 regulation of action potential firing in DRG neurons. Inhibition of pERK1/2 shifts activation and steady-state fast inactivation of Nav1.7 channels in a depolarized direction, indicating that pERK1/2 has the reverse effect, i.e., making it easier to open this channel in response to weak stimuli. We further show that pERK1/2-mediated regulation of Nav1.7 requires phosphorylation of multiple sites within L1 of the channel that have been shown to be phosphorylated by pERK1/2 in vitro, suggesting a direct phosphorylation of the channel by pERK1/2. Our data demonstrate a physiological link of two important molecules, ERK1/2 and Nav1.7, that regulate electrogenesis in DRG neurons under normal and pathological conditions.
We have shown in this study that pERK1/2 inhibition produces a hyperpolarized shift in resting membrane potential, increases the threshold for action potential firing, and reduces firing frequency of small DRG neurons, consistent with an effect on voltage-gated ion channels. pERK1/2 is known to reduce voltage-gated potassium conductance (Yuan et al., 2002) and increase voltage-gated calcium conductance (Fitzgerald, 2000, 2002; Woodall et al., 2008), but, until now, its effect on voltage-gated sodium conductance was not known. Voltage-gated calcium channels in sensory neurons are tonically upregulated via Ras/ERK1/2 signaling, and this process requires a Cavβ subunit but does not depend on the particular Cavβ isoform (Fitzgerald, 2000, 2002). The neuronal Cav2.2 is directly phosphorylated at two phosphoacceptor sites, S409P and PGS449P within L1 of the channel (Martin et al., 2006). Inhibition of pERK1/2 in DRG neurons causes a 50% reduction of the voltage-gated calcium current in these neurons (Fitzgerald and Dolphin, 1997; Fitzgerald, 2000). Although the regulation of voltage-gated potassium conductances in DRG neurons is not well understood, pERK1/2 has been linked to a reduction in the A-type potassium channel current produced by Kv4.2 in dendrites of hippocampal neurons (Yuan et al., 2002), an effect produced by direct phosphorylation of the channel (Adams et al., 2000). Together with these previous results, the regulation of Nav1.7 by pERK1/2 that we report in this study demonstrates an effect of pERK1/2 on three major voltage-gated ion conductances in neurons, highlighting the acute pleiotropic effect on neuronal excitability of this class of MAPK.
Pro-nociceptive cytokines and neurotrophic factors activate MAPKs, which induce hyperexcitability of DRG sensory neurons (Schäfers et al., 2003; Obata and Noguchi, 2004), and this effect has been linked to an increase in the current density of TTX-R sodium currents in these neurons (Jin and Gereau, 2006; Binshtok et al., 2008). Activated p38 MAPK-mediated increase in the current density of Nav1.8 TTX-R channel requires direct phosphorylation of the channel at two phosphoacceptor serine sites within L1 of the channel, and preventing phosphorylation of either site abrogates pp38 effect on Nav1.8 (Hudmon et al., 2008). Here we show modulation of the Nav1.7 sodium channel by pERK1/2, which requires phosphorylation of four serine/threonine residues within L1. Unlike the modulation by pp38 of Nav1.8, the complete abrogation of the pERK1/2 modulation of Nav1.7 requires alanine substitution of at least three sites. Thus, although pp38 and pERK1/2 phosphorylate similar dipeptides (S/TP), different MAPKs may differentially modulate sodium channel isoforms that contribute to different aspects of DRG neuron excitability (Rush et al., 2007).
The in vitro kinase assays have shown that T531, S535, and S608, but not S712, are phosphorylated by pERK1. It has been shown previously that pERK1/2 can phosphorylate multiple sites of Smad3 but with one preferred site (Matsuura et al., 2005), whereas PKA phosphorylates L1 of Nav1.2 at several serine/threonine residues with specific sites more efficiently phosphorylated compared with others (Cantrell and Catterall, 2001). However, individual substitution of phosphoacceptor sites in L1 of full-length Nav1.7 channel with alanine did not have a detectable effect on pERK1/2 modulation of the channel-gating properties. Interestingly, inclusion of S712A in the triple mutant incrementally reduced the phosphorylation signal, and inhibition of pERK1/2 had no effect on gating properties of the triple-mutant channel carrying S712A, suggesting a contribution of this residue to pERK1/2 modulation of Nav1.7, despite the apparent lack of phosphorylation of this residue in the Intein–L1 fusion protein. The further depolarization of V1/2 of fast inactivation of the triple-mutant Nav1.7 (3MUT1 and 3MUT2) compared with the double-mutant channels is analogous to the graded regulation of potassium channel gating and firing properties by multiple phosphorylations of the channel (Park et al., 2006).
The ERK1/2 phosphoacceptor sites within Nav1.7 do not contribute to a recognizable sequence motif for protein–protein interaction. In contrast, the pp38 phosphorylation sites within Nav1.6 (Wittmack et al., 2005) and Nav1.8 (Hudmon et al., 2008) and the major pERK1/2 phosphoacceptor site within L1 of Cav2.2 (Martin et al., 2006) contribute to a PGSP motif that is the minimal motif for binding proteins with Src homology 3 domains (PXXP) or the type IV WW domain (PXpS/TP) (Zarrinpar and Lim, 2000). Modulation of these three channels by activated MAPK produces reduced current density of Nav1.6 (Wittmack et al., 2005) and increased current densities of Nav1.8 (Hudmon et al., 2008) and Cav2.2 (Martin et al., 2006). Thus, it is reasonable to suggest that pERK1/2 phosphorylation of Nav1.7/L1 promotes a structural effect that regulates gating properties of Nav1.7 rather than regulate binding of channel partners leading to altered trafficking or stability of the channel. Consistent with this view, PKA/PKC phosphorylation of multiple sites within Nav1.2/L1 promotes a structural effect that modulates slow inactivation of the channel (Chen et al., 2006). However, unlike Nav1.2, phosphorylation of multiple sites within L1 of Nav1.7 regulates voltage dependence of activation and fast inactivation, with no effect on slow inactivation. Thus, different protein kinases modulate sodium channels in an isoform-dependent manner, even in the context of an apparent common action involving additional negative charges in L1 of the channels.
We have presented in this study evidence consistent with the direct phosphorylation of Nav1.7 by pERK1/2 and for pERK1/2-induced hyperpolarization of activation and fast inactivation of Nav1.7, a threshold channel in DRG neurons that amplifies weak stimuli (Cummins et al., 1998; Rush et al., 2007). It has also been shown that pERK1/2 increases the current density of voltage-gated calcium channels in DRG neurons (Fitzgerald, 2000, 2002) and reduces the A-type voltage-gated potassium currents produced by Kv4.2 in superficial dorsal horn neurons (Adams et al., 2000; Hu et al., 2003; Agthong et al., 2006). Although the effect of pERK1/2 on voltage-gated potassium currents in DRG neurons is not known, it is not unreasonable to suggest that neurotrophic factors and proinflammatory cytokines acting through the ERK1/2 pathway can regulate DRG neuron firing by modulating multiple voltage-gated ion channels including Nav1.7 and that modulation of voltage-gated ion channels, including Nav1.7, by activated MAPK may provide a tractable therapeutic target for pain treatment.
Footnotes
This work was supported by the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs, and the Erythromelalgia Association. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America and the United Spinal Association with Yale University. We thank Lawrence Macala, Emmanuella Eastman, and Bart Toftness for excellent technical assistance.
References
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- Agthong S, Kaewsema A, Tanomsridejchai N, Chentanez V. Activation of MAPK ERK in peripheral nerve after injury. BMC Neurosci. 2006;7:45. doi: 10.1186/1471-2202-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averill S, Delcroix JD, Michael GJ, Tomlinson DR, Fernyhough P, Priestley JV. Nerve growth factor modulates the activation status and fast axonal transport of ERK 1/2 in adult nociceptive neurones. Mol Cell Neurosci. 2001;18:183–196. doi: 10.1006/mcne.2001.1015. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1β sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Mol Brain Res. 1996;43:117–131. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain. 2004;108:237–247. doi: 10.1016/j.pain.2003.12.035. [DOI] [PubMed] [Google Scholar]
- Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. 2008;64:644–653. doi: 10.1002/ana.21527. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18:9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Rush AM, Estacion M, Dib-Hajj SD, Waxman SG. Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat Protoc. 2009;4:1103–1112. doi: 10.1038/nprot.2009.91. [DOI] [PubMed] [Google Scholar]
- Dai Y, Iwata K, Fukuoka T, Kondo E, Tokunaga A, Yamanaka H, Tachibana T, Liu Y, Noguchi K. Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J Neurosci. 2002;22:7737–7745. doi: 10.1523/JNEUROSCI.22-17-07737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128:1847–1854. doi: 10.1093/brain/awh514. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K. Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res Rev. 2009a;60:65–83. doi: 10.1016/j.brainresrev.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009b;4:1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Fitzgerald EM. Regulation of voltage-dependent calcium channels in rat sensory neurones involves a Ras-mitogen-activated protein kinase pathway. J Physiol. 2000;527:433–444. doi: 10.1111/j.1469-7793.2000.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald EM. The presence of Ca2+ channel beta subunit is required for mitogen-activated protein kinase (MAPK)-dependent modulation of alpha1B Ca2+ channels in COS-7 cells. J Physiol. 2002;543:425–437. doi: 10.1113/jphysiol.2002.022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald EM, Dolphin AC. Regulation of rat neuronal voltage-dependent calcium channels by endogenous p21-ras. Eur J Neurosci. 1997;9:1252–1261. doi: 10.1111/j.1460-9568.1997.tb01480.x. [DOI] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol. 2003;551:741–750. doi: 10.1113/jphysiol.2003.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Glauner KS, Gereau RW., 4th ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J Neurophysiol. 2003;90:1671–1679. doi: 10.1152/jn.00340.2003. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Nav1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Gereau RW., 4th Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- Martin SW, Butcher AJ, Berrow NS, Richards MW, Paddon RE, Turner DJ, Dolphin AC, Sihra TS, Fitzgerald EM. Phosphorylation sites on calcium channel alpha1 and beta subunits regulate ERK-dependent modulation of neuronal N-type calcium channels. Cell Calcium. 2006;39:275–292. doi: 10.1016/j.ceca.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Matsuura I, Wang G, He D, Liu F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry. 2005;44:12546–12553. doi: 10.1021/bi050560g. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K. Activation of extracellular signal-regulated protein kinase in the dorsal root ganglion following inflammation near the nerve cell body. Neuroscience. 2004;126:1011–1021. doi: 10.1016/j.neuroscience.2004.04.036. [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Contribution of Nav1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Kocsis JD, Waxman SG. Slow sodium conductances of dorsal root ganglion neurons: intraneuronal homogeneity and interneuronal heterogeneity. J Neurophysiol. 1994;72:2796–2815. doi: 10.1152/jn.1994.72.6.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103:8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol. 2007;579:1–14. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu JY, Kulhanek DJ, Eckenstein FP. Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp Neurol. 2000;166:392–402. doi: 10.1006/exnr.2000.7508. [DOI] [PubMed] [Google Scholar]
- Shimazu K, Takeda K, Yu ZX, Jiang H, Liu XW, Nelson PG, Guroff G. Multiple acute effects on the membrane potential of PC12 cells produced by nerve growth factor (NGF) J Cell Physiol. 2005;203:501–509. doi: 10.1002/jcp.20309. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Brehm P, Halegoua S, Mandel G. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron. 1995;14:607–611. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol. 1994;72:466–470. doi: 10.1152/jn.1994.72.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci. 2005;25:6621–6630. doi: 10.1523/JNEUROSCI.0541-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall AJ, Richards MA, Turner DJ, Fitzgerald EM. Growth factors differentially regulate neuronal Cav channels via ERK-dependent signalling. Cell Calcium. 2008;43:562–575. doi: 10.1016/j.ceca.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Lim WA. Converging on proline: the mechanism of WW domain peptide recognition. Nat Struct Biol. 2000;7:611–613. doi: 10.1038/77891. [DOI] [PubMed] [Google Scholar]