

Abstract
Although brain-derived neurotrophic factor (BDNF) is linked with an increasing number of conditions causing brain dysfunction, its role in the postnatal CNS has remained difficult to assess. This is because the bdnf-null mutation causes the death of the animals before BDNF levels have reached adult levels. In addition, the anterograde axonal transport of BDNF complicates the interpretation of area-specific gene deletion. The present study describes the generation of a new conditional mouse mutant essentially lacking BDNF throughout the CNS. It shows that BDNF is not essential for prolonged postnatal survival, but that the behavior of such mutant animals is markedly altered. It also reveals that BDNF is not a major survival factor for most CNS neurons and for myelination of their axons. However, it is required for the postnatal growth of the striatum, and single-cell analyses revealed a marked decreased in dendritic complexity and spine density. In contrast, BDNF is dispensable for the growth of the hippocampus and only minimal changes were observed in the dendrites of CA1 pyramidal neurons in mutant animals. Spine density remained unchanged, whereas the proportion of the mushroom-type spine was moderately decreased. In line with these in vivo observations, we found that BDNF markedly promotes the growth of cultured striatal neurons and of their dendrites, but not of those of hippocampal neurons, suggesting that the differential responsiveness to BDNF is part of a neuron-intrinsic program.
Introduction
Because of its lesser complexity and greater accessibility compared with the CNS, the peripheral nervous system (PNS) has extensively been used as a model to understand basic principles of vertebrate neural development and to get insights into relevant molecular mechanisms. For example, peripheral sympathetic and sensory ganglia played a key role in the discovery of nerve growth factor (NGF) and in the delineation of its physiology, which in turn greatly contributed to our current understanding of how target tissues regulate their own degree of innervation (Levi-Montalcini and Hamburger, 1951; Levi-Montalcini and Cohen, 1960; Korsching and Thoenen, 1983). As neuronal survival and the growth of dendrites depend both in the PNS and the CNS on successful target innervation and synaptic contacts (for review, see Purves, 1988), brain-derived neurotrophic factor (BDNF) is often assumed by analogy to be a critical retrograde survival and growth factor for CNS neurons. However, the in vivo role of BDNF has been very difficult to explore in the postnatal brain as bdnf−/− mouse mutants die too early for the role of BDNF to be assessed after its marked postnatal increase caused by neuronal activity (Maisonpierre et al., 1990; Zafra et al., 1990; Castrén et al., 1992).
To circumvent this problem, a number of mouse lines have been generated using Cre-mediated excision of bdnf (Rios et al., 2001; Gorski et al., 2003; Baquet et al., 2004; He et al., 2004; Chan et al., 2006, 2008; Monteggia et al., 2007; Unger et al., 2007). However, exploration on the global role of BDNF in CNS development requires a strategy allowing gene excision in all cells contributing to BDNF levels in the CNS. This is a challenging objective, as BDNF is anterogradely transported throughout the brain (Altar et al., 1997) and into the CNS from peripheral sensory ganglia (Zhou and Rush, 1996; Balkowiec and Katz, 2000).
We report here on the generation of a new mouse line with a bdnf excision mediated by Cre recombinase inserted in the tau locus, a gene expressed in postmitotic neurons (Tucker et al., 2001). This leads to the almost complete disappearance of BDNF in the brain and spinal cord as assessed by immunoassay. Detailed morphological analyses of these conditional mutants revealed an unexpected selectivity in the brain areas requiring BDNF for normal postnatal development.
Materials and Methods
Generation of conditional bdnf knock-out mice.
The mice were maintained in the animal facility of the Biozentrum, University of Basel. Mice were housed for 2 months before the start of experiments under conditions of controlled temperature (21–22°C) and humidity (50%) under a 12 h light/dark cycle (lights on at 6:00 A.M.). The mice had ad libitum access to food and water. All experiments were performed in accordance with the Swiss regulations for animal experimentation. LoxP sites were inserted and flanked exon IX (5′ loxP at bp 906, 5′ loxP at bp 3449 locus AY057907), the single protein coding exon of bdnf. Conditional bdnf knock-out animals (cbdnf ko) were generated by breeding mice carrying two floxed bdnf alleles with mice expressing Cre from one allele of the tau locus (tau::cre line) (Korets-Smith et al., 2004) and also carrying one bdnflox allele. Genotypes of mice were determined by PCR using a tail biopsy. Wild-type and targeted alleles of the transgenic lines were amplified with specific primer combinations: floxed and wild-type BDNF allele: forward, 5′-GTT GCG TAA GCT GTC TGT GCA CTG TGC-3′, and reverse, 5′-CAG ACT CAG AGG GCA CTT TGA TGG CTT G-3′; BDNF null allele: forward, 5′-GTT GCG TAA GCT GTC TGT GCA CTG TGC-3′, and reverse, 5′-CAT GGG CAG TGG AGT GTG AG-3′); Cre allele: forward, 5′-GCC GAA ATT GCC AGG ATC AG-3′, and reverse, 5′-AGC CAG CAG CTT GCA TGA TC-3′. Thirty-five cycles were used to amplify the floxed and wild-type bdnf, bdnf-null, and Cre alleles. The PCR products could be distinguished by size on a 1% agarose gel. Unless specifically mentioned, all experiments with mutant and wild-type animals were performed 8 weeks postnatally. Wild-type animals were either bdnflox/lox or bdnfWT. In ELISA measurements, no difference in the BDNF levels were found in the CNS of bdnflox/lox compared with WT animals. The mice used in the present study were kept on a C57BL/6J-SV129 genetic background.
Immunoassay and immunoprecipitation.
BDNF extraction from tissues and quantification by ELISA was performed as previously described (Kolbeck et al., 1999). The ratio of pro-BDNF versus BDNF was determined in hippocampi of postnatal day 1 (P1), P4, P7, P21, and P84 wild-type animals lysed in Tris-HCl (50 mm), pH 7.4, with NaCl (150 mm), EDTA (1 mm), Triton X-100 (1%), Na-deoxycholate (1%), and SDS (0.1%) in the presence of a protease inhibitor mixture (Roche). The lysates were incubated with the BDNF monoclonal antibody no. 9 for 48 h at 4°C as described by Matsumoto et al. (2008), and the immunoprecipitates analyzed by Western blot developed with the BDNF antibody N20 (Santa Cruz).
Volumetric analyses.
Animals were heavily sedated by intraperitoneal injection of Ketalar (5 mg/kg) and Rompun (100 mg/kg) and perfused transcardially with 4% ice-cold paraformaldehyde (PFA) in 1× PBS. The brains were removed and kept in fixative overnight at 4°C. Serial coronal 30- to 35-μm-thick sections were prepared with a vibratome (Leica; VT 1000 S). Consecutive sections were stained with cresyl violet, dehydrated in graded ethanol and xylene, and coverslipped using Eukitt (Kindler). Sections were examined with a light microscope (Leica; 6× objective). Strict morphological criteria were used consistently in all mice to determine the boundaries of striatum, hippocampus, and cortex (Franklin and Paxinos, 1997). The dorsal boundary of the striatum was defined by the corpus callosum, the lateral boundary by the external capsule, and the medial boundary by the lateral ventricle and the corpus callosum, and its ventral boundary by the anterior commissure, excluding the nucleus accumbens. For the cortex, the primary ventral boundary was the corpus callosum. A line connecting the rhinal fissure to the corpus callosum was used in more medial sections to define the anterior ventral portion of the neocortex. Entorhinal cortex was also included in cortical volume measurements. Hippocampal outlines encompassed the dentate gyrus, the CA1–CA4 fields of Ammon's horn, the subiculum, the presibiculum, and the fimbria of the hippocampus. Starting with one of the most anterior sections, every fourth section was analyzed through the anterior–posterior extent of both hemispheres, and between 15 and 25 sections per brain were analyzed. All volumetric quantifications were performed with a Leica microscope (6× objective) equipped with a camera and volumes determined according to the Cavalieri method.
Immunohistochemistry and cell counts.
Mice were killed by cervical dislocation, and the brains were extracted, embedded in OCT medium (Electron Microscopy Sciences), and stored at −80°C. Serial coronal sections (10 μm) were prepared both from cbdnf ko and wild-type mice using a cryostat (Leica; CM 30505). Neurons and oligodendrocytes were stained after quenching of endogenous peroxidase activity with 0.3% H2O2/MeOH (Sigma-Aldrich; 32338-1) for 30 min. Sections were blocked and permeabilized for 1 h with 5% normal goat serum and 0.5% NP-40 in 1× PBS. Primary antibodies against NeuN (1:500) or Olig-2 (1:500) (Millipore Bioscience Research Reagents; MAB377 and AB9600) were applied in blocking solution overnight at 4°C, followed by incubation with secondary antibodies and staining revealed with the Vectastain Elite ABC kit (Vector Laboratories; PK-6101, PK-6102), and with AEC substrate (Vector Laboratories; SK-4200). To compare the number of neurons and oligodendrocytes in the striatum of cbdnf ko and wild-type mice, the number of NeuN- and Olig-2-immunoreactive cell bodies was counted in eight 10 μm sections, 120 μm apart using a 40× objective. NeuN- and Olig-2-positive cells were visualized using a Leica microscope equipped with a camera. Cell bodies were counted in stored images using the measuring module of the AnalysisD software program. Statistical significance was determined using an unpaired t test, and all values represent mean ± SEM.
Electron microscopy.
For electron-microscopic analysis, mice were deeply anesthetized with Ketalar (5 mg/kg) and Rompun (100 mg/kg) and fixed by transcardiac perfusion with 0.1 m cacodylate buffer, pH 7.4, containing 3% glutaraldehyde and 3% formaldehyde. The optic nerves from 2-month-old wild-type and cbdnf ko mice were dissected and kept overnight for postfixation in 2% glutaraldehyde in 0.1 m cacodylate buffer at 4°C. Tissue sections were extensively washed with washing buffer (0.5% sodium chloride, 0.1 m cacodylate buffer, pH 7.2) and postfixed in 1% osmium tetroxide/1.5% potassium hexanoferrate rinsed in 0.1 m cacodylate buffer for 3 h. After dehydration through graded ethanol solutions (70, 80, 96, and 100%), the optic nerves were soaked in propylene oxide for 1 h. After infiltration for 2 h in propylene oxide and Epon (1:1), samples were infiltrated in propylene oxide and Epon (3:1) at room temperature overnight. Samples were then embedded with plastic molds and polymerized at 37°C for 2 h, followed by 45°C for 24 h. Finally, the samples were polymerized at 65°C for 3 d. Semithin sections (1 μm) were cut in a plane orthogonal to the longitudinal axis of the optic nerve with an ultramicrotome (Reichart-Jung) and were stained with toluidine blue for light microscopy (DMRE; Leica). Selected areas were further sectioned at 60–70 nm (ultrathin sections) for transmission electron microscopy. Ultrathin sections were collected on 200 mesh grids and stained with uranyl acetate and lead citrate. Immediately after staining, the grids were thoroughly washed with water and the grids air-dried before examination. Ultrathin sections were examined with an electron microscope (Philips EM400) at an accelerating voltage of 80 kV. Electron micrographs were randomly taken from the optic nerve without knowing the genotype of the mice. The total area of the optic nerve was measured from semithin sections stained with toluidine blue using AnalysisD software. Counts of axons were made at a final magnification of 8600×. For each probe, the sample consisted of 15 pictures corresponding to a total area of 18.33 μm2. The axon size and thickness of myelin sheaths were measured by using AnalysisD software. The statistical analysis was performed using GraphPad Prism 4. All data are presented as mean ± SEM. Values of p < 0.05 were considered to be significant. All the graphs were generated in GraphPad Prism 4, with the exception of axonal size distribution generated in Excel.
DiOlistics and morphological analysis.
Hippocampal and striatal neurons from cbdnf ko and wild-type mice were labeled using DiOlistic on acute slices. Briefly, the mice were anesthetized and decapitated, and the brain was quickly transferred into ice-cold carbogenated (95% O2, 5% CO2) artificial CSF. For striatal analyses, brains were cut coronally in 300 μm sections throughout the striatum with a vibratome (Leica VT 1000 S). Hippocampi were dissected and cut into 400-μm-thick transversal slices. Vibratome slices were immediately fixed in 4% PFA overnight at 4°C. Tungsten particles (50 mg; 0.7 and 1.7 μm in diameter; Bio-Rad) were spread on a glass slide, and 100 μl of dye solution prepared by dissolving 3 mg of lipophilic dye DiI (Invitrogen) in 100 μl of methylene chloride (Sigma-Aldrich). The dried dye-coated particles were removed from the glass slide, resuspended in 3 ml of distilled water, and sonicated. The dye solution was subsequently diluted 1:60 for the striatum or 1:100 for the hippocampus. To improve the bead attachment, the tube walls were precoated with a solution of PVP (polyvinyl-pyrrolidone) (stock solution: 0.05 mg/ml in ethanol; Bio-Rad), and the bullets were stored at room temperature. Dye-coated particles were delivered to the acute slices using a hand-held gene gun (Bio-Rad; Helios Gene Gun System). A membrane filter (3 μm; Millipore) was inserted between the gene gun and the preparation to prevent clusters of large particles from landing on the tissue. After shooting, brain and slices were kept in PBS overnight at 4°C (striatum) or for 5 d at room temperature (hippocampus) to allow dye diffusion. The slices were postfixed with 4% PFA, washed, and mounted using an antifading water-based mounting medium (Biomeda). The neurons were imaged with an Axioplan 2 imaging microscope (Zeiss) equipped with an ApoTome (Zeiss). Each neuron was first imaged with a 20× objective with a z-sectioning of 0.975 μm. Morphological reconstruction of the neurons and their processes was achieved with the Neurolucida software (MicroBrightField) and the Sholl analysis, number, length, and volume of the dendrites with the Neuroexplorer software (MicroBrightField). Spine density of medium spiny neurons was measured on secondary dendrites. The spine density of pyramidal cells was measured for mid-apical dendrites. The selected dendrite segments were imaged using a 63× objective with 0.475 μm increments. The number of spines was normalized per micrometer of dendritic length. For the analysis of dendritic spine types, the images were acquired with a LSM510 Meta confocal microscope (Zeiss) using a 40× water-immersion objective and a zoom 4 and were z-sectioned at 0.3 μm. The Neurolucida software was used to measure length, as well as head and neck diameter of each spine. The three measurements were used to fit each spine within the three classical spine types (Harris et al., 1992): stubby (type I), mushroom (type II), and thin (type III). The statistical analysis was performed using GraphPad Prism 4. All data shown are presented as mean ± SEM. The data obtained were compared between two different experimental conditions using a two-tailed Student t test. Asterisks indicate the significance levels as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Neuronal cultures.
The hippocampus and striatum of embryonic day 16 (E16) mice were dissected in PBS containing glucose (0.2%) and BSA (0.1%), treated with 0.5% trypsin for 10 min at 37°C followed by mechanically dissociation. After centrifugation (5 min at 1000 rpm), cells were plated on glass coverslips, coated overnight with poly-d-lysine (70–150 kDa; Sigma-Aldrich), and cultured in Neurobasal medium supplemented with B-27 (2%), l-glutamine (200 μm), penicillin (5 μg/μl), and streptomycin (12.5 μg/μl). The initial density was 125,000 cells/cm2 in a final volume of 1 ml/well. Twenty-four hours after plating, cells were transfected with 0.5 μg of the reporter plasmid pLL-SYN-SYN-GFP (Gascón et al., 2008) using Fugene reagent (Roche) according to manufacturer's instructions. On day in vitro 2 (DIV 2), the neurons were treated with cyclosporin A (CsA) (1 μm) or tacrolimus (FK506) (100 nm) 1 h before BDNF (40 ng/ml) application. BDNF (40 ng/ml) was reapplied after 2 d. On DIV 7, cells were fixed for 10 min using 4% PFA in PBS and processed for immunocytochemistry using mouse anti-green fluorescent protein (GFP) (1:1000; Cell Signaling) and rabbit anti-GAD65/67 (1:5000; Sigma-Aldrich) primary antibodies. Appropriate secondary antibodies, fluorochromes, and filters were selected for double immunofluorescence analysis. For the measurements of dendritic morphology and soma area, labeled neurons were visualized using standard epifluorescence under a 5× Neofluar objective. Analyses of soma area and dendrite morphology were performed manually with the help of the NIH ImageJ software. For striatal cell measurements, only double-stained cells were selected (GFP- and GAD65/67-positive cells). Because of the heterogeneity of morphologies typically encountered with such cultures, only neurons with one primary neurite longer than 250 μm were analyzed and selected as GFP-positive, GAD65/67-negative cells. For dendritic measurements, concentric circles centered on the cell soma and separated by 10 μm were projected onto the GFP-labeled neuron. Primary dendritic length and number of intersections were expressed as the number of concentric circles crossed by dendrites. Primary dendrites were identified by their direct association with the cell soma. Statistical analysis was performed with the GraphPad Prism 4 software, and the data were presented as mean ± SEM. Significance was assessed with Student's t test for comparison between differences in soma area, and ANOVA with post hoc Bonferroni's test for comparison of dendrites length. Asterisks indicate the significance levels as follows: *p < 0.05, **p < 0.01, and ***p < 0.001. To assess the efficacy of the cyclosporin A and FK506 treatments, NFATc4 nuclear localization was quantified by confocal microscopy using rabbit polyclonal antibodies against NFATc4 (1:50; Santa Cruz) and MAP2 (1:500; Cell Signaling), and the nuclear stain Hoechst. At least 270 neurons were counted on two different coverslips.
The proportion of neurons expressing tau was quantified in cerebral cortical, hippocampal, and striatal neurons dissected from E16 embryos from tau::gfp mice (Tucker et al., 2001). Cells were fixed after 3 d with 4% PFA in PBS and analyzed for MAP2 immunocytochemistry to identify neurons (1:1000; Cell Signaling) and a GFP monoclonal antibody (1:1000; Cell Signaling). More than 500 cells from two different coverslips were counted, and the results are expressed as the percentage ± SEM of MAP2- and GFP-positive cells.
Results
Generation of cbdnf ko mice
A major objective of our study was to examine the impact of BDNF deprivation after the period of postnatal increase of BDNF expression in the CNS (Maisonpierre et al., 1990; Kolbeck et al., 1999). As recent results indicate that there may be a shift in the ratio of pro-BDNF versus mature BDNF during brain maturation (Yang et al., 2009), we first used immunoprecipitation and Western blot analysis to monitor their respective levels. In the hippocampus, the levels of BDNF were found to increase by 12.05 ± 1.03-fold (SEM) (n = 3; p > 0.001), in line with previous ELISA results (Kolbeck et al., 1999). They remained high until 12 weeks, the latest time point examined. Between P1 and 12 weeks, the levels of BDNF are significantly higher than those of pro-BDNF at all time points (Fig. 1A,B), suggesting that neurons increase their capacity to process pro-BDNF in parallel with increased levels of BDNF expression.
Figure 1.

Determination of BDNF levels. A, The hippocampi of wild-type animals were lysed, incubated with a monoclonal antibody recognizing both pro-BDNF and mature BDNF, and examined by Western blot analysis. Note the marked increase of BDNF levels during the first postnatal weeks. B, Quantification of the ratio mature BDNF over pro-BDNF (±SEM) (n = 3). C, BDNF determination by ELISA at P56 in various CNS areas. All results are presented as a mean determined from the analysis of four mice per genotype (p < 0.001, unpaired t test). The white bars represent wild type, and the black bars represent cbdnf ko mice.
Mice homozygous for bdnflox (bdnflox/lox) are viable and fertile, and at 2 months, BDNF protein levels are indistinguishable from WT in the cerebral cortex, the hippocampus, and the striatum (data not shown). Conditional bdnf knock-out (cbdnf ko) mice were generated by mating tauCre/+; bdnflox/+ mice with the bdnflox/lox line. This mating scheme led to 8% of the progeny with the expected cbdnf ko genotype (tauCre/+; bdnflox/lox) with the remaining 17% being bdnf−/−. Indeed, the levels of Cre seem to be sufficiently high in some embryos to cause deletion of bdnf already in the zygote, as previously observed with some, but not all conditional reporter lines (Korets-Smith et al., 2004). BDNF levels were determined by ELISA and found to be at most 5% of wild-type levels in the cerebral cortex of cbdnf ko animals, and not significantly different from background in all other CNS areas (Fig. 1C). We also quantified the proportion of postmitotic neurons expressing Cre from the tau locus using a mouse line expressing GFP from this promoter (Tucker et al., 2001). E16 brains were dissociated and a small percentage of the MAP2-positive cells failed to express GFP: 2.12 ± 0.14% in the cerebral cortex, 1.06 ± 0.01% in the striatum, and 0.72 ± 0.03% in the hippocampus. These results suggest that, in tau::cre animals, most neurons express Cre shortly after they are born (Giacometti et al., 2007). To assess the tissue specificity of bdnf excision, we also determined the levels of BDNF by ELISA in heart, lung, and skeletal muscle and found these to be indistinguishable from wild-type animals (data not shown).
This profound and widespread reduction of BDNF levels throughout the CNS was compatible with the survival of the animals for up to at least 8 months after birth. In line with previous results reported by others (Ernfors et al., 1994; Jones et al., 1994), we also observed that bdnf−/− littermates die during the first weeks after birth. We also measured by quantitative real time PCR the levels of NGF, NT3, and NT4 mRNA and found them to be similar in the hippocampus of mutant and wild-type animals (data not shown).
At birth, cbdnf ko mice were of equal size as their littermates. During the first 3 weeks after birth, conditional mutants lagged somewhat behind littermates in weight gain and displayed bursts of hyperactivity followed by periods of inactivity that were longer than normal. At 2 months, cbdnf ko could not be distinguished from wild-type animals by body length, suggesting that skeletal growth was not affected. With regard to weight gain, female mutants could not be distinguished from wild-type animals at 4 weeks, but by 6 weeks and beyond they all developed mild to severe obesity with a mean increase of ∼43% in body weight compared with wild type at 2 months. In contrast, males were slightly lighter than wild type by 4 weeks but by 8 weeks could no longer be distinguished by body weight from wild type. We have no explanation for this sex specificity (Lyons et al., 1999; Kernie et al., 2000). In addition, the cbdnf ko animals all exhibited a clasping phenotype at 8 weeks, which is also seen in mice with bdnf deletion in the cerebral cortex (Baquet et al., 2004; Strand et al., 2007). The light/dark exploration behavior of mutant animals was also dramatically impaired (Fig. 2).
Figure 2.

cbdnf ko mice have a markedly decrease exploratory behavior. A, Latency of first entry into the light compartment during the dark/light exploration test. B, Number of dark/light compartment transitions during the dark/light exploration test. C, Total time spent in the light chamber during the dark/light exploration test. Each wild-type (white columns) and mutant animal (black columns) was tested for a period of 6 min. All results are presented as mean ± SEM determined from the analysis of n mice per genotype (**p < 0.01, ***p < 0.001, unpaired t test).
BDNF deprivation unevenly affects postnatal brain growth, not cell numbers and myelination
At 2 months, the brains of cbdnf ko animals were slightly smaller and measurably lighter compared with wild-type animals (0.44 ± 0.01 vs 0.37 ± 0.02 g; unpaired t test, p = 0.0004) (Fig. 3A,B). Comparative volume measurements of all major brain areas using Nissl-stained sections revealed that the volume of the striatum was most affected in cbdnf ko animals (19.98 ± 2.10 vs 13.26 ± 1.21 mm3; unpaired t test, p = 0.001) (Fig. 3D), whereas the hippocampus was unchanged (29.18 ± 2.77 vs 28.85 ± 3.10 mm3; unpaired t test, p = 0.40) (Fig. 3C). At 2 weeks, the striatum of cbdnf ko animals was not significantly different from wild type, indicating that BDNF is needed for the postnatal growth of some, but not all brain areas. No significant differences were found in the number of striatal neurons stained with NeuN (unpaired t test, p = 0.40) (Fig. 4A,B), although as expected, the neuronal density was increased (933 ± 33 vs 1333 ± 88 cells/mm2; p = 0.013) (Fig. 4A,C). The number of oligodendrocytes was unchanged in the striatum of cbdnf ko mutants compared with wild-type animals (unpaired t test, p = 0.33) (Fig. 4D,E) and their density not significantly increased (1480 ± 153 vs 1636 ± 47 cells/mm2; p = 0.32) (Fig. 4B,D, F). As the striatum harbors many more myelinated axons than the hippocampus, we tested the possibility whether the lack of BDNF may affect axonal diameter and myelination as previous results indicate that the optic nerve of bdnf−/− animals is hypomyelinated and axonal diameters reduced (Cellerino et al., 1997). In the cbdnf ko mutants, we did not detect any significant reduction in the cross-sectional area of the optic nerve (73,960 ± 6546 vs 61,480 ± 6837 μm2; unpaired t test, p = 0.25) (Fig. 5A,B) nor in the number of axons (25,210 ± 2865 vs 22,320 ± 2082; unpaired t test, p = 0.44) (Fig. 5D) and axonal density (0.36 ± 0.02 vs 0.37 ± 0.01 axon/μm2; unpaired t test, p = 0.67) (Fig. 5C), a result confirmed by the lack of significant differences in the mean size of myelinated axons (wild type, 0.89 ± 0.06 μm; cbdnf ko, 0.83 ± 0.04 μm; unpaired t test, p = 0.79) (Fig. 5F). In addition, the myelin sheath of the optic nerve appeared to be normal (Fig. 5E), as was its thickness (0.14 ± 0.01 vs 0.15 ± 0.01 μm; unpaired t test, p = 0.78) (Fig. 5G) and the ratio between axonal diameter and myelin sheath thickness (G-ratio, 0.76 ± 0.01 vs 0.74 ± 0.01; unpaired t test, p = 0.33) (Fig. 5H). The distribution of axonal diameter was unimodal both in cbdnf ko and wild-type animals (unpaired t test, p > 0.05) (Fig. 5I). Additional quantitative analyses of myelinated axons in cross-sections of the corpus callosum and the spinal cord similarly indicated that the axonal diameter and myelination were not affected in cbdnf ko animals at 2 months (data not shown). These observations indicate that CNS myelination does not require BDNF in the CNS.
Figure 3.

The pan-CNS BDNF deprivation unequally affects postnatal brain growth. A, Examples of a brain of a wild-type and of a cbdnf ko animal at 2 months. B, The brain wet weight of cbdnf ko mice was significantly different from wild-type mice. C, D, Volumes were determined using Nissl-stained sections of P14 and P56 hippocampi and striata from wild-type and cbdnf ko mice by Cavalieri analysis. All results are presented as mean ± SEM determined from the analysis of n mice per genotype (*p < 0.05, unpaired t test). The white bars represent wild type, and the black bars represent cbdnf ko mice.
Figure 4.
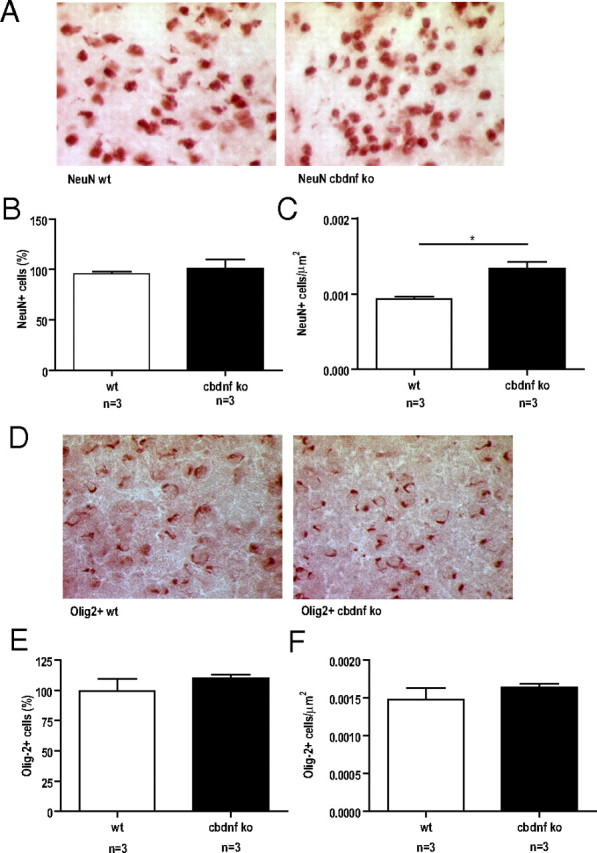
Number and density of neurons and oligodendrocytes in the striatum of 2-month-old mice. Representative images of the striatum of wild-type and cbdnf ko mice immunostained for NeuN (A) and for Olig-2 (D). B, Number of NeuN-positive cells in the striatum of wild-type and cbdnf ko mice. No significant differences were detected between the genotypes. C, Neuronal density in the striatum is higher in cbdnf ko mice compared with wild-type animals. E, Number of Olig-2-positive cells in the striatum of wild-type and cbdnf ko mice. No significant differences were detected between the genotypes. F, Oligodendrocyte density in the striatum of cbdnf ko mice compared with wild-type animals was not significantly different. All results are presented as a mean ± SEM determined from the analysis of n mice per genotype (*p < 0.05, unpaired t test). The white bars represent wild type, and the black bars represent cbdnf ko mice.
Figure 5.
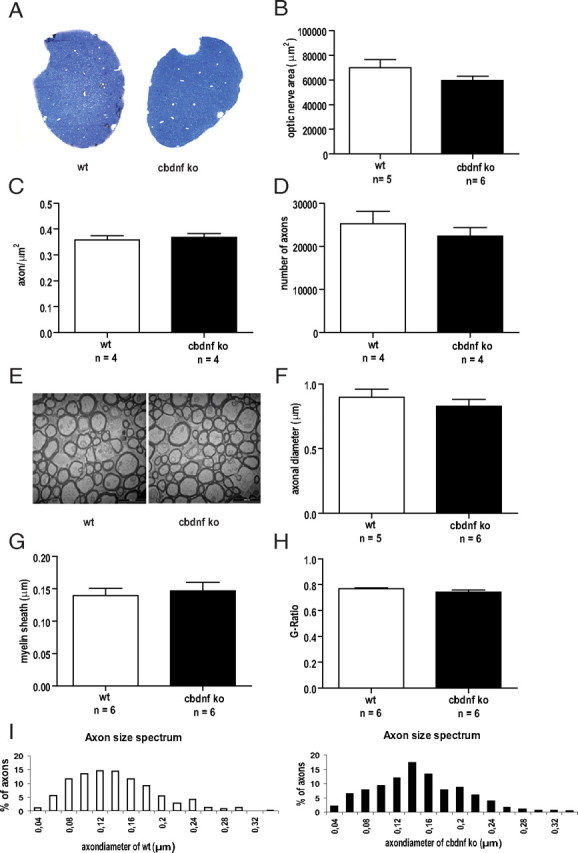
Comparative analysis of the optic nerve from wild-type and cbdnf ko mice. A, Light micrographs of semithin sections stained with toluidine blue of wild-type and cbdnf ko mice. Total area of the optic nerve (B), axonal density (C), and axonal numbers (D) in the optic nerve of cbdnf ko mice were not significantly different from wild-type values. E, Representative EM pictures of myelinated fibers in cross-sections of the optic nerve from wild-type and cbdnf ko animals. Axonal diameters (F), myelin sheath thickness (G), and G ratio (H) of cbdnf ko animals were not significantly different from wild type. Axonal diameters distribution of the optic nerve for a total of 455 axons (I). The frequency histogram indicates a unimodal pattern in both wild-type and cbdnf ko animals. All results are presented as mean ± SEM determined from the analysis of n mice per genotype (unpaired t test). The white bars represent wild-type, and the black bars represent cbdnf ko mice.
cbdnf ko striatal neurons fail to grow postnatally
As the volume reduction of the mutant striatum cannot be accounted for by cell loss or decreased myelination, we then performed detailed analyses of medium spiny neurons using a DiOlistic approach to label single neurons in acute slices (Fig. 6A). In comparison with wild-type neurons (n = 30), mutant neurons (n = 21) were markedly smaller. Total dendritic complexity was determined by Sholl analyses and found to be reduced by ∼50% in cbdnf ko mutants (127.06 ± 6.67 vs 64.76 ± 4.53 μm; unpaired t test, p = 0.001) (Fig. 6B). When the number of intersections was plotted against the distance from the cell body, a significant decrease in dendritic complexity was observed between 30 and 150 μm from the cell body (Fig. 6B) (unpaired t test, p < 0.001). Both the number of dendrites (5.74 ± 0.31 vs 4.09 ± 0.2; unpaired t test, p = 0.001) (Fig. 5C) and the total dendritic length were reduced in cbdnf ko neurons, the latter by ∼50% (1669.05 ± 84.28 vs 793.38 ± 58.27 μm; unpaired t test, p = 0.001) (Fig. 6D). The volume occupied by all branch orders of dendrites was reduced (337.76 ± 61.42 vs 125.97 ± 26.28 μm3; unpaired t test, p = 0.001) (Fig. 6E), whereas the volume of the cell bodies was smaller (99.19 ± 0.69 vs 84.52 ± 0.85 μm3; unpaired t test, p = 0.001) (Fig. 6F). As dendritic spines are the main targets of excitatory synapses formed by cortical afferents, we determined spine densities and total number of spines and found that both were significantly decreased (0.84 ± 0.02 vs 0.70 ± 0.02; unpaired t test, p = 0.001) (Fig. 6G,H).
Figure 6.
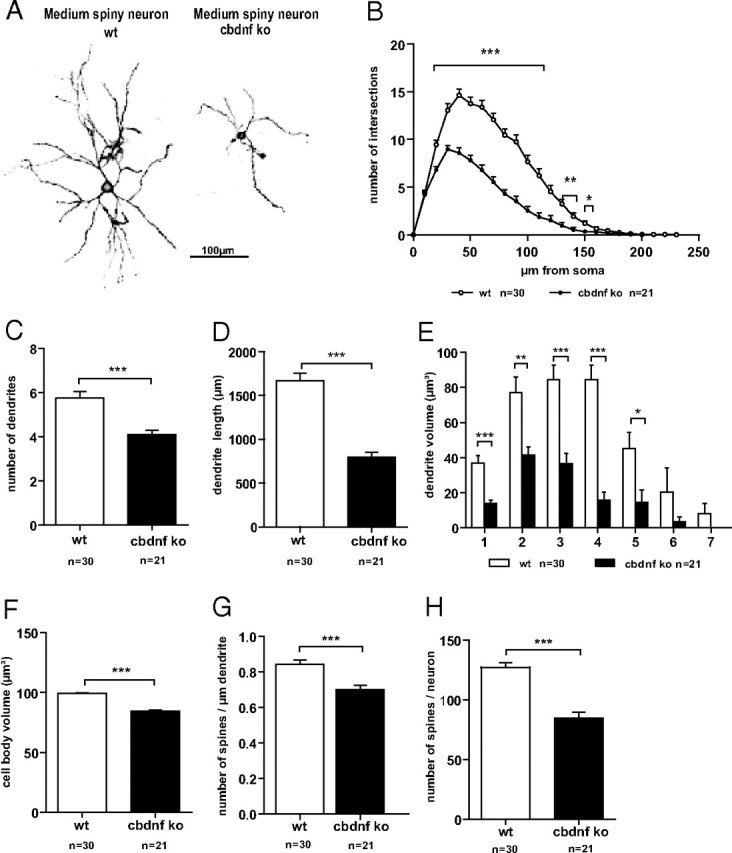
Dendritic morphology of striatal neurons from wild-type and cbdnf ko animals. Confocal image consisting of stacks of multiple optical sections showing the morphology of wild-type and cbdnf ko striatal neurons (A). Note the smaller size of striatal neurons in cbdnf ko mice. B, Sholl analysis comparing dendritic complexity in wild-type and cbdnf ko striatal neurons. Total number of dendrites (C), total dendritic length (D), total volume of the dendrites within the different branch orders (E), total cell body volume (F), spine density (G), and total number of spines per neuron (H) in wild-type and cbdnf ko striatal neurons. All results are presented as mean ± SEM determined from the analysis of n striatal neurons per genotype (*p < 0.05, **p < 0.01, and ***p < 0.001; Student's t test). Scale bar, 100 μm. The open circles/bars represent wild type, and the black circles/bars represent cbdnf ko striatal neurons.
Minimal morphological alterations of cbdnf ko hippocampal neurons
In view of the profound alterations observed in medium spiny neurons in the striatum deprived of BDNF, we also analyzed single pyramidal neurons in the hippocampus even though its overall size was unchanged when compared with wild-type animals (Fig. 3C). We selected CA1 as it is one of the most investigated brain region with regard to the role of BDNF in functional synaptic plasticity. Pyramidal neurons were labeled with DiI in month-old acute hippocampal slices and when qualitatively compared with wild-type CA1 pyramidal neurons, no apparent alterations in dendritic orientation or gross neuronal architecture were observed in cbdnf ko animals (Fig. 7A). This first impression was confirmed by comparing the total dendritic complexity using the Sholl analysis. In view of their different morphology and connectivity, we analyzed the complexity of apical and basal dendrites separately. The apical dendrite complexity was analyzed up to a distance of 400 μm from the cell body. Mutant CA1 neurons revealed only a slight, nonsignificant reduction in complexity when compared with wild-type cells (188.75 ± 12.52 vs 169.41 ± 9.15) (Fig. 7B, left) (wild-type, n = 25; cbdnf ko, n = 29). A more detailed Sholl analysis performed by plotting the number of intersections against the distance from the cell body confirmed these results (Fig. 7C) and revealed a limited, but significant, decrease in complexity of the apical dendrites of cbdnf ko CA1 cells versus CA1 wild-type cells (unpaired t test, p < 0.05, between 220 and 240 μm and between 270 and 290 μm) (Fig. 7C). For the basal dendrites, the same analysis indicated a similar, but even milder trend (wild-type, n = 14; cbdnf ko, n = 16) (Fig. 7D,E). The volume of the cell body revealed no differences between wild-type and mutant CA1 neurons (data not shown). We also compared the density of dendritic spines at the mid-distal portion of the apical dendrite of cbdnf ko animals and found a slight, nonsignificant reduction compared with wild-type neurons (1.41 ± 0.03 vs 1.36 ± 0.02 spines/μm of dendrite) (Fig. 7F). Spines subtypes were also analyzed (Fig. 7G) and a significant reduction in the proportion of mushroom (type II; 53.95 ± 2.34% vs 47.23 ± 1.88%; unpaired t test, p < 0.05; wild-type, n = 26; cbdnf ko, n = 23) spines was observed with a correspondingly higher proportion of thin (type III; 13.87 ± 1.67% vs 19.11 ± 1.66%; unpaired t test, p < 0.05; wild-type, n = 26; cbdnf ko, n = 23) spines. No changes were observed in the proportion of stubby (type I) spines (Harris et al., 1992; Koh et al., 2002). These results thus indicate that BDNF deprivation fails to cause major alterations in the fine structure of 2-month-old CA1 pyramidal neurons, in marked contrast with striatal medium spiny neurons.
Figure 7.
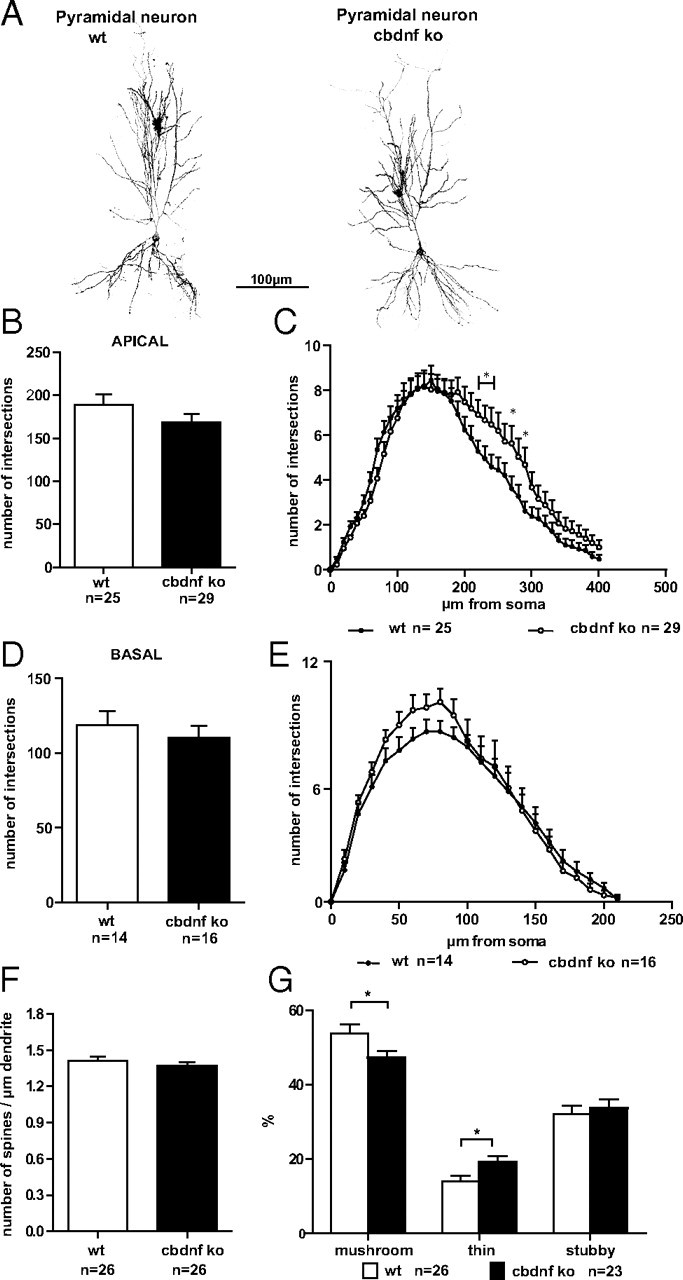
Dendritic morphology of hippocampal CA1 neurons from wild-type and cbdnf ko animals. A, Confocal image consisting of stacks of multiple optical sections showing the morphology of wild-type and cbdnf ko pyramidal neurons. Note the essentially unchanged dendrite complexity and length of pyramidal neurons in cbdnf ko compared with wild-type mice. B, D, Number of apical and basal dendrite intersections in wild-type and cbdnf ko pyramidal neurons. C, E, Sholl analysis comparing both apical and basal dendrites in wild-type and cbdnf ko pyramidal neurons. Spine density (F) and distribution of different spine types (G) in wild-type and cbdnf ko hippocampal CA1 neurons. All results are presented as mean ± SEM determined from the analysis of n CA1 pyramidal neurons per genotype (*p < 0.05, Student's t test). Scale bar, 100 μm. The open circles/bars represent wild-type CA1 pyramidal neurons, and the black circles/bars represent cbdnf ko CA1 pyramidal neurons.
BDNF promotes the growth of cultured striatal, but not hippocampal neurons
To examine whether the differential growth-promoting effects of BDNF on striatal versus hippocampal neurons may reflect intrinsic differences, we next investigated the response to BDNF of cultured neurons dissociated from the corresponding brain regions at E16. These experiments revealed that, after 5 d of exposure to BDNF (40 ng/ml), striatal neurons react with a strong growth response (Fig. 8A,B,E). Quantitative Sholl analysis revealed that BDNF increased the number and length of primary dendrites as well as the total number of intersections with significant differences in the number of branches at a distance between 20 to 120 μm from the cell body (Fig. 8F). When cultured hippocampal neurons were similarly treated, no significant morphological changes were observed after BDNF application. Both cell body size and primary dendritic length, as well as branches remained unchanged (Fig. 9A,B,E,F). In the same cultures, GAD65/67-expressing neurons were observed to have bigger soma sizes in the presence of BDNF compared with control cultures (data not shown).
Figure 8.
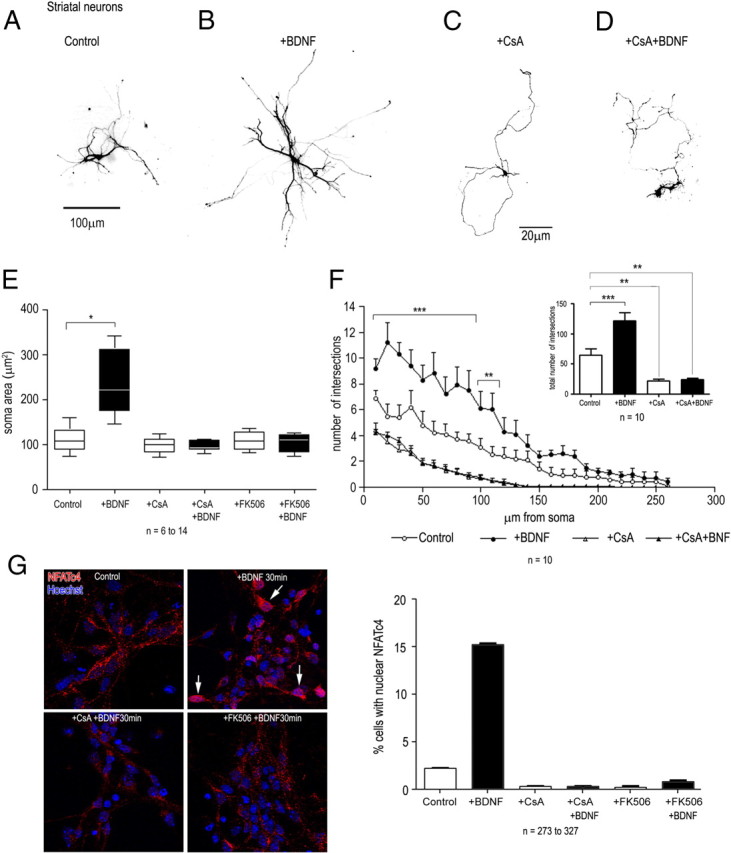
Effects of BDNF, cyclosporin A, and FK506 on the growth of striatal neurons and on NFATc4 nuclear translocation. Neurons were transfected after 1 d in vitro with a GFP reporter plasmid, BDNF (40 ng/ml) was added after 2 d, and the cultures were examined after an additional 5 d. Cyclosporin A (1 μm) or FK506 (100 nm) were added 1 h before BDNF. GFP- and GAD65/67-positive neurons were analyzed by fluorescence microscopy (A, B). BDNF treatment caused a significant increase in soma area (t test, p < 0.001) (E) and in the number and length of dendritic intersections (ANOVA; p < 0.001 from 10 to 90 μm; p < 0.01 from 100 to 110 μm from the cell body), as well as in the total number of dendritic intersections (t test; p < 0.001) (F). Cyclosporin A blocked the growth and arborization of untreated, as well as of BDNF-treated neurons (C–F). G, CsA and FK506 blocked the nuclear transfer of NFATc4 (arrows) observed after BDNF addition to striatal neurons. Nuclear translocation was assessed by counting the number of Hoechst- and NFATc4-positive nuclei. Data represent the mean ± SEM determined from the analysis of n neurons per condition. Statistical analysis were performed with Student's t test and with ANOVA followed by Bonferroni's post hoc test. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: A, B, 100 μm; C, D, 20 μm.
Figure 9.

Effect of BDNF and of cyclosporin A on the growth of hippocampal neurons. Neurons were transfected after 1 d with a GFP reporter vector, BDNF (40 ng/ml) was added after 2 d, and the cultures were examined after an additional 5 d. Cyclosporin A was added 1 h before BDNF. GFP-positive, GAD65/67-negative neurons were analyzed by fluorescence microscopy (A, B). Quantitative analysis failed to reveal any significant differences on the morphology of neurons with BDNF and/or cyclosporin A compared with untreated cultures (C–F). Data represent the mean ± SEM determined from the analysis of 10 neurons per condition. Statistical analysis were performed with Student's t test and with ANOVA followed by Bonferroni's post hoc test. Scale bar, 60 μm.
Previous in vitro and in vivo results have indicated that neurotrophin-dependent elongation of neuronal processes requires an intact calcineurin/nuclear factor of activated T-cells (NFAT) pathway (Graef et al., 2003). In rodents, both striatal and hippocampal neurons express the BDNF receptor TrkB (Altar et al., 1994), and to begin to test the differential response to BDNF downstream of TrkB, we examined whether the growth effects of BDNF observed with striatal neurons would involve a calcineurin-dependent pathway. We found that the calcineurin inhibitor CsA (1 μm) as well as FK506 (100 nm) interfered with the basal growth of the dendrites of striatal, but not of hippocampal neurons, suggesting the presence of a calcineurin-sensitive pathway regulating the growth of striatal neurons, even in the absence of exogenous growth factor (Fig. 8C–F). The BDNF-dependent growth of striatal neurons was completely suppressed by CsA or FK506 (Fig. 8C–E). No significant changes were observed in the growth of processes of hippocampal neurons, either with or without BDNF (Fig. 9C–F). Both CsA and FK506 significantly blocked the nuclear transfer of NFATc4 observed after BDNF addition to both neuronal populations (Fig. 8G).
Discussion
This study reveals that mice survive with barely detectable BDNF levels in their CNS and that, at 2 months, they are behaviorally impaired. The volume of their striatum was markedly reduced, whereas the hippocampus remains mostly unchanged. In vitro experiments reveal differences in the regulation of process extension by BDNF between the corresponding neuronal populations, in line with the results of the in vivo analyses.
BDNF measurements and tau::cre-mediated bdnf excision
Our gene deletion strategy allows the morphological impact of a global deprivation of BDNF to be assessed after BDNF expression has reached its peak in the postnatal brain of wild-type animals (Maisonpierre et al., 1990; Kolbeck et al., 1999). As a recent report suggested a change in the ratio of pro-BDNF versus mature BDNF and also challenged the notion that the levels of BDNF increase postnatally (Yang et al., 2009), we examined this question by immunoprecipitation and Western blot analyses. We found that the ratio of pro-BDNF to mature BDNF remains approximately constant with a large excess of mature BDNF at all ages tested (Fig. 1A,B). At neutral pH, very long incubation times, up to 48 h, were necessary to quantitatively precipitate both pro-BDNF and BDNF and to displace them from binding proteins present in brain extracts. Note that this long incubation time does not cause cleavage of pro-BDNF and that identical results are obtained with BDNF or Myc antibodies when using animals carrying a tagged version of the gene (Matsumoto et al., 2008). The shorter incubation times used by Yang et al. (2009) may explain the discrepancies with our results. Similar difficulties were previously encountered with NGF measurements (Suda et al., 1978).
Previous work with mouse ES cells differentiated in neurons as well as in vivo analyses of mice expressing GFP from the tau locus indicates that it begins to be active as soon as neuronal progenitors drop out of division (Tucker et al., 2001; Bibel et al., 2004). Accordingly, bdnf most likely is not deleted in cycling progenitors in the tau::cre line. This is unlike what has been achieved using a GFAP promoter-driver, which results in a smaller dentate gyrus because of decreased numbers of newborn neurons (Li et al., 2008). Given the profound reduction of BDNF levels in the cbdnf ko animals, our results confirm that postmitotic neurons are the major source of BDNF expression in the brain.
A number of other mouse conditional bdnf mutants have been previously generated using Cre expression driven by promoter constructs in transgenes or by Cre insertion in specific loci (Rios et al., 2001; Gorski et al., 2003; Baquet et al., 2004; He et al., 2004; Chan et al., 2006, 2008; Monteggia et al., 2007; Unger et al., 2007). In most cases, a deliberately restricted pattern of Cre driver expression was part of the objective of the experiment, whereas in others the BDNF reductions were not documented at the protein level. The results of our immunoassays indicate a dramatic reduction in all CNS areas, including the spinal cord, although very small, but detectable quantities could still be detected in the cerebral cortex of cbdnf ko animals, possibly produced by cells others than neurons including endothelial cells (Kermani et al., 2005). Given the near complete overlap of Cre expression with MAP2 labeling (inferred from the GFP staining data) (see Results), it is unlikely that significant numbers of neurons would still express BDNF in our cbdnf ko line.
BDNF deprivation and striatal versus hippocampal neurons
The number of striatal neurons remained unchanged in cbdnf ko animals, as was the number of retinal ganglion cells as inferred from axonal numbers. Although this latter result is in line with a conclusion reached with bdnf−/− mutants (Cellerino et al., 1997), we did not observe any significant decrease in the myelination of optic nerve axons, unlike previously reported (Cellerino et al., 1997). We explain this discrepancy as secondary to reduced postnatal growth and poor health of bdnf−/− analyzed shortly before their death (Cellerino et al., 1997), such as an abnormal development of electrical properties of retinal ganglion cells. The lack of widespread neuronal death should not be taken to imply that BDNF is not necessary for the survival of small neuronal populations in discrete brain nuclei such as subdivisions of the substantia nigra (Baquet et al., 2005) or noradrenergic neurons in a pontine nucleus (Guo et al., 2005).
Measurements of major CNS areas of cbdnf ko animals failed to reveal volume changes in the olfactory bulb, the dentate gyrus (data not shown), and the hippocampus CA1–CA3. A small reduction was observed in the cerebral cortex (data not shown), with the striatum being most affected. Random single-cell dye filling in the latter revealed a highly significant reduction of the total length and volume of dendrites (Fig. 6), indicating that the majority of these neurons—>90% being GABAergic medium spiny neurons—need BDNF for their postnatal dendritic growth. BDNF is most likely delivered by cortical afferents to these neurons as indicated by surgical and pharmacological experiments (Altar et al., 1997). The BDNF mRNA levels in the striatum are ∼2% of those in the cerebral cortex (Hofer et al., 1990), whereas the protein levels only differ by ∼50% (Kolbeck et al., 1999; Baquet et al., 2004). Both this interpretation and the results reported here are fully consistent with a previous study by Baquet et al. (2004) using a emx::cre line to delete bdnf in the cortex, but not in other brain regions. These findings are also in line with reports indicating that the development of GABAergic neurons is regulated by excitatory input thought to release BDNF in an activity-dependent manner (Kohara et al., 2007; Turrigiano, 2007; Hong et al., 2008). Baquet et al. (2004) also noted that, in their emx::cre line, the size of the hippocampus did not significantly differ from control animals after selective bdnf excision, but cautioned the interpretation by a possible anterograde transport of BDNF from other brain areas. Our results now indicate that the morphological development of CA1 neurons does not depend on BDNF, except for small changes in dendritic branches in restricted segments and signs of a modest delay in spine maturation. In line with our findings, we note that the virtually complete elimination of all forms of TrkB in the hippocampus (with a human GFAP promoter driving Cre expression) does markedly affect the growth of postmitotic hippocampal neurons (Luikart et al., 2005). Although changes in spine density, but not in the number of dendritic branches and length, were reported, these may be accounted for by the deletion of TrkB in progenitor cells, now well appreciated to be sensitive to BDNF/TrkB signaling (Bergami et al., 2008; Li et al., 2008).
As decreased duration of long-term potentiation in CA1 is a typical sign of diminished functional plasticity in mice lacking one or both bdnf alleles (Korte et al., 1995; Patterson et al., 1996), we stimulated with high-frequency bursts Schaffer collateral in slices of cbdnf ko mice and found long-term potentiation to be markedly reduced compared with wild-type slices, to a level indistinguishable from that observed in parallel experiments with slices from bdnf−/− animals (data not shown).
Although the clasping phenotype displayed by all mutant animals by 2 months is not surprising in view of the changes observed in the striatum, the reasons for the marked decreased light/dark exploratory behavior are unclear. BDNF has long been known to regulate the development or function of neuronal population critically involved in behavior such as dopamine- or serotonin-secreting neurons (Hyman et al., 1991; Mamounas et al., 2000). As a result, no simple explanation can be proposed for the abnormal exploratory phenotype of our mutant animals (for discussion, see Krishnan et al., 2007).
Mechanisms of growth regulation by BDNF
Our in vitro findings with striatal and hippocampal neurons in response to BDNF (Figs. 8, 9) are consistent with our in vivo observations and suggest cell-intrinsic differences in BDNF responsiveness and a calcineurin-sensitive pathway in striatal, but not in hippocampal neurons. A previous study already indicated a much larger growth response to BDNF of hippocampal GABAergic, compared with pyramidal neurons (Vicario-Abejón et al., 1998), and together these results suggest that TrkB activation by BDNF in GABAergic neurons involves the activation of NFAT and its transfer to the nucleus. Curiously, this transfer is also seen in hippocampal neurons in response to BDNF, but it does not result in readily measurable growth effects, nor does the strong decrease in NFAT nuclear localization after exposure to cyclosporin A or FK506 affect the growth of processes. These results point to a fundamental difference in the regulation of process elongation in these two neuronal populations.
In conclusion, our results indicate that, unlike NGF in the PNS, BDNF is not a major survival factor for most CNS neurons in young adult mice and that its near elimination throughout the CNS primarily affects the postnatal growth of the striatum.
Footnotes
This work was supported by a grant from the Max Planck Society (Hartmut Wekerle, Y.-A.B.), Swiss National Foundation Grants 31003A_124902 (Y.-A.B.) and 3100A0-112583 (N.S.-W.), and Deutsche Forschungsgemeinschaft Grants FOR471 (M.K.) and SFB 581, B4 (M.S., S.W.). We thank Michaela Krug and Dr. Gurumoorthy Krishnamoorthy for help with quantitative RT-PCR experiments.
References
- Altar CA, Siuciak JA, Wright P, Ip NY, Lindsay RM, Wiegand SJ. In situ hybridization of trkB and trkC receptor mRNA in rat forebrain and association with high-affinity binding of [125I]BDNF, [125I]NT-4/5 and [125I]NT-3. Eur J Neurosci. 1994;6:1389–1405. doi: 10.1111/j.1460-9568.1994.tb01001.x. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergami M, Rimondini R, Santi S, Blum R, Götz M, Canossa M. Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc Natl Acad Sci U S A. 2008;105:15570–15575. doi: 10.1073/pnas.0803702105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Richter J, Schrenk K, Tucker KL, Staiger V, Korte M, Goetz M, Barde YA. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat Neurosci. 2004;7:1003–1009. doi: 10.1038/nn1301. [DOI] [PubMed] [Google Scholar]
- Castrén E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci U S A. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Carroll P, Thoenen H, Barde YA. Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci. 1997;9:397–408. doi: 10.1006/mcne.1997.0641. [DOI] [PubMed] [Google Scholar]
- Chan JP, Unger TJ, Byrnes J, Rios M. Examination of behavioral deficits triggered by targeting Bdnf in fetal or postnatal brains of mice. Neuroscience. 2006;142:49–58. doi: 10.1016/j.neuroscience.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Chan JP, Cordeira J, Calderon GA, Iyer LK, Rios M. Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol Cell Neurosci. 2008;39:372–383. doi: 10.1016/j.mcn.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. Ed 2. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Gascón S, Paez-Gomez JA, Díaz-Guerra M, Scheiffele P, Scholl FG. Dual-promoter lentiviral vectors for constitutive and regulated gene expression in neurons. J Neurosci Methods. 2008;168:104–112. doi: 10.1016/j.jneumeth.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Guo H, Hellard DT, Huang L, Katz DM. Development of pontine noradrenergic A5 neurons requires brain-derived neurotrophic factor. Eur J Neurosci. 2005;21:2019–2023. doi: 10.1111/j.1460-9568.2005.04016.x. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, Lindsay RM. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350:230–232. doi: 10.1038/350230a0. [DOI] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermani P, Rafii D, Jin DK, Whitlock P, Schaffer W, Chiang A, Vincent L, Friedrich M, Shido K, Hackett NR, Crystal RG, Rafii S, Hempstead BL. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653–663. doi: 10.1172/JCI200522655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh IY, Lindquist WB, Zito K, Nimchinsky EA, Svoboda K. An image analysis algorithm for dendritic spines. Neural Comput. 2002;14:1283–1310. doi: 10.1162/089976602753712945. [DOI] [PubMed] [Google Scholar]
- Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- Korets-Smith E, Lindemann L, Tucker KL, Jiang C, Kabacs N, Belteki G, Haigh J, Gertsenstein M, Nagy A. Cre recombinase specificity defined by the tau locus. Genesis. 2004;40:131–138. doi: 10.1002/gene.20074. [DOI] [PubMed] [Google Scholar]
- Korsching S, Thoenen H. Nerve growth factor in sympathetic ganglia and corresponding target organs of the rat: correlation with density of sympathetic innervation. Proc Natl Acad Sci U S A. 1983;80:3513–3516. doi: 10.1073/pnas.80.11.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Cohen S. Effects of the extract of the mouse submaxillary salivary glands on the sympathetic system of mammals. Ann N Y Acad Sci. 1960;85:324–341. doi: 10.1111/j.1749-6632.1960.tb49963.x. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Hamburger V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool. 1951;116:321–361. doi: 10.1002/jez.1401160206. [DOI] [PubMed] [Google Scholar]
- Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20:771–782. doi: 10.1523/JNEUROSCI.20-02-00771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Purves D. Cambridge, MA: Harvard UP; 1988. Body and brain. A trophic theory of neural connections. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, Beal MF, Jones L, Kooperberg C, Olson JM, Jones KR. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda K, Barde YA, Thoenen H. Nerve growth factor in mouse and rat serum: correlation between bioassay and radioimmunoassay determinations. Proc Natl Acad Sci U S A. 1978;75:4042–4046. doi: 10.1073/pnas.75.8.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejón C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XF, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–953. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]