

Abstract
Dentate granule cell (DGC) neurogenesis persists throughout life in the hippocampal dentate gyrus. In rodent temporal lobe epilepsy models, status epilepticus (SE) stimulates neurogenesis, but many newborn DGCs integrate aberrantly and are hyperexcitable, whereas others may integrate normally and restore inhibition. The overall influence of altered neurogenesis on epileptogenesis is therefore unclear. To better understand the role DGC neurogenesis plays in seizure-induced plasticity, we injected retroviral (RV) reporters to label dividing DGC progenitors at specific times before or after SE, or used x-irradiation to suppress neurogenesis. RV injections 7 weeks before SE to mark DGCs that had matured by the time of SE labeled cells with normal placement and morphology 4 weeks after SE. RV injections 2 or 4 weeks before seizure induction to label cells still developing during SE revealed normally located DGCs exhibiting hilar basal dendrites and mossy fiber sprouting (MFS) when observed 4 weeks after SE. Cells labeled by injecting RV after SE displayed hilar basal dendrites and ectopic migration, but not sprouting, at 28 d after SE; when examined 10 weeks after SE, however, these cells showed robust MFS. Eliminating cohorts of newborn DGCs by focal brain irradiation at specific times before or after SE decreased MFS or hilar ectopic DGCs, supporting the RV labeling results. These findings indicate that developing DGCs exhibit maturation-dependent vulnerability to SE, indicating that abnormal DGC plasticity derives exclusively from aberrantly developing DGCs. Treatments that restore normal DGC development after epileptogenic insults may therefore ameliorate epileptogenic network dysfunction and associated morbidities.
Introduction
Medial temporal lobe epilepsy (mTLE) is often associated with pharmacoresistant seizures and comorbidities of cognitive dysfunction and depression (Mendez et al., 1986; Helmstaedter et al., 2003; Elger et al., 2004). In addition to neuronal loss and gliosis, human and experimental mTLE pathology involves prominent dentate granule cell (DGC) abnormalities, including mossy fiber sprouting (MFS), DGCs with abnormal dendrites, and dispersed or ectopically located DGCs (Tauck and Nadler, 1985; de Lanerolle et al., 1989; Houser et al., 1990; Mello et al., 1993; Parent et al., 1997; Spigelman et al., 1998; Buckmaster and Dudek, 1999; Ribak et al., 2000; Scharfman et al., 2000; Dashtipour et al., 2001). DGC abnormalities are associated with network hyperconnectivity that may impact mTLE pathogenesis. Timm staining, ultrastructural studies, and electrophysiological evidence from human or experimental mTLE, for example, suggest that mossy fibers form recurrent excitatory synapses onto neighboring DGCs (for review, see Sutula and Dudek, 2007).
In terms of abnormal dendrites, hilar basal dendrites are normally present only transiently on developing rodent DGCs but persist chronically after chemoconvulsant-induced status epilepticus (SE). Hilar basal dendrites in epileptic rats develop mature synapses with ultrastructural evidence of excess excitatory input (Ribak et al., 2000). Hilar ectopic DGCs also arise after SE (Parent et al., 1997). These cells aberrantly integrate into the local circuitry, receive excess excitatory input, exhibit abnormal bursting, and are recruited during spontaneous seizures (Scharfman et al., 2000, 2002; Dashtipour et al., 2001).
Although DGC abnormalities in mTLE are proposed to arise from abnormal plasticity of mature granule neurons, mounting evidence suggests that epileptogenic insults influence developing or newborn DGCs. For example, many hilar ectopic or basal dendrite-containing DGCs found after pilocarpine-induced SE in rats are adult-born (Parent et al., 1997, 2006; Scharfman et al., 2000; Jessberger et al., 2007; Walter et al., 2007). DGCs born after SE, however, have been thought to not contribute to MFS as suppression of neurogenesis after pilocarpine treatment with low-dose irradiation does not inhibit MFS at 4 weeks after SE (Parent et al., 1999). One interpretation of these data is that mature DGCs are responsible for MFS, but other work suggests that developing DGCs generated several weeks before SE can sprout (Jessberger et al., 2007).
We sought to determine the precise developmental stages at which DGCs are vulnerable to aberrant plasticity found in human and experimental mTLE. We labeled dividing DGC progenitors with retroviral (RV) reporters or transiently suppressed neurogenesis before or after SE using low-dose x-irradiation. We found that the developmental stage of DGCs at the time of SE dictates their contribution to seizure-induced plasticity. DGCs still developing during SE or those generated after SE, but not mature DGCs, differentially contribute to the cell populations that develop MFS, hilar basal dendrites, or ectopic migration. These findings indicate that seizure-induced DGC plasticity results from altered development and that DGCs exhibit maturation-dependent vulnerability to specific SE-induced pathologies. Restoring normal DGC development after epileptogenic insults may therefore ameliorate epileptogenic network dysfunction and associated cognitive and mood-related morbidities.
Materials and Methods
Animals.
All animal procedures were performed in accordance with protocols approved by the University Committee on Use and Care of Animals of the University of Michigan. Animals were purchased from Charles River and kept under a constant 12 h light/dark cycle with access to food and water ad libitum.
Pilocarpine-induced SE.
Adult male Sprague Dawley rats [postnatal day 56 (P56)] were pretreated with atropine methylbromide (5 mg/kg, i.p.); Sigma-Aldrich) and 15 min later were given pilocarpine hydrochloride (340 mg/kg, i.p.; Sigma-Aldrich) to induce SE. If convulsive seizure activity was not initiated within 1 h of the initial pilocarpine hydrochloride dose, an additional dose of 170 mg/kg was given. Seizures were monitored behaviorally and then terminated with diazepam (10 mg/kg) after 90 min of SE as previously described (Parent et al., 2002, 2006). Controls received the same treatments as experimental animals except that they were given saline in place of pilocarpine.
Production of high-titer RV.
Replication-incompetent recombinant RV vectors were pseudotyped by cotransfection of GP2-293 packaging cell line (Clontech) with plasmids containing the RV vector (RV-CAG-GFP-WPRE; gift from S. Jessberger and F. Gage, Salk Institute, La Jolla, CA) (Jessberger et al., 2007) and vesicular stomatitis virus-G (VSV-G) envelope protein (Clontech). Transient transfection was performed using calcium phosphate precipitation in 10 cm dishes after 5 × 106 cells/dish were plated in 10 ml of DMEM supplemented with 10% FBS the day before transfection. The cells were incubated at 37°C for 6 h, the medium was replaced, and then the cells were cultured for 65 h. The supernatant containing RV was harvested and filtered through a 0.45 μm pore size filter (Gelman Sciences). The filtered supernatant was then centrifuged in a Sorvall model RC 5C PLUS at 50,000 × g at 4°C for 90 min. The RV-containing pellet was resuspended in 1× PBS (∼0.003× the volume of medium initially harvested), aliquoted, and subsequently stored at −80°C until use. The concentrated RV titer was determined using NIH 3T3 cells and found to be ∼1–5 × 108 colony-forming units/ml.
Intrahippocampal RV injections.
Animals were anesthetized with a ketamine/xylazine mixture and placed on a water-circulating heating blanket. After positioning in a Kopf stereotaxic frame, a midline scalp incision was made, the scalp was reflected by hemostats to expose the skull, and bilateral burr holes were drilled. RV vector (2.5 μl of viral stock solution/site except for P7 pups, in which 1 μl virus/site was injected) was injected into the left and right dentate gyri over 20 min each using a 5 μl Hamilton syringe, and the micropipette was left in place for an additional 2 min. Coordinates for injections (in millimeters from bregma and millimeters depth below the skull) were as follows: at P7, caudal 2.0, lateral 1.5, depth 2.7; at P28, caudal 3.5, lateral 2.1, depth 3.8; at P42, caudal 3.8; lateral 2.3, depth 4.2; at P60, caudal 3.9; lateral 2.3, depth 4.2.
Brain irradiation.
Irradiation was done using an IC-320 Specimen Irradiation System (Kimtron Medical) operated at 300 kV and 10 mA. Rats were anesthetized with ketamine and xylazine, placed in sternal recumbency on a treatment table, and irradiated individually. The radiation dose was centered in a 2 × 2 cm treatment field that covered the hippocampi, with the remainder of the head, neck, and body protected by lead shielding. The corrected dose rate was 1.25 Gy/min. Dosimetry was performed using an ionization chamber connected to an electrometer system that is directly traceable to a National Institute of Standards and Technology calibration. Rats received a 6 Gy total irradiation dose given in two fractions over 3 d. Sham-irradiation controls were handled but not irradiated.
Tissue processing, histology, and immunohistochemistry.
Either 4 or 10 weeks after SE, animals were deeply anesthetized and perfused with 4% paraformaldehyde (PFA). The brains were removed, postfixed for 4–6 h in 4% PFA, cryoprotected in 30% sucrose, and frozen. For some animals, a 0.37% sulfide fixative was also used for subsequent Timm staining. Coronal sections (40 μm thick) were cut with a freezing microtome, and peroxidase and fluorescence (single- and double-label) immunohistochemistry performed on free-floating sections (Parent et al., 1997, 1999). Sections were immunostained with the following antibodies: goat anti-doublecortin (Dcx) (1:1000; Santa Cruz), rabbit (Rb) anti-Prox1 (1:10,000; gift from S. Pleasure, University of California, San Francisco, San Francisco, CA), Rb or mouse (Ms) anti-green fluorescent protein (GFP) (1:1000 or 1:250, respectively; Invitrogen), Ms anti-BrdU (1:1000; Roche), or Ms anti-NF-M (1:500; Millipore Bioscience Research Reagents). MFS was examined by Timm staining. Briefly, coronal sections were mounted onto Superfrost Plus slides (Thermo Fisher Scientific) and then dehydrated, rehydrated, and placed in a staining solution containing gum arabic, citrate, hydroquinone, and silver nitrate, and incubated at 26°C (Parent et al., 1999). After development of the stain, sections were washed, dehydrated, and delipidated in graded ethanols and xylenes, and coverslipped with Permount (Sigma-Aldrich).
Epifluorescence and confocal microscopy.
Single-label images were captured using a Leica DSM-IRB epifluorescence microscope attached to a SPOT-RT digital camera. For double labeling, images were acquired using a Zeiss LSM 510 confocal microscope as 1-μm-thick z-series stacks, imported into Adobe Photoshop, and analyzed for colocalization of immunoreactivity.
Cell quantification after RV reporter injections.
All quantification was performed blind to experimental condition. SE and control groups at all RV reporter injection time points were examined for GFP immunoreactivity. A total of 4978 GFP-expressing granule cells was counted (at least 100 cells from at least three sections were counted per animal). Only cells with clearly demarcated, brightly labeled cell bodies were included. Cells in the DGC were only counted if properly oriented (perpendicular to the granule cell layer) to unambiguously identify hilar basal dendrites. Each cell was analyzed for the presence of a hilar basal dendrite [using criteria of extension into the hilus and presence of clear spines as in the study by Dashtipour et al. (2003)] and for ectopic location, defined as greater than or equal to two cell body widths outside the granule cell layer (n = 3 rats for each control group and n = 4–5 for each experimental group). For Timm staining analysis, a minimum of three sections, spanning the rostral, mid, and caudal dentate gyrus, were examined by densitometry. Pixel density from 10 sampled areas/section were measured in the inner molecular layer using NIH ImageJ, 5 from the lower blade and 5 from the upper blade. The average background level (average of five samples per section) was subtracted and an average mean pixel density calculated for each animal (n = 4–5 per experimental group; n = 6 for each control group). Animals with severely damaged or missing superior blades were excluded from analysis. Statistical comparisons between groups for irradiation and RV studies were made using an ANOVA with post hoc t tests. For animals that were processed 10 weeks after SE, ImageJ was used to determine the percentage Timm-positive area in the lower blade as previously described (Buckmaster and Dudek, 1997) because many animals at this time point had damaged suprapyramidal blades. To accomplish this, the lower blade granule cell layer and molecular layer were carefully outlined and the percentage area stained was calculated in control animals that did not receive irradiation (n = 6) and experimental animals that were irradiated after SE (n = 4). A minimum of three slices were averaged per animal, and a two-tailed Student's t test was performed to compare the two groups. Data are presented as means ± SEM with the significance level set at p < 0.05.
Results
Dentate granule cells born 2–4 weeks before or 4 d after SE form hilar basal dendrites, whereas only DGCs born after SE migrate ectopically
Evidence suggests that both mature and immature or newborn DGCs contribute to seizure-induced plasticity in adult rodents (Parent et al., 1999; Overstreet-Wadiche et al., 2006; Jessberger et al., 2007; Walter et al., 2007). Whether the maturity of DGCs restricts the types of aberrant reorganization they undergo after SE, however, remains unclear. To better determine how cellular maturity influences DGC plasticity induced by SE, we used a RV reporter to label dividing DGC progenitors and their progeny at specific time points before or after SE. This approach offers the advantage of precise temporal control for labeling DGC progenitors, as RV selectively transduces mitotically active cells during S phase, and VSV-G-pseudotyped RV has a half-life of ∼4.5 h at 37°C (Higashikawa and Chang, 2001). Spatial control was achieved by targeting the stereotactic RV reporter injections to the dentate gyrus.
We first focused on DGCs with hilar basal dendrites as previous studies using biocytin cell fills or Golgi stains demonstrated that many more granule neurons with hilar basal dendrites are found after SE than in controls (Spigelman et al., 1998; Buckmaster and Dudek, 1999; Yan et al., 2001). GFP-expressing RV was injected into the dentate gyrus of P7 rat pups and SE was induced with pilocarpine 7 weeks later at P56 (Fig. 1A). Thus, labeled cells were postmitotic for ∼7 weeks by the time of SE. Animals survived for 4 weeks after pilocarpine-induced SE (P84). Because P7 is near the peak of postnatal neurogenesis, hundreds of progenitors were GFP-labeled and matured into DGCs over the subsequent 7 weeks (Fig. 1B,J). However, labeled DGCs showed no structural abnormalities at 1 month after SE, appearing essentially identical with labeled cells in saline-treated controls (Fig. 1B,F,J,N). In contrast, many DGC progenitors labeled at all other time points, including 4 or 2 weeks before SE, or 4 d after SE, showed abnormal hilar basal dendrites at 28 d after pilocarpine treatment (Fig. 1K–M,O–Q, arrowheads) that were not found in controls (Fig. 1C–E,G–I). A small subset of DGCs born at P28 (4 weeks of age at SE) showed robust hilar basal dendrites (Fig. 1K,O, arrowheads), whereas many born on P42 (2 weeks of age at SE) or P60 (4 d after SE) had extensive and very long hilar basal dendrites (Fig. 1L,M,P,Q, arrowheads; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Saline-treated controls receiving RV injections at the three later time points rarely had DGCs with hilar basal dendrites (Fig. 1C–E,G–I; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.
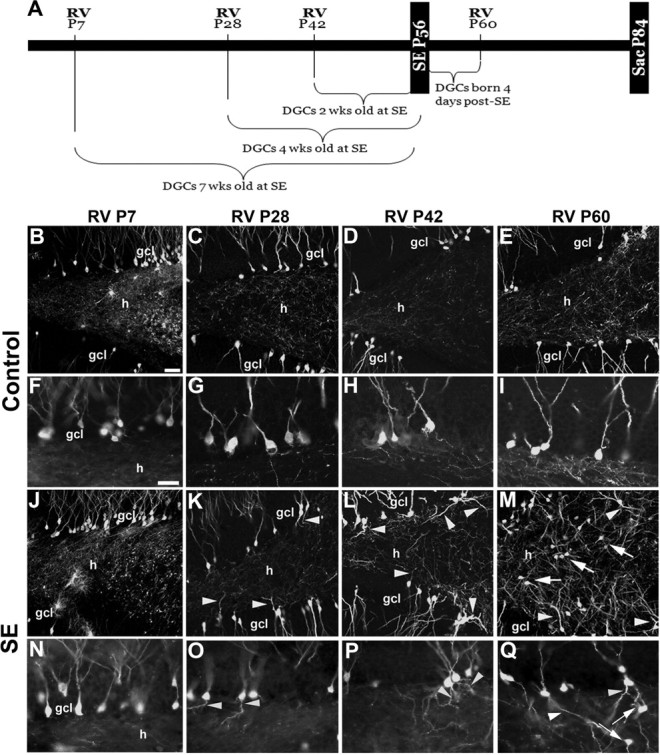
RV-GFP reporter labeling of DGCs generated at specific time points before or after SE. A, Timeline for RV labeling of newborn cells in the dentate gyrus born before or after SE. B–Q, Images of GFP-immunoreactive cells after injection of RV at the designated postnatal ages (P7, P28, P42, or P60) in rats receiving saline (B–I) or pilocarpine (J–Q) at P56. Higher magnification views of RV-GFP-labeled DGCs show granule cells with hilar basal dendrites (arrowheads) or hilar ectopic DGCs (arrows) only at specific RV labeling time points (P28, P42, or P60) with respect to SE at P56. ml, Molecular layer; gcl, granule cell layer; h, hilus. Scale bars: (in B) B–E, J–M, 50 μm; (in F) F–I, N–Q, 25 μm.
When we quantified the percentage of GFP-labeled DGCs with hilar basal dendrites, we found that 4-week-old DGCs (P28 RV injection) exposed to pilocarpine-induced SE were nearly three times more likely to show hilar basal dendrites than DGCs that were fully mature (7 weeks of age) at the time of SE (13.88 ± 2.39% for 4-week-old cells vs 5.10 ± 1.90% for 7-week-old cells) (Fig. 2A). This finding was even more dramatic for cells born 2 weeks before SE, which were ∼6.5 times more likely to form hilar basal dendrites, and cells born 4 d after SE, which were almost 7 times more likely to form basal dendrites than P7-injected rats (Fig. 2A). Animals injected with RV at P28, P42, and P60 that experienced SE also showed significantly greater numbers of DGCs with hilar basal dendrites than their respective controls groups (Fig. 2A). However, mature granule cells (7 weeks of age at SE) exposed to SE did not differ from their age-matched controls (Fig. 2A) (p = 0.25).
Figure 2.

Quantification of the percentage of RV-GFP-labeled DGCs with hilar basal dendrites or ectopic migration to the hilus. A, Hilar basal dendrites were analyzed in GFP-positive cells in the DGC layer. The percentage of DGCs with hilar basal dendrites labeled with RV-GFP at P7 that were mature at the time of SE (P56) was not significantly increased compared with controls (p = 0.25), but cells labeled by RV injections at the three other time points before or after SE did show significantly greater proportions with hilar basal dendrites (*p < 0.005 for all groups vs controls; #p < 0.05 for P28, P42, and P60 vs P7 RV). B, A significantly increased number of hilar ectopic GFP-labeled DGCs were present compared with controls only when RV was injected after SE at P60. *p < 0.005 for the P60 group versus P60 control. Error bars indicate SEM.
Previous work suggests that hilar-ectopic DGCs that appear after SE in adult rodents derive from newborn DGCs (Parent et al., 1997, 2006; Scharfman et al., 2000; Jung et al., 2004; Jessberger et al., 2007; Walter et al., 2007), but other findings raise the possibility that mature DGCs migrate ectopically (Heinrich et al., 2006; Nitta et al., 2008). We therefore examined whether GFP-positive DGCs labeled by RV reporter injection at the time points described above appeared in the hilus 4 weeks after SE. RV-GFP labeling of DGC progenitors at P7 that were mature during SE at P56 failed to show GFP-positive ectopic DGCs in the hilus (Fig. 1J,N). Similarly, DGCs labeled with RV at P28 or P42 and still differentiating at the time of SE did not migrate to the hilus (Fig. 1K,L,O,P). Examination of DGCs born 4 d after SE (P60 injections), however, revealed many GFP-labeled cells in the hilus at 4 weeks after SE (Fig. 1M,Q, arrows). These ectopic cells had a typical DGC morphology but a substantial portion exhibited abnormal dendrites similar to GFP-positive cells with hilar basal dendrites in the DGC layer. The ectopic cells were not included in counts of hilar basal dendrites, however, because the cell bodies were already located in the hilus. Quantification of ectopic GFP-labeled cell numbers showed that only cells labeled after SE were significantly more likely to migrate to the hilus (Fig. 2B). Thus, DGCs generated after SE show multiple structural abnormalities that likely lead to aberrant integration and marked disorganization of dentate gyrus circuitry (Fig. 1, compare E, M).
Effect of DGC maturity on MFS at 4 weeks after SE
The sprouting of granule cell axons, or MFS, is a pathological feature of human mTLE that is replicated by pilocarpine-induced SE and other rodent mTLE models (Sutula et al., 1989; Houser et al., 1990; Mello et al., 1993; Okazaki et al., 1995). Because our RV reporter clearly labels DGC axons, we were able to examine how DGC maturity influences MFS after SE.
In the first series of experiments, we made all observations at P84, 4 weeks after SE (Fig. 1A). Using this time point, we found that, similar to nonseizure controls (Fig. 3E) (data not shown), GFP-labeled cells in rats that received RV at P7 and underwent SE at P56 had axons that projected properly into the hilus and toward stratum lucidum of area CA3 when observed 4 weeks after pilocarpine treatment (Fig. 3A, arrows). In animals receiving RV 2 weeks before SE, in contrast, occasional cells with axons projecting aberrantly into the molecular layer were seen 4 weeks after SE (Fig. 3B, arrowheads). Rats that received RV-GFP injections 4 weeks before pilocarpine treatment frequently exhibited cells with GFP-labeled processes in the DGC layer and extending into the molecular layer that had an axonal morphology (Fig. 3F, arrowheads) or small, bouton-like structures (data not shown).
Figure 3.

Only DGCs still developing and not fully mature at SE display MFS. A, Representative image of DGCs labeled with RV-GFP at P7 that have axons extending normally into the hilus (h) (arrows) 4 weeks after experiencing SE on P56, with no GFP-labeled axons in the inner molecular layer (iml). B, P42 RV-GFP injection labeled a DGC 2 weeks of age at SE with GFP+ axon extending through the granule cell layer (gcl) into the iml (arrowheads). C, D, GFP/NFM double-labeled axon extending from the cell body toward the iml (arrows) 4 weeks after SE in an animal that received RV injection at P28. E, P28 RV-GFP injection into a saline-treated control rat labeled axons only in the hilus. F, P28 RV-GFP injection into an animal that subsequently experienced pilocarpine-induced SE on P56 labeled axonal sprouts in the iml (arrowheads) 4 weeks after SE. Scale bar: A, B, E, F, 75 μm; C, D, 40 μm.
To confirm the axonal nature of the processes, we double-labeled for GFP and NF-M, a medium chain neurofilament that specifically labels axons, but not dendrites, in the hippocampus (Parent et al., 1997). Confocal microscopy was performed to assess colocalization of immunostaining. Double-labeled axons extending from the cell body and projecting into the inner molecular layer were observed at this time point (Fig. 3C,D, arrows), further supporting the idea that DGCs generated 2 or 4 weeks before SE project mossy fibers aberrantly at 4 weeks after SE. No processes double labeled for GFP and NFM appeared in the molecular layer of rats injected with RV-GFP at P7 or P60 when observed 28 d after seizure, suggesting that only cells born several weeks before and still differentiating at the time of SE significantly contributed to the MFS seen 4 weeks after SE. These data are the first evidence that only developing, and not mature, DGCs contribute to MFS and are also consistent with our previous finding that ablating DGC progenitors after SE does not suppress MFS 4 weeks after pilocarpine treatment (Parent et al., 1999).
Transient suppression of neurogenesis at different times with respect to SE attenuates specific forms of aberrant plasticity
As a complementary approach to the RV reporter labeling, we sought a means to transiently suppress DGC neurogenesis before or after SE and determine the effects on aberrant seizure-induced plasticity. Because proliferating DGC progenitors are very susceptible to ionizing irradiation (Parent et al., 1999; Tada et al., 2000), we chose to use low-dose irradiation to accomplish the suppression. We previously irradiated with a 10 Gy dose given in two fractions 1 d before and 4 d after SE to show that DGCs born after SE were not necessary for MFS 4 weeks later (Parent et al., 1999). Here, we used a lower fractionated dose to transiently suppress neurogenesis for only a few weeks. In pilot studies comparing 4, 6, and 9 Gy given over 2 or 3 d, we found that 6 Gy given in two divided 3 Gy doses spaced 1 d apart suppressed DGC neurogenesis for ∼2–3 weeks when given at either P21 and P23 or P56 and P58, as demonstrated by loss of proliferating cells and Dcx-immunoreactive neuroblasts (Fig. 4) (data not shown). Both cell proliferation and Dcx expression returned to baseline levels at 4 weeks after irradiation (Fig. 4).
Figure 4.

Low-dose ionizing irradiation transiently suppresses DGC neurogenesis. A–C, Dcx immunostaining of immature neurons in the DG of rats irradiated (rad) beginning on P21 (P21/P23) shows decreased neurogenesis at 7 d (B, arrows) versus control (A), and recovery 28 d after rad (C). D, E, BrdU labeling shows cell proliferation also recovers 4 weeks after rad at P21/P23 (E) versus control (D). BrdU was given 2 h before killing on P49. F–J, Neurogenesis examined by Dcx immunolabeling is also transiently suppressed after rad beginning on P56 (P56/P58), with knockdown after 2 d (G, arrows) and 7 d (H, arrows), partial recovery at 14 d (I, arrows), and nearly total recovery to control levels (F) after 28 d (J). Scale bar, 100 μm.
We next administered 6 Gy fractionated radiation doses to rats at specific times before or after pilocarpine-induced SE to transiently suppress neurogenesis. Analogous to the RV reporter studies above, we wanted to deplete (instead of label) cohorts of developing DGCs that would otherwise have been a specific age at the time of SE, or those born after SE. Thus, we irradiated or sham-irradiated rats at P21 and P23, P42 and P44, or P60 and P62. Animals received pilocarpine at P56 and were killed 4 weeks after SE at P84 (Fig. 5A), the same time point used in our previous study of irradiation and mossy fiber sprouting (Parent et al., 1999). Radiation at all time points before SE did not impact the latency or severity of the SE episode, although a slightly greater proportion of animals died in irradiated compared with sham-irradiated groups.
Figure 5.
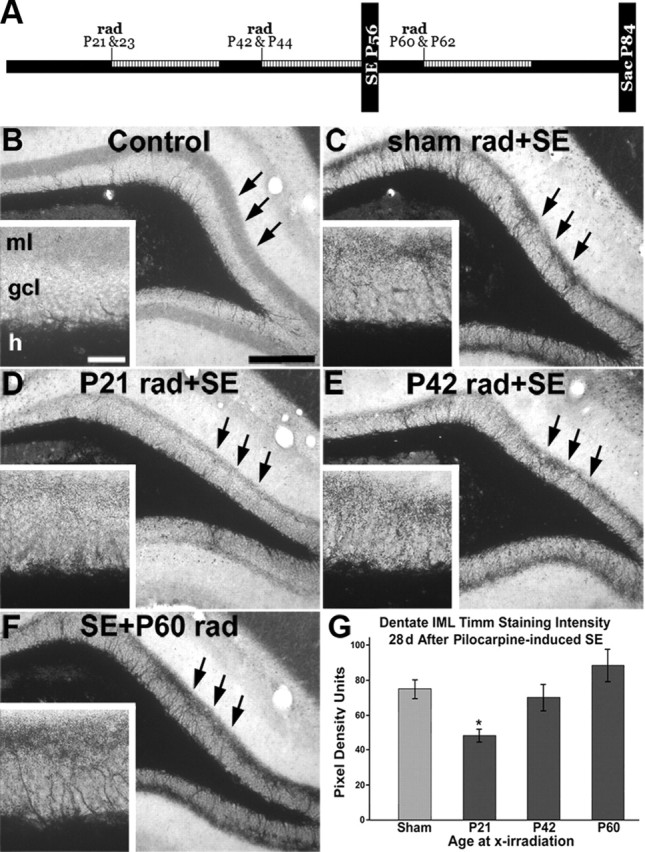
Effect of irradiation before or after SE on MFS. A, Timeline for irradiation studies. The hatched areas show predicted timing of suppressed neurogenesis after 6 Gy x-irradiation (rad). B–F, Representative images of dentate gyrus Timm stain (arrows) in a naive control (B), a control receiving sham rad with SE (C), and rats receiving rad at specific time points before or after SE (D–F). Higher magnification views are shown in the insets. G, Quantitative analysis of pixel density revealed that 6 Gy fractionated irradiation administered 5 weeks before SE (P21) significantly decreased the amount of MFS (*p < 0.05). Irradiation either 2 weeks before or 1 week after SE did not have a significant effect on MFS at 4 weeks after SE. ml, Molecular layer; gcl, granule cell layer; h, hilus. Scale bars: B–F, 100 μm; insets, 25 μm. Error bars indicate SEM.
We examined MFS at 4 weeks after pilocarpine treatment by Timm stain in irradiated and sham-irradiated, pilocarpine-treated rats. Compared with sham-irradiated controls experiencing SE, supragranular Timm staining was visibly reduced by 6 Gy x-irradiation beginning on P21, but not by irradiation beginning at P42 or after SE at P60 when examined 4 weeks after SE induction (Fig. 5B–F, black arrows). When we quantified these data using average pixel density units (PDU), we found that animals irradiated at P21, but not P42, had significantly less MFS at 4 weeks after SE (Fig. 5G) (P21/P23 rad, 48.12 ± 5.09 PDU; P42/P44, 69.85 ± 10.40 PDU; control, 74.96 ± 7.59 PDU; p < 0.05 for P21/P23 vs control). Animals irradiated after SE (P60/P62) did not experience any decrease in MFS at 28 d after SE (P60/P62, 88.18 ± 13.05 PDU; control, 74.96 ± 7.59 PDU) similar to previous results using a higher radiation dose (Parent et al., 1999). Our findings are consistent with the RV-GFP labeling data, as neurogenesis after 6 Gy fractionated irradiation at P21 should be inhibited for several weeks before returning to baseline levels by approximately P49 (Fig. 4A–E). Together, these data suggest that late-differentiating (∼3- to 5-week-old) DGCs, but not those born during the 2 weeks before or for several weeks after SE, generate much of the MFS seen at 28 d after pilocarpine treatment. Therefore, a certain level of maturity (late differentiating) is required for cells to sprout mossy fibers within the first 4 weeks after SE.
To further test the hypothesis that DGCs born after SE migrate ectopically, we immunostained for the DGC-specific marker Prox1 to examine hilar ectopic DGCs in rats receiving 6 Gy fractionated irradiation or sham irradiation on days 4 and 6 (P60/P62) after SE. Irradiated animals showed a marked reduction of hilar Prox-1-immunoreactive cells after SE compared with pilocarpine-treated, sham-irradiated controls (Fig. 6). These results are consistent with our RV-GFP findings and indicate that hilar ectopic DGCs arise from progenitors dividing after SE.
Figure 6.

Effect of irradiation (rad) given after SE on hilar ectopic DGCs. A, A′, Prox1 immunostaining 28 d after SE (on P56) shows many hilar Prox1+ cells in a rat sham irradiated 4 d after SE. B, B′, Representative images from a rat that received 6 Gy rad beginning 4 d after SE (P60/P62) shows few Prox1-immunoreactive cells in the hilus (h). gcl, Granule cell layer. Scale bar, 50 μm.
Cells born after SE show aberrant MFS with longer-term (10 week) survival after SE
Previous studies have demonstrated the progressive nature of MFS, with a peak at ∼100 d after pilocarpine-induced SE (Mello et al., 1993). The results of our initial RV labeling and irradiation experiments, however, suggest that a fairly narrow time window exists during which developing, but not fully mature or newborn, cells respond to SE by sprouting aberrantly. These findings seem inconsistent with the evidence that MFS increases for several months after SE. We therefore reasoned that the 28 d after pilocarpine treatment time point at which we examined GFP labeling and MFS may not have provided sufficient time to observe sprouting from cells generated after SE. Thus, we performed additional experiments in which adult rats underwent pilocarpine-induced SE, were either injected with RV reporter or irradiated 4 d later, and then survived for 10 weeks (instead of 4 weeks).
In the first set of experiments, rats treated with saline or pilocarpine on P56 were injected with RV-GFP on P60 and their dentate gyri analyzed for GFP expression on P126 (10 weeks after SE). Saline-treated animals, as expected, showed many GFP-labeled axons in the hilus but none in the molecular layer (Fig. 7A). Rats that underwent SE on P56, however, showed many horizontally aligned, GFP-immunoreactive processes in the inner molecular layer with the same axonal appearance and small mossy fiber bouton-like structures as those seen in the hilus (Fig. 7B,C, arrows). This finding contrasted with our previous experiments in which rats experiencing SE on P56 and RV-GFP injections on P60 showed no GFP-positive axons in the inner molecular layer by P84 (data not shown). These data indicate that DGCs generated after SE require >24 d to respond to cues in the post-SE environment by sending axons aberrantly into the dentate inner molecular layer; a duration of 10 weeks after SE, in contrast, is sufficient for the DGCs newly generated after SE to exhibit robust MFS.
Figure 7.

Cells born after SE contribute to MFS 10 weeks after pilocarpine treatment. A, GFP immunolabeling of a control injected with RV-GFP 4 d after saline treatment and killed 10 weeks later shows axonal labeling in the hilus (h) but only GFP+ dendrites in the inner molecular layer (iml). B, C, At 10 weeks after SE, animals that received pilocarpine and then RV-GFP 4 d later display many GFP+ axonal processes in the iml (arrows) that appear identical with those seen in the granule cell layer (gcl) and hilus (arrowheads). D, E, Denser iml Timm staining is seen at 10 weeks after SE in a sham-irradiated control (D) than in a rat irradiated beginning 4 d after pilocarpine treatment (E). F, Densitometric analysis of the percentage area of inferior blade gcl and molecular layer that is Timm stain-positive. A dose of 6 Gy fractionated irradiation administered 4 and 6 d after SE significantly decreased the amount of Timm staining (*p < 0.005). Scale bars: (in A) A–C, 50 μm; (in D) D, E, 200 μm; insets, 50 μm. Error bars indicate SEM.
In the second set of experiments, we again used x-irradiation to transiently suppress neurogenesis 4 and 6 d (P60 and P62) after inducing SE on P56 with pilocarpine. This time, however, Timm staining was assessed after a 10 week survival. Controls experienced SE and then sham irradiation. In sham-irradiated controls, we found dense MFS (Fig. 7D) with much darker inner molecular layer Timm staining than in similarly sham-irradiated controls at 4 weeks after SE (compare Figs. 7D, 5C). Unlike the previous experiments in which MFS at 4 weeks after SE was not decreased by post-SE irradiation, however, animals that were irradiated 4 and 6 d after pilocarpine treatment and survived 10 weeks showed a less dense band of inner molecular layer MFS than the respective sham-irradiated controls (Fig. 7D,E). Densitometric quantification of Timm staining in the inferior blade at 10 weeks after SE (Fig. 7F) showed that controls had an average of 46.7 ± 2.7% of the inferior blade granule cell and molecular layers positive for Timm staining, whereas animals irradiated at P60 and P62 after SE had significantly less granule cell and molecular layer Timm staining, with a mean of 30.2 ± 1.4% (p < 0.005). Together with the irradiation and RV reporter studies described above, these findings provide strong evidence that DGCs generated after SE contribute to MFS occurring between 4 and 10 weeks after pilocarpine treatment.
Discussion
In this study, we labeled DGC progenitors in neonatal, juvenile, or adult rats with retroviral reporters such that the cells would be at selected stages of maturity during pilocarpine-induced SE or be born after SE. We also depleted DGC progenitor cohorts by irradiating at specific time points before or after SE. We found that DGCs born 7 weeks before SE were resistant to injury and exhibited no abnormal plasticity at 4 weeks after pilocarpine treatment. RV-GFP-labeled DGCs that were 4 weeks of age at SE developed hilar basal dendrites and MFS when examined 4 weeks after SE, and those generated 2 weeks before pilocarpine treatment showed substantial hilar basal dendrites and some MFS. Many DGCs generated after SE, in contrast, migrated ectopically and developed marked hilar basal dendrites, but did not show MFS, at 4 weeks after SE. When observed 70 d after SE, however, cells born 4 d after pilocarpine treatment showed robust MFS. Along with the irradiation experiments, the data suggest a model in which DGCs show a maturation-specific vulnerability to SE-induced plasticity (Fig. 8). Mature cells are resistant to aberrant plasticity. Late-differentiating, “juvenile” DGCs (∼3–5 weeks of age) respond to injury with MFS and to a lesser extent hilar basal dendrite formation. DGCs that are more immature (≤2 weeks old) at the time of SE show less MFS but pronounced basal dendrites. Finally, DGCs born after SE develop extensive basal dendrites and migrate ectopically to the hilus by 28 d after SE and also show robust MFS by 70 d. Thus, the repertoire of plasticity in response to this epileptogenic insult appears restricted to specific critical periods in DGC development.
Figure 8.

Diagrammatic model showing the influence of granule cell maturity on vulnerability to SE-induced structural plasticity. In this model, fully mature cells (born at P7; top row) are resistant to SE-induced remodeling. Some DGCs born at P28 (4 weeks of age at SE; second row), however, show abnormal hilar basal dendrites (HBDs) (b) and MFS (s). Those born on P42 (2 weeks of age at SE; third row) display extensive HBD formation 4 weeks after SE and to a lesser extent contribute to MFS seen 4 weeks after pilocarpine treatment. DGCs born 4 d after SE (P60 injections; bottom row) exhibit both HBDs and ectopic migration without MFS at 4 weeks after SE, but they develop MFS by 10 weeks. The model assumes that changes seen at 4 weeks remain stable for RV-GFP injections before SE (top 3 rows) as only animals administered RV-GFP after SE (bottom row) were examined at the 10 week time point in this study.
Interestingly, many GFP-labeled cells appeared normal in all groups receiving RV-GFP injections before SE. The proportion of GFP-positive DGCs that either migrated ectopically or showed hilar basal dendrites after SE was on average 5.6% of those labeled at P7, 14% of those labeled at P28, and 33.7% of DGCs labeled at P42 (vs 0–5.9% for the four control groups). When RV was administered after SE (P60), however, the majority of DGCs appeared abnormal (57.4%) when observed 4 weeks after pilocarpine treatment. A single RV injection labels only a small subset of DGCs compared with the total number generated after SE (Parent et al., 1997); therefore, very large numbers of DGCs born after SE probably develop abnormally. Our results also underestimate the degree of abnormal dendrite formation of RV-labeled cells because only DGCs with normal cell body orientation were included. We excluded misoriented cells to identify basal dendrites extending into the hilus unequivocally. Furthermore, dendrites present on ectopic cells were excluded, as they were already located in the hilus. Others have demonstrated that aberrantly developing adult-born neurons stably integrate into the dentate circuitry (Scharfman et al., 2000; McCloskey et al., 2006) and that altered neurogenesis after SE is long lasting, with ectopic cells forming for many months after seizure induction (Rao et al., 2006; Jessberger et al., 2007). Thus, the extent of SE-induced neurogenesis abnormalities is likely even more severe than is suggested by our RV labeling.
The finding of MFS only by developing or newborn, and not mature, DGCs has important mechanistic implications for understanding this form of plasticity. It also helps resolve some of the controversial findings from our previous work and studies by others. We originally reported that newborn cells contributed to MFS at 35 d after SE based on double labeling for BrdU and an axon-specific marker (Parent et al., 1997). This led us to hypothesize that newborn cells played a large role in MFS. However, we subsequently demonstrated that ablating newborn cells did not alter MFS, refuting our original hypothesis (Parent et al., 1999). Our current findings indicate that this refutation was incorrect because the 4 week after SE time point studied in the irradiation experiments was too early to observe MFS by DGCs generated after SE. Our current data instead indicate that a developmental window exists for MFS and that cells born in the first few weeks after SE contribute to inner molecular layer sprouting by 70 d, but not within 28 d, after pilocarpine treatment.
Rather than recapitulating development, SE-induced MFS appears to involve alterations of ongoing development. The timing of MFS from a given DGC in relation to the maturity of that particular cell fits with the idea that DGCs develop vulnerability to a persisting SE-induced signal when they reach 3–5 weeks of age. They respond by aberrantly sprouting axons, and older DGCs appear incapable of responding to these signals. Alternatively, immature DGCs may attain responsiveness to a “sprouting signal” at an earlier age but take longer to sprout. This explanation is unlikely given that adult-born DGCs send mossy fiber axons to area CA3 within a week of birth (Hastings and Gould, 1999; Markakis and Gage, 1999). The idea that successive generations of adult-born DGCs sprout as they develop is consistent with MFS progression for several months after SE (Mello et al., 1993). In fact, the timing of transiently increased neurogenesis for 2–3 weeks after the initial seizures (Parent et al., 1997) followed by a potential chronic suppression of neurogenesis (Hattiangady et al., 2004) fits well with a model in which most or all newborn DGCs eventually sprout, leading to a peak in MFS at 2–3 months after SE.
Our data are consistent with another study that found no MFS from DGCs labeled after SE by RV-GFP injection when hippocampi were examined 4 weeks later (Jessberger et al., 2007). We also confirmed our previous finding that post-SE irradiation did not suppress MFS 4 weeks later (Parent et al., 1999). As in the current study, one group showed that RV-GFP-labeled DGCs that were 4 weeks of age at the time of SE contributed to MFS (Jessberger et al., 2007). In contrast, this group did not observe sprouting from DGCs labeled with RV-GFP 1 week after SE and examined a year later, whereas we found robust MFS from DGCs born after SE when assayed after 10 (but not 4) weeks. They also observed that RV reporter injections 1 week before SE did not label DGCs with morphological alterations. Although we did not inject RV reporter at 1 week before SE, we found that many DGCs born 2 weeks before or 4 d after SE developed hilar basal dendrites. Several possibilities may explain these discrepancies, including our use of a different rat strain and of pilocarpine rather than kainic acid. An unlikely possibility is that the MFS we found from RV-GFP-labeled cells 10 weeks after SE may not persist for a full year. Our hilar basal dendrite findings are similar to data generated in a study of Thy1-GFP transgenic mice showing that DGCs born several months before SE do not develop hilar basal dendrites, whereas many DGCs that are immature during SE do form them (Walter et al., 2007).
We found that irradiation before SE does not change the latency to or severity of SE, but increases SE mortality. This effect probably reflects a tendency for irradiated rats to have more discrete generalized convulsive seizures. Potential nonspecific damage to the dentate gyrus from ionizing irradiation is thus one possible limitation. We therefore interpret the data in the context of our RV reporter labeling strategy, thus combining findings from progenitor labeling and ablation methods to reach our conclusions. We also did not monitor seizure frequency. It will be interesting to determine whether transiently suppressing neurogenesis with low-dose irradiation at specific time points chronically suppresses SE-induced structural abnormalities and whether this will also attenuate epileptogenesis.
Accumulating data support the hypothesis that MFS, hilar ectopic DGCs, and DGCs with hilar basal dendrites contribute to epileptogenesis (Tauck and Nadler, 1985; Ribak et al., 2000; Scharfman et al., 2000, 2002, 2003; Buckmaster et al., 2002; Jung et al., 2004, 2006; Morgan and Soltesz, 2008). The overall contribution of adult-born DGCs to epileptogenesis, however, remains unclear. Adult-born DGCs integrating after SE appear to have complex influences, as recordings from RV-GFP-labeled DGCs in the granular layer after electrically induced SE suggest that they show reduced excitability (Jakubs et al., 2006). These effects may relate to the substantial proportion of RV-GFP-labeled DGCs that appear to integrate normally at the time points we examined using the pilocarpine model, although electrophysiological studies are needed to determine whether reduced excitability of adult-born DGCs also occurs in this model. In support of an epileptogenic role of seizure-induced neurogenesis, one study showed that DGC progenitor ablation with mitotic inhibitors exerts partial antiepileptogenic effects after SE (Jung et al., 2004). Future work should aim to determine the functional implications of suppressing neurogenesis before or after SE by correlating morphological abnormalities with seizures, although such studies may be complicated because ablation methods suppress both aberrant and potentially compensatory neurogenesis. Because the birth of DGCs that migrate ectopically or integrate normally likely temporally overlap, a potential challenge is the ability to ablate one of these populations without also suppressing the other. Interventions that normalize, rather than suppress, neurogenesis should be ideal in this regard.
Footnotes
This work was supported by the Fred Annegers Fellowship from the Epilepsy Foundation (M.M.K.) and the Citizens United for Research in Epilepsy (J.M.P.). We thank F. Gage and S. Jessberger for the viral vector, M. Davis and the Irradiation Core for help with the irradiation experiments, and C. Collins and A. Fedorko for technical assistance.
References
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997;385:385–404. [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurosci. 1999;81:712–721. doi: 10.1152/jn.1999.81.2.712. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Zhang GF, Yamawaki R. Axon sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit. J Neurosci. 2002;22:6650–6658. doi: 10.1523/JNEUROSCI.22-15-06650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashtipour K, Tran PH, Okazaki MM, Nadler JV, Ribak CE. Ultrastructural features and synaptic connections of hilar ectopic granule cells in the rat dentate gyrus are different from those of granule cells in the granule cell layer. Brain Res. 2001;890:261–271. doi: 10.1016/s0006-8993(00)03119-x. [DOI] [PubMed] [Google Scholar]
- Dashtipour K, Wong AM, Obenaus A, Spigelman I, Ribak CE. Temporal profile of hilar basal dendrite formation on dentate granule cells after status epilepticus. Epilepsy Res. 2003;54:141–151. doi: 10.1016/s0920-1211(03)00082-2. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. 2004;3:663–672. doi: 10.1016/S1474-4422(04)00906-8. [DOI] [PubMed] [Google Scholar]
- Hastings NB, Gould E. Rapid extension of axons into the CA3 region by adult-generated granule cells. J Comp Neurol. 1999;413:146–154. doi: 10.1002/(sici)1096-9861(19991011)413:1<146::aid-cne10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol Dis. 2004;17:473–490. doi: 10.1016/j.nbd.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Heinrich C, Nitta N, Flubacher A, Müller M, Fahrner A, Kirsch M, Freiman T, Suzuki F, Depaulis A, Frotscher M, Haas CA. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J Neurosci. 2006;26:4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter C, Kurthen M, Lux S, Reuber M, Elger CE. Chronic epilepsy and cognition: a longitudinal study in temporal lobe epilepsy. Ann Neurol. 2003;54:425–432. doi: 10.1002/ana.10692. [DOI] [PubMed] [Google Scholar]
- Higashikawa F, Chang L. Kinetic analyses of stability of simple and complex retroviral vectors. Virology. 2001;280:124–131. doi: 10.1006/viro.2000.0743. [DOI] [PubMed] [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV. Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci. 1990;10:267–282. doi: 10.1523/JNEUROSCI.10-01-00267.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron. 2006;52:1047–1059. doi: 10.1016/j.neuron.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Zhao C, Toni N, Clemenson GD, Jr, Li Y, Gage FH. Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J Neurosci. 2007;27:9400–9407. doi: 10.1523/JNEUROSCI.2002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KH, Chu K, Kim M, Jeong SW, Song YM, Lee ST, Kim JY, Lee SK, Roh JK. Continuous cytosine-b-d-arabinofuranoside infusion reduces ectopic granule cells in adult rat hippocampus with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Eur J Neurosci. 2004;19:3219–3226. doi: 10.1111/j.0953-816X.2004.03412.x. [DOI] [PubMed] [Google Scholar]
- Jung KH, Chu K, Lee ST, Kim J, Sinn DI, Kim JM, Park DK, Lee JJ, Kim SU, Kim M, Lee SK, Roh JK. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol Dis. 2006;23:237–246. doi: 10.1016/j.nbd.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Markakis EA, Gage FH. Adult-generated neurons in the dentate gyrus send axonal projections to field CA3 and are surrounded by synaptic vesicles. J Comp Neurol. 1999;406:449–460. [PubMed] [Google Scholar]
- McCloskey DP, Hintz TM, Pierce JP, Scharfman HE. Stereological methods reveal the robust size and stability of ectopic hilar granule cells after pilocarpine-induced status epilepticus in the adult rat. Eur J Neurosci. 2006;24:2203–2210. doi: 10.1111/j.1460-9568.2006.05101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Cummings JL, Benson DF. Depression in epilepsy. significance and phenomenology. Arch Neurol. 1986;43:766–770. doi: 10.1001/archneur.1986.00520080014012. [DOI] [PubMed] [Google Scholar]
- Morgan RJ, Soltesz I. Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc Natl Acad Sci U S A. 2008;105:6179–6184. doi: 10.1073/pnas.0801372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta N, Heinrich C, Hirai H, Suzuki F. Granule cell dispersion develops without neurogenesis and does not fully depend on astroglial cell generation in a mouse model of temporal lobe epilepsy. Epilepsia. 2008;49:1711–1722. doi: 10.1111/j.1528-1167.2008.01595.x. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bromberg DA, Bensen AL, Westbrook GL. Seizures accelerate functional integration of adult-generated granule cells. J Neurosci. 2006;26:4095–4103. doi: 10.1523/JNEUROSCI.5508-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Tada E, Fike JR, Lowenstein DH. Inhibition of dentate granule cell neurogenesis with brain irradiation does not prevent seizure-induced mossy fiber synaptic reorganization in the rat. J Neurosci. 1999;19:4508–4519. doi: 10.1523/JNEUROSCI.19-11-04508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Valentin VV, Lowenstein DH. Prolonged seizures increase proliferating neuroblasts in the adult rat subventricular zone-olfactory bulb pathway. J Neurosci. 2002;22:3174–3188. doi: 10.1523/JNEUROSCI.22-08-03174.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol. 2006;59:81–91. doi: 10.1002/ana.20699. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Reddy DS, Shetty AK. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. 2006;83:1088–1105. doi: 10.1002/jnr.20802. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Tran PH, Spigelman I, Okazaki MM, Nadler JV. Status epilepticus-induced hilar basal dendrites on rodent granule cells contribute to recurrent excitatory circuitry. J Comp Neurol. 2000;428:240–253. doi: 10.1002/1096-9861(20001211)428:2<240::aid-cne4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci. 2000;20:6144–6158. doi: 10.1523/JNEUROSCI.20-16-06144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Goodman JH. Spontaneous recurrent seizures after pilocarpine-induced status epilepticus activate calbindin-immunoreactive hilar cells of the rat dentate gyrus. Neuroscience. 2002;111:71–81. doi: 10.1016/s0306-4522(01)00599-1. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AE, Berger RE, Goodman JH, Pierce JP. Perforant path activation of ectopic granule cells that are born after pilocarpine-induced seizures. Neuroscience. 2003;121:1017–1029. doi: 10.1016/s0306-4522(03)00481-0. [DOI] [PubMed] [Google Scholar]
- Spigelman I, Yan XX, Obenaus A, Lee EY, Wasterlain CG, Ribak CE. Dentate granule cells form novel basal dendrites in a rat model of temporal lobe epilepsy. Neuroscience. 1998;86:109–120. doi: 10.1016/s0306-4522(98)00028-1. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Dudek FE. Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog Brain Res. 2007;163:541–563. doi: 10.1016/S0079-6123(07)63029-5. [DOI] [PubMed] [Google Scholar]
- Tada E, Parent JM, Lowenstein DH, Fike JR. X-irradiation causes a prolonged reduction in cell proliferation in the dentate gyrus of adult rats. Neuroscience. 2000;99:33–41. doi: 10.1016/s0306-4522(00)00151-2. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter C, Murphy BL, Pun RY, Spieles-Engemann AL, Danzer SC. Pilocarpine-induced seizures cause selective time-dependent changes to adult-generated hippocampal dentate granule cells. J Neurosci. 2007;27:7541–7552. doi: 10.1523/JNEUROSCI.0431-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan XX, Spigelman I, Tran PH, Ribak CE. Atypical features of rat dentate granule cells: recurrent basal dendrites and apical axons. Anat Embryol (Berl) 2001;203:203–209. doi: 10.1007/s004290000150. [DOI] [PubMed] [Google Scholar]