

Abstract
Rearrangement of the actin cytoskeleton is essential for dynamic cellular processes. Decreased actin turnover and rigidity of cytoskeletal structures have been associated with aging and cell death. Gelsolin is a Ca2+-activated actin-severing protein that is widely expressed throughout the adult mammalian brain. Here, we used gelsolin-deficient (Gsn−/−) mice as a model system for actin filament stabilization. In Gsn−/− mice, emigration of newly generated cells from the subventricular zone into the olfactory bulb was slowed. In vitro, gelsolin deficiency did not affect proliferation or neuronal differentiation of adult neural progenitors cells (NPCs) but resulted in retarded migration. Surprisingly, hippocampal neurogenesis was robustly induced by gelsolin deficiency. The ability of NPCs to intrinsically sense excitatory activity and thereby implement coupling between network activity and neurogenesis has recently been established. Depolarization-induced [Ca2+]i increases and exocytotic neurotransmitter release were enhanced in Gsn−/− synaptosomes. Importantly, treatment of Gsn−/− synaptosomes with mycotoxin cytochalasin D, which, like gelsolin, produces actin disassembly, decreased enhanced Ca2+ influx and subsequent exocytotic norepinephrine release to wild-type levels. Similarly, depolarization-induced glutamate release from Gsn−/− brain slices was increased. Furthermore, increased hippocampal neurogenesis in Gsn−/− mice was associated with a special microenvironment characterized by enhanced density of perfused vessels, increased regional cerebral blood flow, and increased endothelial nitric oxide synthase (NOS-III) expression in hippocampus. Together, reduced filamentous actin turnover in presynaptic terminals causes increased Ca2+ influx and, subsequently, elevated exocytotic neurotransmitter release acting on neural progenitors. Increased neurogenesis in Gsn−/− hippocampus is associated with a special vascular niche for neurogenesis.
Introduction
Cytoskeletal remodeling in response to stress constitutes a fundamental adaptive mechanism in eukaryotic cells: a less dynamic actin cytoskeleton is unable to respond effectively to changing internal and external demands (Gourlay and Ayscough, 2005). Whereas increased turnover of filamentous (F)-actin promotes longevity, decreased actin turnover and rigidity of cytoskeletal structures have been associated with aging and cell death (Wang and Gundersen, 1984; Prinjha et al., 1994; Gonos et al., 1998; Gourlay and Ayscough, 2005). Defects of the cytoskeleton have also been implicated in the etiopathogenesis of neurodegenerative conditions such as Alzheimer's dementia, Huntington's disease, fragile X mental retardation syndrome, severe epileptic seizures, and stroke (Furukawa et al., 1997; Endres et al., 1999; Fulga et al., 2007; Lynch et al., 2008).
Gelsolin is a Ca2+-activated actin binding protein that severs F-actin filaments by breaking noncovalent bonds between actin monomers in a polymer. This results in high-affinity complexes of gelsolin, which remains bound to the barbed ends of filaments inhibiting extension (“capped” filaments) (Janmey and Stossel, 1987; Kinosian et al., 1998). On a reduction in free intracellular Ca2+ levels and in the presence of polyphosphoinositides, gelsolin is released from the barbed ends, providing sites for rapid actin filament extension (Janmey and Stossel, 1987). Gelsolin is the most potent actin filament severing protein identified to date (Sun et al., 1999). Gsn−/− mice therefore represent an excellent model system for dissecting the physiological sequelae of actin filament stabilization on key aspects of cellular plasticity in the brain. So far, histological analyses of the brain of Gsn−/− mice have yielded no gross or microscopic anatomic abnormalities (Endres et al., 1999). We have previously demonstrated antiapoptotic and neuroprotective effects of gelsolin, which can also be harnessed pharmacologically (e.g., by inhibition of histone deacetylation) (Endres et al., 1999; Harms et al., 2004; Meisel et al., 2006; Yildirim et al., 2008).
In mammals, including humans, neurons are continually generated in two distinct brain regions during adulthood. Hippocampal neurogenesis is locally restricted to the dentate gyrus where newly generated cells remain relatively stable in their position in the granule cell layer (Kempermann et al., 2003). In contrast, olfactory bulb (OB) neurogenesis occurs in a long-distance migratory system spanning several millimeters in the adult rodent forebrain (Ninkovic and Götz, 2007). This neurogenic system originates in the subventricular zone (SVZ) of the lateral ventricle wall from where neuroblasts migrate via the rostral migratory stream (RMS) to differentiate into interneurons in the olfactory bulb.
We here show that emigration of newly generated cells from the SVZ into the olfactory bulb is reduced in Gsn−/− mice. Similarly, in vitro, neural progenitor cells (NPCs) derived from Gsn−/− mice showed reduced migratory capacity, whereas neuronal differentiation and proliferation kinetics did not differ between genotypes. In contrast, and unexpectedly, we found that hippocampal neurogenesis is mostly increased in Gsn−/− mice. This finding prompted additional investigation into the effects of impaired actin filament turnover on the extracellular milieu, specifically on neurotransmission and on key aspects of the vascular microenvironment, in which neurogenesis takes place.
Materials and Methods
Animals and treatments
All experimental procedures conformed to institutional guidelines and were approved by an official committee. Mice expressing green fluorescent protein (GFP) under nestin gene regulatory elements have been described in detail previously (Yamaguchi et al., 2000; Fukuda et al., 2003; Kronenberg et al., 2003; Glass et al., 2005). Gsn−/− mice were generated as described previously (Witke et al., 1995). The gsn-null state is not viable in either pure C57BL/6 or BALB/c backgrounds (Witke et al., 1995). We therefore used gender-matched littermates from a mixed 129/SV × BALB/c background (Endres et al., 1999; Harms et al., 2004). Tail snips were used to prepare DNA by standard methods for genotyping (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). All animals were kept on a 12 h light/dark schedule with ad libitum access to food and water. Nucleoside analog 5-bromo-2-deoxyuridine (BrdU) was administered intraperitoneally at a dose of 50 μg/g body weight at a concentration of 10 mg/ml (Kronenberg et al., 2003).
Cell cultures
Culture methods for neural precursors and the G261 cell line were essentially as described previously (Glass et al., 2005). Briefly, the SVZ of 2- to 4-week-old Gsn−/− and Gsn+/+ mice was microdissected, and neurospheres were cultured in Neurobasal medium supplemented with B27 (both from Invitrogen), 20 ng/ml recombinant human epidermal growth factor (EGF), and 20 ng/ml human recombinant fibroblast growth factor 2 (both from R&D Systems). The ability of cells to generate neurons and glia was routinely checked. For differentiation, neurospheres were spun down, triturated, and dissociated. Dissociated cells were plated on poly-l-lysine-coated glass coverslips at a density of 32,000 cells/cm2 and cultured for 7 d without growth factors.
Matrigel assay
Gsn+/+ and Gsn−/− neurospheres were seeded into 24-well cell culture plates at the same density. Briefly, neurospheres were spun down and resuspended in 150 μl of NeuroCult NSC Basal Medium (StemCell Technologies) with 10% proliferation supplement, 0.0002% heparin, 20 ng/ml EGF, and 10 ng/ml FGF, and embedded in Matrigel (1:1 dilution). After incubation at 37°C for 30 min, an additional 200 μl of medium per well was added. Cells were cultured for 20 h. Images of individual neurospheres were taken directly after embedding (0 h) as well as at 20 h. The rim of each neurosphere was defined by the image taken at 0 h. The greatest distance a cell had migrated out of the sphere at 20 h was recorded using LAS software (Leica).
Modified Boyden chamber assay
G261 glioblastoma cells were seeded into 24-well plates at a density of 4 × 104/cm2. After 24 h, FluoroBlok inserts (pore size, 8 μm; BD Biosciences) were placed into the wells and NPCs (26,000) were added to each top chamber. After 24 h of incubation, the membranes of the inserts were stained with 50 μm CFSE dye (Sigma-Aldrich), and then fixed with 4% paraformaldehyde (PFA) and counterstained with 2 μm 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Migrated cells below the FluoroBlok membranes were visualized using an inverted fluorescence microscope (Leica DMI3000). Rate of NPC migration was calculated by counting cells in 16 random microscope fields of each well (at 200× magnification).
Tissue preparation
After an overdose of anesthetics, animals were transcardially perfused with physiological saline followed by 4% PFA in 0.1 m phosphate buffer, pH 7.4. Brains were dissected from the skulls and postfixed overnight. Before sectioning from a dry ice-cooled copper block on a sliding microtome (Leica), the brains were transferred to 30% sucrose in 0.1 m phosphate buffer, pH 7.4, until they sank. Brains were cut in the coronal plane in 40-μm-thick sections. Sections were stored at −20°C in cryoprotectant solution (25% ethylene glycol, 25% glycerol, and 0.05 m phosphate buffer).
Histological procedures
Sections were stained using free-floating immunohistochemistry and prepared for BrdU detection by incubation with 2N HCl for 30 min at 37°C as described in detail previously (Kronenberg et al., 2003). Neurospheres embedded in Matrigel were stained with a preformed complex of primary and secondary antibodies. Primary antibodies were applied in the following concentrations: anti-BrdU (rat; 1:500; Harlan Sera-Lab), anti-calretinin (CR) (rabbit; 1:250; Swant), anti-doublecortin (DCX) (goat; 1:200; Santa Cruz Biotechnology), anti-gelsolin (rabbit; 1:250) (Azuma et al., 1998), anti-GFAP (rabbit; 1:1000; Dako), anti-S100β (rabbit; 1:2500; Swant), anti-NeuN (mouse; 1:100; Millipore Bioscience Research Reagents), and anti-TuJ1 (rabbit; 1:1000; Covance).
Immmunohistochemistry.
Immmunohistochemistry followed the peroxidase method with biotinylated secondary antibodies (all 1:500; Jackson ImmunoResearch Laboratories), ABC Elite reagent (Vector Laboratories), and DAB (diaminobenzidine) (Sigma-Aldrich) as chromogen.
Immunofluorescence.
For immunofluorescence, FITC-, RhodX-, or Cy5-conjugated secondary antibodies were all used at a concentration of 1:250. Fluorescent sections were coverslipped in polyvinyl alcohol with DABCO (diazabicyclooctane) as antifading agent.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining was performed with the In Situ Cell Death Detection kit, TMR red (Roche Applied Science), according to the manufacturer's protocol.
Quantification and imaging
Hippocampal cell counts.
Hippocampal cell counts were determined in one-in-six series of sections covering the entire hippocampus in its rostrocaudal extension as described previously (Kronenberg et al., 2003, 2007). Briefly, cells located in the granule cell layer and adjacent subgranular zone, defined as a two-cell bodies-wide zone of the hilus along the base of the granule cell layer were counted (Brown et al., 2003). Cells in the uppermost focal plane were excluded to avoid oversampling.
BrdU+ cells in the lateral ventricle wall.
Similarly, BrdU+ cells in the lateral ventricle wall were determined in every sixth section from the appearance of the third ventricle to the disappearance of the anterior commissure.
BrdU+ cells in the olfactory bulb.
BrdU+ cells in the olfactory bulb were quantified bilaterally at every third section using the Fractionator probe as implemented in StereoInvestigator software (MicroBrightField). A counting frame of 100 × 100 μm and a grid of 250 × 250 μm were used.
TUNEL+ cells in the rostral migratory stream.
TUNEL+ cells in the rostral migratory stream were sampled in parasagittal brain sections under a fluorescent microscope, and numerical density was calculated by dividing the number of TUNEL+ nuclei by the reference space. The reference space was delineated by DCX immunoreactivity. Phenotypic analysis of BrdU-labeled cells was performed using a spectral confocal microscope (TCS SP2; Leica). Appropriate gain and black level settings were determined on control slices stained with secondary antibodies alone.
[Ca2+]i measurements in neocortical and hippocampal synaptosomes
Synaptosomes were prepared as described previously (Fink et al., 2002a,b) from the neocortex or hippocampus of adult Gsn+/+ or Gsn−/− mice. Briefly, brain tissue was homogenized using a Potter–Elvehjem glass homogenizer (800 rpm) in 40 vol (w/v) of 320 mm sucrose. The homogenate was centrifuged (10 min; 1000 × g at 4°C) to remove nuclei and debris. The supernatant was then centrifuged at 12,000 × g for 10 min. The buffy layer of pelleted synaptosomes was resuspended by gentle agitation in Ca2+-free physiological salt solution (PSS) [composition (in mm): 133 NaCl, 4.8 KCl, 10 HEPES, 1.2 Na2HPO4, 1.2 MgSO4, 10 glucose, pH 7.4]. The synaptosomal suspension (2 ml; ∼3 mg of protein/ml) was incubated with fura-2 AM (5 μm) for 40 min at 37°C with gentle shaking. Fura-2-loaded synaptosomes were centrifuged at 1300 × g, and the pellet was washed with Ca2+-free PSS and centrifuged again. The pellet was then resuspended and stored on ice until use. Aliquots (200 μl) of the washed synaptosomal suspension were diluted with 1.8 ml of PSS (final protein concentration, 125 ± 4 μg/ml) containing 1.3 mm CaCl2 and placed in a quartz cuvette at 37°C. The synaptosomes were kept in suspension with a magnetic stirrer and incubated for 6 min before K+ was elevated by 30 mm for membrane depolarization. Fluorescence (λex = 340/380 nm; λem = 510 nm) was measured with a spectrofluorometer (PerkinElmer LS50B). [Ca2+]i was calculated according to Grynkiewicz et al. (1985) as described in detail previously (Fink et al., 2002a,b).
Stimulated release of glutamate and norepinephrine
Brain slices (0.3 mm thick; diameter in neocortex, 2 mm; in hippocampus, 1.5 mm; in striatum, 1 mm) were prepared and incubated for 30 min in a buffer consisting of the following (in mm): 118 NaCl, 4.8 KCl, 25 NaHCO3, 1.2 KH2PO4, 1.3 CaCl2, 1.2 MgSO4, 11 glucose, 0.06 ascorbic acid, 0.03 disodium EDTA, equilibrated with 95% O2 and 5% CO2. Synaptosomes were prepared by homogenization in 320 mm sucrose, centrifugation at 1000 × g for 10 min, and incubation of 9 ml of the supernatant with 6 ml of Krebs' buffer for 7 min at 37°C. After addition of tritium-labeled norepinephrine ([3H]NE) (50 nm; specific activity, 46.8 Ci/mmol), incubation was continued for 7 min. Labeled synaptosomes were pelleted at 600 × g for 10 min and resuspended in 2.25 ml of ice-cold Krebs' buffer (final protein content, 4656 ± 155 μg/ml). Slices or aliquots of synaptosomal suspensions were layered on Whatman GF/C or GF/B filters in chambers and superfused at 0.6 ml/min with Krebs' buffer. Neurotransmitter release was evoked by K+ elevation (15 mm to induce glutamate release from neocortical, hippocampal, or striatal slices; 9–30 mm to induce [3H]NE release from neocortical synaptosomes) for 2 min. The superfusate was continuously collected in 4 min (synaptosomes) or 5 min (slices) fractions. Tritium content of the superfusate fractions was determined by liquid scintillation counting and stimulation-evoked tritium overflow calculated as described previously (Fink et al., 1989, 1990). On completion of measurements, slices were removed from the chambers and weighed. Samples and glutamate standards were subjected to precolumn o-phthaldialdehyde derivatization and fluorometrically determined after HPLC separation.
Western blotting
Tissue was homogenized in 10 μl/mg tissue lysis buffer [50 mm Tris/HCl, pH 7.5, 120 mm NaCl, 5 mm EDTA, 0.5% NP-40, 10 mm Na4P2O7, 2 mm Na3VO4, 100 mm NaF, 1× Protease Inhibitor Complete (Roche), 10 μg/ml PMSF, 1 mm DTT], incubated on ice for 20 min, and centrifuged at 10 min, 14,000 rpm at 4°C. For NOS-III Western blotting tissue was homogenized in 50 mm Tris-HCl lysis buffer, pH 7.5, containing 1 mm EDTA, 0.25 m sucrose, 20 mm CHAPS [3-(3-chloramidopropyl)-dimethylammonio-1-propanesulfonate], and Protease Inhibitor Complete (Roche). Crude membranes were prepared by centrifugation at 4°C and 1000 × g for 10 min followed by spinning the supernatant at 100,000 × g for 60 min. Cytosolic fractions were collected, and the final pellet containing the membrane fraction was resuspended in buffer. Protein concentrations were determined by BCA Protein Assay (Pierce). Equal amounts of protein were loaded on 4–20% or, for NOS-III, 7.5% Tris-HEPES gels (Pierce) and blotted onto PVDF (polyvinylidene difluoride) membrane. Blots were probed with the following antibodies: goat anti-actin (Santa Cruz; 1:2000), rabbit anti-cofilin (Cell Signaling; 1:750), rabbit anti-phospho-cofilin (Cell Signaling; 1:200), rabbit anti-gelsolin (1:10,000) (Azuma et al., 1998), rabbit anti-serum response factor (SRF) (Abcam; 1:1000), monoclonal anti-eNOS (BD Biosciences Transduction Laboratories; 1:2500), monoclonal anti-GAPDH (Millipore Bioscience Research Reagents; 1:80,000), monoclonal anti-α-tubulin (Sigma-Aldrich; 1:5000), donkey anti-goat HRP (Santa Cruz; 1:5000), donkey anti-rabbit HRP (GE Healthcare; 1:5000), goat anti-mouse HRP (Bio-Rad; 1:10,000), and rabbit anti-mouse HRP (Sigma-Aldrich; 1:10,000).
Quantification of cerebral neurotransmitter levels
Neurotransmitter levels were analyzed using methods described previously (Chourbaji et al., 2008; Kronenberg et al., 2008). Briefly, hippocampus, striatum, and olfactory bulb were dissected on a cold plate (−16°C) according to Franklin and Paxinos (1997). Serotonin (5-HT) and 5-hydroxyindoleacetic acid (5-HIAA) were analyzed using HPLC with electrochemical detection as described in detail previously (Sperk, 1982). NE was measured by HPLC with electrochemical detection after extraction to alumina according to a previously published protocol (Felice et al., 1978) with minor modifications (Sperk et al., 1981). Glutamate, GABA, and taurine were precolumn derivatized with o-phthalaldehyde-2-mercaptoethanol using a refrigerated autoinjector and then separated on an HPLC column (ProntoSil C18 ace-EPS; 50 × 3 mm inner diameter; VDS Optilab) at a flow rate of 0.6 ml/min and a column temperature of 40°C. The mobile phase was 50 mm sodium acetate, pH 5.7, in a linear gradient from 5 to 21% acetonitrile. Derivatized amino acids were detected by their fluorescence at 450 nm after excitation at 330 nm (Piepponen and Skujins, 2001).
Determination of neurotrophin levels
After decapitation, brains were quickly removed and tissue samples stored at −80°C until homogenization. Frozen tissue samples were homogenized by ultrasonication in 20–50 vol of lysing buffer containing 0.1 m Tris-HCl, pH 7.0, 0.4 m NaCl, 0.1% NaN3, and a variety of protease inhibitors as contained in Protease Inhibitor Tablets (Complete; purchased from Roche Diagnostics) and was stored at −80°C until analysis. Endogenous levels of NGF were measured in the rethawed homogenates using a highly sensitive and specific fluorometric two-site ELISA as described in detail previously (Hellweg et al., 1989, 2003). Endogenous levels of BDNF were measured using commercial ELISA kits in principle according to the manufacturer's instructions (Promega) but adapted to the fluorometric technique also used for NGF measurements. Neurotrophin levels are given as picograms per milligram of tissue (wet weight).
mRNA isolation and PCRs
Dissected brain regions were stored at −80°C. Tissues were homogenized, and total RNA was extracted using TRIzol reagent (Invitrogen). For PCR amplification, we used gene-specific primers (Table 1) and Light Cycler FastStart DNA Master SYBR Green I (Roche Diagnostics). PCR conditions were as follows: preincubation, 95°, 10 min; 95°, 15 s, primer specific annealing temperature, 10 s, 72°, 15 s (45 cycles). Crossing points of amplified products were determined using the Second Derivative Maximum Method (Light Cycler, version 3.5; Roche). Quantification of mRNA expression was relative to GAPDH. Specificity of PCR products was checked using melting curve analysis and electrophoresis in a 1.5% agarose gel.
Table 1.
Primer sequences
| Gene | Sense | Antisense |
|---|---|---|
| SRF | GCTACACGACCTTCAGCAAGAG | CAGGTAGTTGGTGATGGGGAAG |
| rodAct (β-actin) | ACCCACACTGTGCCCATCTA | GCCACAGGATTCCATACCCA |
| GAPDH | AGATTGTCAGCAATGCATCCTGC | CCTTCTTGATGTCATCATACTTGG |
| eNos3 | CAGGACTGCACAGGAAATGTTC | AGCACATCAAAGCGGCCATTTC |
| bFGF | CAACCGGTACCTTGCTATGAAG | CGTTTCAGTGCCACATACCAAC |
| Primer sequences for genotype analysis | ||
| Gsn+/+: 280 bp (Grohé et al., 2004) | ||
| Gsn+1/+267 | GTGGAGCACCCCGAATT | CTCAGTTCAGGTATATCCATATCCATACAG |
| Gsn−/−: 480 bp (Witke et al. 1995) | ||
| Neo-3/-4 | ATTGAACAAGATGGATTGCAC | CGTCCAGATCATCCTGAT |
Cerebral blood flow measurements
Cerebral blood flow (CBF) measurements were performed using the 14C-iodoantipyrine technique under etomidate anesthesia (0.03 mg/kg body weight per minute; Braun) as described previously (Endres et al., 2003; Gertz et al., 2006).
Density of perfused vessels
Evans blue (Sigma-Aldrich; 2% in saline) was administered intravenously and allowed to circulate for 5 min under etomidate anesthesia. Animals were decapitated and brains cut into 10 μm coronal cryostat sections and digitized with a cooled CCD camera (Dage-MTI), which was attached to a fluorescence microscope. Images of whole-brain sections at microscopic resolution were obtained by joining together single camera images using tiled-field mapping software (MCID Elite; InterFocus). Regions of interest were specified with the technique of density slicing, including the setting of target acceptance criteria (Göbel et al., 1990).
Statistical analyses
Experiments were performed in a blinded manner. Numerical analyses were performed with Statview 5.0.1 for MacIntosh. Values are presented as means ± SEM. Groups were compared by ANOVA with level of significance set at 0.05 and two-tailed p values.
Results
Gelsolin is expressed in adult neural progenitor cells
Gelsolin is widely expressed throughout the CNS, and its expression by neurons is well established (Tanaka et al., 1993; Furukawa et al., 1997; Star et al., 2002; Yildirim et al., 2008). Here, gelsolin immunoreactivity was studied in transgenic mice expressing the GFP under elements of the nestin promoter that restrict reporter gene expression to neural tissue (Yamaguchi et al., 2000; Fukuda et al., 2003; Kronenberg et al., 2003). Nestin is an intermediate filament protein of neural stem and progenitor cells (Lendahl et al., 1990). Nestin-GFP mice have been used extensively as a tool to characterize NPCs in the adult brain (Filippov et al., 2003; Fukuda et al., 2003; Kronenberg et al., 2003, 2005; Tozuka et al., 2005). Immunohistochemistry of hippocampus and of subventricular zone showed gelsolin expression in nestin-GFP-expressing cells in both neurogenic areas (Fig. 1A,B). Western blot analysis verified lack of gelsolin in Gsn−/− cortex and hippocampus (Fig. 1C). Immunocytochemistry of cultured NPCs derived from Gsn+/+ mice revealed gelsolin immunoreactivity in processes and cell bodies. In contrast, NPCs from Gsn−/− mice lacked gelsolin expression (Fig. 1D).
Figure 1.
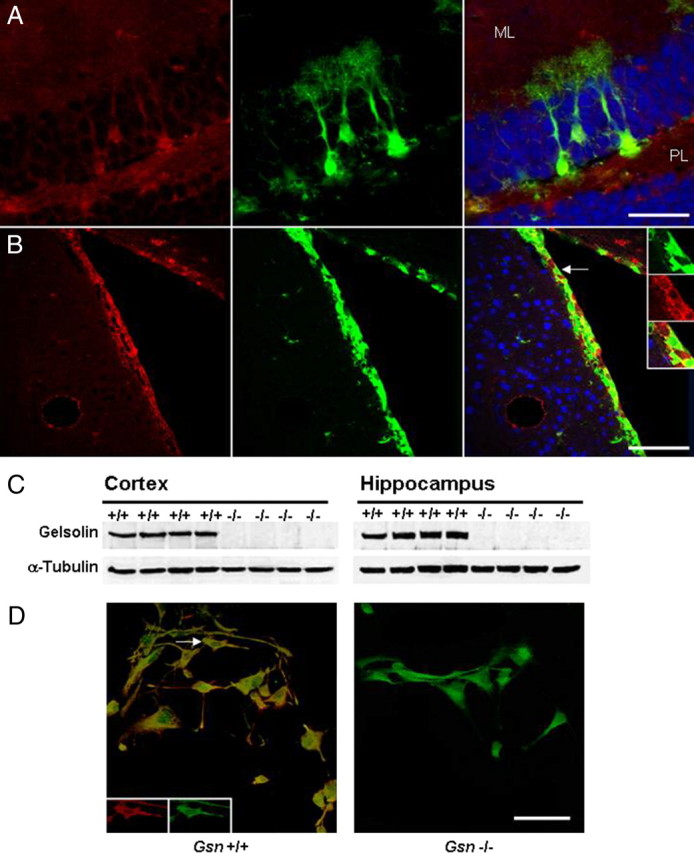
Gelsolin expression in adult neural progenitor cells. Characterization of gelsolin expression (red) in the hippocampal dentate gyrus (A) and in the subventricular zone (B) of 3- to 4-month-old nestin-GFP (green) reporter mice. Blue, Neuronal marker NeuN. A, Confocal image demonstrating gelsolin expression in the cell bodies and processes of radial glia-like nestin-GFP cells in the hippocampal dentate gyrus. Note that, in the granule cell layer, gelsolin is also expressed in neurons. Furthermore, there is widespread gelsolin staining in the neuropil of the molecular layer (ML) and of the polymorphic layer (PL). B, Gelsolin expression in neural progenitors of the subventricular zone. The insets show higher magnification of the nestin-GFP cell marked by arrow. C, Western blot analysis of protein extracts with gelsolin antibody confirms lack of gelsolin in Gsn−/− mice. D, Whereas cultures of neural progenitors derived from Gsn+/+ mice show gelsolin immunoreactivity, neural progenitors from Gsn−/− mice lack gelsolin. Green, Nestin protein. The insets show cell marked by arrow in single channels with separate wavelengths. Scale bars: A, 50 μm; B, 100 μm; D, 50 μm.
Increased hippocampal neurogenesis in Gsn−/− mice
Net hippocampal neurogenesis was assessed 4 weeks after a 7 d course of daily intraperitoneal BrdU injections in young adult (4–5 months) Gsn−/− mice and littermate controls. In this injection paradigm, BrdU counts reflect a combination of initial proliferation and subsequent survival of newly generated cells. BrdU cell counts in the hippocampal dentate gyrus were significantly increased in Gsn−/− compared with wild-type mice (319.2 ± 33 vs 85.2 ± 10.8; F(1,18) = 45.3; p < 0.0001). Since the overall number of BrdU-labeled cells was very low, only 25 (wild type) or 50 (Gsn−/−) BrdU+ cells were used for additional phenotypic analysis, respectively (Fig. 2A,B). The majority of cells analyzed showed colabeling for neuronal marker NeuN with no apparent difference between groups (Gsn−/−, 77 ± 3.1%; wild type, 72.6 ± 4.5%). Net neurogenesis, gliogenesis, and production of undetermined cells are derived by multiplying the numbers of BrdU-labeled cells by the ratio of phenotypes for each animal. Since BrdU cell counts were significantly increased in Gsn−/− mice, net neurogenesis and net gliogenesis (i.e., generation of S100β+ astrocytes) were also significantly increased in Gsn−/− animals (F(1,18) = 31.5, p < 0.0001; F(1,18) = 11, p < 0.01, respectively).
Figure 2.
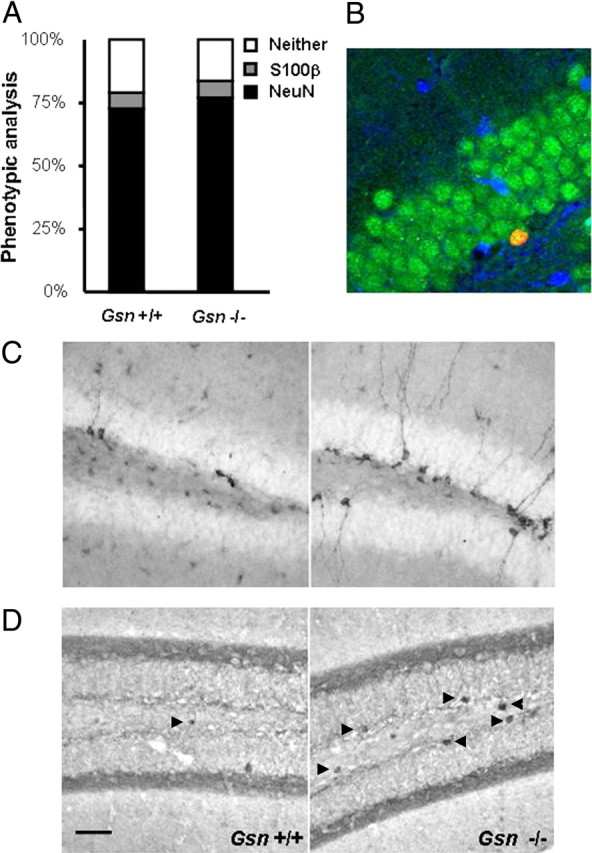
Increased hippocampal neurogenesis in Gsn−/− mice. A, B, Hippocampal neurogenesis was assessed in 4- to 5-month-old mice after a 4 week delay between BrdU injections and killing. Whereas the number of BrdU+ cells was strongly increased in Gsn−/− animals (more than threefold), neuronal versus glial fate commitment did not differ significantly between genotypes. B, Confocal image illustrating newly generated neuron in the dentate gyrus of Gsn−/− mouse. Green, Neuronal marker NeuN. Red, BrdU. Blue, Astrocytic marker S100β. C, D, The number of DCX- and CR-expressing cells was used as a surrogate marker to assess hippocampal neurogenesis in 14- to 16-month-old mice. Representative images of DCX immunoreactivity (C) and CR immunoreactivity (D) in the hippocampal dentate gyrus of Gsn−/− mice and wild-type controls. Scale bar: (in D) B, 23 μm; C, D, 50 μm.
DCX represents a marker of neuronal lineage determination (Rao and Shetty, 2004; Couillard-Despres et al., 2005). In line with the results obtained for adult net neurogenesis, the number of DCX+ cells was significantly increased (approximately fivefold) in Gsn−/− mice compared with wild-type controls (F(1,18) = 151.7; p < 0.0001) and, across groups, showed a strong positive correlation with net neurogenesis (r = 0.9; F(1,18) = 79.1; p < 0.0001).
The increase in hippocampal neurogenesis is preserved in aged Gsn−/− mice
Animals were 14–16 months at the time of killing. DCX-immunoreactive cells and the number of CR-immunoreactive cells were used as surrogate markers of neurogenesis (Fig. 2C,D) (Kempermann et al., 2004; Kronenberg et al., 2007, 2008). Both the number of DCX+ cells and the number of CR+ cells were robustly increased in gelsolin-deficient animals compared with wild-type controls (Fig. 2C,D) (DCX: F(1,20) = 29.6, p < 0.0001; CR: F(1,20) = 39.6, p < 0.0001).
Slowed migration of newly generated cells through the rostral migratory stream in Gsn−/− mice
Olfactory bulb neurogenesis occurs in a migratory system that originates in the subventricular zone of the lateral ventricle wall from where neuroblasts migrate via the RMS into the OB.
BrdU pulse chase experiments yielded qualitative evidence of slowed in vivo migration of newly generated cells through the rostral migratory stream in Gsn−/− mice (Fig. 3A–D). At 10 d after a 5 d course of daily intraperitoneal BrdU injections, the majority of newly generated cells had already left the first half of the RMS close to the lateral ventricle in Gsn+/+ mice (Fig. 3A). In contrast, in Gsn−/− mice investigated at this time point, many BrdU+ cells were still detectable at the beginning of the RMS (Fig. 3B). Similarly, 17 d after a 5 d course of daily intraperitoneal BrdU, only few BrdU+ cells were still detectable in the horizontal limb of the RMS in Gsn+/+ mice (Fig. 3C). Again, in Gsn−/− mice investigated at this later time point, more newly generated cells were observed at the beginning and in the horizontal limb of the RMS (Fig. 3D) (n = 3 animals per genotype per time point; not quantitatively analyzed).
Figure 3.
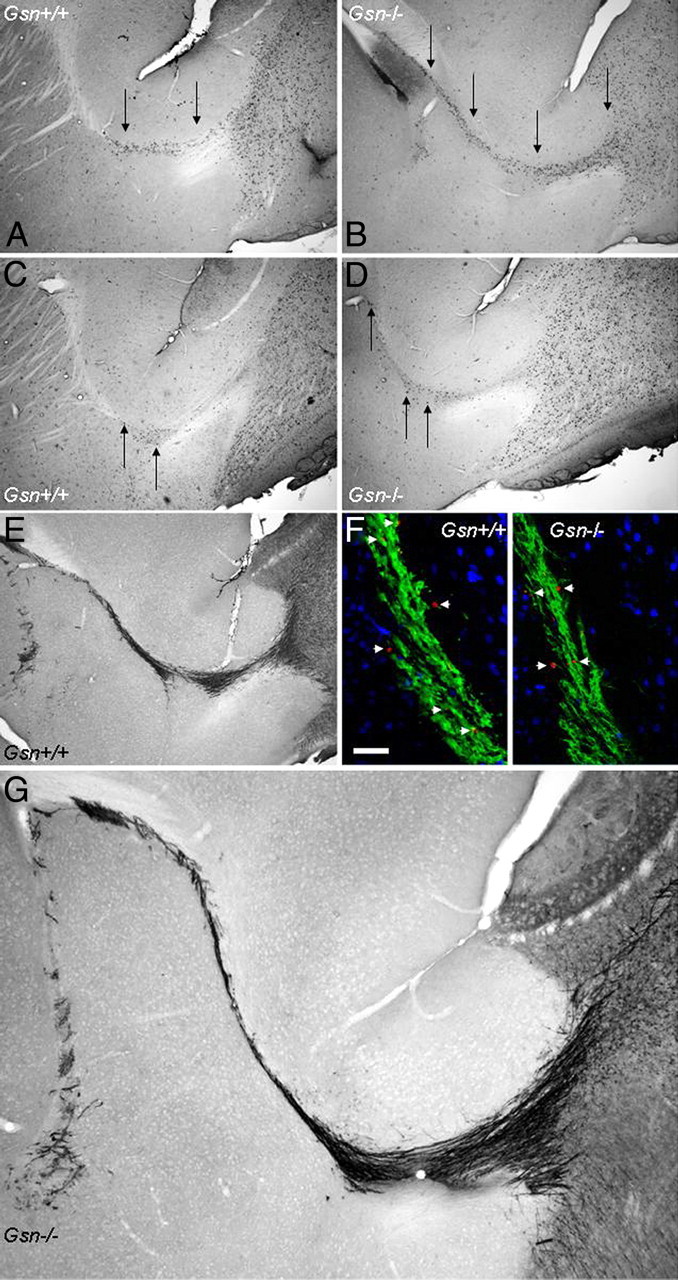
Impairment of RMS migration in Gsn−/− mice. A–D, Migration of newly generated cells through the rostral migratory stream was analyzed in a BrdU pulse chase experiment. Representative parasagittal images of the distribution of BrdU immunoreactivity at 10 (A, B) and 17 (C, D) days after a 5 d course of daily intraperitoneal BrdU demonstrates that BrdU+ cells of Gsn−/− mice (B, D) remain in the RMS longer and travel more slowly compared with BrdU+ cells of Gsn+/+ mice (A, C). The arrows mark BrdU+ cells in the rostral migratory stream. E, G, There were no apparent differences in the pattern of DCX staining in the RMS between genotypes. F, Furthermore, TUNEL staining (red) of the RMS (DCX, green) in parasagittal brain sections did not reveal significant differences between Gsn+/+ and Gsn−/− mice. Arrowheads, TUNEL+ nuclei. Blue, NeuN. Scale bar: (in F) 50 μm.
The majority of newly generated neurons in the olfactory bulb are GABAergic interneurons (Lois and Alvarez-Buylla, 1994; Gage, 2002; Hack et al., 2005; Kohwi et al., 2007; Parrish-Aungst et al., 2007). We therefore measured GABA tissue concentrations in olfactory bulb of Gsn+/+ and Gsn−/− mice (n = 5 animals per group). GABA levels (picomoles/milligram tissue) did not differ significantly between genotypes (Gsn+/+, 7.99 ± 0.26; Gsn−/−, 7.31 ± 0.38). Furthermore, we did not detect gross anatomic abnormalities of the olfactory bulb or of DCX immunoreactivity in the rostral migratory stream in Gsn−/− compared with Gsn+/+ mice (Fig. 3E,G). To examine whether the level of apoptosis in the rostral migratory stream was affected by gelsolin deficiency, we quantified the number of TUNEL+ cells in Gsn+/+ and Gsn−/− mice. Numerical density of TUNEL+ nuclei (×103/mm3) in the RMS did not differ significantly between genotypes (Gsn+/+, 3.7 ± 0.6; Gsn−/−, 3.9 ± 0.4; n = 4 animals per group) (Fig. 3F).
Finally, 30 d after a 7 d course of daily intraperitoneal BrdU injections, using an unbiased stereological approach, BrdU+ cells were quantified in the olfactory bulb and in the wall of the lateral ventricle of young adult Gsn−/− mice and littermate controls. In Gsn−/− mice, the number of BrdU-labeled cells was significantly increased in the lateral ventricle wall (Fig. 4A,B) in parallel with a significantly reduced number of BrdU+ cells in the olfactory bulb (Fig. 4C,D).
Figure 4.

More BrdU+ cells in the lateral ventricle wall and fewer BrdU+ cells in the olfactory bulb of Gsn−/− mice. A, B, BrdU+ cells in the lateral ventricle wall (marked by arrows) were determined in every sixth section from the appearance of the third ventricle to the disappearance of the anterior commissure. Animals were killed 30 d after BrdU treatment. C, D, BrdU+ cells in the olfactory bulb were quantified bilaterally at every third section using unbiased stereology as described in Materials and Methods. The number of BrdU+ cells was significantly reduced in Gsn−/− animals at 30 d after a series of BrdU injections. Scale bar: D, insets, 100 μm. n = 4 animals per genotype. *p < 0.05. Error bars indicate SEM.
Gelsolin-deficient neural progenitor cells exhibit reduced migratory capacity in culture
Additional investigation of the role of gelsolin in mediating NPC motility led to a series of in vitro assays. We measured cell migration out of adult neurospheres seeded in 50% Matrigel during a 20 h period (Fig. 5). Immunohistochemistry confirmed nestin expression in cells migrating outward at this time point (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). NPCs derived from Gsn−/− compared with Gsn+/+ mice exhibited significantly reduced migration distances (in micrometers) (Gsn+/+, 206.0 ± 14.1; Gsn−/−, 59.0 ± 9.9; F(1,23) = 70.4; p < 0.0001) (Fig. 5).
Figure 5.

Gelsolin deficiency impairs neural progenitor cell migration in vitro. A–D, Representative micrographs illustrating slowed migration of Gsn−/− compared with Gsn+/+ cells out of neurospheres seeded in Matrigel during a 20 h period. The greatest distance a cell had migrated out of the sphere at 20 h was recorded using LAS software. Scale bar: (in C) 50 μm. Quantitative data of migration distances in the Matrigel assay are given in the text.
Attraction of neural progenitors to glioma cells is well established (Glass et al., 2005; Kendall et al., 2008; Walzlein et al., 2008). We used the gliotropism of NPCs to also study neural progenitor cell migration in a Boyden chamber assay. Twenty-four hours after G261 cells had been seeded into 24-well plates, Transwell inserts were placed into the wells and NPCs added to the upper chamber. Again, rate of NPC migration across the Transwell assessed 24 h after incubation was significantly reduced by gelsolin deficiency (in number of migrated cells) (Gsn+/+, 389.2 ± 41.7; Gsn−/−, 192.7 ± 25.3; F(1,10) = 16.3; p < 0.01).
Together, these results indicate that lack of gelsolin directly impairs the migratory capacity of adult neural progenitors. In contrast, we did not detect significant differences in the growth and differentiation characteristics of adult neural progenitors derived from Gsn+/+ and Gsn−/− mice (Fig. 6). The percentage of cells expressing neuronal marker TuJ-1 at 7 d after dissociation and plating in differentiation conditions did not differ significantly between genotypes (Fig. 6A,B). Furthermore, gelsolin deficiency did not affect proliferation kinetics of NPCs in vitro (summarized in Fig. 6C).
Figure 6.

Gelsolin deficiency does not affect neuronal differentiation or proliferation kinetics of NPCs in vitro. A–C, Seven days after dissociation and culturing under differentiation conditions, cells were analyzed for expression of neuronal marker TuJ-1 (red). The percentage of cells expressing TuJ-1 did not differ significantly between genotypes (Gsn+/+, 26 ± 3.2; Gsn−/−, 28.8 ± 6.9; analysis of at least 3 × 200 cells per genotype). The images in A and B represent projections of confocal z-series (stack depth, 15 μm each). Blue, DAPI counterstain. C, Summary of growth characteristics. Scale bar: (in A) 50 μm.
Gelsolin deficiency confers increased synaptosomal Ca2+ influx and increased exocytotic neurotransmitter release
In the hippocampus, excitation directly promotes neurogenesis from adult neural progenitor cells (“excitation–neurogenesis coupling”) (Deisseroth et al., 2004; Tozuka et al., 2005; Suzuki et al., 2006; Whitney et al., 2008). Here, we investigated Ca2+ influx in neocortical and hippocampal synaptosomes (Fig. 7). Under control conditions, basal [Ca2+]i before K+ depolarization was not affected by gsn genotype (neocortical synaptosomes: 299 ± 8 nm in Gsn+/+, n = 52; and 308 ± 10 nm in Gsn−/−, n = 41; hippocampal synaptosomes: 272 ± 8 nm in Gsn+/+, n = 41; and 285 ± 11 nm in Gsn−/−, n = 38). After elevation of the K+ concentration in the PSS to 30 mm, [Ca2+]i increased instantly and reached a plateau within 10 s in Gsn+/+ neocortical and hippocampal synaptosomes. K+-induced [Ca2+]i increase in Gsn−/− synaptosomes was steeper and reached a higher plateau (33.5% higher increase in neocortical and 39.1% higher increase in hippocampal synaptosomes; p < 0.05 and p < 0.05, respectively) within ∼3 s after depolarization (Fig. 7A,B, insets). Omission of Ca2+ from the buffer almost completely abolished the K+-induced [Ca2+]i increase in neocortical (reduction by 99% in Gsn+/+ and 97% in Gsn−/−; p < 0.0001 and p < 0.0001, respectively) as well as in hippocampal synaptosomes (reduction by 98% in Gsn+/+ and 99% in Gsn−/−; p < 0.0001 and p < 0.0001, respectively). N-type Ca2+ channel blocker ω-conotoxin GVIA (0.1 μm) and P/Q-type Ca2+ channel blocker ω-agatoxin IVA (0.2 μm) significantly reduced the K+-induced [Ca2+]i increase in both neocortical and hippocampal synaptosomes from either genotype. In contrast, L-type Ca2+ channel blocker nifedipine (1 μm) had no effect on K+-induced [Ca2+]i increase (Fig. 7A,B).
Figure 7.

Gelsolin deficiency confers increased synaptosomal Ca2+ influx. Gelsolin deficiency boosts K+-induced [Ca2+]i increase in Fura-2-loaded neocortical (A) or hippocampal (B) synaptosomes. For depolarization, K+ was elevated to 30 mm. The K+-induced [Ca2+]i increase is presented as percentage of basal cytosolic Ca2+ concentrations. The insets in A and B show representative fluorescence traces of control experiments. Note that, in Gsn−/− synaptosomes, [Ca2+]i increases faster and to a higher level than in wild-type synaptosomes. Although L-type voltage-dependent Ca2+ channel blocker nifedipine (1 μm) does not exert an effect on K+-induced [Ca2+]i increase both N-type voltage-dependent Ca2+ channel blocker ω-conotoxin GVIA (ω-CTx GVIA) (100 nm) and P/Q-type voltage-dependent Ca2+ channel blocker ω-agatoxin IVA (ω-AgaTx IVA) (200 nm) markedly reduce K+-induced [Ca2+]i increase independent of genotype. In contrast, cytochalasin D (Cyto-D) (1 μm), which mimics the effect of endogenous gelsolin, reverts the increased Ca2+ influx in Gsn−/− hippocampal synaptosomes to wild-type levels (B). Values given in A and B represent the means of five to nine experiments performed in duplicate. Error bars indicate SEM. *p < 0.05 compared with the corresponding wild-type controls within each experimental condition. +p < 0.05 compared with the same genotype within the control condition. ΔF represents the changes of fluorescence ratio excited at 340/380 nm.
Cytochalasin D is a cell-permeable mycotoxin that, similar to gelsolin, disrupts actin filaments and inhibits actin polymerization. Cytochalasin D (1 μm) treatment decreased the enhanced Ca2+ influx observed in Gsn−/− hippocampal synaptosomes under control conditions to the levels observed in Gsn+/+ synaptosomes (Fig. 7B). These results indicate that the effects of gelsolin deficiency on synaptosomal Ca2+ influx are mediated by actin filament stabilization.
Next, we analyzed the functional consequences of increased Ca2+ influx into gelsolin-deficient presynaptic terminals. Synaptosomal NE release represents a reliable and robust parameter for the analysis of exocytotic function (Fink et al., 1990; Dry et al., 1991; Pittaluga and Raiteri, 1992). We here monitored tritium-labeled norepinephrine release from neocortical synaptosomes. Independent of genotype, K+ induced [3H]NE release in a concentration-dependent manner. However, at the K+ concentrations studied, the amount of [3H]NE released from Gsn−/− synaptosomes was 18–42% higher than the amount released from Gsn+/+ synaptosomes (Fig. 8A). Importantly, K+-induced [3H]NE release was almost completely Ca2+-dependent in either genotype (Fig. 8A, inset). Again, the enhanced [3H]NE release observed in Gsn−/− compared with Gsn+/+ synaptosomes was completely reversed by incubation with actin disruptor agent cytochalasin D (Fig. 8B). Together, these results show that reduced F-actin turnover in gelsolin-deficient presynaptic terminals causes increased Ca2+ influx and, subsequently, elevated exocytotic neurotransmitter release.
Figure 8.

Gelsolin deficiency confers increased exocytotic neurotransmitter release. A, B, Gelsolin deficiency boosts K+-induced release of tritium-labeled norepinephrine ([3H]NE) from neocortical synaptosomes. For depolarization, K+ was elevated to 9–30 mm (A) or to 15 mm (A, inset; B). K+-induced [3H]NE release is presented as percentage of total tissue tritium. The inset in A shows the results of Ca2+-free experiments. B, Cytochalasin D (Cyto-D) (1 μm) reduces the increased [3H]NE release in Gsn−/− synaptosomes to wild-type levels. Shown are means ± SEM of four to six experiments in quadruplicate. *p < 0.05 compared with the corresponding wild-type synaptosomes. C–E, K+-induced glutamate release in neocortical, hippocampal, or striatal slices is higher in Gsn−/− brains. For depolarization, K+ was elevated to 15 mm. Endogenous glutamate in the superfusate was collected in 5 min fractions and determined by HPLC. Glutamate release is presented in picomoles per milligram of brain tissue (wet weight). Shown are means ± SEM of 5–18 experiments. *p < 0.05 compared with the corresponding wild-type controls.
Glutamate is the most abundant excitatory neurotransmitter in hippocampus. Changes in glutamate release and reuptake are therefore indicative of profound alterations of activity in hippocampal networks in general. We studied whether reduced actin turnover by gelsolin deficiency would impact the efflux of glutamate from superfused brain slices (Fig. 8C–E) after K+ depolarization (15 mm). Indeed, K+-induced release of glutamate from neocortical, hippocampal, and striatal brain slices was significantly increased by gelsolin deficiency.
We also investigated global tissue levels of a variety of neurotransmitters such as glutamate, GABA, dopamine, NE, 5-HT, and degradation products in hippocampus of Gsn+/+ and Gsn−/− mice. We did not detect significant differences between genotypes in any of the neurotransmitters investigated (Table 2), indicating that the biosynthesis of neurotransmitters is not majorly affected by gelsolin deficiency in vivo.
Table 2.
Hippocampal neurotransmitter concentrations
| Gelsolin+/+ | Gelsolin−/− | |
|---|---|---|
| Norepinephrine (pg/mg tissue) | 385.01 ± 12.63 | 381.76 ± 8.19 |
| Dopamine (pg/mg tissue) | 309.26 ± 95.53 | 201.28 ± 58.01 |
| DOPAC (pg/mg tissue) | 33.71 ± 3.31 | 35.98 ± 4.36 |
| HVA (pg/mg tissue) | 104.64 ± 29.77 | 70.64 ± 9.11 |
| 5-HT (pg/mg tissue) | 991.09 ± 13.70 | 1077.91 ± 22.03 |
| 5-HIAA (pg/mg tissue) | 629.62 ± 69.61 | 640.15 ± 24.40 |
| GABA (pmol/mg tissue) | 2.83 ± 0.04 | 2.82 ± 0.12 |
| Glutamate (pmol/mg tissue) | 10.86 ± 0.29 | 12.14 ± 0.55 |
| Taurine (pmol/mg tissue) | 10.60 ± 0.10 | 10.45 ± 0.43 |
DOPAC, 3,4-Dihydroxyphenylacetic acid; HVA, homovanillic acid. Values represent mean ± SEM. N = 5 animals per group.
Dephosphorylation of cofilin in Gsn−/− mice
Remodeling of the actin cytoskeleton is essential for cellular processes such as membrane trafficking, morphogenesis, and migration (Giganti and Friederich, 2003; Gourlay and Ayscough, 2005). Like gelsolin, cofilin is an actin-binding protein that promotes disassembly of actin filaments (Loisel et al., 1999). In line with a previous study, we found that total cofilin levels in brain did not differ between genotypes (Furukawa et al., 1997). However, we here show that phosphorylated cofilin, which lacks actin depolymerizing activity (Bamburg, 1999), is reduced in Gsn−/− mice (Fig. 9). Dephosphorylation of cofilin in Gsn−/− mice was not related to changes of either SRF or actin protein levels. mRNA levels of SRF target genes actin and srf also did not differ between Gsn−/− and Gsn+/+ mice (data not shown).
Figure 9.

Dephosphorylation of cofilin in Gsn−/− mice. Western blot of protein extracts from hippocampus and frontal cortex of control (+/+) and gelsolin-deficient animals (−/−) probed with antibodies for actin, cofilin, phospho-cofilin, and SRF. Comparable loading of protein is confirmed by GAPDH staining.
Increased NGF content in hippocampus of Gsn−/− mice
NGF plays a key role in neuronal motility and migration. Specifically, extracellular NGF has been shown to increase F-actin levels and to lead to dephosphorylation of cofilin (Melamed et al., 1995; Meberg et al., 1998; Yoshizawa et al., 2005; Endo et al., 2007). NGF has also been demonstrated to increase adult hippocampal neurogenesis (Frielingsdorf et al., 2007). We therefore studied NGF expression at the protein level using an ELISA (Fig. 10). Tissue concentrations of NGF were significantly increased in brains of Gsn−/− mice. In contrast, hippocampal BDNF concentrations and basic fibroblast growth factor (bFGF) mRNA expression in hippocampus did not differ between Gsn+/+ and Gsn−/− mice (data not shown).
Figure 10.

Increased NGF levels in Gsn−/− mice. Tissue content of NGF determined at the protein level by ELISA is increased in Gsn−/− brain. N = 10 animals per genotype. *p < 0.05. Error bars indicate SEM.
Increased endothelial nitric oxide (NOS-III) expression, increased regional cerebral blood flow, and increased density of perfused microvessels in hippocampus of Gsn−/− mice
The microanatomy of hippocampal neurogenesis in rodents suggests that a distinct neurovascular environment contributes to the growth of new neurons (the “vascular niche”) (Palmer et al., 2000; Fabel et al., 2003a,b). NOS-III is a key regulator of vascular tone, remodeling, and angiogenesis (Sessa, 2004). In vitro, absence of NOS-III decreases neurosphere formation and progenitor cell proliferation (Chen et al., 2005). Hippocampal NOS-III expression was increased in Gsn−/− mice both at the mRNA and protein levels (Fig. 11A,B). Importantly, membrane-bound NOS-III was significantly increased in hippocampal homogenates of Gsn−/− mice (Fig. 11B). In parallel, both absolute regional CBF (rCBF) and density of perfused microvessels were significantly increased in hippocampus of Gsn−/− compared with Gsn+/+ mice (Fig. 11C).
Figure 11.

Increased NOS-III expression and increased density of perfused microvessels in hippocampus of Gsn−/− mice. Relative NOS-III mRNA expression in hippocampus and in frontal cortex (A) is reported as the value × 1000 normalized to GAPDH for each sample (n = 4–5 animals per genotype). *p < 0.05. B, Hippocampal NOS-III protein expression. Western blots show similar NOS-III levels in cytosolic fractions (left) and increased levels in the membrane fractions (right) of Gsn−/− mouse hippocampus. Top panels, Representative blots of two different adult mice of either genotype. Bottom panels, Densitometrical quantification of the detected NOS-III bands, presented as ratios of NOS-III optical density (O.D.) over α-tubulin O.D. Error bars indicate SEM. C, Representative images of Evans blue staining in hippocampus of Gsn+/+ and Gsn−/− mice (10 μm cryostat sections). The density of perfused microvessels as determined using a tiled-field mapping technique and computer-assisted analysis was significantly increased by 14 microvessels/mm2 in Gsn−/− mice (F(1,16) = 4.6; p < 0.05). Correspondingly, absolute cerebral blood flow in hippocampus (milliliters · 100 g−1 · min−1) as assessed by the 14C-iodoantipyrine technique was also significantly increased in Gsn−/− mice (36.5 ± 2.9 vs 26.1 ± 1.9; F(1,16) = 8.7; p < 0.01). Scale bar: (in C) 1000 μm.
A specialized vascular niche in which dividing stem cells and their transit-amplifying progeny are tightly apposed to the vasculature has recently also been described in the adult SVZ (Tavazoie et al., 2008). Interestingly, absolute regional cerebral blood flow in the vicinity of the lateral ventricle wall was also significantly increased in Gsn−/− mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Discussion
This study yielded the following major findings: (1) Actin filament stabilization elicited a strong increase in net hippocampal neurogenesis. (2) Migration of neural progenitors from the SVZ into the olfactory bulb was slowed in Gsn−/− mice. However, the level of apoptosis in the RMS was not affected by gelsolin deficiency. (3) In vitro, NPCs derived from Gsn−/− mice showed impaired migratory capacity, whereas neuronal differentiation and proliferation kinetics did not differ between genotypes. (4) Depolarization-induced [Ca2+]i levels were increased in presynaptic terminals derived from Gsn−/− mice. Consequently, exocytotic neurotransmitter release was enhanced by gelsolin deficiency, which is in line with the hypothesis that excitation stimuli promote neurogenesis (Deisseroth et al., 2004; Tozuka et al., 2005). (5) Neurogenesis occurs in association with neovasculature (the “vascular niche for neurogenesis”) (Palmer et al., 2000; Louissaint et al., 2002; Newton et al., 2003; Tavazoie et al., 2008). In Gsn−/− animals, increased numbers of BrdU+ cells in the lateral ventricle wall and in the dentate gyrus were observed in the context of elevated regional cerebral blood flow. In hippocampus, this was accompanied by significantly increased NOS-III transcription and, at the protein level, significantly increased expression of NOS-III in membrane fractions.
In our genetic system, gelsolin deficiency resulted in a relatively mild phenotype. The gsn-null state thus did not involve ventricular enlargement or a severe broadening of the subventricular zone such as have been described in the case of conditional srf deletion (Alberti et al., 2005). A special role for NGF and cofilin in regulating neuronal actin dynamics has been described (Collins, 1984; Meberg et al., 1998; Lanier and Gertler, 2000; Montani et al., 2009). Specifically, extracellular NGF promotes neuronal migration (Yoshizawa et al., 2005) and leads to actin filament depolymerization via cofilin dephosphorylation (Endo et al., 2007). Increased levels of extracellular NGF have also been shown to promote hippocampal neurogenesis (Frielingsdorf et al., 2007). We detected dephosphorylation (i.e., activation) (Niwa et al., 2002) of cofilin and increased NGF levels in Gsn−/− brains. In contrast, hippocampal BDNF concentrations and bFGF mRNA expression did not differ between genotypes. Although gelsolin is a target gene of SRF (Alberti et al., 2005), gelsolin deficiency did not cause a counterregulatory upregulation of SRF protein expression. Also, transcription of SRF target genes srf and β-actin was not induced in Gsn−/− mice.
Our observation of reduced progenitor migration fits well with previous reports that demonstrate reduced migratory capacity of Gsn−/− fibroblasts and neutrophils (Witke et al., 1995; Azuma et al., 1998). Whereas impaired actin filament turnover resulted in impaired migration of NPCs in vitro, our data indicate that gelsolin deficiency does not directly affect proliferation and neuronal differentiation of NPCs. NPCs make up only a small proportion of the cells in the adult CNS. We therefore investigated more general mechanisms of how gelsolin deficiency might shape the extracellular milieu such as to promote hippocampal neurogenesis. We propose two complementary mechanisms: (1) increased exocytotic neurotransmitter release onto neural progenitor cells and (2) a special vascular microenvironment.
While providing structural support, the actin cytoskeleton also modulates ion channel function. For example, voltage-dependent Ca2+ and NMDA channel activities decrease on actin depolymerization (Johnson and Byerly 1993; Rosenmund and Westbrook 1993; Akopian et al., 2006). Conversely, primary hippocampal neurons cultured from mice lacking gelsolin exhibit enhanced Ca2+ influx after exposure to glutamate (Furukawa et al., 1997). We here used K+-induced [Ca2+]i increase in synaptosomes to monitor Ca2+ influx through presynaptic voltage-dependent calcium channels (VDCCs). Synaptosomes have been used extensively to analyze presynaptic mechanisms of Ca2+ influx and neurotransmitter release because they retain the machinery for the uptake, storage, and exocytosis of neurotransmitters (Fink et al., 2002a; Baldwin et al., 2003). Ca2+ release from intrasynaptosomal stores does not significantly contribute to depolarization-induced [Ca2+]i increase (Mulkey and Zucker, 1991). [Ca2+]i increase after depolarization was elevated in Gsn−/− synaptosomes. Importantly, disruption of F-actin by cytochalasin D reduced this additional Ca2+ influx in Gsn−/− synaptosomes to the level observed in untreated synaptosomes derived from Gsn+/+ mice. Next, we show that gelsolin deficiency increases exocytotic neurotransmitter release from presynaptic terminals. Again, the effects of gelsolin deficiency were directly attributable to impaired actin filament turnover as demonstrated by the fact that treatment with cytochalasin D reduced the enhanced norepinephrine release observed in Gsn−/− synaptosomes to wild-type levels. Since exocytotic neurotransmitter release is primarily dependent on Ca2+ influx (Pastuszko et al., 1984; Hori et al., 1985; Nicholls and Sihra, 1986; Duarte et al., 1993; Engisch and Nowycky, 1996), our observations are likely generalizable to other neurotransmitter systems in Gsn−/− brain. We note that actin filament stabilization may potentially also interfere with neuronal or glial reuptake mechanisms, which might further contribute to increased extracellular neurotransmitter concentrations on stimulation.
Considerable evidence has accumulated to support the concept that adult neurogenesis is regulated by network activity (Cameron et al., 1995; Blümcke et al., 2001; Bruel-Jungerman et al., 2006). Specifically, the ability of neural progenitors to sense excitation and thereby implement coupling between network activity and neurogenesis has been established. However, so far, different neurotransmitters have been implicated in this process (Deisseroth et al., 2004; Tozuka et al., 2005; Suzuki et al., 2006; Whitney et al., 2008). In an elaborate in vitro system favoring neuronal differentiation of adult NPCs, Deisseroth et al. (2004) found glutamatergic excitation driving neuronal differentiation. In contrast, electrophysiological recordings in fresh murine hippocampal slices recently demonstrated that transiently proliferating neural progenitors receive direct GABAergic, but not glutamatergic, input from hilar interneurons (Tozuka et al., 2005; Wang et al., 2005). Our data suggest that, in Gsn−/− mice, increased amounts of neurotransmitter vesicles are exocytotically released onto NPCs resulting in their subsequent enhanced excitation. Importantly, since release of GABA from hippocampal GABAergic interneurons is induced by multiple neurotransmitters including glutamate (Matsuyama et al., 1997; Katona et al., 1999; Nishikawa et al., 2005; Tozuka et al., 2005), the ultimate effects of gelsolin deficiency on excitation of NPCs may integrate individual effects on a number of different neurotransmitter systems. Our results highlight the importance of increased neurotransmission onto NPCs in Gsn−/− hippocampus. An additional contributing mechanism not further explored here may be that, in situ, because of reduced rundown of voltage-dependent calcium currents, Gsn−/− NPCs themselves also display special susceptibility to excitation inputs from the extracellular milieu.
NOS-III is a key regulator of vascular tone, remodeling, and angiogenesis (Sessa, 2004). In vitro, an absence of NOS-III decreases neurosphere formation and progenitor cell proliferation (Chen et al., 2005). Similarly, a decrease in hippocampal neurogenesis has been demonstrated in NOS-III-deficient mice (Reif et al., 2004). Here, we show that hippocampal NOS-III expression is increased in Gsn−/− mice. For activation, NOS-III is cotranslationally and posttranslationally acylated, which allows membrane association. Membrane-bound NOS-III was also significantly increased in hippocampal homogenates of Gsn−/− mice, indicating a higher amount of fully functional NOS-III in hippocampus. In parallel, both absolute rCBF and density of perfused microvessels were significantly increased in hippocampus of Gsn−/− compared with Gsn+/+ mice. Interestingly, an additional analysis of rCBF in the vicinity of the lateral ventricle wall also showed an inducing effect of gelsolin deficiency on blood flow in this area.
Disruption of the actin cytoskeleton has been shown to increase NOS-III activity in cultured pulmonary artery endothelia (Kondrikov et al., 2006). Similarly, we have previously demonstrated increased NOS-III expression and activity in mouse aorta on treatment with cytochalasin D (Laufs et al., 2000). The observation of increased NOS-III expression in hippocampus of Gsn−/− mice is counterintuitive and in apparent discrepancy to these previous reports. Indeed, NOS-III expression in aorta is also decreased in Gsn−/− mice (Laufs et al., 2000). However, tissue- and cell-specific effects of actin filament stabilization have to be considered. In addition to vascular endothelium, neurons may serve as a major source of NOS-III in the brain. Notably, nitric oxide contributes widely to synaptic plasticity (Garthwaite and Boulton, 1995; Hopper and Garthwaite, 2006). Activation of VDCC couples excitation to gene expression. In neurons, cAMP response element-binding protein (CREB) plays a crucial role in this “excitation–transcription” coupling (Dolmetsch et al., 2001; Lonze and Ginty, 2002; Carlezon et al., 2005). The NOS-III promoter contains a cis-acting sequence for CREB (Niwano et al., 2003). Accordingly, actin stabilization will likely contribute to increased NOS-III expression via reduced current rundown in hippocampal neurons of Gsn−/− mice. In line with this interpretation, increased neuronal NOS-III expression has previously been demonstrated at early time points after kainic acid-induced seizures (Kim et al., 2000). Similarly, excitation–transcription coupling may also contribute to enhanced hippocampal NGF content in Gsn−/− mice. A critical role for CREB in mediating NGF gene expression in the CNS has recently been established (McCauslin et al., 2006).
In summary, actin stabilization by gelsolin deficiency causes slowed emigration of progenitors from the SVZ into the olfactory bulb but increased hippocampal neurogenesis. We here identify increased exocytotic neurotransmitter release and a special vascular environment characterized by enhanced hippocampal NOS-III expression as key factors underlying increased hippocampal neurogenesis in Gsn−/− mice.
Footnotes
This work was supported by VolkswagenStiftung (Lichtenberg Program) (M.E.), Deutsche Forschungsgemeinschaft (K.B.F., M.E.), Bundesministerium für Bildung und Forschung (Center for Stroke Research Berlin, Berlin, Germany) (M.E.), and Schilling Foundation (M.E.). Part of the research reported here was performed as a doctoral thesis (S.E.) (in preparation) funded by Deutsche Forschungsgemeinschaft (R.H.). We thank Wolfgang Kuschinsky (Institut für Physiologie und Pathophysiologie, Universität Heidelberg, Heidelberg, Germany) for his generous support. We are indebted to David Kwiatkowski (Harvard University, Cambridge, MA) for the provision of gelsolin knock-out mice and for helpful advice. We thank Annemarie Bunge, Maria Harlacher, Bettina Herrmann, Mathilde Lorenz, and Anke Sänger for excellent technical assistance.
References
- Akopian A, Szikra T, Cristofanilli M, Krizaj D. Glutamate-induced Ca2+ influx in third-order neurons of salamander retina is regulated by the actin cytoskeleton. Neuroscience. 2006;138:17–24. doi: 10.1016/j.neuroscience.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Krause SM, Kretz O, Philippar U, Lemberger T, Casanova E, Wiebel FF, Schwarz H, Frotscher M, Schütz G, Nordheim A. Neuronal migration in the murine rostral migratory stream requires serum response factor. Proc Natl Acad Sci U S A. 2005;102:6148–6153. doi: 10.1073/pnas.0501191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma T, Witke W, Stossel TP, Hartwig JH, Kwiatkowski DJ. Gelsolin is a downstream effector of rac for fibroblast motility. EMBO J. 1998;17:1362–1370. doi: 10.1093/emboj/17.5.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin ML, Rostas JA, Sim AT. Two modes of exocytosis from synaptosomes are differentially regulated by protein phosphatase types 2A and 2B. J Neurochem. 2003;85:1190–1199. doi: 10.1046/j.1471-4159.2003.01779.x. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Blümcke I, Schewe JC, Normann S, Brüstle O, Schramm J, Elger CE, Wiestler OD. Increase of nestin-immunoreactive neural precursor cells in the dentate gyrus of pediatric patients with early-onset temporal lobe epilepsy. Hippocampus. 2001;11:311–321. doi: 10.1002/hipo.1045. [DOI] [PubMed] [Google Scholar]
- Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci. 2003;17:2042–2046. doi: 10.1046/j.1460-9568.2003.02647.x. [DOI] [PubMed] [Google Scholar]
- Bruel-Jungerman E, Davis S, Rampon C, Laroche S. Long-term potentiation enhances neurogenesis in the adult dentate gyrus. J Neurosci. 2006;26:5888–5893. doi: 10.1523/JNEUROSCI.0782-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, McEwen BS, Gould E. Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J Neurosci. 1995;15:4687–4692. doi: 10.1523/JNEUROSCI.15-06-04687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Chen J, Zacharek A, Zhang C, Jiang H, Li Y, Roberts C, Lu M, Kapke A, Chopp M. Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J Neurosci. 2005;25:2366–2375. doi: 10.1523/JNEUROSCI.5071-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourbaji S, Vogt MA, Fumagalli F, Sohr R, Frasca A, Brandwein C, Hörtnagl H, Riva MA, Sprengel R, Gass P. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008;22:3129–3134. doi: 10.1096/fj.08-106450. [DOI] [PubMed] [Google Scholar]
- Collins F. An effect of nerve growth factor on the parasympathetic ciliary ganglion. J Neurosci. 1984;4:1281–1288. doi: 10.1523/JNEUROSCI.04-05-01281.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, Malenka RC. Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron. 2004;42:535–552. doi: 10.1016/s0896-6273(04)00266-1. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Dry KL, Phillips JH, Dart AM. Catecholamine release from bovine adrenal chromaffin cells during anoxia or metabolic inhibition. Circ Res. 1991;69:466–474. doi: 10.1161/01.res.69.2.466. [DOI] [PubMed] [Google Scholar]
- Duarte CB, Ferreira IL, Carvalho AP, Carvalho CM. Relation of exocytotic release of gamma-aminobutyric acid to Ca2+ entry through Ca2+ channels or by reversal of the Na+/Ca2+ exchanger in synaptosomes. Pflugers Arch. 1993;423:314–323. doi: 10.1007/BF00374411. [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Mizuno K. LIM kinase and slingshot are critical for neurite extension. J Biol Chem. 2007;282:13692–13702. doi: 10.1074/jbc.M610873200. [DOI] [PubMed] [Google Scholar]
- Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, Azuma T, Mattson MP, Kwiatkowski DJ, Moskowitz MA. Neuroprotective effects of gelsolin during murine stroke. J Clin Invest. 1999;103:347–354. doi: 10.1172/JCI4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schröck H, Nickenig G, Kuschinsky W, Dirnagl U, Laufs U. Mechanisms of stroke protection by physical activity. Ann Neurol. 2003;54:582–590. doi: 10.1002/ana.10722. [DOI] [PubMed] [Google Scholar]
- Engisch KL, Nowycky MC. Calcium dependence of large dense-cored vesicle exocytosis evoked by calcium influx in bovine adrenal chromaffin cells. J Neurosci. 1996;16:1359–1369. doi: 10.1523/JNEUROSCI.16-04-01359.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabel K, Toda H, Fabel K, Palmer T. Copernican stem cells: regulatory constellations in adult hippocampal neurogenesis. J Cell Biochem. 2003a;88:41–50. doi: 10.1002/jcb.10377. [DOI] [PubMed] [Google Scholar]
- Fabel K, Fabel K, Tam B, Kaufer D, Baiker A, Simmons N, Kuo CJ, Palmer TD. VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur J Neurosci. 2003b;18:2803–2812. doi: 10.1111/j.1460-9568.2003.03041.x. [DOI] [PubMed] [Google Scholar]
- Felice LJ, Felice JD, Kissinger PT. Determination of catecholamines in rat brain parts by reverse-phase ion-pair liquid chromatography. J Neurochem. 1978;31:1461–1465. doi: 10.1111/j.1471-4159.1978.tb06573.x. [DOI] [PubMed] [Google Scholar]
- Filippov V, Kronenberg G, Pivneva T, Reuter K, Steiner B, Wang LP, Yamaguchi M, Kettenmann H, Kempermann G. Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Mol Cell Neurosci. 2003;23:373–382. doi: 10.1016/s1044-7431(03)00060-5. [DOI] [PubMed] [Google Scholar]
- Fink K, Göthert M, Molderings G, Schlicker E. N-methyl-d-aspartate (NMDA) receptor-mediated stimulation of noradrenaline release, but not release of other neurotransmitters, in the rat brain cortex: receptor location, characterization and desensitization. Naunyn Schmiedebergs Arch Pharmacol. 1989;339:514–521. doi: 10.1007/BF00167254. [DOI] [PubMed] [Google Scholar]
- Fink K, Göthert M, Schlicker E. Veratridine and other depolarizing agents counteract the inhibitory effect of Mg2+ ions on N-methyl-d-aspartate (NMDA)-induced noradrenaline release in vitro. Naunyn Schmiedebergs Arch Pharmacol. 1990;342:53–60. doi: 10.1007/BF00178972. [DOI] [PubMed] [Google Scholar]
- Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, Göthert M. Inhibition of neuronal Ca2+ influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology. 2002a;42:229–236. doi: 10.1016/s0028-3908(01)00172-1. [DOI] [PubMed] [Google Scholar]
- Fink KB, Paehr M, Djoufack PC, Weissbrich C, Bösel J, Endres M. Effects of cytoskeletal modifications on Ca2+ influx after cerebral ischemia. Amino Acids. 2002b;23:325–329. doi: 10.1007/s00726-001-0145-z. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Frielingsdorf H, Simpson DR, Thal LJ, Pizzo DP. Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol Dis. 2007;26:47–55. doi: 10.1016/j.nbd.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Kato F, Tozuka Y, Yamaguchi M, Miyamoto Y, Hisatsune T. Two distinct subpopulations of nestin-positive cells in adult mouse dentate gyrus. J Neurosci. 2003;23:9357–9366. doi: 10.1523/JNEUROSCI.23-28-09357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Fu W, Li Y, Witke W, Kwiatkowski DJ, Mattson MP. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J Neurosci. 1997;17:8178–8186. doi: 10.1523/JNEUROSCI.17-21-08178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Gertz K, Priller J, Kronenberg G, Fink KB, Winter B, Schröck H, Ji S, Milosevic M, Harms C, Böhm M, Dirnagl U, Laufs U, Endres M. Physical activity improves long-term stroke outcome via endothelial nitric oxide synthase-dependent augmentation of neovascularisation and cerebral blood flow. Circ Res. 2006;99:1132–1140. doi: 10.1161/01.RES.0000250175.14861.77. [DOI] [PubMed] [Google Scholar]
- Giganti A, Friederich E. The actin cytoskeleton as a therapeutic target: state of the art and future directions. Prog Cell Cycle Res. 2003;5:511–525. [PubMed] [Google Scholar]
- Glass R, Synowitz M, Kronenberg G, Walzlein JH, Markovic DS, Wang LP, Gast D, Kiwit J, Kempermann G, Kettenmann H. Glioblastoma-induced attraction of endogenous neural precursor cells is associated with improved survival. J Neurosci. 2005;25:2637–2646. doi: 10.1523/JNEUROSCI.5118-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göbel U, Schröck H, Seller H, Kuschinsky W. Glucose utilization, blood flow and capillary density in the ventrolateral medulla of the rat. Pflugers Arch. 1990;416:477–480. doi: 10.1007/BF00382679. [DOI] [PubMed] [Google Scholar]
- Gonos ES, Derventzi A, Kveiborg M, Agiostratidou G, Kassem M, Clark BF, Jat PS, Rattan SI. Cloning and identification of genes that associate with mammalian replicative senescence. Exp Cell Res. 1998;240:66–74. doi: 10.1006/excr.1998.3948. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing. Nat Rev Mol Cell Biol. 2005;6:583–589. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- Grohé C, Kann S, Fink L, Djoufack PC, Paehr M, van Eickels M, Vetter H, Meyer R, Fink KB. 17β-Estradiol regulates nNOS and eNOS activity in the hippocampus. Neuroreport. 2004;15:89–93. doi: 10.1097/00001756-200401190-00018. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hack MA, Saghatelyan A, de Chevigny A, Pfeifer A, Ashery-Padan R, Lledo PM, Götz M. Neuronal fate determinants of adult olfactory bulb neurogenesis. Nat Neurosci. 2005;8:865–872. doi: 10.1038/nn1479. [DOI] [PubMed] [Google Scholar]
- Harms C, Bösel J, Lautenschlager M, Harms U, Braun JS, Hörtnagl H, Dirnagl U, Kwiatkowski DJ, Fink K, Endres M. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol Cell Neurosci. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Hock C, Hartung HD. An improved rapid and highly sensitive enzyme immunoassay for nerve growth factor. Tech J Methods Cell Mol Biol. 1989;1:43–49. [Google Scholar]
- Hellweg R, von Arnim CA, Büchner M, Huber R, Riepe MW. Neuroprotection and neuronal dysfunction upon repetitive inhibition of oxidative phosphorylation. Exp Neurol. 2003;183:346–354. doi: 10.1016/s0014-4886(03)00127-4. [DOI] [PubMed] [Google Scholar]
- Hopper RA, Garthwaite J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci. 2006;26:11513–11521. doi: 10.1523/JNEUROSCI.2259-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori N, Ffrench-Mullen JM, Carpenter DO. Kainic acid responses and toxicity show pronounced Ca2+ dependence. Brain Res. 1985;358:380–384. doi: 10.1016/0006-8993(85)90989-8. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Stossel TP. Modulation of gelsolin function by phosphatidylinositol 4,5-biphosphate. Nature. 1987;325:362–364. doi: 10.1038/325362a0. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+ Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130:391–399. doi: 10.1242/dev.00203. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Jessberger S, Steiner B, Kronenberg G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 2004;27:447–452. doi: 10.1016/j.tins.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Kendall SE, Najbauer J, Johnston HF, Metz MZ, Li S, Bowers M, Garcia E, Kim SU, Barish ME, Aboody KS, Glackin CA. Neural stem cell targeting of glioma is dependent on phosphoinositide 3-kinase signaling. Stem Cells. 2008;26:1575–1586. doi: 10.1634/stemcells.2007-0887. [DOI] [PubMed] [Google Scholar]
- Kim IS, Huh YB, Park C, Kang MJ, Lee JR, Kim JH, Yoo JH, Ahn HK. Expressional change of endothelial nitric oxide synthase in rat cerebral cortex after kainic acid-induced seizure. Korean J Anat. 2000;33:471–478. [Google Scholar]
- Kinosian HJ, Newman J, Lincoln B, Selden LA, Gershman LC, Estes JE. Ca2+ regulation of gelsolin activity: binding and severing of F-actin. Biophys J. 1998;75:3101–3109. doi: 10.1016/S0006-3495(98)77751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, Petryniak MA, Long JE, Ekker M, Obata K, Yanagawa Y, Rubenstein JL, Alvarez-Buylla A. A subpopulation of olfactory bulb GABAergic interneurons is derived from Emx1- and Dlx5/6-expressing progenitors. J Neurosci. 2007;27:6878–6891. doi: 10.1523/JNEUROSCI.0254-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrikov D, Han HR, Block ER, Su Y. Growth and density-dependent regulation of NO synthase by the actin cytoskeleton in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L41–L50. doi: 10.1152/ajplung.00444.2004. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G. Subpopulations of proliferating cells of the adult hippocampus respond differently to physiologic neurogenic stimuli. J Comp Neurol. 2003;467:455–463. doi: 10.1002/cne.10945. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Wang LP, Synowitz M, Gertz K, Katchanov J, Glass R, Harms C, Kempermann G, Kettenmann H, Endres M. Nestin-expressing cells divide and adopt a complex electrophysiologic phenotype after transient brain ischemia. J Cereb Blood Flow Metab. 2005;25:1613–1624. doi: 10.1038/sj.jcbfm.9600156. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Lippoldt A, Kempermann G. Two genetic rat models of arterial hypertension show different mechanisms by which adult hippocampal neurogenesis is increased. Dev Neurosci. 2007;29:124–133. doi: 10.1159/000096217. [DOI] [PubMed] [Google Scholar]
- Kronenberg G, Harms C, Sobol RW, Cardozo-Pelaez F, Linhart H, Winter B, Balkaya M, Gertz K, Gay SB, Cox D, Eckart S, Ahmadi M, Juckel G, Kempermann G, Hellweg R, Sohr R, Hörtnagl H, Wilson SH, Jaenisch R, Endres M. Folate deficiency induces neurodegeneration and brain dysfunction in mice lacking uracil DNA glycosylase. J Neurosci. 2008;28:7219–7230. doi: 10.1523/JNEUROSCI.0940-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LM, Gertler FB. From Abl to actin: Abl tyrosine kinase and associated proteins in growth cone motility. Curr Opin Neurobiol. 2000;10:80–87. doi: 10.1016/s0959-4388(99)00058-6. [DOI] [PubMed] [Google Scholar]
- Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, Huang PL, Böhm M, Schoen FJ, Moskowitz MA, Liao JK. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Long-distance neuronal migration in the adult mammalian brain. Science. 1994;264:1145–1148. doi: 10.1126/science.8178174. [DOI] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Louissaint A, Jr, Rao S, Leventhal C, Goldman SA. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron. 2002;34:945–960. doi: 10.1016/s0896-6273(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Nei K, Tanaka C. Regulation of GABA release via NMDA and 5-HT1A receptors in guinea pig dentate gyrus. Brain Res. 1997;761:105–112. doi: 10.1016/s0006-8993(97)00318-1. [DOI] [PubMed] [Google Scholar]
- McCauslin CS, Heath V, Colangelo AM, Malik R, Lee S, Mallei A, Mocchetti I, Johnson PF. CAAT/enhancer-binding protein delta and cAMP-response element-binding protein mediate inducible expression of the nerve growth factor gene in the central nervous system. J Biol Chem. 2006;281:17681–17688. doi: 10.1074/jbc.M600207200. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Meisel A, Harms C, Yildirim F, Bösel J, Kronenberg G, Harms U, Fink KB, Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem. 2006;98:1019–1031. doi: 10.1111/j.1471-4159.2006.04016.x. [DOI] [PubMed] [Google Scholar]
- Melamed I, Turner CE, Aktories K, Kaplan DR, Gelfand EW. Nerve growth factor triggers microfilament assembly and paxillin phosphorylation in human B lymphocytes. J Exp Med. 1995;181:1071–1079. doi: 10.1084/jem.181.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani L, Gerrits B, Gehrig P, Kempf A, Dimou L, Wollscheid B, Schwab ME. Neuronal Nogo-A modulates growth cone motility via Rho-GTP/LIMK1/cofilin in the unlesioned adult nervous system. J Biol Chem. 2009;284:10793–10807. doi: 10.1074/jbc.M808297200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Zucker RS. Action potentials must admit calcium to evoke transmitter release. Nature. 1991;350:153–155. doi: 10.1038/350153a0. [DOI] [PubMed] [Google Scholar]
- Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, Duman RS. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci. 2003;23:10841–10851. doi: 10.1523/JNEUROSCI.23-34-10841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Sihra TS. Synaptosomes possess an exocytotic pool of glutamate. Nature. 1986;321:772–773. doi: 10.1038/321772a0. [DOI] [PubMed] [Google Scholar]
- Ninkovic J, Götz M. Signaling in adult neurogenesis: from stem cell niche to neuronal networks. Curr Opin Neurobiol. 2007;17:338–344. doi: 10.1016/j.conb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Kubo K, Ishizeki J, Takazawa T, Saito S, Goto F. The interaction of noradrenaline with sevoflurane on GABAA receptor-mediated inhibitory postsynaptic currents in the rat hippocampus. Brain Res. 2005;1039:153–161. doi: 10.1016/j.brainres.2005.01.060. [DOI] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- Niwano K, Arai M, Tomaru K, Uchiyama T, Ohyama Y, Kurabayashi M. Transcriptional stimulation of the eNOS gene by the stable prostacyclin analogue beraprost is mediated through cAMP-responsive element in vascular endothelial cells: close link between PGI2 signal and NO pathways. Circ Res. 2003;93:523–530. doi: 10.1161/01.RES.0000091336.55487.F7. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Parrish-Aungst S, Shipley MT, Erdelyi F, Szabo G, Puche AC. Quantitative analysis of neuronal diversity in the mouse olfactory bulb. J Comp Neurol. 2007;501:825–836. doi: 10.1002/cne.21205. [DOI] [PubMed] [Google Scholar]
- Pastuszko A, Wilson DF, Erecińska M. Effects of kainic acid in rat brain synaptosomes: the involvement of calcium. J Neurochem. 1984;43:747–754. doi: 10.1111/j.1471-4159.1984.tb12796.x. [DOI] [PubMed] [Google Scholar]
- Piepponen TP, Skujins A. Rapid and sensitive step gradient assays of glutamate, glycine, taurine and gamma-aminobutyric acid by high-performance liquid chromatography-fluorescence detection with o-phthalaldehyde-mercaptoethanol derivatization with an emphasis on microdialysis samples. J Chromatogr B Biomed Sci Appl. 2001;757:277–283. doi: 10.1016/s0378-4347(01)00156-6. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Raiteri M. N-methyl-d-aspartic acid (NMDA) and non-NMDA receptors regulating hippocampal norepinephrine release. I. Location on axon terminals and pharmacological characterization. J Pharmacol Exp Ther. 1992;260:232–237. [PubMed] [Google Scholar]
- Prinjha RK, Shapland CE, Hsuan JJ, Totty NF, Mason IJ, Lawson D. Cloning and sequencing of cDNAs encoding the actin cross-linking protein transgelin defines a new family of actin-associated proteins. Cell Motil Cytoskeleton. 1994;28:243–255. doi: 10.1002/cm.970280307. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Reif A, Schmitt A, Fritzen S, Chourbaji S, Bartsch C, Urani A, Wycislo M, Mössner R, Sommer C, Gass P, Lesch KP. Differential effect of endothelial nitric oxide synthase (NOS-III) on the regulation of adult neurogenesis and behaviour. Eur J Neurosci. 2004;20:885–895. doi: 10.1111/j.1460-9568.2004.03559.x. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- Sperk G. Simultaneous determination of serotonin, 5-hydroxindoleacetic acid, 3,4-dihydroxyphenylacetic acid and homovanillic acid by high performance liquid chromatography with electrochemical detection. J Neurochem. 1982;38:840–843. doi: 10.1111/j.1471-4159.1982.tb08708.x. [DOI] [PubMed] [Google Scholar]
- Sperk G, Berger M, Hörtnagl H, Hornykiewicz O. Kainic acid-induced changes of serotonin and dopamine metabolism in the striatum and substantia nigra of the rat. Eur J Pharmacol. 1981;74:279–286. doi: 10.1016/0014-2999(81)90046-7. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Sun HQ, Yamamoto M, Mejillano M, Yin HL. Gelsolin, a multifunctional actin regulatory protein. J Biol Chem. 1999;274:33179–33182. doi: 10.1074/jbc.274.47.33179. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Nelson AD, Eickstaedt JB, Wallace K, Wright LS, Svendsen CN. Glutamate enhances proliferation and neurogenesis in human progenitor cell cultures derived from the fetal cortex. Eur J Neurosci. 2006;24:645–653. doi: 10.1111/j.1460-9568.2006.04957.x. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Kira M, Subue K. Gelsolin is localized in neuronal growth cones. Dev Brain Res. 1993;76:268–271. doi: 10.1016/0165-3806(93)90217-x. [DOI] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. 2005;47:803–815. doi: 10.1016/j.neuron.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Walzlein JH, Synowitz M, Engels B, Markovic DS, Gabrusiewicz K, Nikolaev E, Yoshikawa K, Kaminska B, Kempermann G, Uckert W, Kaczmarek L, Kettenmann H, Glass R. The antitumorigenic response of neural precursors depends on subventricular proliferation and age. Stem Cells. 2008;26:2945–2954. doi: 10.1634/stemcells.2008-0307. [DOI] [PubMed] [Google Scholar]
- Wang E, Gundersen D. Increased organization of cytoskeleton accompanying the aging of human fibroblasts in vitro. Exp Cell Res. 1984;154:191–202. doi: 10.1016/0014-4827(84)90679-7. [DOI] [PubMed] [Google Scholar]
- Wang LP, Kempermann G, Kettenmann H. A subpopulation of precursor cells in the mouse dentate gyrus receives synaptic GABAergic input. Mol Cell Neurosci. 2005;29:181–189. doi: 10.1016/j.mcn.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Whitney NP, Peng H, Erdmann NB, Tian C, Monaghan DT, Zheng JC. Calcium-permeable AMPA receptors containing Q/R-unedited GluR2 direct human neural progenitor cell differentiation to neurons. FASEB J. 2008;22:2888–2900. doi: 10.1096/fj.07-104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin-promoter GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol. 2008;210:531–542. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M, Kawauchi T, Sone M, Nishimura YV, Terao M, Chihama K, Nabeshima Y, Hoshino M. Involvement of a Rac activator, P-Rex1, in neurotrophin-derived signaling and neuronal migration. J Neurosci. 2005;25:4406–4419. doi: 10.1523/JNEUROSCI.4955-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]