

Abstract
The involvement of reactive oxygen species (ROS) in morphine-induced analgesia and tolerance has been suggested, yet how and where ROS take part in these processes remains largely unknown. Here, we report a novel role for the superoxide-generating enzyme NOX1/NADPH oxidase in the regulation of analgesia and acute analgesic tolerance. In mice lacking Nox1 (Nox1−/Y), the magnitude of the analgesia induced by morphine was significantly augmented. More importantly, analgesic tolerance induced by repeated administration of morphine was significantly suppressed compared with that in the littermates, wild-type Nox1+/Y. In a membrane fraction obtained from the dorsal spinal cord, no difference was observed in morphine-induced [35S]GTPγS-binding between the genotypes, whereas morphine-stimulated GTPase activity was significantly attenuated in Nox1−/Y. At 2 h after morphine administration, a significant decline in [35S]GTPγS-binding was observed in Nox1+/Y but not in Nox1−/Y. No difference in the maximal binding and affinity of [3H]DAMGO was observed between the genotypes, but the translocation of protein kinase C isoforms to the membrane fraction following morphine administration was almost completely abolished in Nox1−/Y. Finally, the phosphorylation of RGS9-2 and formation of a complex by Gαi2/RGS9-2 with 14-3-3 found in morphine-treated Nox1+/Y were significantly suppressed in Nox1−/Y. Together, these results suggest that NOX1/NADPH oxidase attenuates the pharmacological effects of opioids by regulating GTPase activity and the phosphorylation of RGS9-2 by protein kinase C. NOX1/NADPH oxidase may thus be a novel target for the development of adjuvant therapy to retain the beneficial effects of morphine.
Introduction
Opioids are among the most effective analgesics currently in use. However, the efficacy of morphine is limited by the rapid development of tolerance. Up to the present, roles of reactive oxygen and nitrogen species in the development of morphine antinociceptive tolerance have been documented (Rokyta et al., 2003; Muscoli et al., 2007; Doyle et al., 2009). Of particular interest is the fact that morphine-induced tolerance was associated with activation of spinal NADPH oxidase (Doyle et al., 2010). This finding introduced the possibility that the enzyme may be a critical source of reactive oxygen species mediating nociceptive signaling. NADPH oxidase is a superoxide-generating flavoenzyme comprising a membrane-bound catalytic subunit NOX and several cytosolic regulatory subunits. NOX has several homologs, including NOX2 implicated in neurodegenerative and psychiatric disorders (Sorce and Krause, 2009). NOX2 was also reported to take part in the development of neuropathic pain induced by nerve injury (Kim et al., 2010). NOX1 is not well understood, and although its role in some organs is being elucidated (Matsuno et al., 2005, Cui et al., 2011), its function in the nervous system is still unclear. Previously, we demonstrated the involvement of NOX1 in hyperalgesia using mice lacking the Nox1 gene (Nox1−/Y) (Ibi et al., 2008).
While the molecular mechanisms underlying the development of opioid-induced tolerance have not been fully clarified, desensitization appears to contribute to tolerance. Morphine desensitizes the receptor via a protein kinase C (PKC)-dependent pathway (Johnson et al., 2006), while a synthetic opioid agonist desensitizes MOR μ-opioid receptor by G-protein-coupled receptor kinase (GRK) dependent internalization and the resultant downregulation of MOR expression (Zhang et al., 1998). Regulator of G-protein signaling (RGS) proteins were identified as taking part in the receptor's desensitization independent of its internalization and downregulation (Garzón et al., 2001). In fact, a study in primary cultured neurons showed that desensitization occurs independent of internalization (Arttamangkul et al., 2006). RGS proteins share a conserved domain with GTPase-activating protein (GAP) activity, and accelerate the hydrolysis of guanosine triphosphate (GTP) by Gα, thus limiting the duration of G-protein coupled receptor (GPCR) signaling. RGS9-2 is a splice variant of the rgs9 gene with a unique pattern of localization in regions mediating responses to opiates (Gold et al., 1997; Rahman et al., 1999). Acute morphine administration increased expression of RGS9-2 in the nervous system, and mice lacking RGS9 show increased morphine analgesia with delayed tolerance (Zachariou et al., 2003). Intriguingly, phosphorylation of the RGS protein by various kinases including PKC has been documented to regulate GAP activity (Willars, 2006). In preceding communication, we reported that activation of PKC by ROS derived from NOX1 was the key mechanism underlying the development of hyperalgesia (Ibi et al., 2008). As a logical extension of these findings, a possible correlation may exist between NOX1/NADPH oxidase and desensitization of MOR.
This led us to undertake the investigation of whether NOX1/NADPH oxidase is the source of ROS-mediating opiate responses. We report here a novel role for NOX1/NADPH oxidase in morphine-induced analgesia and acute analgesic tolerance by regulating GTPase activity and phosphorylation of RGS9-2 by PKC.
Materials and Methods
Reagents.
Morphine-HCl was obtained from Takeda Pharmaceutical Company. The antibodies against RGS9, 14-3-3, Gαi2, PKCδ, PKCε, and PKCγ were purchased from Santa Cruz Biotechnology. The anti-PKCα antibody was obtained from BD Biosciences. The phospho-(Ser) PKC substrate antibody was obtained from CST. EZ-Link-sulfo-NHS-LC-Biotin, NeutrAvidin-agarose was obtained from Pierce. Hydroethidine was from Invitrogen and L012 from Wako Pure Chemicals Industries. The PrimeScript RT reagent Kit and SYBR Premix EX Taq II were purchased from Takara. The RNeasy Micro Kit was obtained from Qiagen. The protease inhibitor cocktail, phosphatase inhibitor cocktail, and CanGet Signal were obtained from Nacalai Tesque.
Animals.
Male mice deficient in Nox1 (Nox1−/Y) and control littermates (Nox1+/Y) were housed in a temperature-controlled room (21–23°C) with a 12 h light/dark cycle. For the present experiments, 11–12-week-old mice were used. All procedures were done in accordance with the policies and recommendations of the International Association for the Study of Pain and were approved by the Committee for Animal Research at Kyoto Prefectural University of Medicine.
Assessment of nociception.
Mechanical nociception and thermal nociception were evaluated as described previously (Ibi et al., 2008). In brief, thermal nociception was determined as the latency to paw licking and tapping in the hot plate test (54°C). The cutoff time was 30 s. Pressure (20 g/s) was gradually applied to the tail of mice and nociceptive thresholds were determined as the pressure (g) required to elicit tail withdrawal. The cutoff weight was 500 g. The morphine-induced analgesia was evaluated as the latency to mechanical and thermal nociceptive stimuli. The test was performed 30–90 min after the morphine administration. To investigate the development of tolerance following the repeated administration of morphine, mice were given morphine (10 mg/kg, i.p.) once a day for 7 consecutive days. Antinociception was calculated as a percentage of the maximum possible effect (MPE) according to the following formula: MPE = (latency at 30 min after morphine injection − latency at preinjection)/(cutoff time − latency at preinjection) × 100.
Primary culture.
Primary cultures of neurons and glia in the spinal cord were prepared as described previously (Urushitani et al., 2006) with minor modifications. The dorsal spinal cords of fetal mice at 14 d of gestation were dissected, minced, and treated with 0.25% trypsin-EDTA for 30 min at 37°C. Cell suspensions plated on 0.1% polyethylenimine-coated dishes were maintained in Eagle's MEM (EMEM) supplemented with heat-inactivated FBS and horse serum. After 3 d, neuronal cells were incubated in EMEM containing 10 μm cytosine-β-d-arabinofuranoside for 48 h for subsequent mRNA analyses. Glial cells were recovered from cell suspensions plated on noncoated dishes and incubated in 5% CO2 for 30 min to allow adhesion to the bottom of dishes. The second passage of these cells was used for mRNA analyses. Total RNA was isolated using the RNeasy Micro Kit according to its instructions.
Analysis of mRNA.
Total RNA was isolated from DRG by the acid guanidinium thiocyanate/phenol/chloroform method. Reverse transcription was performed with the PrimeScript RT reagent Kit according to the instructions. Real-time PCR was performed by use of the GeneAmp 5700 Sequence Detection System (Applied Biosystems) with the SYBR Premix EX Taq II. The primer sequences used were the same as described previously (Cui et al., 2009), as follows: for NOX3, 5′-ctcgttgcctacgggatagc-3′ and 5′-ccttcagcattccttggcct-3′; Duox1, 5′-ggctgacccatcacctctac-3′ and 5′-taattgcagggaccaggaag-3′; Duox2, 5′-ccaaggacgagttcttcacc-3′ and 5′-ccagactcccggaacataga-3′; 18s rRNA, 5′-ttcgccatcactgccattaag-3′ and 5′-ctggctgtacttcccatcctt-3′; RGS9-2, 5′-ggcagctggaagaagaagagaa-3′ and 5′-gagggctctctgttctcagtga-3′; RGSZ1, 5′-cgtcttctggatggcctgtg-3′ and 5′-atgtgttgggatggctccac-3′; RGSZ2, 5′-cttctggctggcctgtgaag-3′ and 5′-tcatacatgtgtaggctggg-3′; GAIP, 5′-cctcatgcaccgggctcgta-3′ and 5′-ctgtgggccctaaagggtgttgttc-3′; and Hprt, 5′-aaactttgctttccctggtta-3′ and 5′-aggctttgtatttggcttttc-3′.
Immunohistochemistry.
The spinal cord was dissected and quickly embedded in OCT compound. Cryosections were fixed in 4% paraformaldehyde for 1 h and treated with 1% H2O2 to quench endogenous peroxidase. After being blocked with 1% goat serum for 1 h, sections were incubated with an anti-PKCγ antibody (1:2000) overnight at 4°C, washed with PBS, and incubated with biotinylated anti-rabbit IgG (1:1000) for 1 h. Washed sections were then incubated in avidin-biotin complex solution (1:1000) for 1 h, and developed in diaminobenzidine-H2O2 solution (Winsky-Sommerer et al., 2004) for 30 min.
Dihydroethidium staining and detection of superoxide production.
Detection of superoxide production was performed by dihydroethidium (DHE) staining and chemiluminescence using L012 as previously described (Cui et al., 2009; Matsuno et al., 2005) with minor modifications. Frozen spinal cord samples embedded in OCT compound were cut at 10 μm and placed on a glass slide. Sections were incubated with 1 μm DHE for 20 min at 37°C and then washed three times with PBS. For detection, a 543 nm He-Ne laser combined with a 560 nm long-pass filter was used.
Superoxide production in the dorsal spinal cord was measured with the L012 chemiluminescence assay using a Luminescensor (Atto). Tissue homogenate was prepared in Krebs-HEPES buffer, pH 7.4. Preincubation was performed for 20 min at 37°C and the measurement was started by the addition of 10 μm L012. After 15 min, 30 μm DPI, an inhibitor of NADPH oxidase, was added and the measurement was continued for another 2 min. Chemiluminescence of L012 was expressed as relative light units (RLU; counts/mg protein/min), calculated by subtracting the count obtained in the presence of DPI.
Guanosine-5′-o-(3-[35S]thio)triphosphate-binding assay.
Guanosine-5′-o-(3-[35S]thio)triphosphate ([35S]GTPγS)-binding was assayed as described previously (Narita et al., 2001) with minor modifications. The dorsal spinal cord was dissected at 2 or 24 h after morphine administration. The crude membrane homogenate was incubated at 30°C for 1 h in binding assay buffer (5 mm MgCl2, 1 mm EGTA, and 100 mm NaCl in 50 mm Tris-HCl, pH7.4) with various concentrations of the agonist, 30 μm guanosine-5′-diphosphate (GDP) and 50 pm [35S]GTPγS (specific activity, 1250 Ci/mmol; PerkinElmer Life Sciences). The reaction was terminated by filtrating through Whatman GF/B glass filters presoaked in 50 mm Tris-HCl, pH7.4, and 5 mm MgCl2 at 4°C. Filters were washed with ice-cold buffer and the radioactivity was determined by a liquid scintillation counter. Nonspecific binding was measured in the presence of 10 μm unlabeled GTPγS. The binding of [35S]GTPγS to the membrane was calculated as a percentage of basal [35S]GTPγS-binding measured in the presence of GDP and absence of the agonist.
Measurement of GTPase activity.
GTPase activity of spinal cord membrane was determined by measuring [γ-32P]GTP (5000 Ci/mmol; Institute of Isotope, Budapest, Hungary) hydrolysis as described previously (Nakagawa et al., 1999). The reaction was started by the addition of 20 μl of the membrane fraction to 80 μl of morphine (0.1–10 μm) or vehicle-containing prewarmed reaction mixture [0.4 nm [γ-32P]GTP (4 × 105 cpm), 0.1 mm EDTA, 2 mm dithiothreitol, 100 mm NaCl, 5 mm MgCl2, 1 mm ATP, 10 mm creatine phosphate, 2.5 units of creatine phosphokinase, 0.25 mm App(NH)p, 1 mm ouabain, and 10 mm Tris-HCl, pH 7.4]. Unlabeled GTP was added at 0.3 or 1 μm. The reaction was continued for 30 min at 30°C and terminated by the addition of 900 μl of an ice-cold suspension of 5% (w/v) activated charcoal (Norit A) in 20 mm H3PO4, pH 2.3. After 15 min of centrifugation at 11,000 × g, 500 μl of the supernatant was removed, and the amount of 32Pi released by the hydrolysis of GTP was determined by liquid scintillation counter. Nonspecific GTPase was assessed in the presence of 100 μm GTP. Data were corrected for the background and expressed as a percentage of basal GTPase activity.
[Tyrosyl-3,5-3H(N)]DAMGO-binding assay.
A selective MOR agonist, [D-Ala(2)-N-MePhe(4)-Gly-ol(5)]enkephalin (DAMGO)-binding was assayed as described previously (Minami et al., 1995) with minor modifications. The crude membrane was resuspended in the buffer for the binding assay. Incubations of membranes with [3H]DAMGO were performed at 25°C for 2 h and the reaction was terminated by filtrating through Whatman GF/B glass filters presoaked with 0.1% polyethylenimine. The filters were washed with ice-cold buffer and the radioactivity was measured by liquid scintillation counting. Nonspecific binding was determined in the presence of 10 μm unlabeled DAMGO.
Detection of membrane translocation of PKC isoforms.
Apocynin (2 mg/ml in tap water), an inhibitor of NADPH oxidase and an antioxidant (Heumüller et al., 2008), was administered to Nox1+/Y for 18 d as described previously (Harraz et al., 2008). TEMPOL (2.9 μmol), a membrane-permeable radical scavenger, was injected intrathecally (Schwartz et al., 2008). Morphine (10 mg/kg, i.p.) was administered immediately after TEMPOL injection. At 2 h after morphine administration, the dorsal spinal cord dissected from mice was homogenized and centrifuged at 1000 × g for 5 min. Supernatants were centrifuged at 10,000 × g for 10 min to remove the mitochondrial fraction, and further centrifuged at 100,000 × g for 30 min at 4°C to obtain the membrane fraction. The resultant pellet dissolved in lysis buffer (25 mm β-glycerophosphate, 2 mm EGTA, 1% Triton X-100, 1 mm PMSF, and 2 mm DTT in 25 mm Tris-HCl, pH7.2) was subjected to SDS-PAGE and immunoblot analyses using anti-PKC antibodies.
Immunoprecipitation experiments.
The dorsal spinal cord was homogenized in ice-cold 25 mm Tris-HCl, pH7.2, buffer containing 1 mm EGTA and 0.32 m sucrose supplemented with protease and phosphatase inhibitor cocktails. Homogenate was centrifuged at 1000 × g for 5 min and the supernatant was centrifuged at 20,000 × g for 20 min to obtain a crude synaptosomal (P2) fraction. The pellet was washed twice and suspended in immunoprecipitation (IP) buffer (50 mm NaCl, 40 mm octyl-β-d-glucopyranoside, 0.5 mm NaF, and 2 mm sodium orthovanadate supplemented with the protease inhibitor cocktail in 50 mm Tris-HCl buffer, pH7.4). The P2 fraction was then incubated overnight at 4°C with biotinylated IgGs (1 μg) raised against the RGS9 or 14-3-3 proteins. These antibodies were first reacted with protein A/G, and purified antibodies were obtained by elution with glycine-HCl, pH2.5, and subsequent neutralization with Tris-HCl, pH9.5. The biotinylation of antibodies was performed using EZ-Link-sulfo-NHS-LC-Biotin according to the manufacturer's instructions. Following the addition of NeutrAvidin-agarose (30 μl) and incubation for 60 min at 4°C, samples were centrifuged at 1000 × g for 1 min, and supernatant fractions were removed. The agarose pellets were washed five times, centrifuged, and resuspended in IP buffer. The proteins were eluted with Laemmli sample buffer and analyzed by SDS-PAGE, followed by immunoblotting using specific antibodies.
Immunoblotting.
Membrane fractions were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were incubated overnight at 4°C with anti-PKCα, anti-PKCε, and anti-PKCγ antibodies (1:1000); and anti-Gαi2, anti-14-3-3, anti-RGS9-2 antibodies, and phospho-(Ser)PKC substrate antibody (1:2000). After being washed, the membrane was incubated with HRP-conjugated secondary antibodies for 1 h at room temperature, and subsequently with Immobilon Western Chemiluminescent HRP substrate (Millipore). The density of the band was analyzed using ImageJ software.
Statistical analyses.
All data are expressed as the mean ± SEM. The statistical significance of differences between groups was assessed with a two-way ANOVA followed by the Bonferroni–Dunn test.
Results
Morphine-induced analgesia was potentiated in Nox1−/Y
Previously, no difference in locomotor activity and coordinated movement was found between Nox1+/Y and Nox1−/Y (Ibi et al., 2008). In untreated mice, no significant difference in the threshold for thermal (9.64 ± 0.52 s latency in Nox1+/Y vs 8.43 ± 0.34 s in Nox1−/Y, N = 9–10) and mechanical (107.32 ± 7.55 g in Nox1+/Y vs 99.30 ± 6.45 g in Nox1−/Y, N = 10) nociceptive stimuli was observed between the two genotypes. In mice treated with intraperitoneal injections of 3 mg/kg morphine, no difference in analgesic effects was observed between the genotypes (data not shown). On the other hand, the effects of 5–10 mg/kg morphine were significantly potentiated in Nox1−/Y, which was sustained until 90 min after the injection (F(1,52) = 17.40) (Fig. 1b). Similar effects were demonstrated in Nox1−/Y treated with intrathecal injections of 0.5 μg of morphine (45.08 ± 1.92 g in Nox1+/Y vs 84.65 ± 5.34 g in Nox1−/Y, p < 0.01, N = 6).
Figure 1.

Morphine-induced analgesia was potentiated in Nox1−/Y. Time course of the effects of morphine (5 mg/kg or10 mg/kg) administered intraperitoneally. Top and bottom demonstrate morphine-induced analgesia against thermal and mechanical nociception, respectively. WT and KO represent control littermates (Nox1+/Y) and Nox1-deficient mice (Nox1−/Y), respectively. Values were obtained from 8 to 9 animals per group. *p < 0.05 compared with Nox1+/Y.
NOX1 mRNA was expressed in spinal cord neurons as well as in glia
Localization of NOX1 was next investigated in the spinal cord using anti-NOX1 antibodies of different sources. When compared with the staining pattern obtained in Nox1−/Y, however, no specific signal for NOX1 was detected. Expression of NOX1 mRNA was therefore examined in primary cultures prepared from fetal mouse spinal cord. As shown in Figure 2a, the NOX1 transcript was detected not only in neuronal cells but also in glia. Except for NOX3, all other NOXs, p47phox, p67phox, the organizer subunit NOXO1, and the activator subunit NOXA1 were detected in both cell lineages (data not shown). Similar levels of NOX1 mRNA were expressed in these cells when verified by quantitative real-time PCR (NOX1/18S rRNA = 4.7 × 10−3 ± 0.5 × 10−3 in neuron vs 5.2 × 10−3 ± 0.7 × 10−3 in glia, N = 4–6).
Figure 2.

Expression of NOX1 mRNA and superoxide production in the dorsal spinal cord. a, Representative RT-PCR of NOX isoforms expressed in cultured neurons and glia isolated from the spinal cord. b, Superoxide production in the dorsal spinal cord. Left panels demonstrate immunohistochemical staining of PKCγ (scale bar, 100 μm), DHE staining, and merged DHE/phase contrast (scale bar, 25 μm).
Superoxide production in the dorsal spinal cord was reduced in Nox1−/Y
MOR is known to occur in laminae I and II of the spinal cord (Polgár et al., 1999). When superoxide production in the dorsal horn was examined by DHE staining, the fluorescence in Nox1−/Y appeared slightly less intense than that in Nox1+/Y (Fig. 2b). In accord with these findings, superoxide production assessed by DPI-sensitive L012 chemiluminescence (RLU) was significantly attenuated in Nox1−/Y (4299.15 ± 559.41 in Nox1+/Y vs 2231.00 ± 235.48 in Nox1−/Y, p < 0.01, N = 6–8).
Morphine-induced activation of GTPase was suppressed in Nox1−/Y
To elucidate the molecular mechanisms underlying the augmented effects of morphine in Nox1−/Y, we next measured G-protein activation based on [35S]-GTPγS-binding. Morphine dose-dependently increased [35S]-GTPγS-binding to the dorsal spinal cord membrane obtained from untreated mice. As shown in Figure 3a, there was no difference in the morphine-induced [35S]-GTPγS-binding between the genotypes (F(1,168) = 0.53). Opioid receptor signaling involves the intrinsic GTPase activity of the Gα subunit dissociated from the receptor following interaction with GTP. Hydrolysis of GTP dictates the duration of Gα-GTP- and Gβγ-mediated signaling. We therefore examined the GTPase activity in the presence of 0.3 μm GTP or 1 μm GTP, at a concentration at which opioid agonists activate the GTPase activity of the Gi/o and Gz/q/11 protein, respectively (Garzón et al., 1997). As shown in Figure 3b, addition of morphine dose-dependently increased GTP hydrolysis in the membrane fraction obtained from Nox1+/Y, but not from Nox1−/Y (F values of 0.3 μm GTP and 1 μm GTP are F(1,67) = 8.35, F(1,56) = 9.61). On the other hand, mRNA levels of RGS proteins that regulate MOR agonist signaling were similar in both genotypes (Fig. 3c). These findings suggest that NOX1/NADPH oxidase may modulate opioid receptor signaling downstream of G-protein activation, without affecting the level of RGS expression.
Figure 3.
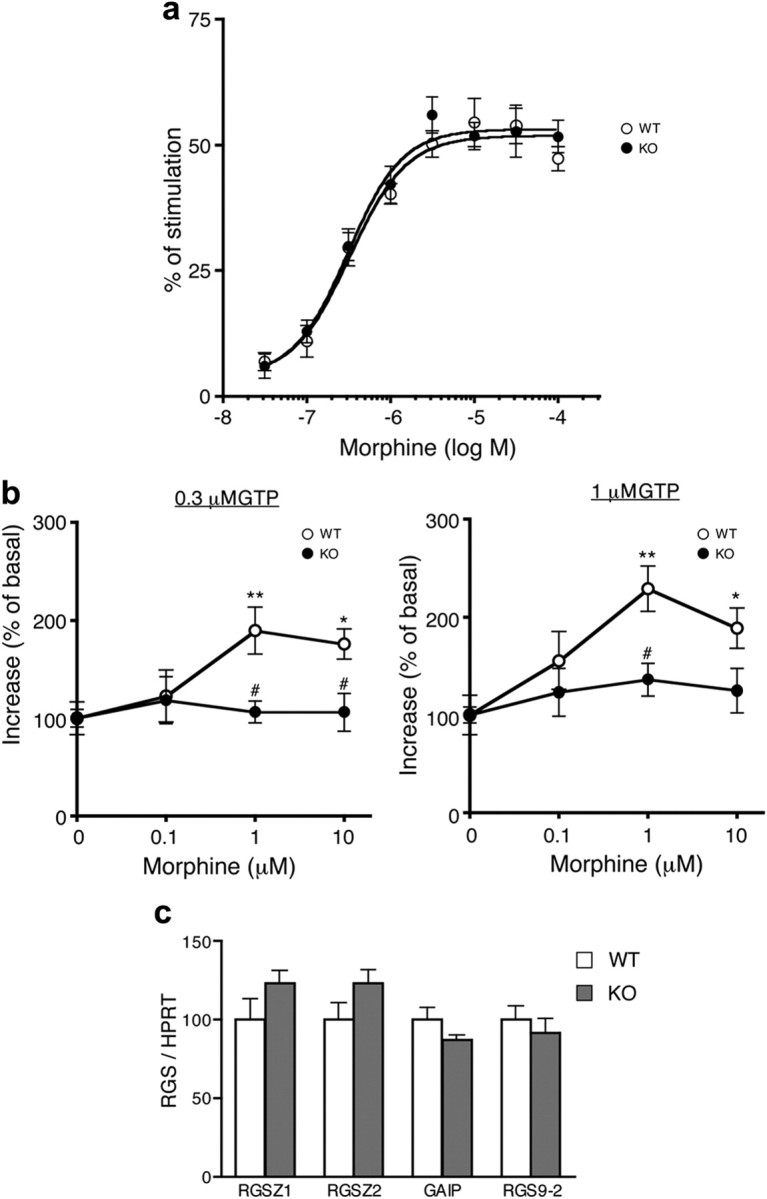
Morphine-induced activation of GTPase was suppressed in Nox1−/Y. a, Activation of [35S]GTPγS-binding induced by increasing the concentration of morphine in the dorsal spinal cord membrane obtained from untreated mice. Values were obtained from 10 to 11 animals per group. b, Effects of increasing the concentration of morphine on GTP hydrolysis in the presence of 0.3 μm (left) or 1 μm GTP (right). The membrane fraction was obtained from the dorsal spinal cord. *p < 0.05, **p < 0.01 compared with control. #p < 0.05 compared with Nox1+/Y. Values were obtained from 9 to 10 animals per group. c, Expression of RGS mRNA in the dorsal spinal cord. Values were obtained from 4 animals per group.
Morphine-induced analgesic tolerance was suppressed in Nox1−/Y
Next, we investigated the role of NOX1 in morphine-induced tolerance. At 24 h after the initial administration of morphine (10 mg/kg), mice were treated with the same amount of morphine, and analgesic effects against thermal stimuli were determined. As shown in Figure 4a, a significant reduction in morphine-induced analgesia was demonstrated in Nox1+/Y, indicating the development of acute tolerance. In contrast, similar levels of analgesia were maintained in Nox1−/Y (F(3,88) = 59.75). Following the daily administration of morphine for 7 d, the analgesic effect of morphine was markedly diminished in both genotypes, whereas the magnitude of decrease was less in Nox1−/Y compared with Nox1+/Y(F(1,80) = 11.49) (Fig. 4b). Intriguingly, these findings were in line with a previous study demonstrating that NADPH oxidase inhibitors blocked the development of chronic morphine-induced tolerance (Doyle et al., 2010). Since the difference in analgesic effects between the genotypes was apparent as early as 24 h after the first administration of morphine, we subsequently focused on the regulatory mechanisms of NOX1/NADPH oxidase in the induction of acute analgesic tolerance to morphine.
Figure 4.

Morphine-induced analgesic tolerance was suppressed in Nox1−/Y. a, Acute tolerance developed 24 h after the morphine injection. Latency for thermal nociceptive stimuli was determined. Analgesic effect of morphine (10 mg/kg, i.p.) was demonstrated by first (first inj.) and second (second inj.) administration in Nox1+/Y and Nox1−/Y. **p < 0.01 compared with the first administration. Values were obtained from 7 to 8 animals per group. b, Development of tolerance during daily administration of morphine (10 mg/kg/d, i.p.). Analyses were performed at 30 min after the last injection. Vertical axis indicates percentage of MPE versus day 1. Values were obtained from 11 animals per group. **p < 0.01 compared with the first administration; #p < 0.05 compared with Nox1−/Y on day 7.
[35S]-GTPγS-binding was preserved in Nox1−/Y treated with morphine
In saline-treated Nox1+/Y, addition of DAMGO as well as morphine dose-dependently increased [35S]GTPγS-binding to the membrane isolated from the dorsal spinal cord. At 30 min after the morphine injection, no difference in [35S]GTPγS-binding was observed between the genotypes (data not shown). At 2 h after the injection, however, [35S]GTPγS-binding stimulated with morphine or DAMGO was significantly attenuated in Nox1+/Y, but not in Nox1−/Y (F values of morphine and DAMGO are F(3,312) = 14.98, F(3,304) = 16.86) (Fig. 5a). At 24 h after the injection, [35S]GTPγS-binding was further attenuated in Nox1+/Y, but significantly preserved in Nox1−/Y (F values of morphine and DAMGO are F(3,263) = 17.73, F(3,312) = 74.29). On the other hand, there was no difference in [35S]GTPγS-binding between the genotypes when stimulated with DPDPE, a selective agonist for the δ-opioid receptor (DOR), or U69593, a selective agonist for the κ-opioid receptor (KOR) (F values of DPDPE and U69593 are F(3,98) = 2.76, F(3,102) = 1.17) (Fig. 5b). These findings suggest the involvement of MOR in NOX1/NADPH oxidase-mediated development of acute analgesic tolerance to morphine.
Figure 5.

[35S]GTPγS-binding was preserved in Nox1−/Y treated with morphine. a, Effects of increasing the concentration of DAMGO (left) or morphine (right) on [35S]GTPγS-binding to the membrane obtained from the dorsal spinal cord of mice at 2 h after morphine administration (10 mg/kg, i.p.). *p < 0.05, **p < 0.01 compared with saline-injected mice. #p < 0.05, ##p < 0.01 compared with morphine (Mor.)-injected Nox1−/Y. Values were obtained from 11 to 12 animals per group. b, Effects of increasing the concentrations of opioid receptor agonists on [35S]GTPγS-binding at 24 h after morphine administration. *p < 0.05, **p < 0.01 compared with saline-injected mice. #p < 0.05, ##p < 0.01 compared with morphine-injected Nox1−/Y. Values were obtained from 6 to 12 animals per group.
[3H]DAMGO-binding was unaltered in mice treated with morphine
To examine whether the altered [35S]GTPγS-binding in morphine-treated mice is due to a decrease in the number of MOR in the membrane, we performed saturation binding analyses using [3H]DAMGO. The maximal binding of [3H]DAMGO in spinal cord membrane was unaltered following morphine treatment. Furthermore, there was no difference in the affinity (Kd) of [3H]DAMGO among untreated and treated animal groups (Table 1).
Table 1.
[3H]DAMGO-binding to the dorsal spinal cord membrane
| Bmax (fmol/mg protein) | KD (nm) | |
|---|---|---|
| WT, saline | 117.36 ± 7.72 | 1.09 ± 0.14 |
| KO, saline | 112.77 ± 2.28 | 0.95 ± 0.10 |
| WT, morphine | 108.95 ± 4.24 | 1.12 ± 0.17 |
| KO, morphine | 119.77 ± 5.24 | 1.34 ± 0.17 |
The values (the mean ± SEM) based on the Scatchard plot analyses were obtained from 7–8 independent experiments. Bmax and KD were determined by fitting the data to a one-site binding model using nonlinear regression. The values of two-way ANOVA in the Bmax and KD values are F(1,26) = 2.02 and F(1,26) = 1.45, respectively.
Morphine-induced translocation of PKC was attenuated in Nox1−/Y
Since previous studies showed the involvement of PKC isoforms in tolerance to morphine (Bilsky et al., 1996; Bailey et al., 2006), we next examined the activation of PKC following morphine administration. Among PKC isoforms, PKCα and ε in the dorsal root ganglion, as well as PKCα and γ in the spinal cord, were known to take part in morphine-induced tolerance (Smith et al., 2007). At 2 h after the morphine injection, the translocation of PKCα, PKCε, and PKCγ to the membrane fraction of the dorsal spinal cord was markedly increased in Nox1+/Y, whereas it was significantly suppressed in Nox1−/Y (F values of PKCα, PKCε, and PKCγ are F(1,20) = 26.31, F(1,20) = 10.76, F(1,20) = 15.23) (Fig. 6a). Consistent with these findings, the translocation of PKC isoforms was significantly attenuated in Nox1+/Y administered with an antioxidant apocynin for 18 d (F values of PKCα, PKCε, and PKCγ are F(1,15) = 11.03, F(1,15) = 7.05, F(1,15) = 4.57), or with TEMPOL before morphine administration (F values of PKCα, PKCε, and PKCγ are F(1,15) = 6.58, F(1,15) = 5.71, F(1,15) = 7.78) (Fig. 6b,c). No apparent abnormal behavior was observed in mice during chronic administration of apocynin. These findings suggest that ROS derived from NOX1/NADPH oxidase modulate the translocation of PKC isoforms in the dorsal spinal cord induced by morphine.
Figure 6.

Morphine-induced translocation of PKC was attenuated in Nox1−/Y and in Nox1+/Y treated with antioxidants. a, The membrane was isolated from the dorsal spinal cord of mice 2 h after morphine administration (10 mg/kg, i.p.). Left panels demonstrate representative immunoblots of PKCα, PKCε, PKCγ, and Na+ K+-ATPase. Right panels demonstrate quantitative measurements of the density of PKCα, PKCε, and PKCγ relative to that of Na+ K+-ATPase. **p < 0.01 compared with saline-injected Nox1+/Y; #p < 0.05, ##p < 0.01. Values were obtained from 6 animals per group. b, Effects of apocynin on morphine-induced translocation of PKC. Apocynin (2 mg/ml in water) was administered to mice for 18 d. Values were obtained from 4 to 6 animals per group. *p < 0.05, **p < 0.01 compared with saline-injected mice; #p < 0.05. c, Effects of TEMPOL on morphine-induced translocation of PKC. TEMPOL (2.9 μmol) was intrathecally administered immediately before morphine administration. Values were obtained from 4 to 6 animals per group. *p < 0.05, **p < 0.01 compared with saline-injected mice; #p < 0.05, ##p < 0.01.
Morphine-induced phosphorylation of RGS9-2, formation of Gαi2/RGS9-2/14-3-3, and degradation of Gαi2 were attenuated in Nox1−/Y
The results obtained in [35S]GTPγS and [3H]DAMGO-binding assays suggested the uncoupling of the MOR and Gα protein, rather than the receptor's internalization, to be involved in the desensitization induced by morphine. One of the mechanisms documented for uncoupling is the formation of a Gα, RGS9-2, and 14-3-3 proteins complex (Garzón et al., 2005). The formation of this complex is accompanied by serine phosphorylation of RGS9-2. RGS9-2 has the putative 14-3-3-binding motif, K(K/S)DpSY(P/A), and the PKC consensus sequence, (K/R)XXS. We therefore examined the phosphorylation of the target serine residue of PKC in RGS9-2 using a phospho-(Ser)PKC substrate antibody. As shown in Figure 7a, phosphorylation of RGS9-2 was significantly increased at 1 h after morphine administration in Nox1+/Y, whereas it was significantly suppressed in Nox1−/Y (F(1,32) = 4.24). In accordance with these findings, increased coprecipitation of RGS9-2 and Gαi2 with 14-3-3 proteins depicted in Nox1+/Y was significantly blunted in Nox1−/Y treated with morphine (F values of RGS9-2 and Gαi2 are F(1,28) = 9.24, F(1,24) = 9.95) (Fig. 7b). Moreover, a significant decrease in Gαi2 at 24 h after morphine administration in the crude membrane homogenate was abolished in Nox1−/Y, suggesting that the dissociation of Gαi2 from the membrane was attenuated by Nox1 deficiency (F(1,28) = 6.77) (Fig. 7c).
Figure 7.

Morphine-induced phosphorylation of RGS9-2, formation of Gαi2/RGS9-2/14-3-3, and degradation of Gαi2 were attenuated in Nox1−/Y. a, Morphine-induced phosphorylation of RGS9-2. The dorsal spinal cord was dissected from mice 1 h after saline or morphine administration (10 mg/kg, i.p.). Phosphorylation of RGS9-2 was detected in samples immunoprecipitated by an anti-RGS9 antibody. **p < 0.01 compared with saline-injected Nox1+/Y; #p < 0.05. Values were obtained from 9 animals per group. b, Coimmunoprecipitation of RGS9-2 and Gαi2 with 14-3-3 proteins. The dorsal spinal cord was dissected from mice 2 h after saline or morphine injection. RGS9-2 and Gαi2 were detected in samples immunoprecipitated by an anti-14-3-3 antibody. **p < 0.01 compared with saline-injected Nox1+/Y; ##p < 0.01. Values were obtained from 7 to 8 animals per group. c, Levels of Gαi2 protein. The dorsal spinal cord was dissected from mice at 24 h after saline or morphine injection, and the synaptosomal fraction was subjected to immunoblotting. **p < 0.01 compared with saline-injected Nox1+/Y. Values were obtained from 8 animals per group.
Discussion
We presently demonstrated a novel role for NOX1/NADPH oxidase in morphine-induced analgesia and analgesic tolerance. The principal findings obtained were as follows: (1) Analgesia induced by the intraperitoneal as well as intrathecal injection of morphine was significantly augmented in Nox1−/Y, while no difference in the threshold for nociceptive stimuli was observed between the untreated genotypes. (2) Superoxide production in the dorsal spinal cord was significantly attenuated in Nox1−/Y. (3) No difference in morphine-induced [35S]GTPγS-binding was observed between the untreated genotypes, whereas the morphine-induced increase in GTPase activity was almost completely abolished in the spinal cord membrane of Nox1−/Y. (4) The analgesic tolerance and reduced [35S]GTPγS-binding caused by morphine administration were significantly ameliorated in Nox1−/Y. (5) The translocation of PKC and phosphorylation of RGS9-2 induced by morphine were significantly attenuated in Nox1−/Y. (6) The formation of a complex by Gαi2 and RGS9-2 with 14-3-3 proteins and subsequent degradation of Gαi2 were significantly attenuated in Nox1−/Y. Based on these findings, it is reasonable to conclude that NOX1-derived ROS negatively regulate the analgesic effects of morphine.
Regardless of the route of administration, morphine-induced analgesia was significantly augmented in Nox1−/Y. Metabolized primarily in the liver, morphine is converted into morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) (Kilpatrick and Smith, 2005). M3G does not bind to the opioid receptor and has no analgesic effect, while M6G binds to MOR and elicits more potent effects than morphine (van Dorp et al., 2006). In our study, the effects of the intrathecal administration of morphine were potentiated in Nox1−/Y similar to the findings obtained in intraperitoneally treated mice. Accordingly, our results suggest that the increased analgesic effects in Nox1−/Y are not attributable to the enhanced metabolic conversion of morphine into M6G.
While no difference in morphine-induced activation of G-protein was observed between the genotypes, GTP hydrolysis was activated by the addition of morphine in the spinal cord membrane of Nox1+/Y, but not of Nox1−/Y. Since previous studies demonstrated the upregulation of RGS2 expression caused by oxidative stress (Zmijewski et al., 2001), we examined whether the expression of RGS mRNAs was affected by the Nox1 deficiency. Gα proteins are inactivated by RGSZ1, RGSZ2, and GAIP (Hollinger and Hepler, 2002), while RGS9-2 associated with Gα has limited GAP activity (Lan et al., 2000). No difference in mRNA levels of these RGS proteins was observed between the genotypes. The mechanisms underlying the NOX1-mediated activation of GTPase by morphine are therefore still unclear. ROS derived from NOX1 may directly accelerate the hydrolysis of GTP by Gα, as documented for small GTPases (Heo, 2011), or augment the GAP activity of the RGS protein through posttranslational modifications (Willars, 2006). In fact, NOX1-derived ROS induced an intramolecular disulfide formation in low molecular weight tyrosine phosphatase to increase phosphorylation of p190RhoGAP, a GTPase-activating protein for Rho (Shinohara et al., 2007). A plausible interpretation of the present data may be that reduced GTPase activity in the spinal cord accounts for the increased analgesic effect of morphine demonstrated in Nox1−/Y.
Administration of morphine significantly decreased MOR agonist-stimulated binding of [35S]GTPγS in Nox1+/Y, while the maximal binding of [3H]DAMGO to the spinal cord membrane was unaffected. These findings indicate that morphine elicits desensitization without an apparent change in the number of MOR. This is in agreement with previous studies, which found that morphine-induced desensitization took place without β-arrestin-mediated MOR internalization (Johnson et al., 2006; Groer et al., 2007). Most relevantly, it was demonstrated that the initial exposure to morphine causes Gα depletion due to the interaction of Gα with RGS, while further exposure to morphine elicits phosphorylation and internalization of MOR (Rodríguez-Muñoz et al., 2007). Since the maximal binding of [3H]DAMGO was determined after a single administration of morphine, our results were in accord with these earlier findings.
The increased phosphorylation of RGS9-2 in morphine-treated Nox1+/Y was significantly suppressed in Nox1−/Y. RGS proteins are not simply GTPase-activating proteins, but are also involved in the regulation of G-protein-effector interactions (Abramow-Newerly et al., 2006). The decrease in the interaction of MOR with Gα and resultant sequestering of MOR-activated Gα could be attributed to the interaction of RGS9-2 with 14-3-3 proteins, which is facilitated by PKC-dependent Ser-phosphorylation of RGS9-2 (Garzón et al., 2005). Consistent with this, the formation of a complex by RGS9-2/Gαi2 with 14-3-3 proteins and dissociation of Gαi2 from the membrane were markedly increased in Nox1+/Y following morphine administration, and suppressed by the Nox1 deficiency. Accordingly, RGS9-2 appears to be a target of PKC affected by ROS derived from NOX1 (Fig. 8). Of interest in this regard, the phenotype of Nox1−/Y treated with morphine resembled that reported in RGS9-downregulated or RGS9-deficient mice that showed increased analgesia and delayed tolerance (Garzón et al., 2001; Zachariou et al., 2003).
Figure 8.

Possible mechanisms involved in analgesic tolerance regulated by ROS derived from NOX1/NADPH oxidase. a, RGS9-2 interacts with GTP-bound Gα and enhances GTP hydrolysis by Gα, thereby terminating MOR signaling. b, phosphorylation of RGS9-2 is enhanced by ROS-mediated activation of PKC derived from NOX1/NADPH oxidase. This may increase the association of Gαi2 and RGS9-2 with 14-3-3 proteins and disrupt the interaction of Gα with MOR, leading to the desensitization of MOR. Adapted from the review by Garzón et al. (2008).
A critical issue to be addressed is the molecular mechanism underlying the NOX1-mediated activation of PKC demonstrated in this study. We previously reported that ROS derived from NOX1/NADPH oxidase in dorsal root ganglia (DRG) neurons modulate transient receptor potential vanilloid 1 (TRPV1) in a PKC-dependent manner.
In DRG neurons, ROS derived from NOX1 enhance the translocation of PKC via modulation of the redox state of cysteine residues located at the C1A site (Ibi et al., 2008). The colocalization of PKCγ immunoreactivity and DHE fluorescence in the dorsal spinal cord suggests that NOX1-mediated activation of PKC also takes place in a way similar to that in DRG neurons. The signaling pathways involved in the recruitment of PKCγ to RGS proteins and MOR have been intensively studied (Rodríguez-Muñoz et al., 2008, 2011). The physical interaction with activated PKC may easily phosphorylate RGS proteins and increase their binding to 14-3-3 proteins (Garzón et al., 2005).
In conclusion, we demonstrated a novel role for NOX1 in morphine-induced analgesia and induction of acute tolerance. NOX1/NADPH oxidase attenuates the analgesic effects of morphine, possibly by accelerating the hydrolysis of GTP by Gα. Furthermore, ROS derived from NOX1 augment analgesic tolerance to morphine by activating PKC and then ensuing the formation of a complex by Gα and RGS9-2 with 14-3-3 proteins. The present study provides the first evidence that NOX1/NADPH oxidase is the source of ROS implicated in the modulation of morphine-related analgesia and tolerance. The NOX1 isoform may thus be a novel target for the development of effective adjuvant therapy to maintain the beneficial effects of morphine.
Footnotes
This work was partly supported by Research for Promoting Technological Seeds (A) from Japan Science and Technology Agency (M. I.).
References
- Abramow-Newerly M, Roy AA, Nunn C, Chidiac P. RGS proteins have a signaling complex; interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal. 2006;18:579–591. doi: 10.1016/j.cellsig.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Torrecilla M, Kobayashi K, Okano H, Williams JT. Separation of mu-opioid receptor desensitization and internalization: endogeneous receptors in primary neuronal cultures. J Neurosci. 2006;26:4118–4125. doi: 10.1523/JNEUROSCI.0303-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G. How important is protein kinase C in mu-opioid receptor desensitization and morphine tolerance? Trends Pharmacol Sci. 2006;27:558–565. doi: 10.1016/j.tips.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Bilsky EJ, Bernstein RN, Wang Z, Sadée W, Porreca F. Effects of naloxone and D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen Thr-NH2 and the protein kinase inhibitors H7 and H8 on acute morphine dependence and antinociceptive tolerance. J Pharmacol Exp Ther. 1996;277:484–490. [PubMed] [Google Scholar]
- Cui W, Matsuno K, Iwata K, Ibi M, Katsuyama M, Kakehi T, Sasaki M, Ikami K, Zhu K, Yabe-Nishimura C. NADPH oxidase isoforms and anti-hypertensive effects of atrovastatin demonstrated in two animal models. J Pharmacol Sci. 2009;111:260–268. doi: 10.1254/jphs.09148fp. [DOI] [PubMed] [Google Scholar]
- Cui W, Matsuno K, Iwata K, Ibi M, Matsumoto M, Zhang J, Zhu K, Katsuyama M, Torok NJ, Yabe-Nishimura C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced from (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fiblosis induced by bile duct ligation. Hepatology. 2011;54:949–958. doi: 10.1002/hep.24465. [DOI] [PubMed] [Google Scholar]
- Doyle T, Bryant L, Batinic-Haberle I, Little J, Cuzzocrea S, Masini E, Spasojevic I, Salvemini D. Supraspinal inactivation of mitochondrial superoxide dismutase is a source of peroxynitrite in the development of morphine antinociceptive tolerance. Neuroscience. 2009;164:702–710. doi: 10.1016/j.neuroscience.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle T, Bryant L, Muscoli C, Cuzzocrea S, Esposito E, Chen Z, Salvemini D. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett. 2010;483:85–89. doi: 10.1016/j.neulet.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzón J, Martínez-Peña Y, Sánchez-Blázquez P. Gx/z is regulated by μ but not δ opioid receptors in stimulation of the low Km GTPase activity in mouse periaqueductal grey matter. Eur J Neurosci. 1997;9:1194–1200. doi: 10.1111/j.1460-9568.1997.tb01474.x. [DOI] [PubMed] [Google Scholar]
- Garzón J, Rodríguez-Díaz M, López-Fando A, Sánchez-Blázquez P. RGS9 proteins facilitate acute tolerance to mu-opioid effects. Eur J Neurosci. 2001;13:801–811. doi: 10.1046/j.0953-816x.2000.01444.x. [DOI] [PubMed] [Google Scholar]
- Garzón J, Rodríguez-Muñoz M, López-Fando A, Sánchez-Blázquez P. Activation of mu-opioid receptors transfers control of Galpha subunits to the regulator of G-protein signaling RGS9-2: role in receptor desensitization. J Biol Chem. 2005;280:8951–8960. doi: 10.1074/jbc.M407005200. [DOI] [PubMed] [Google Scholar]
- Garzón J, Rodríguez-Muñoz M, Sánchez-Blázquez P. Do pharmacological approaches that prevent opioid tolerance target different elements in the same regulatory machinery? Curr Drug Abuse Rev. 2008;1:222–238. doi: 10.2174/1874473710801020222. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Ni YG, Dohlman HG, Nestler EJ. Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J Neurosci. 1997;17:8024–8037. doi: 10.1523/JNEUROSCI.17-20-08024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induced m-opioid receptor-arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schöneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J. Redox control of GTPase: from molecular mechanisms to functional significance in health and disease. Antioxid Redox Signal. 2011;14:689–724. doi: 10.1089/ars.2009.2984. [DOI] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidase but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, Nakagawa T, Sango K, Shirai Y, Yokoyama T, Kaneko S, Saito N, Yabe-Nishimura C. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28:9486–9494. doi: 10.1523/JNEUROSCI.1857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Kilpatrick GJ, Smith TW. Morphine-6-glucuronide: actions and mechanisms. Med Res Rev. 2005;25:521–544. doi: 10.1002/med.20035. [DOI] [PubMed] [Google Scholar]
- Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci U S A. 2010;107:14851–14856. doi: 10.1073/pnas.1009926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan KL, Zhong H, Nanamori M, Neubig RR. Rapid kinetics of regulator of G-protein signaling (RGS)-mediated Galphai and Galphao deactivation. Galpha specificity of RGS4 AND RGS7. J Biol Chem. 2000;275:33497–33503. doi: 10.1074/jbc.M005785200. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- Minami M, Onogi T, Nakagawa T, Katao Y, Aoki Y, Katsumata S, Satoh M. DAMGO, a μ-opioid receptor selective ligand, distinguishes between μ- and κ-opioid receptors at a different regions from that for the distinction between μ- and δ-opioid receptors. FEBS Lett. 1995;364:23–27. doi: 10.1016/0014-5793(95)00340-f. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Ozawa T, Watanabe T, Minami M, Satoh M. Sensitization of the adenylyl cyclase system in cloned kappa-opioid receptor-transfected cells following sustained agonist treatment: A chimeric study using G protein αi2/αq subunits. Jpn J Pharmacol. 1999;81:353–361. doi: 10.1254/jjp.81.353. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Narita M, Nagase H, Suzuki T, Tseng LF. Involvement of spinal protein kinase Cgamma in the attenuation of opioid mu-receptor-mediated G-protein activation after chronic intrathecal administration of [D-Ala2,N-MePhe4,Gly-Ol(5)]enkephalin. J Neurosci. 2001;21:3715–3720. doi: 10.1523/JNEUROSCI.21-11-03715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgár E, Fowler JH, McGill MM, Todd AJ. The types of neuron which contain protein kinase C gamma in rat spinal cord. Brain Res. 1999;833:71–80. doi: 10.1016/s0006-8993(99)01500-0. [DOI] [PubMed] [Google Scholar]
- Rahman Z, Gold SJ, Potenza MN, Cowan CW, Ni YG, He W, Wensel TG, Nestler EJ. Cloning and chanracterization of RGS9-2: a striatal enriched alternative spliced product of the RGS9 gene. J Neurosci. 1999;19:2016–2026. doi: 10.1523/JNEUROSCI.19-06-02016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, de la Torre-Madrid E, Sánchez-Blázquez P, Garzón J. Morphine induces endocytosis of neuronal mu-opioid receptors through the sustained transfer of Galpha subunits to RGSZ2 proteins. Mol Pain. 2007;3:19. doi: 10.1186/1744-8069-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, de la Torre-Madrid E, Sánchez-Blázquez P, Wang JB, Garzón J. NMDAR-nNOS generated zinc recruits PKCgamma to the HINT1-RGS17 complex bound to the C terminus of Mu-opioid receptors. Cell Signal. 2008;20:1855–1864. doi: 10.1016/j.cellsig.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, de la Torre-Madrid E, Sánchez-Blázquez P, Garzón J. NO-released zinc supports the simultaneous binding of Raf-1 and PKCγ cysteine-rich domains to HINT1 protein at the mu-opioid receptor. Antioxid Redox Signal. 2011;14:2413–2425. doi: 10.1089/ars.2010.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta R, Holecek V, Pekárkova I, Krejcová J, Racek J, Trefil L, Yamamotová A. Free radicals after pain stimulation are influenced by antioxidants and analgesics. Neuro Endocrinol Lett. 2003;24:304–309. [PubMed] [Google Scholar]
- Schwartz ES, Lee I, Chung K, Chung JM. Oxidative stress in the spinal cord is an important contributor in capsaicin-induced mechanical secondary hyperalgesia in mice. Pain. 2008;138:514–524. doi: 10.1016/j.pain.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, Miyazaki H, Sumimoto H, Kamata T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007;282:17640–17648. doi: 10.1074/jbc.M609450200. [DOI] [PubMed] [Google Scholar]
- Smith FL, Gabra BH, Smith PA, Redwood MC, Dewey WL. Determination of the role of conventional, novel and atypical PKC isoforms in the expression of morphine tolerance in mice. Pain. 2007;127:129–139. doi: 10.1016/j.pain.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- van Dorp EL, Romberg R, Sarton E, Bovill JG, Dahan A. Morphine-6-glucuronide: morphine's successor for postoperative pain relief? Anesthesia and analgesia. 2006;102:1789–1797. doi: 10.1213/01.ane.0000217197.96784.c3. [DOI] [PubMed] [Google Scholar]
- Willars GB. Mammalian RGS proteins: Multifunctional regulators of cellular signaling. Semin Cell Dev Biol. 2006;17:363–376. doi: 10.1016/j.semcdb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Winsky-Sommerer R, Yamanaka A, Diano S, Borok E, Roberts AJ, Sakurai T, Kilduff TS, Horvath TL, de Lecea L. Interaction between the corticotropin-releasing factor system and hypocretins (orexins): a novel circuit mediating stress response. J Neurosci. 2004;24:11439–11448. doi: 10.1523/JNEUROSCI.3459-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Georgescu D, Sanchez N, Rahman Z, DiLeone R, Berton O, Neve RL, Sim-Selley LJ, Selley DE, Gold SJ, Nestler EJ. Essential role for RGS9 in opiate action. Proc Natl Acad Sci U S A. 2003;100:13656–13661. doi: 10.1073/pnas.2232594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci U S A. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski JW, Song L, Harkins L, Cobbs CS, Jope RS. Oxidative stress and heat shock stimulate RGS2 expression in 1321N1 astrocytoma cells. Arch Biochem Biophys. 2001;392:192–196. doi: 10.1006/abbi.2001.2430. [DOI] [PubMed] [Google Scholar]