

Abstract
The role of prion protein (PrP) is insufficiently understood partially because PrP-deficient (−/−) neurons from C57BL/6J mice seem to differentiate normally and are functionally mildly impaired. Here, we reassessed this notion and, unexpectedly, discovered that PrP−/− hippocampal growth cones were abnormally small and poor in filopodia and cargo-containing vesicles. Based on our findings that PrP-PrP trans-interaction recruits E-cadherin to cell contact sites and reggie microdomains, and that reggies are essential for growth by regulating membrane trafficking, we reasoned that PrP and reggie might promote cargo (N-cadherin) delivery via PrP-reggie-connected signaling upon PrP activation (by PrP-Fc-induced trans-interaction). In wild-type but not PrP−/− neurons, PrP activation led to (1) enhanced PrP-reggie cocluster formation, (2) reggie-associated fyn and MAP kinase activation, (3) Exo70 and N-cadherin (cargo) recruitment to reggie, (4) the preference of the growth cone for PrP-Fc as substrate, and (5) longer neurites. Conversely, PrP-reggie-induced N-cadherin recruitment was blocked by mutant TC10, the GTPase downstream of reggie, triggering exocyst-assisted cargo delivery. This implies that PrP functions in reggie-mediated signaling and cargo trafficking, thus promoting growth cone complexity and vitality and thereby growth cone elongation.
Introduction
The glycosylphosphatidylinositol (GPI)-anchored cellular prion protein (PrP)—prominently expressed in the brain—has become famous for its role in neurodegenerative disorders, while the elucidation of its physiological function is lagging behind (Prusiner, 1982; Aguzzi et al., 2008). Many different functions have, in fact, been attributed to PrP (Linden et al., 2008), and increasingly many PrP interacting proteins are being identified with positive and negative effects on axon growth (Gauczynski et al., 2002; Zanata et al., 2002; Santuccione et al., 2005; Parkyn et al., 2008; Devanathan et al., 2010), myelination (Bremer et al., 2010), synapse formation (Collinge et al., 1994), and long-term-potentiation (LTP) (Khosravani et al., 2008) (for review, see Linden et al., 2008). Connected to these functions are signal transduction pathways that PrP seems to trigger in cooperation with cis-interaction partners and/or lipid rafts (Simons and Ehehalt, 2002). Means of efficiently activating PrP and the ensuing signaling cascades include antibody (Ab)-mediated PrP cross-linking, which triggers src tyrosine kinases in neurons (Mouillet-Richard et al., 2000; Santuccione et al., 2005) and MAPK and Ca2+ signaling in T-cells (Stuermer et al., 2004). A more physiological way is activation of PrP by PrP-PrP trans-interaction, which controls, for instance, cell–cell adhesion in the developing zebrafish embryo (Málaga-Trillo et al., 2009). PrP activation and the recruitment of E-cadherin to cell contact sites in the embryo, as well as the recruitment of the T-cell receptor (TCR) in lymphocytes occur in association with reggie/flotillin microdomains suggesting that PrP communicates with reggie-associated intracellular signaling and trafficking pathways, enabling PrP to participate in events as diverse as cell adhesion, axon growth, T-cell activation and synaptic function (for review, see Solis et al., 2010; Stuermer, 2010).
Reggies, evolutionarily conserved and present in basically every cell type (Stuermer, 2010), are essential for axon growth by their ability to activate fyn, MAPK, and Rho-GTPases (Langhorst et al., 2008; Munderloh et al., 2009) and to modulate cytoskeletal dynamics. In adipocytes, flotillins/reggies participate in plasma membrane-directed delivery of the cargo protein Glut4 (glucose transporter 4), which is an exocyst-, RalA-, and TC10 (cdc-42-related)-dependent pathway (Baumann et al., 2000; Chen et al., 2007).
We hypothesized and here examined the possibility that PrP and reggie would affect trafficking and deployment of neuronal cadherin (N-cadherin) in nerve cells (as opposed to E-cadherin in epithelial cells) and control its polarized delivery to the axon, and thus compared hippocampal neurons from prnp−/− mice (PrP−/− neurons) with wild-type (wt) neurons. Our findings show unexpected and so far unknown abnormalities of PrP−/− growth cones, which are consistent with the suggested role of PrP in reggie-mediated N-cadherin (cargo) trafficking.
Materials and Methods
Animals.
Wild-type and PrP knock-out (prnp−/−) C57BL/6J mice were purchased from Adriano Aguzzi (Zentrum für Klinische Forschung, University of Zurich, Zurich, Switzerland) and maintained in the Konstanz University animal facility.
Antibodies.
The following monoclonal (mAbs) and polyclonal (pAbs) antibodies were used: mAb against reggie-1 (ESA; BD Biosciences Transduction Laboratories), pAb against reggie-1 (Santa Cruz), pAb against PrP (gift from A. Aguzzi), mAbs and pAbs against fyn (Santa Cruz), active fyn (pAb Tyr-527-dephosphorylated; pAb Tyr-416-phosphorylated; Cell Signaling Technology), pAb against N-cadherin, Exo70, MAPK, pMAPK/pErk1,2 (Santa Cruz), and Alexa 488 or 568 phalloidin (Invitrogen). Secondary antibodies against rabbit, and mouse Ig coupled to HRP, Cy2, Cy3, or Cy5 were from Dianova.
DNA constructs.
The Fc and PrP-Fc constructs were generated by inserting in-frame the mouse PrP leader sequence (amino acids 1–22) and the mouse PrP cDNA lacking the GPI signal sequence (amino acids 1–226) into the NheI/XbaI sites of the pIg plus vector (Ott et al., 2001), respectively. The N-cadherin-EGFP vector was kindly provided by Brian McKay (University of Arizona, Tucson, AZ). The CA (Q75L) and DN (T31N) mutants for TC10 were kindly provided by Alan Saltiel (University of Michigan, Ann Arbor, MI). The ECFP-TC10 construct was generated by excising the TC10 cDNA from the original vector and subcloning in-frame into the BglII/EcoRI sites of the pECFP-C1 plasmid (Clontech). The wt PrP vector has been previously described (Schrock et al., 2009). The reggie-siRNA was the same as in the study by Munderloh et al. (2009). The N-cadherin siRNAs were from siGENOME SMARTpool CDH2 (Dharmacon): -CCAAUCAACUUGCCAGAAA, -AAAGGAAAGUUCAGUAUGA, -GGACAUCAAUGGCAAUCAA, and -GAUCAAAGCCUGGGACGUA.
Cultures and transfection of hippocampal neurons.
For immunocytochemical experiments, hippocampal neurons, prepared from 1- to 3-d-old mice of either sex, were grown for 2–4 d in Neurobasal A medium containing B27 supplement (Invitrogen) on glass coverslips coated with 100 μg/ml poly-l-lysine and 20 μg/ml laminin, respectively (Bodrikov et al., 2005). Stimulation of live neurons was performed with 4 μg/ml PrP-Fc, applied for 25 min, and pAb against PrP, applied for 10 min and secondary Ab for 5 min at 37°C in the incubator. In control experiments, PrP-Fc was replaced by L1-Fc (R&D Systems). Neurons and HpL3-4 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Cultures and transfection of HPL3-4 cells.
The HpL3-4-PrP-deficient cell line was kindly provided by Takashi Onodera (University of Tokyo, Tokyo, Japan). HpL3-4 cells were maintained in DMEM (Invitrogen), containing 10% fetal calf serum. All reagents were applied in culture medium at 37°C in an incubator supplied with CO2.
Colocalization analysis.
The analysis of protein colocalization was performed as described previously (Bodrikov et al., 2005). In brief, a PrP or reggie cluster was defined by increased immunofluorescence of at least 30% above background. PrP or reggie clusters were automatically outlined using the threshold function of the Scion Image software (Scion). Within the outlined areas, the mean intensities of fluorescence obtained by Abs against PrP, N-cadherin, Exo70, or Fc/PrP-Fc and associated with reggie or PrP clusters were measured. The same threshold was used for all groups. All experiments were performed two to three times. Colocalization profiles were plotted using NIH ImageJ software.
Immunolabeling.
For immunolabeling, neurons were fixed in 4% PFA (in PBS), pH 7.3, for 15 min at room temperature (RT), washed, and blocked in 3% BSA. PrP pAb was applied in PBS and 3% BSA to fixed and nonpermeabilized cells, 30 min at RT, and the corresponding secondary Abs 30 min at RT. Abs against reggie, N-cadherin, Exo70, and fyn were applied to fixed cells after permeabilization in 0.25% Triton X-100 in PBS, 5 min at RT. Abs were applied in 3% BSA in PBS for 2 h at 37°C and detected with the corresponding secondary Abs applied for 45 min at RT (Bodrikov et al., 2005).
Immunoprecipitation and coimmunoprecipitation.
Brain homogenates were prepared using 50 mm Tris · HCl buffer, pH 7.5, with 1 mm CaCl2, 1 mm MgCl2, and 1 mm NaHCO3. One milligram samples were lysed for 40 min on ice with RIPA buffer [50 mm Tris · HCl buffer, pH 7.5, containing 150 mm NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxycholate, 1 mm Na2P2O7, 1 mm NaF, 2 mm Na3VO4, 0.1 mm PMSF, and EDTA-free protease inhibitor mixture (Roche Diagnostics)]. HpL3-4 cells were washed twice in ice-cold PBS, lysed (30 min on ice, RIPA buffer), and subjected to centrifugation for 15 min at 10,000 × g and 4°C. Supernatants were cleared with protein A/G-agarose beads (Santa Cruz Biotechnology; 3 h at 4°C) and incubated with the corresponding Abs or control Ig (1.5 h at 4°C), followed by precipitation with protein A/G-agarose beads (1 h at 4°C). The beads were washed three times in RIPA buffer, twice in PBS, and analyzed by immunoblotting.
Gel electrophoresis and immunoblotting.
Proteins were separated by 8% SDS-PAGE and electroblotted onto nitrocellulose transfer membrane (PROTRAN; Schleicher & Schuell) overnight at 5 mA. Immunoblots were incubated in primary Abs and peroxidase-conjugated secondary Abs and visualized with SuperSignal (West Pico reagents; Pierce) on BIOMAX film (Sigma-Aldrich). Molecular mass markers were prestained protein standards from Bio-Rad Laboratories. Chemiluminescence was quantified using TINA 2.09 software (University of Manchester, Manchester, UK).
Analysis of N-cadherin in live neurons.
Neurons were transfected with N-cadherin-EGFP 2 d after plating. Recordings of live neurons were performed using a Colibri Cell Observer SD imaging system equipped with an AxioCam HRm (Zeiss). During recordings, neurons were maintained on the microscope stage in an incubator (Zeiss) at 37°C and 5% CO2. Images were acquired at 10 s intervals.
Neurite outgrowth assay.
Neurons were grown on glass coverslips coated either with 100 μg/ml poly-l-lysine or 20 μg/ml laminin, fixed 24 h after Fc or PrP-Fc application, and neurite length was determined using ImageJ software.
Stripe assay.
The silicone matrix (Vielmetter et al., 1990) with parallel channels of 50 and 40 μm width was placed onto a glass coverslip coated with poly-l-lysine and PrP-Fc was filled in through the inlet channel. Unbound protein was removed by three washes with PBS. The hippocampal neurons were seeded onto the striped carpet, were fixed after 5 d, and stained with phalloidin to visualize F-actin or/and with anti-Fc pAb, to detect the PrP-Fc stripes.
Immunofluorescence quantification.
Profiles of distributions of the immunofluorescence signals along neurites, obtained by ImageJ software, were used to calculate mean immunofluorescence intensities along neurites and to analyze the correlation coefficients between distributions of PrP, Exo70, N-cadherin, and reggie.
Data analysis.
Values are expressed as mean values ± SEM (n = number of cells or experiments). One-way ANOVA or paired Student's t test was used for statistical analysis and values of p < 0.05 were considered to be statistically significant.
Results
Activation of PrP enhances its association with reggie, activates fyn and MAPK, and promotes neurite growth
PrP consistently colocalized with reggie along neurites and growth cones even without stimulation by PrP-Fc. However, when activated by PrP Abs (Mouillet-Richard et al., 2000), or soluble PrP-Fc to mimic PrP-PrP trans-interaction (Málaga-Trillo et al., 2009), the number of PrP-reggie coclusters was significantly increased (Fig. 1a–d). No such increase occurred with Fc and nonspecific IgG, respectively (Fig. 1a–d). To make sure that indeed PrP-PrP trans-interaction is important for PrP-reggie complex formation, we analyzed in PrP−/− neurons whether exogenously added PrP-Fc would associate with reggie in the absence of PrP at the cell membrane. PrP-Fc induced the association of PrP with reggie only in wt neurons (Fig. 1e,f). Thus, PrP activation dynamically regulates the association of PrP with reggie. To confirm that PrP recruitment to reggie clusters is indeed effected by activating PrP, we replaced PrP-Fc by the extracellular domains of the cell adhesion protein L1 fused to Fc. The degree of colocalization between PrP and reggie was equal (mean intensity of PrP in reggie clusters: 105 ± 4.5 after Fc and 108 ± 5.5 after L1-Fc) showing that transactivation of the homophilic cell adhesion molecule L1, which is known to promote axon growth (Kamiguchi and Lemmon, 1997), has no measurable influence on the degree of colocalization of PrP with reggie.
Figure 1.
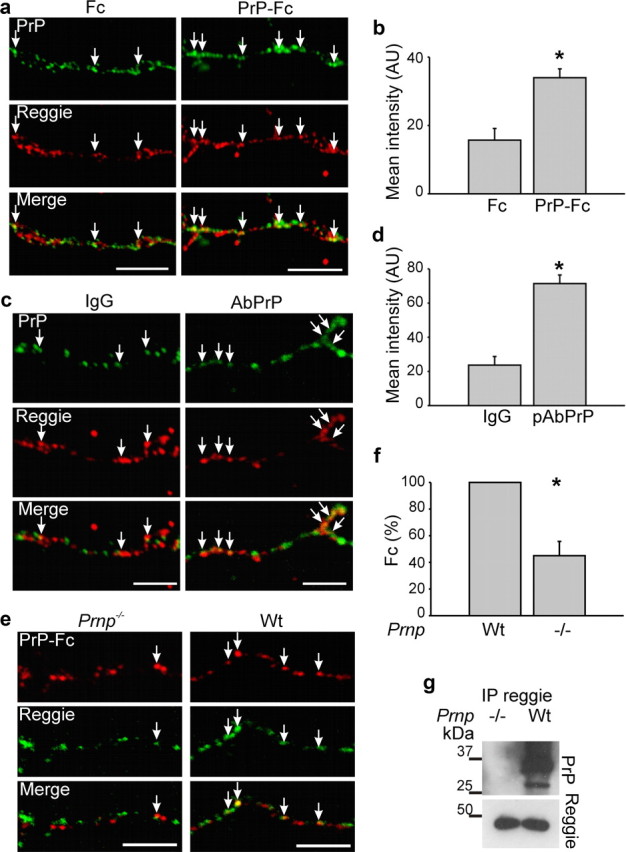
Increased cocluster formation between PrP and reggie after PrP activation. a, c, Hippocampal neurons were incubated live with PrP-Fc or Fc as control or with PrP Abs to activate PrP and labeled with Ab against reggie after fixation. PrP-Fc induced increased coclustering of PrP and reggie along the axon and growth cone (arrows). Scale bars, 10 μm. b, d, The corresponding histograms show mean labeling intensity of PrP in reggie clusters. Mean values ± SEM (n > 50 neurons) are shown in arbitrary units (AU). *Statistical significance, p ≤ 0.05, paired Student's t test. e, Wt and PrP−/− neurons were incubated live with PrP-Fc, fixed, and labeled with anti-Fc Abs. PrP-Fc increased its association with reggie only in wt but not in PrP−/− neurons. Scale bar, 5 μm. f, The histogram shows mean (±SEM; n ≥ 50 neurons) intensity of Fc in reggie clusters. *Statistical significance, p ≤ 0.05, paired Student's t test. g, Reggie Ab coimmunoprecipitates PrP from lysates of wt brain. Immunoprecipitation from lysates of PrP−/− brain served as a control.
To assess whether PrP communicates with reggie in vivo, coimmunoprecipitation was performed in brain homogenates and, indeed, showed PrP in the reggie immunoprecipitate (Fig. 1g) consistent with the PrP-reggie coimmunoprecipitation in T-cells (Stuermer et al., 2004). To determine how PrP activation might affect the association of fyn and MAPK with reggie, we used the PrP-deficient HPL3-4 cell line transfected with PrP. The level of active fyn and MAPK was determined by Abs specific for the active forms of each kinase, which were applied to reggie immunoprecipitates from PrP-expressing HPL3-4 cells after PrP-Fc or Fc application.
Application of PrP-Fc (but not Fc) raised the activation of fyn and MAPK in reggie immunoprecipitates of PrP-expressing HPL3-4 cells (Fig. 2a,b). Whether this signaling occurs likewise in vivo was analyzed by a similar experiment on wt and PrP−/− brains.
Figure 2.
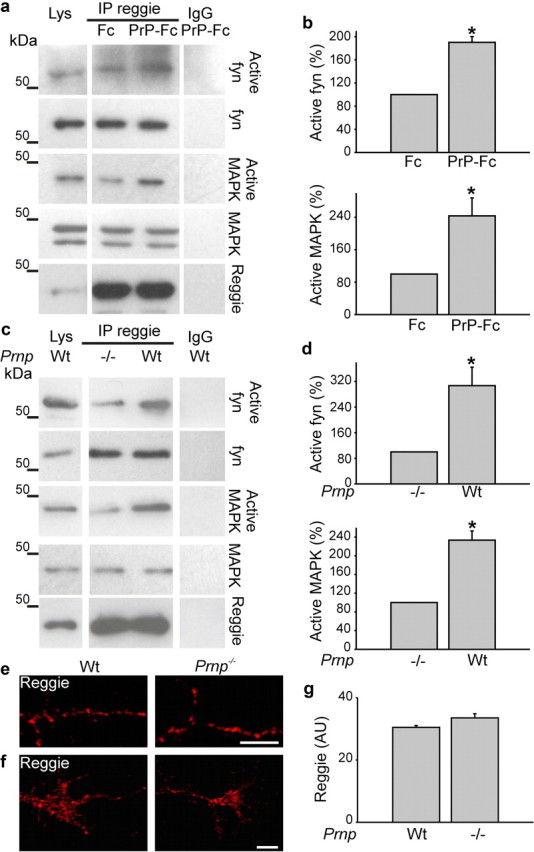
PrP regulates reggie-associated fyn and MAPK activity. a, HPL3-4 cells transfected with PrP were incubated live with Fc or PrP-Fc. Reggie immunoprecipitates from cell lysates (Lys) were probed by Western blot with Abs against total and active forms, respectively, of fyn and MAPK. Nonimmune rabbit IgG was used as control. Western blots for reggie were included as loading control. b, Activated fyn and MAPK were increased in reggie immunoprecipitates when stimulated with PrP-Fc. c, Reggie immunoprecipitates from 0 to 3 d wt and PrP−/− mice were probed with Abs against total and active fyn and MAPK, respectively. Immunoprecipitation with nonspecific IgG was used as control. Active fyn and MAPK were reduced in PrP−/− brains. Histograms (b, d) show quantification of the blots with values for PrP−/− and Fc stimulation, respectively, set to 100%. Mean values ± SEM (n > 6) are shown. *Statistical significance, p ≤ 0.05, paired Student's t test. e, f, PrP−/− and wt hippocampal neurons, when labeled with reggie Ab, show no apparent difference in the distribution of reggie in PrP−/− and wt neurons. Scale bar, 5 μm. g, The histogram corresponding to e and f shows mean intensity of reggie. Mean values ± SEM (n > 50) are shown. *Statistical significance, p ≤ 0.05, paired Student's t test.
Not the level of total but of active fyn and MAPK was reduced in reggie immunoprecipitates from PrP−/− mice compared with wt (Fig. 2c,d), so that reggie is indeed involved in PrP-mediated fyn and MAPK activation in the brain. This and the subsequent results were supported by controls showing that reggie protein expression and distribution were not changed in the absence of PrP (Fig. 2e–g).
To substantiate our finding that PrP promotes signaling in association with reggie, we analyzed fyn activation and its association with reggie in hippocampal neurons from wt and PrP−/− mice by immunostainings. Neurons were exposed to PrP-Fc or Fc and the intensity of immunofluorescence with the Ab against activated fyn was analyzed along neurites of the stimulated neurons. PrP-Fc application significantly increased the staining of activated fyn along neurites, and significantly more in wt than in PrP−/− neurons (Fig. 3a,b).
Figure 3.
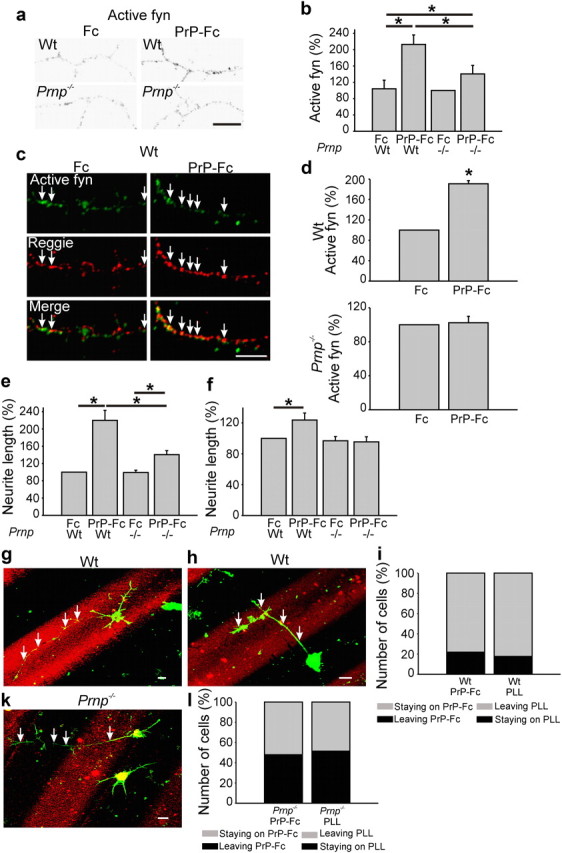
PrP-Fc leads to an increase in neurite length, reggie-associated fyn activation, and preference of neurite extension. a, Wt and PrP−/− hippocampal neurons were incubated live with Fc or PrP-Fc, fixed, and labeled with Abs against active fyn. Immunofluorescence signals are shown in black-and-white to ease recognition of the difference in immunolabeling intensities between groups. Scale bar, 10 μm. b, Application of PrP-Fc increased the level of active fyn in wt and in PrP−/− neurons as indicated in the histogram. Mean values ± SEM (n > 100 neurons). *Statistical significance, p ≤ 0.05, one-way ANOVA test. c, WT and PrP−/− hippocampal neurons were incubated live with Fc or PrP-Fc to cluster PrP. Cells were fixed and labeled with active fyn and reggie Abs. The amount of active fyn in reggie clusters was reduced in PrP−/− neurons. Scale bar, 10 μm. d, The histogram shows mean (±SEM; n ≥ 50 neurons) intensity of active fyn in reggie clusters. *Statistical significance, p ≤ 0.05, paired Student's t test. e, f, PrP−/− and wt hippocampal neurons growing on poly-l-lysine (e) and laminin (f) were incubated with PrP-Fc or Fc as control and lengths of the longest neurites were measured. e, On poly-l-lysine, wt neurons stimulated with PrP-Fc show a 120% increase in neurite length over Fc-treated neurons, and a 64% increase compared with PrP-Fc-treated PrP−/− neurons. f, On laminin, PrP-Fc increased neurite length of wt neurons by 24% over Fc, but had no effect on neurite length in PrP−/− neurons. Mean values ± SEM are shown (n > 100). *Statistical significance, p ≤ 0.05, one-way ANOVA test. g, h, When seeded on a substrate consisting of alternating lanes of poly-l-lysine and PrP-Fc (red), wt neurons showed a preference to reside and to extend neurites (arrows) on the PrP-Fc stripe, whereas neurites (arrows) of PrP−/− neurons (k) did not exhibit a preference for either substrate and crossed the stripes. Scale bar, 10 μm. i, The histograms reflect the number of wt neurons that resided on PrP-Fc and either kept all processes on PrP-Fc (staying on PrP-Fc) or sent processes to poly-l-lysine (leaving PrP-Fc) as opposed to wt neurons that resided on poly-l-lysine and either kept their neurites on poly-l-lysine (staying on PLL) or sent processes to the PrP-Fc lane (leaving PLL). These values are compared with the behavior of PrP−/− neurons (l) as is reflected by the corresponding histogram.
Then we immunostained wt and PrP−/− hippocampal neurons with Abs against active fyn and reggie after PrP activation by PrP-Fc. PrP-Fc application significantly increased the staining of active fyn in reggie clusters along neurites and growth cones in wt yet not in PrP−/− neurons (Fig. 3c,d).
Whether PrP-induced and reggie-associated signaling correlates with increased neurite growth was assessed next by comparing the length of neurites of wt and PrP−/− neurons before and after treatment with PrP-Fc. There was no difference in neurite length between wt and PrP−/− neurons without stimulation; however, when treated with PrP-Fc, wt neurons exhibited an overall 64% increase in neurite length compared with PrP-Fc-treated PrP−/− neurons (Fig. 3e), showing that signaling ensuing from PrP activation in association with reggie promotes neurite growth.
To analyze whether the gain in neurite length by PrP-Fc treatment is substrate dependent, PrP-Fc was applied to neurons grown either on poly-l-lysine or on laminin. Neurite length was again enhanced by PrP-Fc-induced activation of axon-associated PrP but only by 24%. Still, this increase was only seen in wt and not in PrP−/− neurons.
Whether the PrP-Fc-mediated stimulation of axon growth would be so strong that neurons would exhibit a preference for PrP-Fc, we performed a stripe assay (Vielmetter et al., 1990) in which wt and PrP−/− neurons were given a choice between PrP-Fc and poly-l-lysine offered as substrates in alternating lanes. Wt neurons exhibited a clear preference for the PrP-Fc lanes. Seventy-eight percent of wt neurons preferred to reside on and extend their neurites on PrP-Fc as a substrate and 82% of those residing on poly-l-lysine preferred to extend and to retain their neurites on PrP-Fc (Fig. 3g–i). PrP−/− neurons (Fig. 3k,l) did not show a preference for PrP-Fc (51%) or poly-l-lysine (52%). Transposed to the situation in vivo, our results would suggest that neurons in the brain of PrP−/− mice might suffer from axon guidance defects that have so far remained unnoticed since mice appear superficially normal.
PrP and reggie communicate with the GTPase TC10 and the exocyst component Exo70
We determined next whether PrP-reggie-induced fyn and MAPK activation in hippocampal neurons can trigger the GTPase TC10, which lies downstream of reggie (Baumann et al., 2000), binds GTP after EGFR (epidermal growth factor receptor)-induced fyn and MAPK activation, controls neurite growth in PC12 cells (Pommereit and Wouters, 2007) and the polarized exocytosis of the IGF-1 receptor in hippocampal neurons (Dupraz et al., 2009). Moreover, TC10 participates in the exocyst-dependent polarized delivery of cargo to the growth cone (Pommereit and Wouters, 2007) as well as of Glut4 to the adipocyte plasma membrane (Chen et al., 2007). Therefore, we argued that PrP might through reggie communicate with the exocyst to promote the delivery of cargo (N-cadherin) to the growing axon (Stuermer, 2010). First, we analyzed whether reggie communicates with TC10 in hippocampal neurons and whether TC10 can rescue the loss of neurites after reggie downregulation. As before (Munderloh et al., 2009), application of reggie-1 siRNAs severely perturbed axon growth and differentiation of hippocampal neurons. When the TC10 constitutive-active (CA) mutant was overexpressed in cells transfected with reggie-1 siRNAs, the “loss-of-reggie” phenotype was rescued in 62% of the neurons (Fig. 4a,b)—which succeeded in sending out neurites—indicating that TC10 lies indeed in the neuronal reggie pathway required for process formation and axon growth. We tested next whether Exo70 and the exocyst-mediated polarized delivery of cargo is connected to reggie and influenced by PrP (Stuermer, 2010). HPL3-4 cells were transfected with PrP and stimulated with PrP-Fc or Fc as control. In these cells, PrP-Fc application led to a significant increase of Exo70 association with reggie (Fig. 4c,d). To examine whether PrP associates with reggie and Exo70 in primary neurons, the degree of colocalization between PrP and Exo70 after PrP-Fc or PrP Ab application was determined (Fig. 4e–h). PrP-Fc or PrP Ab-mediated clustering of PrP resulted in a twofold increase in the association of reggie and Exo70, suggesting that PrP activation promotes the reggie-associated and exocyst-dependent function connected to axon growth.
Figure 4.
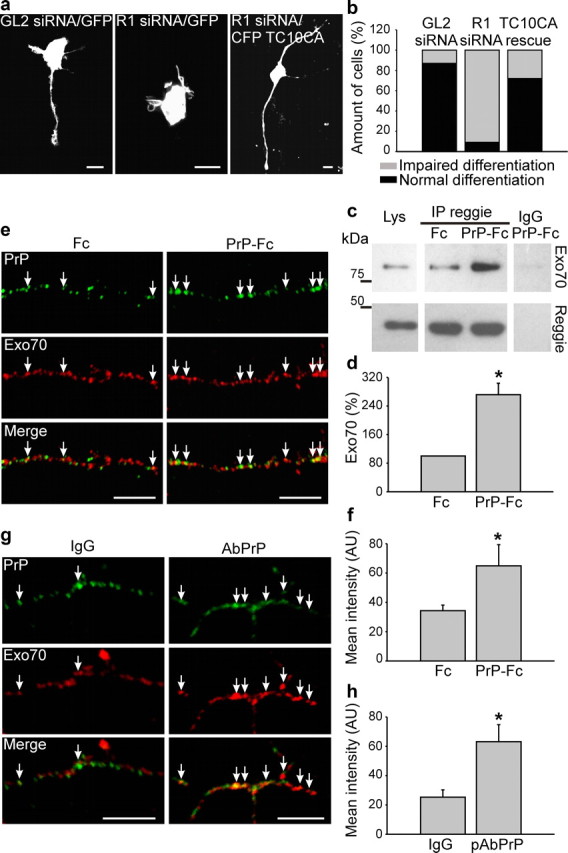
PrP-Fc-induced signaling affects TC10 and Exo70 interaction with reggie. a, b, Neurons were transfected with control (GL2) siRNA, reggie-1 siRNA, or simultaneously transfected with reggie-1 siRNA and CA TC10, respectively. The reggie-1 siRNA phenotype was rescued in 60% of the neurons. Scale bar, 10 μm. c, d, Reggie immunoprecipitates from HPL3-4 cells transfected with wt PrP and incubated live with Fc or PrP-Fc were probed with Abs against Exo70 and reggie. Immunoprecipitation with nonspecific IgG was used as control. The level of Exo70 is increased in reggie immunoprecipitates from PrP-Fc-stimulated cells. Histograms show the quantification of the blots with OD for Fc stimulation set to 100%. Mean values ± SEM are shown (n > 6). *Statistical significance, p ≤ 0.05, paired Student's t test. e, Live neurons, treated with Fc or with PrP-Fc, and labeled with Abs against Exo70 show twice as many coclusters between Exo70 and PrP after exposure to PrP-Fc as opposed to Fc (arrows). Scale bar, 10 μm. f, The histogram shows mean labeling intensity of Exo70 in PrP clusters. Mean values ± SEM (n > 50 neurons) are shown. *Statistical significance, p ≤ 0.05, paired Student's t test. g, Wt hippocampal neurons incubated live with PrP Abs to activate PrP show an increased colocalization of PrP and Exo70 (arrows). Scale bars, 10 μm. h, Histogram shows mean labeling intensity of Exo70 in PrP clusters. Mean values ±SEM (n > 50 neurons) are shown in arbitrary units (AU). *Statistical significance, p ≤ 0.05, paired Student's t test.
PrP activation regulates N-cadherin transport in the axon
Since reggie is necessary for neurite growth and TC10 and Exo70 for the polarized transport of membrane and membrane proteins, we reasoned that PrP-reggie-associated signaling might regulate trafficking of N-cadherin into the growth cone (Stuermer, 2010). We first analyzed the degree of colocalization of N-cadherin with PrP after PrP activation by PrP-Fc or PrP Ab. PrP activation induced the recruitment of N-cadherin to the growth cone and to PrP-reggie coclusters (Fig. 5a–d), implying that PrP-reggie-associated signaling drives the translocation of N-cadherin into the axon and growth cone. In PrP−/− neurons, the degree of colocalization between N-cadherin and reggie was reduced to 42% compared with the colocalization values in wt neurons (Fig. 5e). To verify biochemically that the PrP-reggie-dependent recruitment of N-cadherin involves TC10, we cotransfected HPL3-4 cells with PrP and the TC10 dominant-negative (DN) mutant, stimulated with PrP-Fc or Fc and analyzed reggie-N-cadherin cocluster formation. PrP-Fc application increased the amount of N-cadherin in reggie immunoprecipitates 2.5-fold (Fig. 5f,g). This effect, however, was blocked by DN TC10, indicating that PrP signals in association with reggie in a TC10-dependent manner for the recruitment of N-cadherin.
Figure 5.
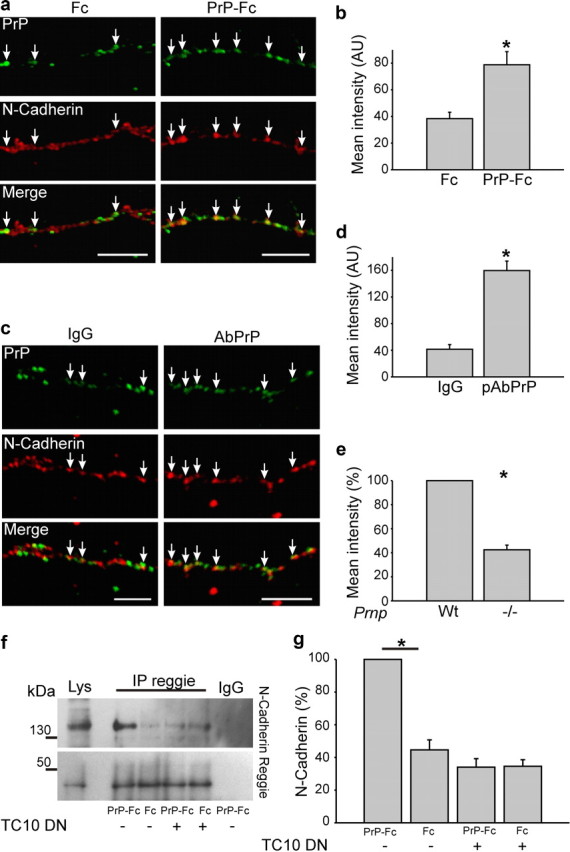
PrP activation by PrP-Fc recruits N-cadherin to PrP clusters in a reggie-TC10-dependent manner. a, Live hippocampal neurons treated with PrP-Fc (as opposed to Fc) show an increase in the colocalization of PrP and N-cadherin. Scale bar, 10 μm. b, The histogram shows mean labeling intensity of N-cadherin in PrP clusters. Mean values ± SEM (n > 50 neurons) are shown. *Statistical significance, p ≤ 0.05, paired Student's t test. c, Wt hippocampal neurons incubated live with PrP Abs to activate PrP show an increased colocalization of PrP and N-cadherin (arrows). Scale bars, 10 μm. d, Histogram shows mean labeling intensity of N-cadherin in PrP clusters. Mean values ± SEM (n > 50 neurons) are shown in arbitrary units (AU). *Statistical significance, p ≤ 0.05, paired Student's t test. e, Histogram shows the mean labeling intensity of N-cadherin in reggie clusters, which is in PrP−/− neurons only 42% of the value in wt neurons. f, HPL3-4 cells were transfected with PrP, DN TC10, and stimulated with Fc or PrP-Fc. The level of N-cadherin in reggie immunoprecipitates was analyzed by Western blots. Overexpression of DN TC10 reduced the amount of N-cadherin in reggie immunoprecipitates. g, The histogram shows the quantification of the blots with the OD for PrP-Fc stimulation of cells transfected with PrP set to 100%. Mean values ± SEM (n > 6) are shown. *Statistical significance, p ≤ 0.05, one-way ANOVA test.
Since PrP regulates in zebrafish embryos the delivery of E-cadherin to the cell surface at cell contact sites (Málaga-Trillo et al., 2009), we asked whether PrP after PrP-Fc addition regulates the recruitment of N-cadherin to the plasma membrane. HPL3-4 cells were cotransfected with N-cadherin-EGFP and wt PrP or empty vector as control and the level of N-cadherin-EGFP on the plasma membrane was analyzed (Fig. 6a–e). PrP deficiency resulted in decreased N-cadherin-EGFP fluorescence over the plasma membrane, suggesting that PrP is indeed involved in trafficking and plasma membrane exposure of N-cadherin.
Figure 6.
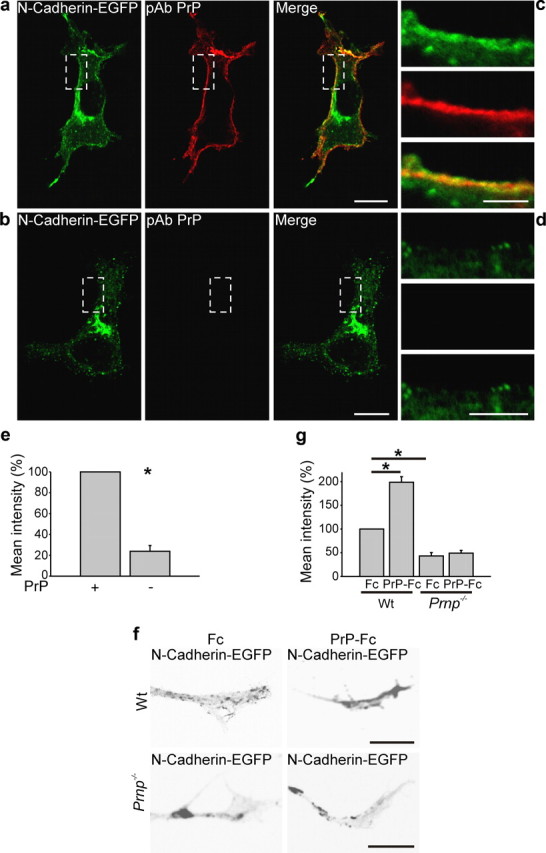
PrP regulates the delivery of N-cadherin to the plasma membrane. a, b, PrP-deficient HPL3-4 cells were transfected with PrP and N-cadherin-EGFP and the level of N-cadherin at the plasma membrane was compared with cells cotransfected with N-cadherin-EGFP and empty vector as control. Immunofluorescence and the corresponding histograms show that the mean intensity of N-cadherin-EGFP at the plasma membrane is four times lower in PrP-deficient cells compared with PrP-transfected cells. The boxed region in a is enlarged in c, and that of b in d. Scale bar: a, b, 20 μm; magnified regions, 10 μm. e, The histogram shows mean intensity of N-cadherin-EGFP. Mean values ± SEM (n > 100) are shown. *Statistical significance, p ≤ 0.05, paired Student's t test. f, Wt and PrP−/− neurons were transfected with N-cadherin-EGFP and treated with Fc or PrP-Fc. Note that PrP-Fc increased the level of N-cadherin at the periphery of the growth cones in wt but not in PrP−/− hippocampal neurons. g, The histogram shows mean intensity of N-cadherin-EGFP. Mean values ± SEM (n > 100 neurons) are shown. *Statistical significance, p ≤ 0.05, one-way ANOVA test.
To extend this analysis to primary neurons, we transfected wt and PrP−/− hippocampal neurons with N-cadherin-EGFP and analyzed after PrP-Fc addition the redistribution of N-cadherin from central to plasma membrane-near aspects of the growth cones. In wt neurons, application of PrP-Fc resulted in a twofold increase of N-cadherin-EGFP at the growth cone plasma membrane compared with the Fc-treated group. In contrast, PrP-Fc failed to shift N-cadherin-EGFP to the plasma membrane in PrP−/− cells compared with the Fc-stimulated group. The combined observations indicate that the increased association of PrP with reggie promotes the transfer of N-cadherin toward the plasma membrane of the growth cone through the participation of TC10 and Exo70 (Fig. 6f,g).
Absence of PrP impairs growth cone development and trafficking of N-cadherin-containing vesicle
Morphological defects in PrP-deficient neurons have so far not been reported. Interestingly, however, we noted that growth cones of N-cadherin-EGFP-transfected PrP−/− neurons were abnormally small and translucent due to the relative absence of N-cadherin-containing cargo vesicles (Fig. 7a). Quantification over 200 cells showed that 72% of growth cones in PrP−/− neurons were small and abnormally translucent (Fig. 7b) as opposed to 18% in wt mice. Furthermore, the number of N-cadherin-EGFP vesicles was 60% smaller in PrP−/− growth cones than in their wt counterparts (Fig. 7g,h), and the vesicle mobility was significantly reduced (Fig. 7g,i). To determine how these defects might compare with potential abnormalities in N-cadherin-deficient neurons, we analyzed growth cones of N-cadherin siRNA-treated cells. Like growth cones of PrP−/− neurons, N-cadherin siRNA-treated growth cones were significantly smaller in area than their control siRNA-transfected counterparts (Fig. 7f).
Figure 7.
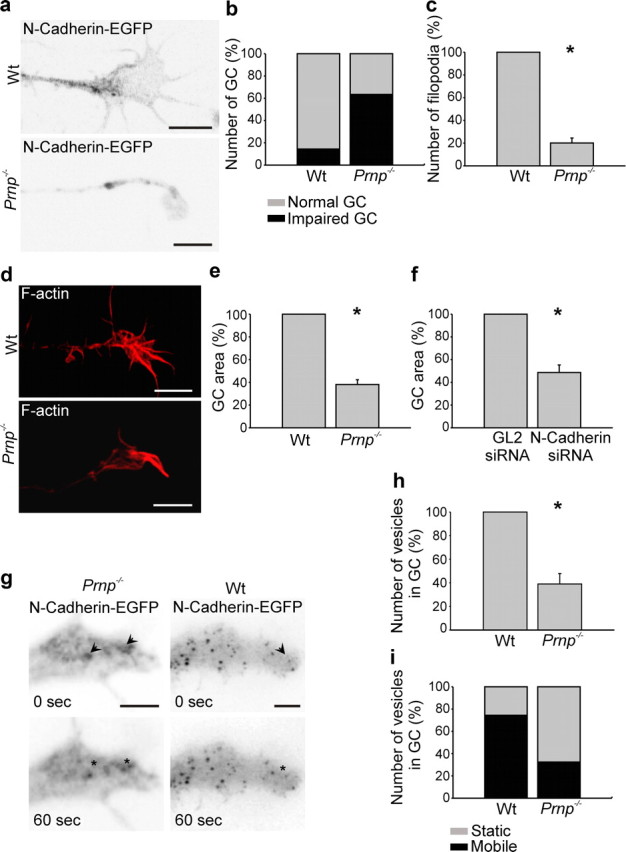
PrP−/− hippocampal neurons exhibit abnormal growth cones. a–h, Wt and PrP−/− neurons were transfected with N-cadherin-EGFP or stained with phalloidin, to visualize the cell and growth cone morphology. Growth cones of PrP−/− neurons were abnormally small (a, b, d, e), had significantly fewer filopodia (c), and were poor in N-cadherin cargo vesicles (h). e, f, Growth cone size was significantly reduced in PrP−/− neurons much as in siRNA N-cadherin-treated neurons. When monitored by time-lapse recordings (g, i), the N-cadherin vesicles were by far less motile in PrP−/− neurons than in their wt counterparts (i). Scale bar, 5 μm. Mean values ± SEM (n > 100) are shown. *Statistical significance, p ≤ 0.05, paired Student's t test.
Thus, PrP−/− growth cones suffer from an impaired supply with N-cadherin vesicles. This can account for their relative failure, compared with their wt counterparts, to increase elongation upon PrP stimulation. These findings demonstrate, for the first time, that neurons from PrP-deficient mice show specific defects in their growth cones. To foster this impression, we compared growth cones of phalloidin-stained wt and PrP−/− neurons. PrP−/− growth cones were significantly smaller in area than their wt counterparts and had significantly fewer filopodia (Fig. 7c,d). Together, our data suggest that the lack of PrP impairs the association of reggie with N-cadherin, implying that PrP activates cargo delivery to the growth cone in a reggie-dependent manner and thus participates in aspects of neuronal differentiation needed for the intricate functional interconnections in the fully functional brain.
Discussion
The key observation in our study, the existence of developmentally impaired growth cones in PrP−/− hippocampal neurons, has revealed a so-far-unknown facet of the physiological function of PrP. PrP seems to regulate—in association with reggie—trafficking of cargo (exemplified by N-cadherin) to the growth cone (Stuermer, 2010) and to participate thereby in the differentiation of neurons. This function is comparable with the role that PrP plays in E-cadherin deployment at cell contact sites (Málaga-Trillo et al., 2009), which is indispensable for embryonic development. Polarized trafficking of N-cadherin into the axon is driven by PrP-reggie-dependent signal transduction cascades and includes, by the present evidence, the activation of fyn and MAPK, as well as TC10 and the exocyst, which both are known to function in cargo supply to the growth cone (Fig. 8). PrP activation by PrP-Fc or Abs increases the association of PrP with reggie and promotes cargo delivery by the PrP-induced and reggie-dependent signaling cascade resulting in the emergence of larger and filopodia-rich growth cones equipped with cargo (N-cadherin) containers and in an overall 120% increase in neurite length. Conversely, the notion that continuous cargo delivery and turnover are required for growth cone integrity and neurite growth (Shao et al., 2002) compellingly complies with our discovery that PrP-deficient growth cones are abnormally small and translucent in consequence of their reduced cargo content and relatively immobile cargo (N-cadherin) vesicles. That N-cadherin is crucial for the formation of growth cones is demonstrated by their abnormally small size in N-cadherin siRNA-transfected neurons.
Figure 8.

Model of the PrP-reggie-mediated recruitment of N-cadherin (cargo) to the growth cone. Before PrP activation, PrP and reggie are partially associated with one another along neurites and in growth cones. PrP activation enhances PrP-reggie complex formation, induces reggie-dependent activation of fyn and MAPK, which mobilizes N-cadherin and the TC10- and Exo70-dependent delivery of the cargo to the growth cone. This promotes growth cone formation and neurite outgrowth.
The beneficial effect of PrP on growth cone complexity and elongation suggests that PrP might play a role in wiring the neural network in the brain to a larger extent than previously thought. The present observation that hippocampal neurons show a preference for PrP-Fc over poly-l-lysine in the stripe assay at least points to a potential function of PrP in axon guidance. Our data lead to the speculation that PrP−/− mice might possess axon guidance defects, which seem to have been overlooked or are compensated by a (other) GPI-anchored protein(s).
The documented importance of reggies for axon growth (Munderloh et al., 2009) is entirely in agreement with their suggested influence on membrane trafficking and cargo delivery (Stuermer, 2010). The idea that PrP might promote neuronal differentiation through reggie-dependent signaling and cargo delivery originated, on the one hand, from the emergence of poorly developed PrP−/− growth cones and, on the other hand, from the striking PrP-reggie coclusters at strategically important cellular sites in wt cells. On top came the evidence that PrP activates MAPK and Ca2+ signaling in conjunction with reggie for the recruitment of the TCR to the T-cell cap (Stuermer et al., 2004) and the finding from zebrafish embryos that PrP-PrP trans-interaction drives the delivery of E-cadherin from the Rab11 compartment to cell contact sites in association with reggie (Málaga-Trillo et al., 2009).
The fact that PrP activation led to an increased density of PrP-reggie-coclusters pointed to a joined PrP-reggie function in the growth cone. Our results imply that the present defects of PrP−/− growth cones originate indeed from an impairment of the PrP-induced, reggie-associated signaling cascade. This is supported by the fact that each of the direct or indirect signaling associates of reggie (fyn, MAPK, Exo70, TC10) contributes to axon growth and to the polarized delivery of cargo (Chen et al., 2007; Pommereit and Wouters, 2007; Dupraz et al., 2009; Stuermer, 2010). The link between PrP, reggie, and TC10 is further emphasized by the fact that neurite elongation is aborted in siRNA reggie-treated neurons yet rescued, as shown here, by CA TC10. TC10 and RalA are involved in the exocyst-dependent trafficking of cargo vesicles (Vega and Hsu, 2001; Dupraz et al., 2009), which corresponds to our results showing that the positive effect of PrP activation on cocluster formation between N-cadherin and reggie in HPL3-4 cells is blocked by DN TC10. Thus, the outcome of our present work is in line with the hypothesis suggesting that PrP and reggie regulate the recruitment and targeted delivery of cargo (N-cadherin) in developing neurons (Stuermer, 2010).
Cargo recruitment is not restricted to the growing tip but also occurs along the axon where PrP and reggie form striking coclusters and recruit N-cadherin to the plasma membrane for side branch formation and filopodia extension/retraction for growth cone reorientation as well as for interstitial growth and thus neuronal complexity. Immunostainings show the striking concentrations of PrP and reggie in the growth cone before stimulation (Stuermer, 2010; present study) that may be crucial for particularly strong trans-interaction and signaling induced by PrP-Fc in vitro, and by growth cone encounters with PrP-expressing neighboring cells in vivo, and is important for the events at growth cones and cell contacts that undergo dynamic membrane turnover (Shao et al., 2002; D'Souza-Schorey, 2005). This suggests that coclusters of PrP and reggie might drive recruitment as well as cargo turnover.
The proposed role of PrP and reggie in this locally highly dynamic membrane trafficking events finds support in studies showing that plasma membrane-exposed PrP is subject to remarkably rapid recycling (Shyng et al., 1993), that PrP-trafficking containers in neurons possess continuous association of kinesin and dynein assemblies (Encalada et al., 2011), and that reggie-decorated vesicles in HeLa cells are trafficking rapidly between the plasma membrane and intracellular compartments, and interact with the cytoskeleton and with the specific molecular motor, KIF9 (Langhorst et al., 2007; Munderloh et al., 2009; Cornfine et al., 2011). Defects in leukocyte leading edge formation and migration in the flotillin-1 (reggie-2) −/− mouse (Ludwig et al., 2010) confirm this view. These results are in line with a role of PrP and reggie in cargo transport, delivery, and turnover, which might be equally crucial at the synapse where PrP is reportedly accumulated (Moya et al., 2000) (for review, see Linden et al., 2008), where reggie was also detected (Suzuki, 2002; Kokubo et al., 2003; Solis et al., 2010) and to where the glutamate and NMDA receptors are recruited as molecular scaffolds for LTP, learning, and memory—being reportedly impaired in PrP−/− mice (Khosravani et al., 2008) (for review, see Linden et al., 2008).
How GPI-anchored proteins and PrP transduce signals into the cell is incompletely understood. In some cases, PrP interacts in cis with transmembrane proteins (for review, see Linden et al., 2008) for signal transduction. However, PrP as well as Thy-1 tend to preferentially associate with lipid rafts/reggie microdomains, an association that is enhanced by Ab- or Fc-induced clustering and sufficient for signal transduction (Morozova et al., 2011), implying that transmembrane proteins may not be absolutely necessary for PrP signaling. A computer-assisted simulation approach has shown that clustering of GPI-anchored proteins in one leaflet of the bilayer favors cross-membrane signaling of membrane proteins in the other half by local pertubations of the lipid bilayer (Morozova et al., 2011). In other words, clustering of PrP by PrP trans-interaction or Ab-mediated cross-linking is highly efficient in locally perturbing the membrane, in particular when PrP clusters join reggies that form clusters on their own through oligomerization (Solis et al., 2007). The PrP-reggie coclusters can attain properties in cross-membrane signaling like transmembrane proteins. Thereby membrane-mediated oligomerization (i.e., coclustering) could serve as a preselection or sorting mechanism that facilitates signaling events (Morozova et al., 2011).
Together, by showing that PrP activation stimulates reggie-dependent signal transduction and cargo recruitment, we would predict that PrP-reggie not only promote growth cone elongation and neuronal differentiation but also synaptic functions through their effect on glutamate and NMDA receptor deployment (Solis et al., 2010). In more general terms, the role of PrP seems to consist in the initiation and coordination of the reggie-dependent signaling and the ensuing highly dynamic cargo recruitment (Stuermer; 2010) and thereby in the creation of the morphological and functional complexity of neurons.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft. We thank Adriano Aguzzi for the polyclonal PrP antibodies and PrP−/− mice, and Takashi Onodera for the HpL3-4-PrP deficient cells.
References
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- Bodrikov V, Leshchyns'ka I, Sytnyk V, Overvoorde J, den Hertog J, Schachner M. RPTPalpha is essential for NCAM-mediated p59fyn activation and neurite elongation. J Cell Biol. 2005;168:127–139. doi: 10.1083/jcb.200405073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein myo1c. Dev Cell. 2007;13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- Cornfine S, Himmel M, Kopp P, El Azzouzi K, Wiesner C, Krüger M, Rudel T, Linder S. The kinesin KIF9 and reggie/flotillin proteins regulate matrix degradation by macrophage podosomes. Mol Biol Cell. 2011;22:202–215. doi: 10.1091/mbc.E10-05-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanathan V, Jakovcevski I, Santuccione A, Li S, Lee HJ, Peles E, Leshchyns'ka I, Sytnyk V, Schachner M. Cellular form of prion protein inhibits Reelin-mediated shedding of Caspr from the neurona cell surface to potentiate Caspr-mediated inhibition of neurite outgrowth. J Neurosci. 2010;30:9292–9305. doi: 10.1523/JNEUROSCI.5657-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey C. Disassembling adherens junctions: breaking up is hard to do. Trends Cell Biol. 2005;15:19–26. doi: 10.1016/j.tcb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Dupraz S, Grassi D, Bernis ME, Sosa L, Bisbal M, Gastaldi L, Jausoro I, Cáceres A, Pfenninger KH, Quiroga S. The TC10-Exo70-TC10 complex is essential for membrane expansion and axonal specification in developing neurons. J Neurosci. 2009;29:13292–13301. doi: 10.1523/JNEUROSCI.3907-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encalada SE, Szpankowski L, Xia CH, Goldstein LS. Stable kinesin and dynein assembles drive the axonal transport of mammalian prion protein vesicles. Cell. 2011;144:551–565. doi: 10.1016/j.cell.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauczynski S, Krasemann S, Bodemer W, Weiss S. Recombinant human prion protein mutants huPrP D178N/M129 (FFI) and huPrP+9OR (fCJD) reveal proteinase K resistance. J Cell Sci. 2002;115:4025–4036. doi: 10.1242/jcs.00086. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. Neural cell adhesion molecule L1: signaling pathways and growth cone motility. J Neurosci Res. 1997;49:1–8. doi: 10.1002/(sici)1097-4547(19970701)49:1<1::aid-jnr1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo H, Helms JB, Ohno-Iwashita Y, Shimada Y, Horikoshi Y, Yamaguchi H. Ultrastructural localization of flotillin-1 to cholesterol-rich membrane microdomains, rafts, in rat brain tissue. Brain Res. 2003;965:83–90. doi: 10.1016/s0006-8993(02)04140-9. [DOI] [PubMed] [Google Scholar]
- Langhorst MF, Solis GP, Hannbeck S, Plattner H, Stuermer CA. Linking membrane microdomains to the cytoskeleton: regulation of the lateral mobility of reggie-1/flotillin-2 by interaction with actin. FEBS Lett. 2007;581:4697–4703. doi: 10.1016/j.febslet.2007.08.074. [DOI] [PubMed] [Google Scholar]
- Langhorst MF, Jaeger FA, Mueller S, Sven Hartmann L, Luxenhofer G, Stuermer CA. Reggies/flotillins regulate Rho GTPase signalling during axon outgrowth and axon regeneration. Eur J Cell Biol. 2008;87:921–931. doi: 10.1016/j.ejcb.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Otto GP, Riento K, Hams E, Fallon PG, Nichols BJ. Flotillin microdomains interact with the cortical cytoskeleton to control uropod formation and neutrophil recruitment. J Cell Biol. 2010;191:771–781. doi: 10.1083/jcb.201005140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7:e55. doi: 10.1371/journal.pbio.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova D, Guigas G, Weiss M. Dynamic structure of peripheral membrane proteins. PLoS Comput Biol. 2011;7:e1002067. doi: 10.1371/journal.pcbi.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- Moya KL, Salès N, Hässig R, Créminon C, Grassi J, Di Giamberardino L. Immunolocalization of the cellular prion protein in normal brain. Microsc Res Tech. 2000;50:58–65. doi: 10.1002/1097-0029(20000701)50:1<58::AID-JEMT9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Munderloh C, Solis GP, Bodrikov V, Jaeger FA, Wiechers M, Málaga-Trillo E, Stuermer CA. Reggies/flotillins regulate retinal axon regeneration in the zebrafish optic nerve and differentiation of hippocampal and N2a neurons. J Neurosci. 2009;29:6607–6615. doi: 10.1523/JNEUROSCI.0870-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott H, Diekmann H, Stuermer CA, Bastmeyer M. Function of Neurolin (DM-RASP/SC-1) in guidance of motor axons during zebrafish development. Dev Biol. 2001;235:86–97. doi: 10.1006/dbio.2001.0278. [DOI] [PubMed] [Google Scholar]
- Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, Morris RJ. LRP1 controls biosynthetic trafficking of neuronal prion protein. J Cell Sci. 2008;121:773–783. doi: 10.1242/jcs.021816. [DOI] [PubMed] [Google Scholar]
- Pommereit D, Wouters FS. An NGF-induced Exo70-TC10 complex locally antagonises Cdc42-mediated activation of N-WASP to modulate neurite outgrowth. J Cell Sci. 2007;120:2694–2705. doi: 10.1242/jcs.03475. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock Y, Solis GP, Stuermer CA. Regulation of cell spreading, focal adhesion formation and filopodia extension by the cellular prion protein 5014. FEBS Lett. 2009;583:389–393. doi: 10.1016/j.febslet.2008.12.038. [DOI] [PubMed] [Google Scholar]
- Shao Y, Akmentin W, Toledo-Aral JJ, Rosenbaum J, Valdez G, Cabot JB, Hilbush BS, Halegoua S. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J Cell Biol. 2002;157:679–691. doi: 10.1083/jcb.200201063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem. 1993;268:15922–15928. [PubMed] [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis GP, Hoegg M, Munderloh C, Schrock Y, Malaga-Trillo E, Rivera-Milla E, Stuermer CA. Reggie/flotillin proteins are organized into stable tetramers in membrane microdomains. Biochem J. 2007;403:313–322. doi: 10.1042/BJ20061686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis GP, Málaga-Trillo E, Plattner H, Stuermer CA. Cellular roles of the prion protein in association with reggie/flotillin microdomains. Front Biosci. 2010;15:1075–1085. doi: 10.2741/3662. [DOI] [PubMed] [Google Scholar]
- Stuermer CA. The reggie/flotillin connection to growth. Trends Cell Biol. 2010;20:6–13. doi: 10.1016/j.tcb.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Stuermer CA, Langhorst MF, Wiechers MF, Legler DF, Von Hanwehr SH, Guse AH, Plattner H. PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. FASEB J. 2004;18:1731–1733. doi: 10.1096/fj.04-2150fje. [DOI] [PubMed] [Google Scholar]
- Suzuki T. Lipid rafts at postsynaptic sites: distribution, function and linkage to postsynaptic density. Neurosci Res. 2002;44:1–9. doi: 10.1016/s0168-0102(02)00080-9. [DOI] [PubMed] [Google Scholar]
- Vega IE, Hsu SC. The exocyst complex associates with microtubules to mediate vesicle targeting and neurite outgrowth. J Neurosci. 2001;21:3839–3848. doi: 10.1523/JNEUROSCI.21-11-03839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielmetter J, Stolze B, Bonhoeffer F, Stuermer CA. In vitro assay to test the differential substrate affinities of growing axons and migratory cells. Exp Brain Res. 1990;81:283–287. doi: 10.1007/BF00228117. [DOI] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell curface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]