

Abstract
This study was aimed to examine the role of iron in causing periventricular white matter (PWM) damage following a hypoxic injury in the developing brain. Along with iron, the expression of iron regulatory proteins (IRPs) and transferrin receptor (TfR), which are involved in iron acquisition, was also examined in the PWM by subjecting 1-d-old Wistar rats to hypoxia. Apart from an increase in iron levels in PWM, Perls' iron staining showed an increase of intracellular iron in the preponderant amoeboid microglial cells (AMCs) in the tissue. In response to hypoxia, the protein levels of IRP1, IRP2, and TfR in PWM and AMCs were significantly increased. In primary microglial cultures, administration of iron chelator deferoxamine reduced the generation of iron-induced reactive oxygen and nitrogen species and proinflammatory cytokines such as tumor necrosis factor-α and interleukin-1β. Primary oligodendrocytes treated with conditioned medium from hypoxic microglia exhibited reduced glutathione levels, increased lipid peroxidation, upregulated caspase-3 expression, and reduced proliferation. This was reversed to control levels on treatment with conditioned medium from deferoxamine treated hypoxic microglia; also, there was reduction in apoptosis of oligodendrocytes. The present results suggest that excess iron derived primarily from AMCs might be a mediator of oligodendrocyte cell death in PWM following hypoxia in the neonatal brain.
Introduction
Iron is essential for proper development of the brain in the fetal and early neonatal period. Several conditions such as hypoxic–ischemic injuries result in alterations in iron metabolism. An increased level of iron in the serum and cerebrospinal fluid in infants (Shouman et al., 2008) and the neonatal rat brain in hypoxic–ischemic injuries has been reported (Palmer et al., 1999). Hypoxia–ischemia causes damage to selected regions of the brain, the periventricular white matter (PWM) being selectively vulnerable to damage in premature infants (Johnston, 1997; Volpe, 2003). Oligodendrocyte death and axon degeneration are characteristic features of hypoxia-induced PWM damage (PWMD). The role of iron, known to accumulate in tissues in hypoxic injuries (Palmer et al., 1999; Yang et al., 2008), in causing PWMD has remained elusive.
Amoeboid microglial cells (AMCs) (Kaur et al., 2006, 2009), the resident immune cells in the developing PWM, are known to accumulate iron following a hypoxic injury (Kaur and Ling, 1999); this was suggested as an effort by the cells to sequester excess iron. In vitro studies have shown that iron induces activation of reactive oxygen species (ROS), which are involved in the pathogenesis of PWMD. However, it is not known whether iron accumulation in the AMCs would cause oligodendrocyte death through increased production of ROS following a hypoxia/reoxygenation injury.
Iron storage and intake in a cell is regulated by iron regulatory proteins (IRPs) (IRP1 and IRP2) through interaction with the iron-responsive elements (IREs) (Rouault et al., 1989) on the mRNAs of the iron storage and iron uptake proteins. IRPs are responsible for the post-transcriptional regulation of transferrin receptor (TfR) protein expression and determine the iron storage capacity of cells by regulating translation of both H- and L-ferritin mRNA (Eisenstein, 2000). IRPs are also influenced by numerous factors including hypoxia/reoxygenation (Schneider and Leibold, 2003).
Hypoxia differentially regulates IRP1 and IRP2 RNA binding activities, inactivating IRP1 and increasing IRP2 (Hanson et al., 1999), which is reversed on reoxygenation (Schneider and Leibold, 2003). IRP1 activity has also been reported to be increased by hypoxia in Hep3B cells (Toth et al., 1999). In addition to IRP1 and IRP2, TfR is also involved in cellular uptake of iron through binding and internalization of the diferric transferrin. Although TfR has been found to be localized in AMCs (Kaur and Ling, 1995), expression of IRPs in these cells is not known.
In the light of the above, we aimed to determine the iron concentration in the hypoxic PWM and to confirm its localization in the AMCs. The expression of IRP1, IRP2, and TfR was also examined to identify the possible mechanism of iron uptake in AMCs and its involvement in the production of ROS and inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which cause oligodendrocyte and axon damage in hypoxia (Deng et al., 2008, 2010). The mechanisms by which ROS, TNF-α, and IL-1β might damage the oligodendrocytes were also investigated. Iron chelator deferoxamine was used to elucidate the role of increased microglial iron in causing hypoxia-induced PWMD.
Materials and Methods
Animals.
One-day-old Wistar rats were used for this study as the development of white matter in these rats has been reported to be equivalent to midgestation in humans (Sheldon et al., 1996). Forty-eight rats were subjected to hypoxic exposure by placing them in a multigas chamber (Sanyo Biomedical Electrical; model MCO-18M) filled with 95% N2 and 5% O2 for 2 h. The animals were then allowed to recover under normoxic conditions for 3 and 24 h, 3, 7, and 14 d before killing. Another group of 48 rats served as age-matched controls and were placed outside the chamber. A total of 56 1-d-old Wistar rats were used for primary microglial and oligodendroglial cultures. All the experiments were approved by the Institutional Animal Care and Use Committee, National University of Singapore.
Primary cell culture: microglial cell culture.
One-day-old postnatal rats were used to set up mixed glial culture according to the method of Giulian and Baker (1986). In brief, the cerebral cortex excised from the rat brain was subjected to dissociation by trypsinization, after the removal of meninges. The dissociated cells were seeded in a 75 cm2 flask at a density of 1.2 × 106 cells/ml of DMEM (Sigma-Aldrich) supplemented with 10% fetal calf serum (HyClone/Thermo Fisher Scientific), 10 ml/L antibiotic–antimycotic solution (Sigma-Aldrich), 0.1 mm nonessential amino acids, and 1 ml/L insulin and placed in a humidified chamber at 37°C with 5% CO2 and 95% air. The culture medium was replaced after 24 h and then on alternate days.
Microglial purification.
After 2 weeks, the microglial cells were purified from the mixed glial cells culture by means of mild trypsinization (0.05–0.12%) in the presence of 0.2–0.5 mm EDTA for 10 min as described before by Saura et al. (2003). Purified microglial cells were then detached from the 75 cm2 flask by means of trypsinization, and the detached cells were plated at a density of 2.5 × 105 cells/well in a 24-well plate for immunocytochemistry and 1 × 106 cells/flask for protein extraction and ROS analysis. On the following day, the cells were subjected to various treatments as described below.
The purity of microglia was assessed by immunocytochemical detection using the antibody OX42 (1:100) (catalog #O-MAS 3706; Harlan-Sera Laboratory), a specific marker for rat microglia, and DAPI (20 μg/ml; Sigma-Aldrich), a nuclear marker for all cells. The purity of the microglial cultures was ∼96%.
Treatment of microglial culture.
The microglial cultures were divided into three groups—control (C), hypoxia (H), and hypoxia with deferoxamine (H+D). Deferoxamine (Sigma-Aldrich; catalog #D9533), an iron chelator, was used to study the role of iron in causing PWMD. For all the experiments, the microglial cells were exposed to hypoxia in a chamber (model 18M; multigas incubator; Sanyo) filled with mixture of gases containing 1% oxygen, 5% CO2 and 94% nitrogen at 37°C for 4 h. The control groups were incubated in an incubator with 95% air and 5% CO2 at 37°C. The amount of deferoxamine added to the cultures was 100 μm (Zhang et al., 2006). Immediately after hypoxic exposure, the hypoxic microglia along with the matching controls were fixed and processed for double-immunofluorescence labeling for OX42 and IRP1, IRP2, and TfR as described below. Furthermore, the culture medium from each group was collected for measuring TNF-α and IL-1β released by microglia, for assessing the effect of conditioned medium on glutathione (GSH) levels and lipid peroxidation (LPO) in oligodendrocytes, for examining the activation of caspase-3 in oligodendrocytes by TNF-α, for estimating the effect of IL-1β on proliferation of oligodendrocytes, and for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
Oligodendrocyte culture.
From the confluent mixed glial cultures, the oligodendrocytes were purified by shaking, a method previously described by McCarthy and de Vellis (1980). The purified cells were plated at a density of 2.5 × 105 cells/well on poly-l-lysine-coated coverslips in 24-well plate for studying the activation of caspase-3 by TNF-α and for TUNEL assay, 1 × 106 cells/flask for extracting protein for GSH assay and LPO, and 1 × 104 cells/well in 96-well plates for proliferation assay. The plated cells were allowed to grow for 5 d in culture medium containing 1 mm sodium pyruvate (Sigma-Aldrich; catalog #P2256), 5 μg/ml insulin, 50 μg/ml apotransferrin (Sigma-Aldrich; catalog #T1147), 30 nm sodium selenite (Sigma-Aldrich; catalog #S5261), 30 nm triiodothyronine (Sigma-Aldrich; catalog #T2752), and 0.5% fetal bovine serum as previously described by Armstrong (1998). The purity of oligodendrocytes was assessed by labeling with CC1 (1:20) (EMD Chemicals; catalog #OP80), a monoclonal antibody raised against adenomatous polyposis coli that labels the cell body of oligodendrocytes (Bhat et al., 1996; Fuss et al., 2000). The purity of oligodendrocyte cultures was ∼96%.
Iron assay.
The iron content in the PWM of rats at 3, 24 h, 3, 7, and 14 d (n = 3 at each time point) after hypoxic exposure and the corresponding age matched controls was estimated using an iron assay kit (BioVision; catalog #K 390-100). The iron content in control, hypoxic, and hypoxia plus deferoxamine groups of microglial cell cultures was also determined using the same kit. Homogenates of PWM and cell lysates were prepared as described below for Western blot, and the assay was performed as per manufacturer's instructions. The concentration of iron was calculated using the standard curve obtained.
Perls' iron staining.
Perls' iron staining was performed on rats at 3 d after hypoxic exposure and their corresponding controls (n = 3 for each group). The rats were perfused under 6% pentobarbital anesthesia with 2% paraformaldehyde and the brains were removed. Frozen brain sections of 40 μm thickness containing PWM were cut using a cryostat (model 3050; Leica Microsystems). Sections were immersed in Perls' solution (1:2; 10% HCl and 10% potassium ferrocyanide) at room temperature for 30 min, a method previously described by Wang et al. (2002). The sections were then rinsed in deionized water and immersed for 15 min in 0.005% 3,3′-diaminobenzidine with 0.5 ml of 1% hydrogen peroxide as described previously by Hill and Switzer (1984). The sections were then incubated for 1 h with FITC-conjugated lectin from tomato (Lycopersicon esculentum; 1:100; Sigma-Aldrich) to label the microglia. The sections were washed in PBS and mounted using a fluorescent mounting medium (Dako). Perls' iron staining was performed in control, hypoxic, and hypoxia + deferoxamine group of primary microglial cultures according to a method previously described by Dang et al. (2010). The cells were then stained with lectin and processed as described above.
Western blotting.
The PWM tissue immediately above the lateral ventricle (area shown in Fig. 1B) was carefully removed with the help of a dissecting microscope from the rats at 3 and 24 h, 3, 7, and 14 d (n = 5 at each time point) after hypoxic exposure and their corresponding age-matched controls. The dissected tissue was immediately frozen in liquid nitrogen and was kept at −80°C until protein extraction. Protein extraction was done by homogenizing the tissue with tissue protein extraction reagent (Pierce/Thermo Fisher Scientific; catalog #78510) containing protease inhibitors. Mammalian protein extraction reagent (Pierce/Thermo Fisher Scientific; catalog #78501) was used to extract protein from microglial cultures. All procedures were performed at 4°C. Homogenates were centrifuged at 13,000 rpm for 20 min and the supernatant was collected. Bradford's method (Bradford, 1976) was used to estimate the protein concentration, using bovine serum albumin (Sigma-Aldrich) as a standard. Samples of supernatants containing 40 μg of protein were heated to 95°C for 5 min and were separated by SDS-PAGE in 10% SDS gels, in a Mini-Protean 2 apparatus (Bio-Rad Laboratories). Protein bands were electroblotted onto 0.45 μm polyvinylindene difluoride membranes (Bio-Rad) and were blocked with 5% (w/v) nonfat dried milk overnight at 4°C. The membranes were then incubated with dilutions of anti-IRP1 (1:1000) (Alpha Diagnostic International; catalog #IRP11-A), anti-IRP2 (1:200) (Millipore Bioscience Research Reagents; catalog #AB15508), and anti-TfR (1:200) (AbD Serotec; catalog #MCA155G) antibodies. They were then incubated with horseradish peroxidase-conjugated secondary antibodies (Pierce/Thermo Fisher Scientific). Specific binding was revealed by an enhanced chemiluminescence kit (Pierce/Thermo Fisher Scientific) following the manufacturer's instructions.
Figure 1.
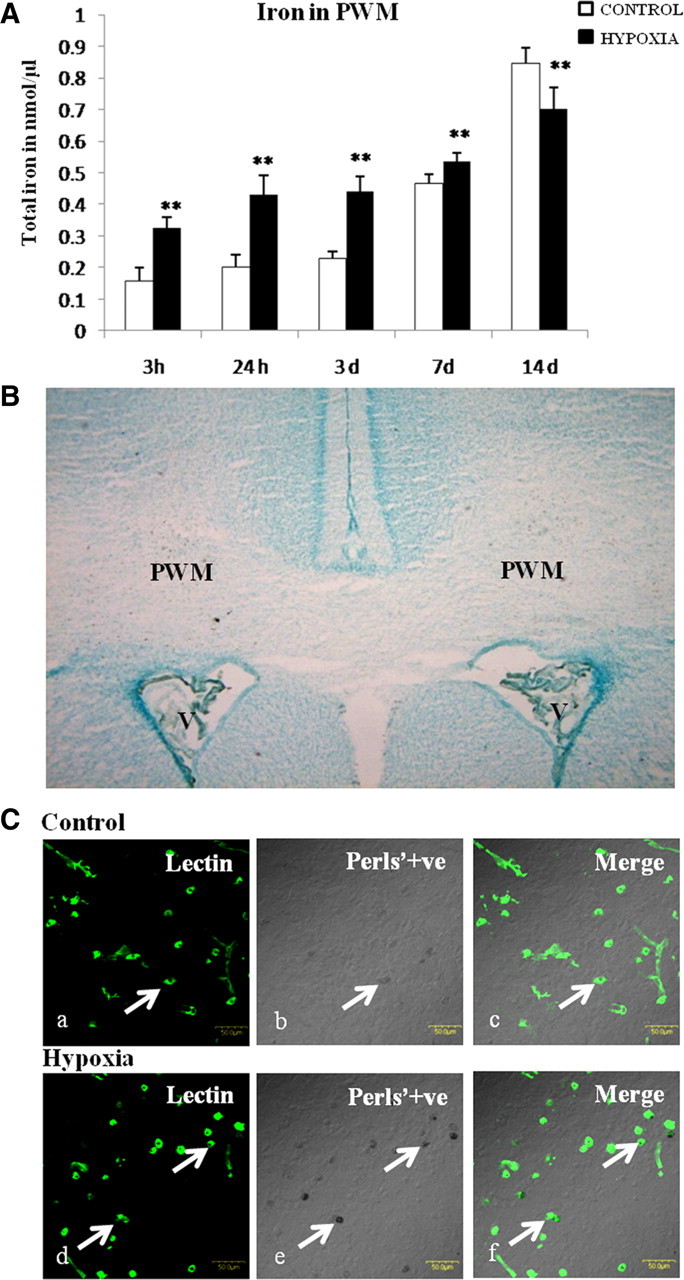
A, Iron content in the PWM of postnatal rats at 3 and 24 h, 3, 7, and 14 d after hypoxic exposure and their corresponding controls. Significant differences in total iron level between hypoxic and control groups are expressed as **p < 0.01. B, Light-microscopic image of a brain section stained with Perls' solution and methyl green, showing the region of interest, PWM just peripheral to the lateral ventricles (V). C, Confocal images of Perls' iron-stained sections showing iron localization in the PWM at 3 d after hypoxic exposure and their corresponding controls. Distribution of lectin (marker for microglia) (Ca, Cd; green) and iron localization (Cb, Ce; black) is seen in AMCs (arrows) in the PWM in overlays Cc and Cf (merge). Scale bars, 50 μm. The experiments were performed in triplicate.
Double immunofluorescence.
For the cellular localization of IRP1, IRP2, and TfR, the rats were killed at 3 d after hypoxic exposure with their age-matched controls (n = 5 for each group). Rats were anesthetized with 6% sodium pentobarbital and the brains were removed following perfusion of the rats with 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were then placed in the same fixative for 4 h following which they were placed in 0.1 m phosphate buffer containing 15% sucrose at 4°C overnight. Brain sections of 40 μm thickness containing the PWM were cut using a cryostat (Leica Microsystems). Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol for 30 min and the sections were subsequently washed with PBS. The sections were then divided into two groups. Group 1 was incubated with the mixture of primary antibodies [IRP1 (Santa Cruz Biotechnology; catalog #SC25535)/IRP2 (Millipore Bioscience Research Reagents) and OX42; 1:100 dilution in PBS] overnight at room temperature and subsequently incubated with respective secondary antibodies; Cy3-conjugated goat anti-rabbit IgG and FITC-conjugated sheep anti-mouse IgG (1:100; Sigma-Aldrich). The sections in Group 2 were incubated with OX42 overnight, subsequently with secondary antibody tagged with Cy3 for 1 h followed by incubation with FITC-conjugated anti-TfR antibody (1:50; Abcam; catalog #ab33927). The sections were then washed in PBS and mounted using a fluorescent mounting medium (Dako). Cellular localization was then examined under a confocal microscope (FV1000; Olympus). The primary microglial cells were fixed with 4% paraformaldehyde for 20 min and processed as described above for localization of IRP1, IRP2, and TfR. A few sections were incubated with isotype control antibody rabbit anti-rat IgG (Pierce Biotechnology) to confirm the specificity of the antibodies and processed as described above.
For studying the effect of intracellular iron on TNF-α and IL-1β production, fixed microglial cultures of control, hypoxic, and hypoxia + deferoxamine groups were subjected to blocking with 3% normal goat serum for 1 h followed by incubation with mixture of primary antibodies [OX42 + TNF-α (1:200; Millipore Bioscience Research Reagents; catalog #AB1837P), OX42 + IL-1β (1:200; Millipore Bioscience Research Reagents; catalog #AB1832P)] overnight at 4°C. They were then incubated with a mixture of secondary antibodies; Cy3-conjugated goat anti-rabbit IgG and FITC-conjugated sheep anti-mouse IgG (1:100; Sigma-Aldrich) and processed as described above.
Flow-cytometric measurement of ROS in microglia.
Fluoroprobe 5-(and 6)-choloromethyl-2′,7′-dicholoro-dihydrofluorescein diacetate (CM-H2DCFDA) (catalog #C6827; Molecular Probes) was used to estimate the intracellular ROS production in microglial cells. The control, hypoxia, and hypoxia + deferoxamine groups of cultured microglial cells grown on 60 cm2 culture dishes were detached by mild trypsinization and washed with PBS. The cells were then resuspended in 0.5 ml of prewarmed PBS containing CM-H2DCFDA (10 μm; diluted in DMSO) and incubated for 40 min at 37°C in the dark. Following incubation, the cells were immediately subjected to flow-cytometric analysis (CyAn ADP; Beckman Coulter). The fluorescence intensity of 2′,7′-dichlorofluorescein, the oxidized product of CM-H2DCFDA was measured at an excitation of 488 nm and emission at 535 nm. The measured fluorescence intensity was considered to be directly proportional to the amount of ROS produced.
Extracellular reactive oxygen and nitrogen species released by microglia.
The conditioned medium derived from control, hypoxia, and deferoxamine-treated hypoxic microglia was used to measure the total ROS/reactive nitrogen species (RNS) levels released by these cells. The concentration of ROS/RNS liberated was measured using Oxiselect in vitro ROS/RNS assay kit (catalog #STA-347; Cell Biolabs) according to the manufacturer's instructions. The relative fluorescence of the samples and the standards were read at 480 nm excitation/530 nm emissions using SpectraMaxM5 microplate reader (Molecular Devices). The concentration of the total ROS/RNS liberated by microglia was calculated using the 2′,7′-dichlorodihydrofluorescein standard curve.
Measurement of TNF-α and IL-1β released by microglia.
The conditioned medium was obtained from control, hypoxic, and hypoxia + deferoxamine groups of microglial cell cultures. The concentration of cytokines TNF-α and IL-1β liberated by the microglial cells into the medium was measured using the TNF-α (catalog #BE45471) and IL-1β (catalog #27193) ELISA kits (IBL) according to the manufacturer's instructions. The concentration of TNF-α and IL-1β was calculated using the assay standard curve.
Measurement of glutathione and lipid peroxidation in primary oligodendrocytes.
Antioxidant such as GSH protects a cell against oxidative stress (Bobyn et al., 2002). GSH depletion in oligodendrocytes is implicated in their susceptibility to free radical-mediated injury (Back et al., 1998). Activated microglia have been reported to release ROS (Block et al., 2007), which induces LPO (Su et al., 2011). On the basis of these observations, the primary cultured oligodendrocytes were divided into six groups and treated as described below for GSH and LPO measurement. Group 1 cells served as control, Group 2 cells were treated with conditioned medium from control microglial cultures, Group 3 cells were subjected to treatment with deferoxamine, Group 4 cells were subjected to hypoxia in a chamber (model MCO18M; multigas incubator; Sanyo) containing 1% oxygen, 5% CO2, and 94% nitrogen at 37°C for 4 h, Group 5 cells were treated with conditioned medium from microglial cultures subjected to hypoxia, and Group 6 cells were treated with conditioned medium from hypoxic microglial cultures treated with deferoxamine. After 4 h treatment, protein from all of the above-mentioned groups was extracted using mammalian protein extraction reagent containing protease inhibitors. The extract was further centrifuged at 14,000 rpm for 20 min and the supernatant was used to measure GSH and LPO.
Glutathione assay.
QuantiChrom Glutathione assay kit (DIGT-250; BioAssay Systems) was used to determine the GSH content in all six groups of oligodendrocytes. The concentration of GSH was determined from the optical density of samples and calibrator read at 412 nm with a precision microplate reader (Molecular Devices) as per manufacturer's instructions.
Lipid peroxidation assay.
The LPO content in all six groups of oligodendrocytes was estimated using the lipid peroxidation calorimetric assay kit, BioAssay (catalog #2496-30; US Biological) as per the manufacturer's instructions. The samples were read at 586 nm using a precision microplate reader (Molecular Devices) and the concentration of LPO in the samples was calculated using the malondialdehyde (MDA) standard curve.
Caspase-3 labeling in primary oligodendrocytes.
TNF-α was previously reported to mediate apoptosis by inducing caspase-3 activation (Utaisincharoen et al., 2000). Hence, TNF-α-induced caspase-3 activation in primary oligodendrocytes was studied by exposing the cells to recombinant rat TNF-α (100 ng/ml; Millipore Bioscience Research Reagents; catalog #GF046) as described previously (Yeh et al., 2000). In the present study, the activation of caspase-3 in oligodendrocytes by TNF-α in the microglial conditioned medium was assessed by dividing the primary cultured oligodendrocytes into six groups. Group 1 cells served as control; Group 2 cells were treated with conditioned medium from control microglial cultures; Group 3 cells were subjected to treatment with deferoxamine; Group 4 cells were treated with conditioned medium from hypoxic microglial cultures; Group 5 cells were treated with conditioned medium from hypoxic microglial cultures treated with deferoxamine; Group 6 cells were treated with medium from hypoxic microglial cells containing anti-TNF-α (10 μg/ml; Millipore Bioscience Research Reagents) to test the neutralization effects of TNF-α as described by us previously (Sivakumar et al., 2011). After 4 h of the above treatment, the cells were fixed in 4% paraformaldehyde for 20 min and blocked with 3% normal goat serum for 1 h. They were then incubated with mixture of primary antibodies directed against cleaved caspase-3 (1:200; Cell Signaling Technology) and CC1 overnight at 4°C. They were then incubated with mixtures of secondary antibodies and processed as described above under the section on double immunofluorescence.
Oligodendrocyte proliferation assay.
Proliferation of oligodendrocytes was previously reported to be inhibited when IL-1β was added to the culture medium (Vela et al., 2002). Hence, the effect of IL-1β on proliferation of oligodendrocytes was studied using the Quick Cell Proliferation assay kit (Abcam; catalog #ab65473) according to the manufacturer's instructions. The primary cultured oligodendrocytes were divided into Group 1 untreated control cells; Group 2 cells were treated with conditioned medium from control microglial cultures; Group 3 cells were treated with deferoxamine; Group 4 cells were treated with IL-1β peptide (100 pg/ml; IBL; catalog #27193), as described previously (Palmer et al., 1995), and were neutralized with anti-IL-1β (10 μg/ml; Millipore Bioscience Research Reagents); Group 5 cells were treated with conditioned medium from hypoxic microglial cultures; Group 6 cells were treated with conditioned medium from hypoxic microglial cultures treated with deferoxamine; Group 7 cells were treated with medium from hypoxic microglial cells containing anti-IL-1β (10 μg/ml; Millipore Bioscience Research Reagents). After 4 h of treatment, 10 μl of WST-1/ECS solution was added to each well and the reaction was allowed to occur in standard culture condition for 4 h. The optical density was measured at 440 nm using a precision microplate reader (Molecular Devices). The results are expressed as inhibition of proliferation with respect to control cells without treatment. For neutralization studies, the concentration of anti-IL-1β used was based on the cell viability assay performed, which showed an increased viability of oligodendrocytes at 10 μg/ml of anti-IL-1β (data not shown).
TUNEL assay for oligodendrocytes.
The TUNEL assay of primary cultured oligodendrocytes was performed using an in situ cell death detection kit (Roche Diagnostics/Roche Applied Science; catalog #11684795910) according to the manufacturer's instructions. The primary cultured oligodendrocytes were divided into six groups as mentioned under the measurement of glutathione and lipid peroxidation sections. The total number of TUNEL-positive oligodendrocytes was obtained by taking into account seven random microscopic fields for each slide at an objective magnification of 40× by a blinded observer. The percentage of TUNEL-positive oligodendrocytes against the total number of oligodendrocytes was calculated and averaged.
Statistical analysis.
The data were presented as mean ± SD. Statistical significance of differences between control, hypoxic, and treatment groups were calculated using paired Student's t test. Statistical significance between the groups was represented as *p < 0.05 and **p < 0.01. With respect to cells subjected to hypoxia/hypoxic conditioned medium, statistical significance was represented as #p < 0.05 and ##p < 0.01.
Results
Iron concentration in PWM
The iron concentration in the PWM was significantly increased in hypoxic rats killed at different time points extending from 3 h to 7 d when compared with their corresponding controls (Fig. 1A); it was significantly decreased at 14 d. This indicates that hypoxia leads to an increase in iron content in the PWM, which is sustained over a period of 7 d.
Iron localization in AMCs
When compared with control rats, many more cells in the PWM (area shown in Fig. 1B) in hypoxic rats exhibited intense staining for iron by Perls' method. The iron stained cells were identified to be the AMCs because they were also labeled by lectin (a widely used marker for microglia) (Fig. 1C). Hence the results indicate that iron accumulation specifically localized in the AMCs is increased by hypoxia.
Western blot analysis of IRP1, IRP2, and TfR protein expression
Protein expression of IRP1, IRP2, and TfR showed a significant difference between the control and hypoxic groups. Immunoreactive band for IRP1 appeared at 97 kDa (Fig. 2A) and was significantly increased at 3 and 7 d after the hypoxic exposure (Fig. 2B). Immunoreactive band for IRP2 appeared at 105 kDa (Fig. 2A) and was found to be significantly increased at 24 h, 3, 7, and 14 d (Fig. 2C). Immunoreactive band for TfR (Fig. 2A) at 95 kDa was found to be significantly increased from 3 h to 7 d but was significantly downregulated at 14 d (Fig. 2D). All in all, hypoxic exposure induces an upregulated protein expression for all three biomarkers notably at 3 and 7 d after hypoxia.
Figure 2.

Western blot showing the protein expression of IRP1, IRP2, and TfR in the PWM of postnatal rats at 3 and 24 h, 3, 7, and 14 d after hypoxic exposure and their corresponding controls. A shows the immunoreactive bands of IRP1 (97 kDa), IRP2 (105 kDa), TfR (95 kDa), and β-actin (43 kDa). B–D are their corresponding bar graphs showing significant changes in the optical density following hypoxic exposure (given as mean ± SD). The experiment was repeated five times, and a representative blot is shown here. Significant differences in protein levels between hypoxic and control groups are expressed as follows: *p < 0.05; **p < 0.01.
Hypoxia increases IRP1, IRP2, and TfR immunoexpression in AMCs
Double immunofluorescence showed the expression of IRP1, IRP2, and TfR in the OX42 labeled AMCs in PWM. IRP1 and IRP2 immunofluorescence was found to be evidently enhanced at 3 d following hypoxic exposure when compared with that of controls (Fig. 3Aa–f,Ba–f). As in IRPs, TfR immunoexpression was augmented at 3 d following hypoxic exposure in AMCs (Fig. 3Ca–f). Thus, the immunoexpression of IRP1, IRP2, and TfR was upregulated following hypoxic exposure.
Figure 3.
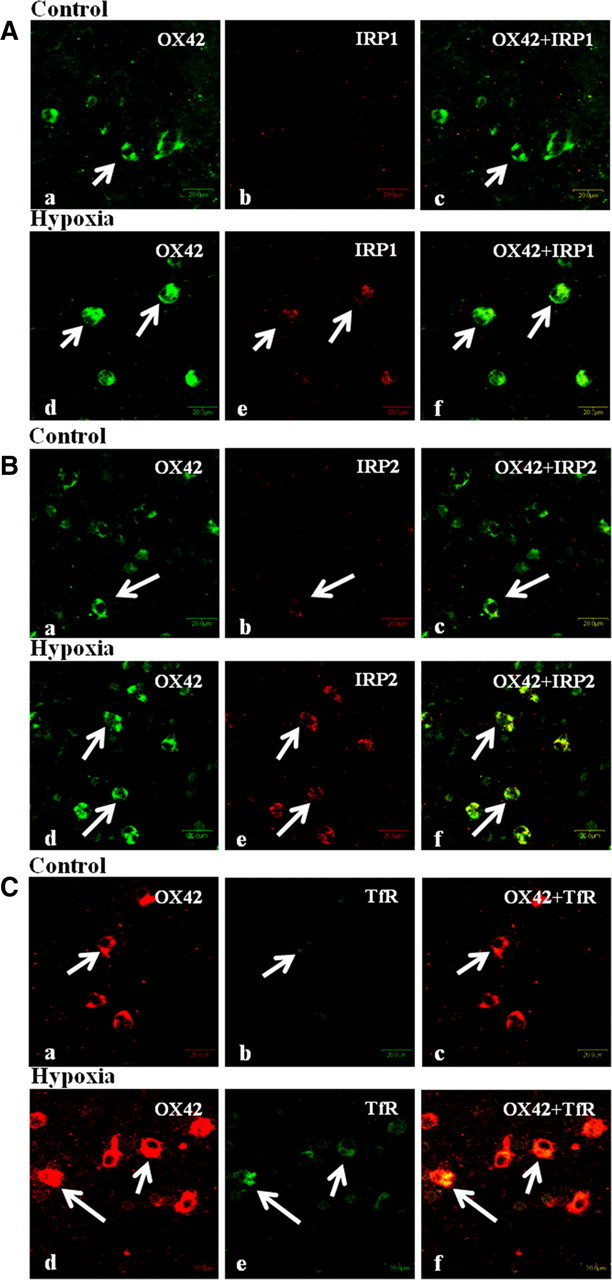
Confocal images showing the expression of OX42, IRP1, IRP2, and TfR in the PWM at 3 d after hypoxic exposure (Ad–f, Bd–f, Cd–f) and their corresponding control (Aa–c, Ba–c, Ca–c). Expression of OX42 (A, Ba,d: green; Ca,d: red), IRP1 (Ab,e: red), IRP2 (Bb,e: red), TfR (Cb,e: green), and a colocalization of OX42 with IRP1, IRP2, and TfR (A–Cc,f). Scale bars: A–C, 20 μm. The experiment was repeated five times.
The sections incubated with isotype controls did not show any immunoreactivity confirming the specificity of the antibodies used (data not shown).
Hypoxia increases iron content in microglial cells in culture
The iron content was significantly increased in microglial cells subjected to hypoxia (Fig. 4A) when compared with that of control. However, the increased level of iron content as elicited by hypoxia was suppressed by deferoxamine. Perls' iron staining also showed an increase in iron content in hypoxic microglia (Fig. 4B). This was manifested by a significant increase in the percentage of cells that were Perls' positive in hypoxic group when compared with control group. Deferoxamine reduced the incidence of Perls' positive microglia when compared with hypoxic microglia not treated with deferoxamine (Fig. 4C). The results thus indicate that hypoxia increases the iron content in microglial cells that may be suppressed by deferoxamine.
Figure 4.
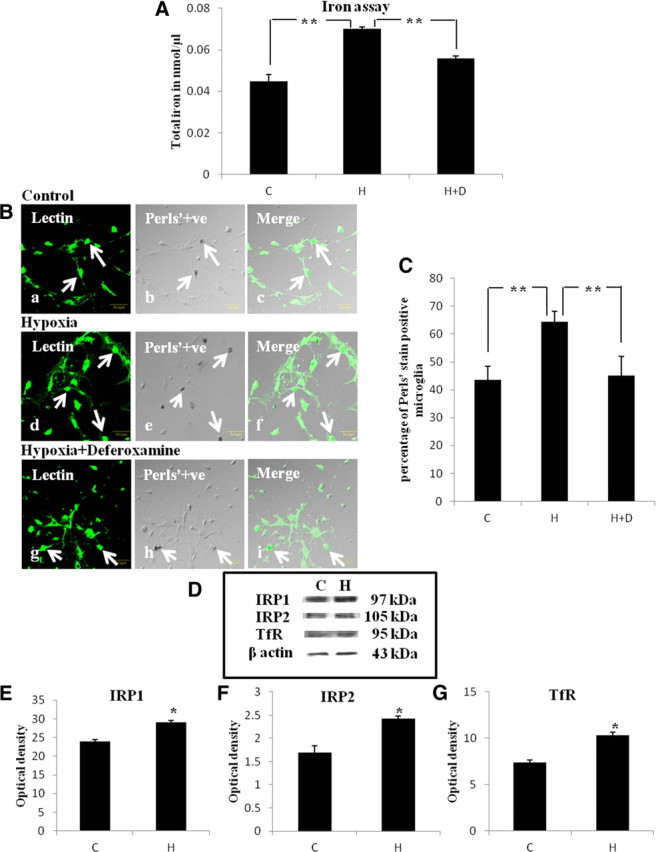
A, Bar graph in A shows significant changes in the total iron content in microglial culture. Note the reduction of iron in hypoxic microglial cells when treated with deferoxamine (H+D). Significant differences in total iron level between control (C), hypoxic (H), and deferoxamine treated groups are expressed as follows: *p < 0.05; **p < 0.01. B, Confocal images of Perls' iron-stained primary microglial cells in control, hypoxic, and deferoxamine (Hyp+Def)-treated hypoxic microglia. Distribution of lectin (a, d, g: green) and iron localization (b, e, h: black) is seen in microglia (arrows). Scale bars, 50 μm. C, Bar graph in C shows the significant changes in the percentage of microglia that are Perls' positive. Significant differences between the groups are expressed as follows: *p < 0.05; **p < 0.01. The experiment was repeated in triplicate. D, Western blot showing the protein expression of IRP1, IRP2, and TfR in the primary microglial culture. E–G are their corresponding bar graphs showing significant changes in the optical density between control (C) and hypoxia (H) (given as mean ± SD). Significant differences in protein levels between hypoxic and control groups are expressed as follows: *p < 0.05; **p < 0.01. The experiment was repeated five times.
Hypoxia increases IRPs and TfR expression in microglial cells in culture
Protein expression of IRP1, IRP2, and TfR was significantly increased in microglial cells subjected to hypoxia when compared with control groups (Fig. 4D–G). By immunofluorescence labeling IRP1, IRP2, and TfR immunofluorescence was noticeably enhanced in hypoxic microglial cells (Fig. 5Aa–f,Ba–f,Ca–f).
Figure 5.
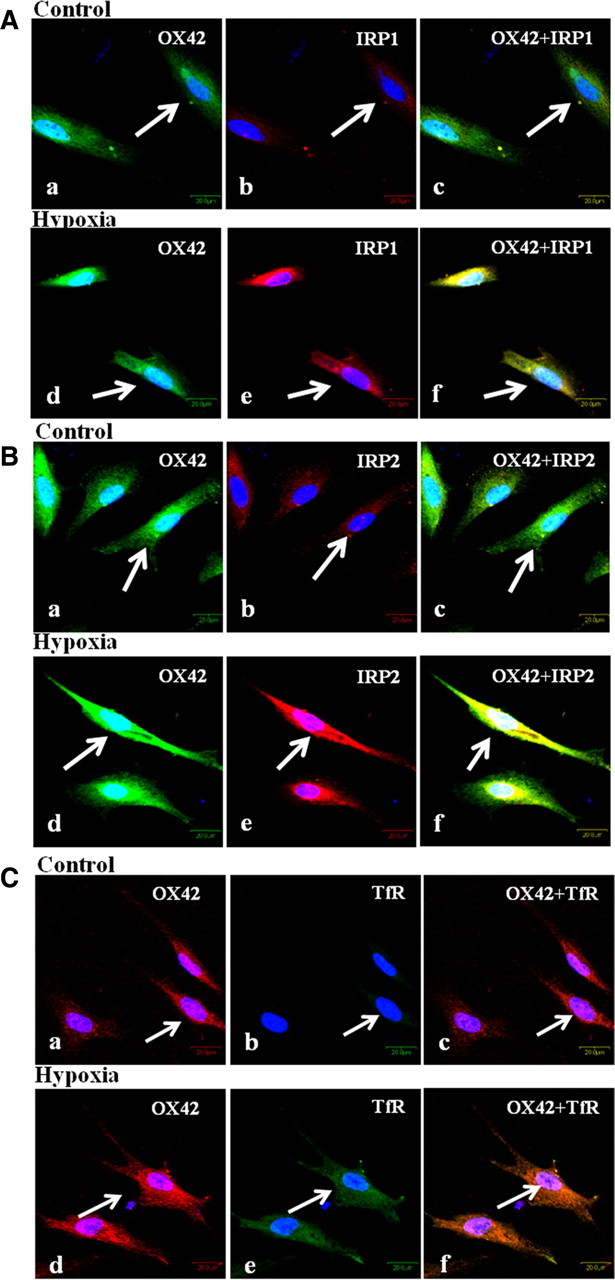
Confocal images showing the colocalization of IRP1 (Ab,e: red), IRP2 (Bb,e: red), TfR (Cb,e: green), and OX42 (Aa,d, Ba,d: green; Ca,d: red) in primary microglial culture. Note the enhanced IRP1, IRP2, and TfR immunofluorescence intensity in the hypoxic microglia when compared with the corresponding control cells. The cells were subjected to 4 h of hypoxic exposure and processed immediately thereafter for immunolabeling. Scale bars, 20 μm. The experiment was repeated in triplicate.
Iron is responsible for ROS production in microglia
The ROS levels in microglia were increased by ∼30% when the cells were exposed to hypoxia for 4 h compared with that of control group (Fig. 6A). However, when treated with deferoxamine, the hypoxia-induced increase in ROS production was reduced by ∼20% when compared with the hypoxic microglia not treated with deferoxamine.
Figure 6.
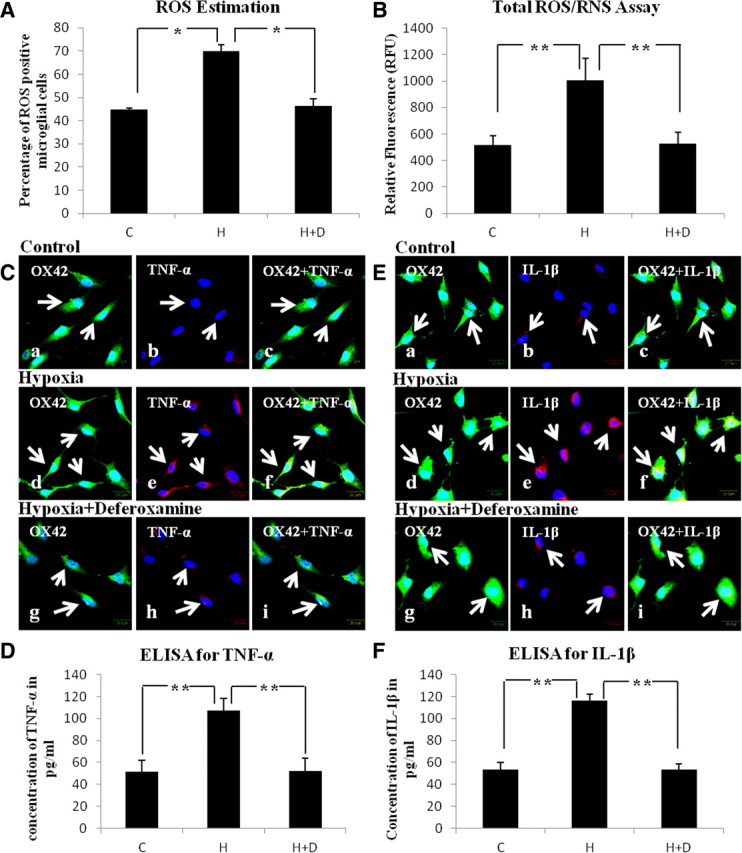
A, Bar graph represents the intracellular ROS estimation using 10 μm CM-H2DCFDA in primary microglial cells. Note a marked increase in ROS level following hypoxia (H) for 4 h when compared with control (C). The hypoxia-induced increase in ROS is suppressed by ∼30% with deferoxamine (H+D) when compared with that of hypoxic microglia. B, Bar graph in B represents the total ROS/RNS released by control (C), hypoxia (H), and hypoxia + deferoxamine (H+D) microglia. C, Confocal images showing the colocalization of OX42 (a, d, g: green) and TNF-α (b, e, h: red) in control (C), hypoxia (H), and hypoxia + deferoxamine (H+D) microglia. Note enhanced TNF-α immunofluorescence is attenuated with deferoxamine treatment. The bar graph in D shows the concentration of TNF-α released into the medium by control (C), hypoxia (H), and hypoxia + deferoxamine (H+D) microglia. E, Confocal images showing the colocalization of OX42 (a, d, g: green) and IL-1β (b, e, h: red) in control (C), hypoxia (H), and hypoxia + deferoxamine (H+D) microglia. Note hypoxia enhanced IL-1β immunofluorescence is attenuated by deferoxamine. Bar graph in F shows the concentration of IL-1β released into the medium in control (C), hypoxia (H), and hypoxia + deferoxamine (H+D) microglia. Scale bars, 20 μm. Significant differences between the groups are expressed as follows: *p < 0.05; **p < 0.01. Each experiment was performed in triplicate.
Extracellular ROS/RNS released by microglia in response to iron accumulation
The amount of total ROS/RNS released by hypoxic microglial cells was significantly increased (Fig. 6B) when compared with that of control microglia. It was, however, markedly decreased when the hypoxic microglia were treated with deferoxamine compared with the hypoxic microglia not treated with deferoxamine. The results indicate that iron might be responsible for hypoxia-induced ROS/RNS production in microglia.
Iron is responsible for production of cytokines in microglia
TNF-α (Fig. 6Cd–f) and IL-1β (Fig. 6Ed–f) immunoexpression in hypoxic microglial cells was upregulated when compared with control (Fig. 6Ca–c,Ea–c) but was attenuated in hypoxic microglial cells treated with deferoxamine (Fig. 6Cg–i,Eg–i). Likewise, hypoxic microglial cells showed a significant increase in the amount of TNF-α (Fig. 6D) and IL-1β (Fig. 6F) released. When treated with deferoxamine, the amount of cytokines released into the medium by the hypoxic microglia was significantly reduced when compared with the hypoxic group not treated with deferoxamine. These results suggest that iron mediates the production of TNF-α and IL-1β in hypoxic microglia.
Effect of microglial conditioned medium on glutathione levels in oligodendrocytes
GSH was significantly decreased in hypoxic oligodendrocytes and in oligodendrocytes treated with conditioned medium from hypoxic microglial cells when compared with that of control group (Fig. 7A). However, the GSH in cells treated with medium from deferoxamine treated hypoxic microglia was significantly increased when compared with hypoxic oligodendrocytes and those treated with medium derived from hypoxic microglia (Fig. 7A). The results indicate that conditioned medium from hypoxic microglia could reduce the glutathione levels in primary oligodendrocytes.
Figure 7.
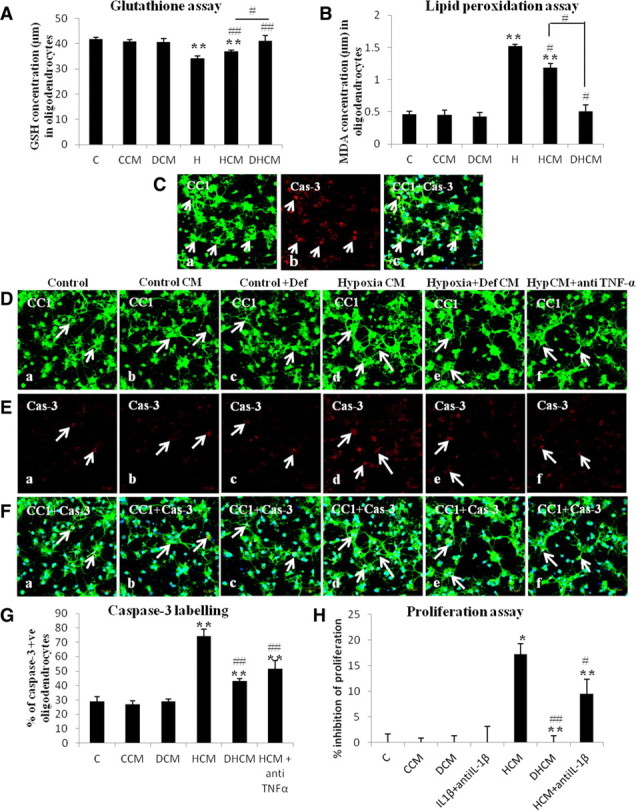
A, Bar graph represents the glutathione content in primary oligodendrocytes in control group (C), oligodendrocytes treated with conditioned medium from control microglia (CCM), oligodendrocytes treated with deferoxamine (DCM), hypoxic oligodendrocytes (H), oligodendrocytes treated with conditioned medium from hypoxic microglia (HCM), and with conditioned medium from deferoxamine treated hypoxic microglial cells (DHCM). B, Bar graph represents the MDA concentrations in oligodendrocyte cultures in all six groups. Significant differences between the groups are expressed as follows: *p < 0.05, **p < 0.01 with respect to control; and #p < 0.05, ##p < 0.01 with respect to hypoxia. C, Confocal images showing caspase-3 (Cas-3)-positive oligodendrocytes on treatment with TNF-α [Ca, CC1 (green); Cb, Cas-3 (red); Cc, CC1 + Cas-3 + DAPI (blue)]. D–F, Caspase-3 labeling of oligodendrocytes showing a significant increase in cells treated with conditioned medium from hypoxic microglial cells. D shows the confocal images of CC1-stained oligodendrocytes in all groups, E shows the confocal images of cells that are caspase-3 positive in all groups, F shows the colocalization of CC1, caspase-3, and DAPI-positive cells (green, CC1; red, Cas-3; blue, DAPI). Scale bars, 50 μm. The bar graph in G shows the percentage of caspase-3-positive cells in all groups including HCM+ anti-TNF-α (treated with conditioned medium from hypoxic microglia containing 10 μg/ml anti-TNF-α). The bar graph in H represents the proliferation of oligodendrocytes in all groups including IL-1β + anti-IL-1β (treated with 100 pg/ml IL-1β peptide neutralized with 10 μg/ml anti-IL-1β) and HCM + anti-IL-1β (treated with conditioned medium from hypoxic microglia containing 10 μg/ml anti-IL-1β). See text for detailed description of changes. Significant differences between the various groups are expressed as follows: *p < 0.05, **p < 0.01 with respect to control; and #p < 0.05, ##p < 0.01 with respect to HCM. Each experiment was performed in triplicate.
Effect of microglial conditioned medium on lipid peroxidation in oligodendrocytes
The LPO levels as manifested by MDA concentrations in hypoxic oligodendrocyte cultures and in oligodendrocytes treated with medium from hypoxic microglial cells were significantly increased when compared with that of control oligodendrocytes (Fig. 7B). However, in cells treated with conditioned medium from deferoxamine treated hypoxic microglial cells, MDA concentrations were significantly reduced compared with hypoxic oligodendrocytes not treated with deferoxamine (Fig. 7B). These results suggest that LPO in oligodendrocytes increases on treatment with conditioned medium from hypoxic microglia.
Caspase-3 activation in oligodendrocytes by TNF-α
On treatment with exogenous TNF-α, many oligodendrocytes in culture were caspase-3 positive (Fig. 7C), indicating that the cytokine causes oligodendrocytes cell death. We next examined the effect of microglial conditioned medium on expression of caspase-3 in oligodendrocytes as well as to ascertain the involvement of TNF-α in this. In the control group (Fig. 7Da,Ea,Fa), in the group treated with conditioned medium from control microglia (Fig. 7Db,Eb,Fb) or with deferoxamine (Fig. 7Dc,Ec,Fc), only a few oligodendrocytes were caspase-3 positive. Oligodendrocytes subjected to treatment with conditioned medium from hypoxic microglial cells showed an increased caspase-3 immunolabeling (Fig. 7Dd,Ed,Fd). A significant reduction in caspase-3 labeling was observed when oligodendrocytes were treated with medium from hypoxia + deferoxamine microglial cells (Fig. 7De,Ee,Fe) or from the medium that was neutralized with TNF-α antiserum (Fig. 7Df,Ef,Ff). The percentage of caspase-3-positive cells was significantly increased when oligodendrocytes were treated with medium from hypoxic microglial cells. However, this was significantly reversed on treatment of the cells with medium from hypoxia + deferoxamine microglial cells or with medium that was neutralized with TNF-α antiserum when compared with that of cells treated with medium from hypoxic microglial cells not treated with deferoxamine or TNF-α antiserum (Fig. 7G). Hence, TNF-α in the conditioned medium from hypoxic microglia induces caspase-3 activation in primary oligodendrocytes.
IL-1β on proliferation of oligodendrocytes
Cleavage of tetrazolium salt WST-1 to formazan is effected by cellular mitochondrial dehydrogenases in viable cells. The cleavage of WST-1 increases in cell proliferation resulting in an increase in the amount of formazan dye formed. On the basis of this and using the proliferation assay, it was found that the proliferation of oligodendrocytes treated with conditioned medium from hypoxic microglial cells was inhibited by >15% (Fig. 7H). In oligodendrocytes treated with medium from deferoxamine treated hypoxic microglial cells, inhibition of cell proliferation was low or negligible (Fig. 7H). Remarkably, inhibition of proliferation of oligodendrocytes was significantly reduced when the cells were treated with conditioned medium from hypoxic microglia with added IL-1β antiserum (Fig. 7H). The results indicate that proliferation of oligodendrocytes was reduced by the presence of IL-1β in the conditioned medium from hypoxic microglia.
Microglial iron induces apoptosis of oligodendrocytes
TUNEL assay was performed in primary oligodendrocytes in cultures to examine their apoptosis following hypoxia and by treating them with conditioned medium from hypoxic microglia. In the control group (Fig. 8Aa,Ba,Ca) and in cells treated with conditioned medium from control microglia (Fig. 8Ab,Bb,Cb) or with deferoxamine (Fig. 8Ac,Bc,Cc), only a few oligodendrocytes showed TUNEL. The number of TUNEL-positive oligodendrocytes was significantly increased in hypoxic group (Fig. 8Ad,Bd,Cd) and on treatment with conditioned medium from hypoxic microglial cells (Fig. 8Ae,Be,Ce). However, there was a significant reduction in apoptosis when the oligodendrocytes were treated with conditioned medium from hypoxia + deferoxamine microglial cells (Fig. 8Af,Bf,Cf) when compared with that of hypoxic group not treated with deferoxamine (Fig. 8D). These results suggest that conditioned medium from hypoxic microglia containing increased iron could cause apoptosis of oligodendrocytes.
Figure 8.

TUNEL assay of oligodendrocytes showing a significant increase in apoptosis following hypoxia and on treatment with conditioned medium from microglial cells subjected to hypoxia. A shows the confocal images of DAPI-stained nucleus in all of the groups, B shows the confocal images of cells that are TUNEL positive in all of the groups, and C shows the colocalization of DAPI- and TUNEL-positive cells. Bar graph in D shows the percentage of cells that are TUNEL positive in each group (C, control; CCM, treated with conditioned medium control microglia; DCM, treated with deferoxamine; H, hypoxia; HCM, treated with conditioned medium from hypoxic microglia; DHCM, treated with conditioned medium from hypoxic microglial cells plus deferoxamine). Scale bars, 50 μm. Significant differences between the various groups are expressed as follows: *p < 0.05, **p < 0.01 with respect to control; and #p < 0.05, ##p < 0.01 with respect to hypoxia. The experiment was repeated in triplicate.
Discussion
The developing brain has a high requirement of iron, as it is essential for the normal myelination, neurotransmitter release, and energy metabolism (Beard and Connor, 2003). However, excess iron in hypoxic–ischemic conditions has been reported to cause neurodegeneration (Shadid et al., 1998; Li et al., 2009). In neonatal animals, hypoxia–ischemia increases the brain iron content (Adcock et al., 1996; Palmer et al., 1999). Because excess iron can cause brain damage, it seems justified to suggest that it may exacerbate hypoxic injuries in neonatal brain. In the latter, it was surmised that the PWM, which is highly vulnerable to hypoxia–ischemia in preterm infants (Johnston, 1997; Volpe, 2003), may be damaged by excess iron in hypoxia. PWMD is characterized by selective loss of oligodendrocytes, astrogliosis, microglial activation, and axonal swelling (Skoff et al., 2001; Folkerth, 2006; Haynes et al., 2008). We show here that hypoxia induced a drastic increase in iron content in the PWM, indicating an iron imbalance in the tissue. In this connection, a major finding was the increased iron accumulation in the AMCs that are widely distributed between the immature oligodendrocytes.
Another major finding was the increased expression of IRP1, IRP2, and TfR in AMCs following the hypoxic exposure, suggesting that the microglial cells actively regulate iron homeostasis in hypoxia. It has been reported that the cellular iron homeostasis is regulated by two cytosolic proteins, the IRPs, and the dysregulation of these IRPs is responsible for the pathology of various neurodegenerative diseases (Cairo et al., 2002). Hypoxia is known to alter the expression of IRPs (Hanson et al., 1999; Christova and Templeton, 2007) and TfR (Kaur and Ling, 1999). The role of IRPs and TfR in causing iron accumulation in microglial cells is supported by the increase in iron localization, IRPs, and TfR in these cells following a hypoxic exposure. IRPs were previously reported to be upregulated in HEK293 cell lines due to hypoxia (Schneider and Leibold, 2003; Christova and Templeton, 2007).
TfR is a major source of cellular iron uptake (Taylor and Morgan, 1990; Roberts et al., 1993). IRPs binding to IRE on TfR mRNA leads to stabilization of the TfR mRNA (Kühn and Hentze, 1992; Henderson, 1996) and thereby increases the TfR expression in human hepatoma cells (Toth et al., 1999). The present results show an upregulated TfR protein expression in the PWM following hypoxic exposure. The increased TfR protein expression could be due to the post-transcriptional regulation exerted by the IRPs binding to the IREs on 3′-UTR of the TfR mRNA. Concomitant to the increase in TfR protein expression, there was an increase in iron concentration in the PWM and also in cultured microglial cells. It is therefore suggested that increased TfR expression might be one of the mechanisms leading to iron accumulation in microglia following the hypoxic exposure. This is supported by our previous work, which suggested that increased expression of TfR was involved in increased uptake of iron for storage in microglial cells (Kaur and Ling, 1999).
Excess iron is implicated in overproduction of ROS and in increasing oxidative stress (Zecca et al., 2004). Hypoxia/reoxygenation is known to enhance the production of free radicals (Palmer et al., 1999), which have a role in the pathogenesis of PWMD (Haynes et al., 2005) as they may be toxic to the immature oligodendrocytes. The developing brain is at a greater risk of free radical-mediated injury as there is an imbalance between antioxidant- and oxidant-generating systems (Buonocore et al., 2001; Welin et al., 2005). Hypoxia is known to reduce the levels of the antioxidant enzymes such as GSH in the neonatal PWM (Kaur et al., 2010). We have demonstrated a significant decrease in GSH in primary cultured oligodendrocytes when exposed to hypoxia and also when exposed to conditioned medium from hypoxic microglial cells. In hypoxic–ischemic conditions, GSH depletion has been implicated in the vulnerability of developing oligodendrocytes (Back et al., 2002), leading to their death by free radical-mediated mechanisms (Juurlink, 1997). Free radical production in the developing white matter in response to hypoxic–ischemic conditions has been reported (Welin et al., 2005) and microglial cells are known to be involved in free radical generation (Colton et al., 1996). We show here that microglial cultures subjected to hypoxia released increased levels of ROS. It is suggested that this would be attributed to the increased iron accumulation in the cells. This is supported by the decrease in ROS production in hypoxic microglial cells treated with the iron chelator deferoxamine.
ROS causes selective degeneration of oligodendrocytes (Griot et al., 1990) by promoting LPO (Su et al., 2011). Following a hypoxic–ischemic injury, the white matter showed increased levels of MDA (Kaur et al., 2010) and 4-HNE, which are toxic to axons and oligodendrocytes (Lin et al., 2004). In the present study, cultured oligodendrocytes exposed to hypoxia and conditioned medium from hypoxic microglial cells exhibited high MDA levels, which were significantly reduced in cells treated with conditioned medium from deferoxamine treated hypoxic microglial cells. This would be attributed to the reduced iron induced ROS/RNS released by deferoxamine treated hypoxic microglial cells.
In addition to ROS, activated microglia secrete cytokines such as TNF-α and IL-1β that can induce the apoptosis of oligodendrocytes (Zhang et al., 2005; Deng et al., 2008). We have shown that, under hypoxic conditions, iron mediates the release of proinflammatory cytokines TNF-α and IL-1β. The relationship between iron and cytokine production was supported by the fact that iron chelator deferoxamine effectively reduced the cytokine production. Further evidence to the role of iron in augmenting the production of cytokines is an increase in TNF-α and IL-1β secretion by microglia when treated with lipopolysaccharide supplemented with iron (Zhang et al., 2006). These results collectively suggest that iron plays an important role in mediating cytokine production by microglia. Previous studies have reported TNF-α induced caspase-3 expression in septohippocampal cultures (Zhao et al., 2001) and caspase-3 is activated only during apoptosis (Armstrong et al., 1996; Wang et al., 1996). Evidence for TNF-α-induced apoptosis of oligodendrocytes comes from the present results where caspase-3 expression was increased in oligodendrocytes treated with conditioned medium from hypoxic microglial cells. Furthermore, caspase-3 expression was reduced in oligodendrocytes treated with medium from deferoxamine treated hypoxic microglia, or with medium from hypoxic microglia, which was neutralized with TNF-α antibody. IL-1β was reported to inhibit cell growth by inhibiting growth factor induced p42/p44 MAPK pathway activation (Vela et al., 2002). In the present study, interaction of IL-1β with oligodendrocytes was derived from the proliferation assay, which showed that proliferation of oligodendrocytes was reduced when treated with medium from hypoxic microglial cells. Proliferation of oligodendrocytes was significantly increased when treated with conditioned medium that was neutralized with IL-1β antiserum as evidenced by the reduced inhibition of proliferation by proliferation assay (Fig. 7H). This is in accord with a previous study that demonstrated the inhibition of proliferation of cultured oligodendrocyte progenitors on addition of IL-1β to the culture medium (Vela et al., 2002). The damaging role of TNF-α and IL-1β on oligodendroyctes is supported by our previous study, which reported an increased expression of TNF receptor 1 and IL-1 receptor 1 in oligodendrocytes in hypoxia (Deng et al., 2008).
Additional evidence to the damaging role of excess accumulation of microglial iron on oligodendrocytes is derived from the TUNEL assay, which showed an increase in apoptosis of oligodendrocytes when treated with medium from hypoxic microglial cells. It is suggested that this may be linked to microglial iron, which mediates production of inflammatory cytokines and ROS. This is supported by the fact that the incidence of apoptosis was significantly reduced in oligodendrocytes treated with conditioned medium derived from deferoxamine treated hypoxic microglia when compared with those subjected to conditioned medium from hypoxic microglia but not treated with deferoxamine. Arising from the above results, we suggest that IRPs are involved in the TfR protein upregulation in hypoxic microglial cells. This would result in increased iron accumulation leading to production of ROS and cytokines such as TNF-α and IL-1β and causing apoptosis and inhibited proliferation of oligodendrocytes in the PWM.
While it is unequivocal that increase in iron in hypoxic microglial cells can cause oligodendrocyte death/apoptosis, the possibility that elevated iron in hypoxic oligodendrocytes can also partake in their cell death is considered. To determine this, primary oligodendrocytes were subjected to hypoxic exposure. Interestingly, increase in oligodendrocyte iron was remarkably low compared with the hypoxic microglia, 0.009 nmol/μl of cell homogenate in hypoxic oligodendrocytes versus 0.07 nmol/μl in hypoxic microglia (Rathnasamy et al., unpublished data). Additionally, the glutathione levels in oligodendrocytes treated with conditioned medium from hypoxic oligodendrocytes were comparable with the control oligodendrocytes (data not shown), suggesting that oligodendrocyte iron plays only a minor role, if any, in oligodendrocyte death in hypoxia.
The present results have shown an increase in iron levels in the PWM in neonatal brain following hypoxia. The increased iron accumulation was specifically localized in AMCs, which was coupled by increased IRPs and TfR expression. Furthermore, intracellular iron increase in AMCs was accompanied by increase in ROS, TNF-α, and IL-1β generation, which was reduced when the cells were treated with the iron chelator deferoxamine. Associated with this was a reduction in apoptosis of oligodendrocytes when treated with conditioned medium from hypoxia + deferoxamine microglia. It is suggested that intracellular iron increase in microglia is a major contributing factor to the enhanced release of ROS and cytokines that cause apoptosis of oligodendrocytes in hypoxia. The role of iron and its regulatory proteins in this would involve mechanisms that it exerts on oligodendrocytes such as reduction of their antioxidant defenses, increased caspase-3 expression, and decreased proliferation. This is manifested by the reduced GSH and increased MDA in oligodendrocytes when treated with conditioned medium derived from hypoxic microglia. The phenomenon, however, was reversed when oligodendrocytes were treated with conditioned medium from hypoxic microglia with added deferoxamine.
Footnotes
This work was supported by National Medical Research Council (Singapore) Research Grant R-181-000-120-213. We thank Dr. Viswanathan Sivakumar and Madhuvika Murugan for their technical assistance.
The authors declare no competing financial interests.
References
- Adcock LM, Yamashita Y, Goddard-Finegold J, Smith CV. Cerebral hypoxia-ischemia increases microsomal iron in newborn piglets. Metab Brain Dis. 1996;11:359–367. doi: 10.1007/BF02029496. [DOI] [PubMed] [Google Scholar]
- Armstrong RC. Isolation and characterization of immature oligodendrocyte lineage cells. Methods. 1998;16:282–292. doi: 10.1006/meth.1998.0685. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas-induced activation of the cell death-related protease CPP32 Is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr. 2003;23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bobyn PJ, Franklin JL, Wall CM, Thornhill JA, Juurlink BH, Paterson PG. The effects of dietary sulfur amino acid deficiency on rat brain glutathione concentration and neural damage in global hemispheric hypoxia-ischemia. Nutr Neurosci. 2002;5:407–416. doi: 10.1080/1028415021000055952. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Buonocore G, Perrone S, Bracci R. Free radicals and brain damage in the newborn. Biol Neonate. 2001;79:180–186. doi: 10.1159/000047088. [DOI] [PubMed] [Google Scholar]
- Cairo G, Recalcati S, Pietrangelo A, Minotti G. The iron regulatory proteins: targets and modulators of free radical reactions and oxidative damage. Free Radic Biol Med. 2002;32:1237–1243. doi: 10.1016/s0891-5849(02)00825-0. [DOI] [PubMed] [Google Scholar]
- Christova T, Templeton DM. Effect of hypoxia on the binding and subcellular distribution of iron regulatory proteins. Mol Cell Biochem. 2007;301:21–32. doi: 10.1007/s11010-006-9393-2. [DOI] [PubMed] [Google Scholar]
- Colton C, Wilt S, Gilbert D, Chernyshev O, Snell J, Dubois-Dalcq M. Species differences in the generation of reactive oxygen species by microglia. Mol Chem Neuropathol. 1996;28:15–20. doi: 10.1007/BF02815200. [DOI] [PubMed] [Google Scholar]
- Dang TN, Bishop GM, Dringen R, Robinson SR. The putative heme transporter HCP1 is expressed in cultured astrocytes and contributes to the uptake of hemin. Glia. 2010;58:55–65. doi: 10.1002/glia.20901. [DOI] [PubMed] [Google Scholar]
- Deng Y, Lu J, Sivakumar V, Ling EA, Kaur C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. 2008;18:387–400. doi: 10.1111/j.1750-3639.2008.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YY, Lu J, Ling EA, Kaur C. Microglia-derived macrophage colony stimulating factor promotes generation of proinflammatory cytokines by astrocytes in the periventricular white matter in the hypoxic neonatal brain. Brain Pathol. 2010;20:909–925. doi: 10.1111/j.1750-3639.2010.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstein RS. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu Rev Nutr. 2000;20:627–662. doi: 10.1146/annurev.nutr.20.1.627. [DOI] [PubMed] [Google Scholar]
- Folkerth RD. Periventricular leukomalacia: overview and recent findings. Pediatr Dev Pathol. 2006;9:3–13. doi: 10.2350/06-01-0024.1. [DOI] [PubMed] [Google Scholar]
- Fuss B, Mallon B, Phan T, Ohlemeyer C, Kirchhoff F, Nishiyama A, Macklin WB. Purification and analysis of in vivo-differentiated oligodendrocytes expressing the green fluorescent protein. Dev Biol. 2000;218:259–274. doi: 10.1006/dbio.1999.9574. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griot C, Vandevelde M, Richard A, Peterhans E, Stocker R. Selective degeneration of oligodendrocytes mediated by reactive oxygen species. Free Radic Res Commun. 1990;11:181–193. doi: 10.3109/10715769009088915. [DOI] [PubMed] [Google Scholar]
- Hanson ES, Foot LM, Leibold EA. Hypoxia post-translationally activates iron-regulatory protein 2. J Biol Chem. 1999;274:5047–5052. doi: 10.1074/jbc.274.8.5047. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–233. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BR. Iron regulatory proteins 1 and 2. Bioessays. 1996;18:739–746. doi: 10.1002/bies.950180909. [DOI] [PubMed] [Google Scholar]
- Hill JM, Switzer RC., 3rd The regional distribution and cellular localization of iron in the rat brain. Neuroscience. 1984;11:595–603. doi: 10.1016/0306-4522(84)90046-0. [DOI] [PubMed] [Google Scholar]
- Johnston MV. Hypoxic and ischemic disorders of infants and children. Brain Dev; Lecture for 38th Meeting of Japanese Society of Child Neurology; July 1996; Tokyo. 1997. pp. 235–239. [DOI] [PubMed] [Google Scholar]
- Juurlink BH. Response of glial cells to ischemia: roles of reactive oxygen species and glutathione. Neurosci Biobehav Rev. 1997;21:151–166. doi: 10.1016/s0149-7634(96)00005-x. [DOI] [PubMed] [Google Scholar]
- Kaur C, Ling EA. Transient expression of transferrin receptors and localisation of iron in amoeboid microglia in postnatal rats. J Anat. 1995;186:165–173. [PMC free article] [PubMed] [Google Scholar]
- Kaur C, Ling EA. Increased expression of transferrin receptors and iron in amoeboid microglial cells in postnatal rats following an exposure to hypoxia. Neurosci Lett. 1999;262:183–186. doi: 10.1016/s0304-3940(99)00075-0. [DOI] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Ang LS, Sundaresan A. Hypoxic damage to the periventricular white matter in neonatal brain: role of vascular endothelial growth factor, nitric oxide and excitotoxicity. J Neurochem. 2006;98:1200–1216. doi: 10.1111/j.1471-4159.2006.03964.x. [DOI] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Yip GW, Ling EA. Expression of syndecan-2 in the amoeboid microglial cells and its involvement in inflammation in the hypoxic developing brain. Glia. 2009;57:336–349. doi: 10.1002/glia.20764. [DOI] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Ling EA. Melatonin protects periventricular white matter from damage due to hypoxia. J Pineal Res. 2010;48:185–193. doi: 10.1111/j.1600-079X.2009.00740.x. [DOI] [PubMed] [Google Scholar]
- Kühn LC, Hentze MW. Coordination of cellular iron metabolism by post-transcriptional gene regulation. J Inorg Biochem. 1992;47:183–195. doi: 10.1016/0162-0134(92)84064-t. [DOI] [PubMed] [Google Scholar]
- Li L, Li YW, Zhao JY, Liu YZ, Holscher C. Quantitative analysis of iron concentration and expression of ferroportin 1 in the cortex and hippocampus of rats induced by cerebral ischemia. J Clin Neurosci. 2009;16:1466–1472. doi: 10.1016/j.jocn.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Lin S, Rhodes PG, Lei M, Zhang F, Cai Z. alpha-Phenyl-n-tert-butyl-nitrone attenuates hypoxic-ischemic white matter injury in the neonatal rat brain. Brain Res. 2004;1007:132–141. doi: 10.1016/j.brainres.2004.01.074. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Menzies SL, Roberts RL, Pavlick G, Connor JR. Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J Neurosci Res. 1999;56:60–71. doi: 10.1002/(SICI)1097-4547(19990401)56:1<60::AID-JNR8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest. 1995;95:2555–2564. doi: 10.1172/JCI117956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RL, Fine RE, Sandra A. Receptor-mediated endocytosis of transferrin at the blood-brain barrier. J Cell Sci. 1993;104:521–532. doi: 10.1242/jcs.104.2.521. [DOI] [PubMed] [Google Scholar]
- Rouault TA, Hentze MW, Haile DJ, Harford JB, Klausner RD. The iron-responsive element binding protein: a method for the affinity purification of a regulatory RNA-binding protein. Proc Natl Acad Sci U S A. 1989;86:5768–5772. doi: 10.1073/pnas.86.15.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Schneider BD, Leibold EA. Effects of iron regulatory protein regulation on iron homeostasis during hypoxia. Blood. 2003;102:3404–3411. doi: 10.1182/blood-2003-02-0433. [DOI] [PubMed] [Google Scholar]
- Shadid M, Buonocore G, Groenendaal F, Moison R, Ferrali M, Berger HM, van Bel F. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci Lett. 1998;248:5–8. doi: 10.1016/s0304-3940(98)00303-6. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Chuai J, Ferriero DM. A rat model for hypoxic-ischemic brain damage in very premature infants. Biol Neonate. 1996;69:327–341. doi: 10.1159/000244327. [DOI] [PubMed] [Google Scholar]
- Shouman BO, Mesbah A, Aly H. Iron metabolism and lipid peroxidation products in infants with hypoxic ischemic encephalopathy. J Perinatol. 2008;28:487–491. doi: 10.1038/jp.2008.22. [DOI] [PubMed] [Google Scholar]
- Sivakumar V, Foulds WS, Luu CD, Ling EA, Kaur C. Retinal ganglion cell death is induced by microglia derived proinflammatory cytokines in the hypoxic neonatal retina. J Pathol. 2011;224:245–260. doi: 10.1002/path.2858. [DOI] [PubMed] [Google Scholar]
- Skoff RP, Bessert DA, Barks JD, Song D, Cerghet M, Silverstein FS. Hypoxic-ischemic injury results in acute disruption of myelin gene expression and death of oligodendroglial precursors in neonatal mice. Int J Dev Neurosci. 2001;19:197–208. doi: 10.1016/s0736-5748(00)00075-7. [DOI] [PubMed] [Google Scholar]
- Su MY, Huang HY, Li L, Lu YH. Protective effects of 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone to PC12 cells against cytotoxicity induced by hydrogen peroxide. J Agric Food Chem. 2011;59:521–527. doi: 10.1021/jf104408d. [DOI] [PubMed] [Google Scholar]
- Taylor EM, Morgan EH. Developmental changes in transferrin and iron uptake by the brain in the rat. Brain Res Dev Brain Res. 1990;55:35–42. doi: 10.1016/0165-3806(90)90103-6. [DOI] [PubMed] [Google Scholar]
- Toth I, Yuan L, Rogers JT, Boyce H, Bridges KR. Hypoxia alters iron-regulatory protein-1 binding capacity and modulates cellular iron homeostasis in human hepatoma and erythroleukemia cells. J Biol Chem. 1999;274:4467–4473. doi: 10.1074/jbc.274.7.4467. [DOI] [PubMed] [Google Scholar]
- Utaisincharoen P, Tangthawornchaikul N, Ubol S, Chaisuriya P, Sirisinha S. TNF-alpha induces caspase 3 (CPP 32) dependent apoptosis in human cholangiocarcinoma cell line. Southeast Asian J Trop Med Public Health. 2000;31(Suppl 1):167–170. [PubMed] [Google Scholar]
- Vela JM, Molina-Holgado E, Arévalo-Martín A, Almazán G, Guaza C. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Mol Cell Neurosci. 2002;20:489–502. doi: 10.1006/mcne.2002.1127. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Cerebral white matter injury of the premature infant—more common than you think. Pediatrics. 2003;112:176–180. doi: 10.1542/peds.112.1.176. [DOI] [PubMed] [Google Scholar]
- Wang KK, Nath R, Raser KJ, Hajimohammadreza I. Maitotoxin induces calpain activation in SH-SY5Y neuroblastoma cells and cerebrocortical cultures. Arch Biochem Biophys. 1996;331:208–214. doi: 10.1006/abbi.1996.0300. [DOI] [PubMed] [Google Scholar]
- Wang XS, Ong WY, Connor JR. Increase in ferric and ferrous iron in the rat hippocampus with time after kainate-induced excitotoxic injury. Exp Brain Res. 2002;143:137–148. doi: 10.1007/s00221-001-0971-y. [DOI] [PubMed] [Google Scholar]
- Welin AK, Sandberg M, Lindblom A, Arvidsson P, Nilsson UA, Kjellmer I, Mallard C. White matter injury following prolonged free radical formation in the 0.65 gestation fetal sheep brain. Pediatr Res. 2005;58:100–105. doi: 10.1203/01.PDR.0000163388.04017.26. [DOI] [PubMed] [Google Scholar]
- Yang J, Khong PL, Wang Y, Chu AC, Ho SL, Cheung PT, Wu EX. Manganese-enhanced MRI detection of neurodegeneration in neonatal hypoxic-ischemic cerebral injury. Magn Reson Med. 2008;59:1329–1339. doi: 10.1002/mrm.21484. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zhang X, Haaf M, Todorich B, Grosstephan E, Schieremberg H, Surguladze N, Connor JR. Cytokine toxicity to oligodendrocyte precursors is mediated by iron. Glia. 2005;52:199–208. doi: 10.1002/glia.20235. [DOI] [PubMed] [Google Scholar]
- Zhang X, Surguladze N, Slagle-Webb B, Cozzi A, Connor JR. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia. 2006;54:795–804. doi: 10.1002/glia.20416. [DOI] [PubMed] [Google Scholar]
- Zhao X, Bausano B, Pike BR, Newcomb-Fernandez JK, Wang KK, Shohami E, Ringger NC, DeFord SM, Anderson DK, Hayes RL. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res. 2001;64:121–131. doi: 10.1002/jnr.1059. [DOI] [PubMed] [Google Scholar]