

Abstract
Astrocytes are now distinguished as major regulators of neuronal growth and synaptic development. Recently, they have been identified as key players in the progression of a number of developmental disorders; however, in fragile X syndrome (FXS), the role of astrocytes is not known. Using a coculture design, we found that hippocampal neurons exhibited abnormal dendritic morphology and a decreased number of presynaptic and postsynaptic protein aggregates when they were grown on astrocytes from a fragile X mouse. Moreover, we found that normal astrocytes could prevent the development of abnormal dendrite morphology and preclude the reduction of presynaptic and postsynaptic protein clusters in neurons from a fragile X mouse. These experiments are the first to establish a role for astrocytes in the altered neurobiology of FXS. Our results support the notion that astrocytes contribute to abnormal dendrite morphology and the dysregulated synapse development in FXS.
Introduction
Fragile X syndrome (FXS) is a neurodevelopmental disorder, caused by a mutation in the fragile X mental retardation 1 (FMR1) gene (Kremer et al., 1991). Affecting ∼1 of 2500 individuals (both male and female with the full mutation) (Hagerman, 2008), FXS is the most common cause of inherited mental retardation. Children with FXS exhibit a wide spectrum of cognitive and behavioral features including the following: mild to severe cognitive impairment, attention deficit, anxiety, social withdrawal, susceptibility to seizures, motor disorders, and autistic behaviors (Beckel-Mitchener and Greenough, 2004). To date, the mutation in the FMR1 gene is the leading known inherited cause of autism (Reddy, 2005).
FMR1 codes for the fragile X mental retardation protein (FMRP) (Devys et al., 1993), a regulator of mRNA translation (Brown et al., 2001; Miyashiro et al., 2003; Liao et al., 2008), which plays an important role modulating the translation of a number of mRNAs that are important for dendritic growth (Brown et al., 2001; Lee et al., 2003) and the development of synapses (Greenough et al., 2001; Miyashiro et al., 2003; Liao et al., 2008). In FXS, there is an abnormal number (>200) of CGG repeats in the 5` noncoding region of the FMR1 gene (Kremer et al., 1991), which results in hypermethylation, transcriptional silencing, and a lack of FMRP. Without FMRP, there is a disturbance of translational regulation and a disruption in the composition of the normal protein milieu.
The Fmr1 knock-out (Fmr1 KO) mouse is a well established model of FXS. As in the human, the Fmr1 KO mouse displays a lack of FMRP (Bakker et al., 1994). This model recapitulates many characteristics of FXS, both behaviorally and at the cellular level, including the following: learning impairments (Bakker et al., 1994), hyperactivity (Qin et al., 2002), susceptibility to seizures (Chen and Toth, 2001), characteristics associated with attention deficits (Nielsen et al., 2002), predictors of autistic behaviors (Frankland et al., 2004), and abnormal neuronal dendritic spine development (Comery et al., 1997; Nimchinsky et al., 2001). Although preclinical and clinical studies have suggested a neuronal pathology in FXS, there is new evidence that glial–neuronal interactions may be important in FXS. In the past 10 years, FMRP has also been found in oligodendrocytes and their precursors (Wang et al., 2004; Castrén et al., 2005). We recently reported that FMRP is also expressed in developing mouse astrocytes (Pacey and Doering, 2007). In fact, it appears that the expression of FMRP is developmentally regulated. Pacey and Doering (2007) found that FMRP was expressed in early development in cells of the glial lineage both in vitro and in vivo, but not in adult astrocytes. Wang et al. (2004) also demonstrated a developmental expression of FMRP in glial cells. In their experiments, they found that FMRP was expressed in oligodendrocyte progenitors but not in mature oligodendrocytes (Wang et al., 2004). However, the effect of a lack of FMRP in the glial lineage on neuronal development has not been determined experimentally.
Materials and Methods
Animals.
All animal experiments were performed in accordance with the guidelines set out by the Canadian Council on Animal Care and were approved by the McMaster University Animal Research Ethics Board. The FMRP mouse colony was established from breeding pairs of FVB.129P2(B6)-Fmr1tm1Cgr mice originally obtained from Dr. Carl Dobkin at the New York State Institute for Basic Research in Developmental Disabilities (Staten Island, NY). The Fmr1 targeted insertion (KO) mutation was originally engineered in embryonic day 14 (E14) embryonic stem cells derived from the 129/Ola strain (Bakker et al., 1994). FVB/N-129 hybrid mice carrying this mutation were repeatedly backcrossed (n = 11 FVB/N backcross generations). The congenic Fmr1 KO mice used in these experiments were also regularly genotyped. The wild-type (WT) and KO mice were maintained as individual strains, and both male and female mice were used in the experiments. The FMRP knock-out mice [FVB.129P2(B6)-Fmr1tm1Cgr] were housed and bred at the McMaster University Central Animal Facility.
Cell culture.
Primary hippocampal neurons with astrocytes were grown in coculture conditions as detailed previously by our laboratory (Jacobs and Doering, 2009). Briefly, astrocytes were isolated from either WT or Fmr1 KO postnatal day 0 (P0) to P1 pups and grown on poly-l-lysine (1 mg/ml)- and laminin (0.1 mg/ml)-coated coverslips in MEM (Invitrogen) supplemented with 6% glucose and 10% horse serum (Invitrogen) for 1 week. Primary hippocampal neurons were then isolated from embryonic day 17 (day of sperm plug counted as E1) animals and seeded on the astrocytes and maintained in MEM supplemented with N2 (Invitrogen), sodium pyruvate (Invitrogen), and 6% glucose, for the duration of the experiments. For each experiment, separate litters of mice were used to generate both the neurons and astrocytes.
Immunocytochemistry.
After 7 d in vitro (DIV), the cells were fixed with ice-cold (−20°C) methanol and processed for immunocytochemistry. After the appropriate serum block, the cells were incubated with primary antibodies overnight at 4°C. Secondary antibodies were applied for 3 h at room temperature. The following antibodies, diluted in 1% BSA, were used: chicken microtubule-associated protein 2 (MAP2) (1:20,000; Neuromics), mouse monoclonal synaptophysin (clone SVP-38; 1:250; Sigma-Aldrich), mouse monoclonal postsynaptic density 95 (PSD-95) (clone 6G6-1C9; 1:200; Millipore Bioscience Research Reagents), anti-mouse Alexa Fluor 594 (1:1500; Invitrogen), and anti-chicken FITC (1:100; Jackson ImmunoResearch Laboratories). Coverslips were mounted with Vectashield fluorescent mounting medium with 4`,6-diamidino-2-phenylindole (DAPI).
Image acquisition and quantification.
Images were captured using a Zeiss Axioskop 2 epifluorescence and Axiovision image acquisition software. The quantification of dendritic arbor morphology and puncta analysis were performed using plug-ins for ImageJ (http://rsbweb.nih.gov/ij/). Morphological measurements of isolated neurons were obtained using Neuronmetrics (http://www.ibridgenetwork.org/arizona/UA07-56-Neuronmetrics) (Narro et al., 2007). Puncta analysis was performed on isolated cells using a custom-written plug-in (Christopherson et al., 2005, Eroglu et al., 2009).
For each experiment, the same culture conditions were established in multiple wells. For analysis, two or three coverslips per experiment were used to obtain the sample of isolated neurons. For the morphometric analyses, ∼100 cells were selected at random from three coverslips, from each of three independent experiments (with the exception of the KO/KO condition in which only two experiments were performed). For the synaptic protein analysis, ∼50 isolated neurons were selected at random per experiment, over two coverslips for each of three independent experiments (with the exception of the KO/KO condition as above).
Statistical analyses.
For all analyses, the observer was blind to the identity of the sample. Statistical analyses were conducted using SPSS17, with an α level set to 0.05. Data are presented as mean ± SEM.
N represents the number of independent culture preparations per condition, and n represents the total number of cells analyzed from those combined experiments. For multiple comparisons, a one-way ANOVA with post hoc Tukey's tests were performed. Where data had significantly different variances (as determined by Levene's test for equal variances) and deviated significantly from a normal distribution (as determined by the Shapiro–Wilk test for normality), Mann–Whitney U (two-tailed) tests were used. Effect size is indicated by Cohen's f2, or Cohen's d using a pooled SD, where appropriate.
Results
In these experiments, we investigated the role of astrocytes in the development of the abnormal dendrite morphology and synaptic aberrations seen in FXS. To examine whether astrocytes from an FMRP-deficient animal could affect normal neuron growth, we used a coculture procedure in which neurons are grown at low densities (400 cells/μl) in serum-free media (Jacobs and Doering, 2009). In these cultures, the neurons are dependent on a feeder layer of astrocytes for survival (Banker, 1980). Consistent with previously reported findings, hippocampal neurons from both WT (normal) and Fmr1 KO (fragile X) mice grown in the absence of an astrocyte feeder layer did not survive. Both WT and Fmr1 KO neurons grown on WT astrocytes exhibited normal neuronal growth at 7 DIV (Fig. 1a, top row). In stark contrast, when Fmr1 KO neurons were grown on Fmr1 KO astrocytes, the neurons exhibited a distinct abnormal morphology (Fig. 1a, bottom row). The dendritic arbors were more complex and individual processes showed random meandering on the astrocyte surface.
Figure 1.

Effects of astrocytes on the growth of hippocampal neurons in coculture at 7 DIV. E17 primary hippocampal neurons were cocultured with P0–P1 primary cortical astrocytes for 7 DIV in each of four coculture conditions. a, Immunofluorescent images of neurons in each of the four culture combinations. Neurons are stained with an antibody directed against the neuronal dendritic marker, MAP2. Scale bar, 100 μm. b, Quantification of percentage of surviving neurons at 7 DIV in each of the four culture conditions. Data shown are mean values ± SEM from two or three independent experiments (10–15 regions of 1.5 mm2 from 2 coverslips per experiment). Significant differences revealed by post hoc Tukey's tests are indicated (p < 0.001).
The survival of Fmr1 KO neurons (grown on either WT or Fmr1 KO astrocytes) was decreased compared with WT neurons (p = 0.004; f2 = 0.12) (Fig. 1b). However, survival was not significantly affected by growth on Fmr1 KO astrocytes compared with WT, for either WT (p = 0.977) or Fmr1 KO neurons (p = 0.382) (Fig. 1b). Therefore, the results that we present are not attributable to a density effect or selective neuron survival, and the morphological differences seen are a result of some quality of the different astrocyte populations. This difference could not be attributed to a significant variance in the purity of the astrocyte populations because the astrocytes isolated from both WT and Fmr1 KO mice were 98.7 ± 0.5% GFAP positive. Therefore, the observed alterations in the neuronal phenotype can be concluded to result from the characteristics of the astrocytes imparted by the genotype of their origin animal.
Fmr1 KO astrocytes alter the morphology of WT neurons
To evaluate more precisely how the Fmr1 KO astrocytes affected the growth of the neurons, we performed a detailed morphological analysis of the dendritic arbors. After staining with an antibody targeting the dendrite marker, MAP2, isolated neurons were selected at random from a minimum of two coverslips per experiment, and the morphology of their dendritic arbors was analyzed. WT neurons exhibited altered morphology when grown on astrocytes isolated from an Fmr1 KO mouse (Fig. 2a,b). Comparison of the dendritic arbor morphology of WT neurons grown on Fmr1 KO astrocytes with those grown on WT astrocytes showed the following: the length of the longest primary dendrite and the extent of the area covered by the dendritic arbor were decreased by 15.5% (p < 0.001; d = 0.32) and 31.8% (p < 0.001; d = 0.47), respectively (Fig. 2c,d); the area covered per micrometer of dendrite was decreased by 17.3% (p < 0.001; d = 0.38) (Fig. 2g); the number of branches per cell was increased by 13.0% (p = 0.046; d = 0.22) (Fig. 2e); and the branch density (number of dendritic branches per square micrometer) was increased by 57.4% (p < 0.001; d = 0.54) (Fig. 2h). These observations indicate that growth on Fmr1 KO astrocytes appeared to alter the normal dendritic arborization of WT neurons.
Figure 2.
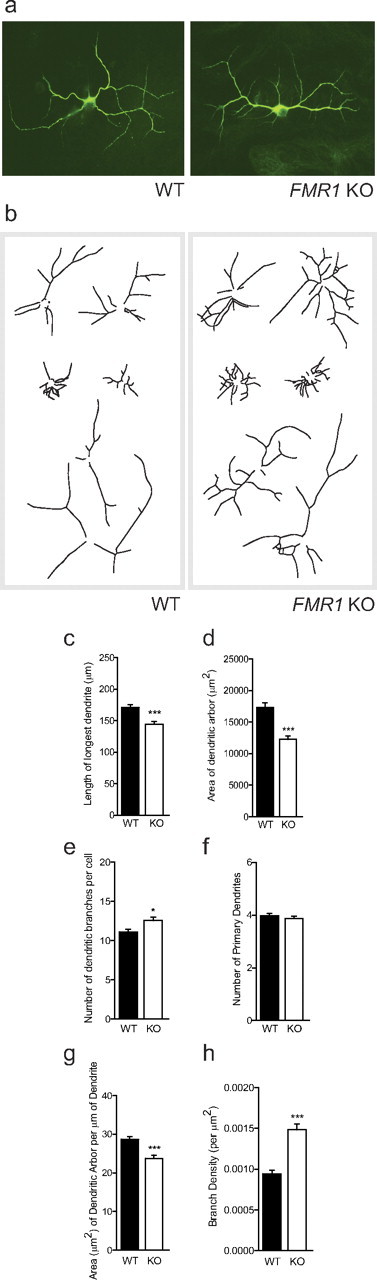
Effects of Fmr1 KO astrocytes on dendritic morphology of WT neurons. WT E17 primary hippocampal neurons were cocultured for 7 DIV with primary cortical astrocytes from either WT or Fmr1 KO mice. a, Immunofluorescent images of neurons at 7 DIV stained with an antibody directed against the neuronal dendritic marker, MAP2. The left and right images are representative WT neurons cultured on WT and Fmr1 KO astrocytes, respectively. Scale bar, 50 μm. b, Representative dendritic arbor skeletons of WT neurons grown on WT astrocytes (left panel) or Fmr1 KO astrocytes (right panel). c–h, Quantification of dendritic arbor morphology. Data shown are mean values ± SEM. WT: n = 297, N = 3; Fmr1 KO: n = 282, N = 3. Significant differences revealed by Mann–Whitney U tests (two-tailed) are indicated (*p < 0.05; ***p < 0.001).
WT astrocytes prevent the abnormal dendrite morphology of Fmr1 KO neurons
Given that Fmr1 KO astrocytes impacted the dendrite morphology of the cultured WT hippocampal neurons, we next tested whether WT astrocytes are able to prevent the development of the abnormal neuron morphology in Fmr1 KO hippocampal neurons. We analyzed the morphology of Fmr1 KO neurons grown on both Fmr1 KO and WT astrocytes. Notably, Fmr1 KO neurons grown on WT astrocytes exhibited morphological alterations toward a normal neuronal dendritic phenotype (Fig. 3a–h). When grown on WT astrocytes, Fmr1 KO neurons exhibited an increase in the length of the longest dendrite (16.7%; p < 0.001; d = 0.39) (Fig. 3c), the total area covered by the dendritic arbor (36.9%; p < 0.001; d = 0.61) (Fig. 3d), and the area covered per micrometer of dendrite (24.9%; p < 0.001; d = 0.68) (Fig. 3g). Conversely, when grown on WT astrocytes, Fmr1 KO neurons showed a decrease in the number of branches (Fig. 3e), number of primary dendrites (Fig. 3f), and branch density (Fig. 3h), by 6.9% (p = 0.381; d = 0.13), 13.8% (p < 0.001; d = 0.42), and 33.3% (p < 0.001; d = 0.51), respectively. However, for all parameters, growth on WT astrocytes restored the Fmr1 KO phenotype closer to the WT neuron morphology. In fact, growth of Fmr1 KO neurons on WT astrocytes restored the phenotype by as much as 71.7% (relative difference of Fmr1 KO neurons grown on WT astrocytes vs Fmr1 KO astrocytes compared with WT neurons grown on WT astrocytes) (Fig. 3c–h, dotted line). These observations support the notion that astrocytes from a WT mouse prevent the development of abnormal dendritic morphology of Fmr1 KO neurons.
Figure 3.
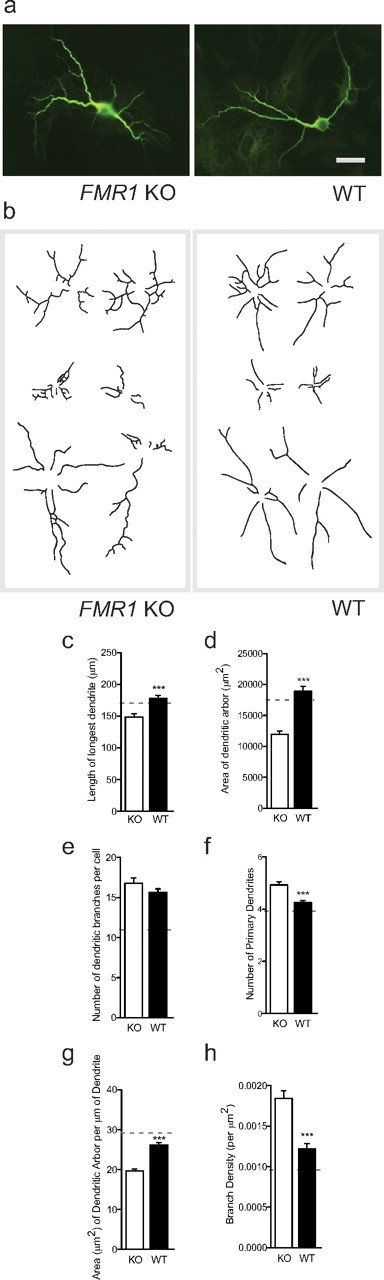
WT astrocytes rescue the dendritic morphology of Fmr1 KO neurons. Fmr1 KO E17 primary hippocampal neurons were cultured for 7 DIV with primary cortical astrocytes from either WT or Fmr1 KO mice. a, Neurons stained with an antibody directed against the neuronal dendritic marker, MAP2. The left and right images are representative of Fmr1 KO neurons cultured on WT or Fmr1 KO astrocytes, respectively. Scale bar, 50 μm. b, Representative dendritic arbor skeletons of Fmr1 KO neurons grown on WT astrocytes (left panel) or Fmr1 KO astrocytes (right panel). c–h, Quantification of dendritic arbor morphology. Data shown are mean values ± SEM. WT: n = 318, N = 3; Fmr1 KO: n = 201, N = 2. Significant differences revealed by Mann–Whitney U tests (two-tailed) are indicated (***p < 0.001). The horizontal dotted line indicates mean value for WT neurons grown on WT astrocytes (from Fig. 2c–h).
Fmr1 KO astrocytes alter the synaptic protein expression in WT neurons
Next, we sought to determine whether the initial synaptic protein packaging was altered in the WT neurons grown on Fmr1 KO astrocytes. Early in neuron development before synaptic formation, presynaptic and postsynaptic protein complexes are packaged for transport to future synaptic sites. These synaptic protein aggregates can be visualized as puncta (spots of intense staining) with immunocytochemistry. We used antibodies directed at synaptophysin (a presynaptic protein that exists as part of the synaptic vesicle membrane) and PSD-95 (a postsynaptic protein that forms part of the postsynaptic density) to determine whether the early packaging of synaptic proteins was altered.
At 7 DIV, the clustering of both presynaptic and postsynaptic proteins was altered in WT neurons grown on Fmr1 KO astrocytes (Fig. 4a,b). We found a 34.8% decrease (p < 0.001; d = 0.38) (Fig. 4c) in the number of synaptophysin-immunoreactive (IR) puncta per cell, and a 13.9% reduction (p < 0.001, d = 0.15) (Fig. 4d) in the number of PSD-95-IR puncta per cell. These results demonstrate that the initial synaptic protein clustering is impaired when WT neurons are grown on Fmr1 KO astrocytes.
Figure 4.
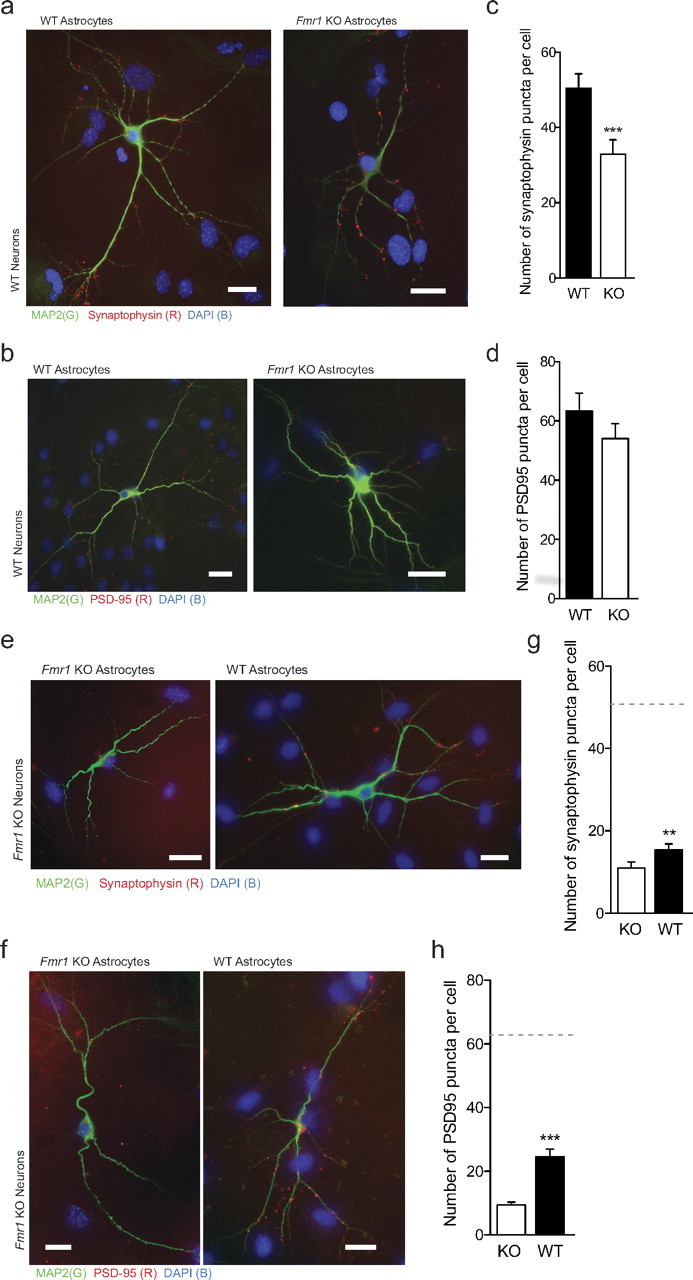
WT astrocytes rescue the synaptic protein aggregation in Fmr1 KO neurons. a–d, WT E17 primary hippocampal neurons were cultured for 7 DIV with primary cortical astrocytes from either WT or Fmr1 KO mice. a, b, Immunofluorescent images of WT neurons stained with antibodies directed against MAP2 (green) and synaptophysin (a) or PSD-95 (b) (red). Nuclei are stained with DAPI (blue). The images on the left and right are representative WT neurons grown on WT astrocytes and Fmr1 KO astrocytes, respectively. Scale bars, 50 μm. c, Quantification of synaptophysin puncta per cell. Data shown are mean values ± SEM. WT: n = 147, N = 3; Fmr1 KO: n = 143, N = 3. d, Quantification of PSD-95 puncta per cell. Data shown are mean values ± SEM. WT: n = 111, N = 3; Fmr1 KO: n = 119, N = 3. e–h, Fmr1 KO E17 primary hippocampal neurons were cultured for 7 DIV with primary cortical astrocytes from either WT or Fmr1 KO mice. e, f, Immunofluorescent images of neurons stained with antibodies directed against MAP2 (green) and synaptophysin (e) or PSD-95 (f) (red). Nuclei are stained with DAPI (blue). The images on the left and right are representative Fmr1 KO neurons grown on WT and Fmr1 KO astrocytes, respectively. Scale bars, 50 μm. g, Quantification of synaptophysin puncta per cell. Data shown are mean values ± SEM. WT: n = 112, N = 3; Fmr1 KO: n = 94, N = 2. h, Quantification of PSD-95 puncta per cell. Data shown are mean values ± SEM. WT: n = 143, N = 3; Fmr1 KO: n = 94, N = 2. Significant differences revealed by Mann–Whitney U tests (two-tailed) are indicated (**p < 0.05; ***p < 0.001).
WT astrocytes prevent the alterations of synaptic proteins in Fmr1 KO neurons
Given that the dendritic morphology of Fmr1 KO neurons approached normal when grown on WT astrocytes, we examined whether astrocytes from a WT mouse could also influence the synaptic protein distribution of Fmr1 KO neurons. At 7 DIV, the clustering of both presynaptic and postsynaptic proteins was increased in Fmr1 KO neurons grown on WT astrocytes (Fig. 4e,f). We found a 40.3% (p = 0.003; d = 0.30) (Fig. 4g) increase in the number of synaptophysin-IR puncta in Fmr1 KO neurons grown on WT astrocytes. This equated to 30.6% of the number seen in WT neurons on WT astrocytes (compared with 22.8% of normal). Similarly, we observed an increase in the number of PSD-95-IR puncta per cell. In fact, there was almost three times (p < 0.001; d = 0.70) (Fig. 4h) the number of PSD-95-IR puncta per cell, increasing the quantity to 39.0% of normal numbers (compared with 14.7%). Therefore, these results support the concept that WT astrocytes can increase the number of both presynaptic and postsynaptic protein clusters, contributing to the recovery of the alterations in synaptic protein expression in Fmr1 KO neurons.
Discussion
These experiments are novel and exciting as they are the first to establish a role for astrocytes in the altered neurobiology of FXS. Our data show that astrocytes are involved in shaping the dendritic arbors of neurons in FXS. During development, numerous pathways between neurons are made, and then the redundant connections are pared back to create a streamlined neural communication network. Glial cells provide guidance cues that aid in the correct extension (Banker, 1980; Powell and Geller, 1999) and elimination of neural processes (Barres, 2008). In FXS, as in other disorders that manifest with cognitive impairment (Kaufmann and Moser, 2000), the fine-tuning is often impaired. It follows that the astrocytes in FXS (and other disorders of cognitive impairment) could be lacking in their capacity to promote appropriate neurite patterning. Consistent with this, our results revealed more complex dendritic arbors with increased branch density in neurons grown with astrocytes from a fragile X mouse. Furthermore, the abnormal increase in branch density was prevented in fragile X neurons when cultured with normal astrocytes. These results are consistent with the abnormal pruning associated with FXS. Furthermore, the findings imply that FXS astrocytes are deficient in their capacity to effectively regulate the pruning process that is required for efficient neural communication and could contribute to the cognitive impairment seen in individuals with FXS.
The current experiments demonstrated that astrocytes are also involved in the early stages of synapse development. Abnormal levels of synaptic proteins have been shown in FXS (Liao et al., 2008), and synaptic inefficiencies have also been described in a number of developmental disorders (Südhof, 2008). Astrocytes are required for the efficient formation, maturation, and maintenance of synapses (Pfrieger and Barres, 1997; Ullian et al., 2001). Therefore, in addition to lacking the full capacity to promote appropriate dendrite elaborations, astrocytes in FXS could also be deficient in their ability to regulate synapse development. In agreement with this, we found that neurons grown with astrocytes from a fragile X mouse exhibited a decreased number of presynaptic and postsynaptic protein aggregates. Furthermore, this abnormal decrease was prevented in fragile X neurons when they were cultured with normal astrocytes. These findings are in concert with the current understanding of synaptic protein dysregulation in FXS. In addition, these experiments suggest that astrocytes contribute to the erroneous synapse development in FXS and could therefore be a fundamental factor in the development of the behavioral maladaptations seen in individuals with FXS.
Previous research has shown that glial cells contribute to the development of several neurological disorders (for review, see Barres, 2008), and our results support the concept of astrocytes guiding appropriate neurite and synapse development (Pfrieger and Barres, 1997; Mauch et al., 2001; Ullian et al., 2001; Murai et al., 2003; Christopherson et al., 2005; Nishida and Okabe, 2007; Guizzetti et al., 2008). Most recently, Ballas et al. (2009) found that astrocytes lacking the protein responsible for Rett syndrome, MeCP (methyl-CpG-binding protein 2), could not support normal neuron growth. Similar to Rett syndrome, FXS was previously thought to be a disease resulting from a loss of function caused by a lack of a specific protein in neurons only. However, it is possible that the abnormal phenotype in FXS, as in Rett syndrome, results from additional non-neuronal deficiencies. For example, the astrocytes in individuals with FXS may also be dysfunctional, and this deficit could be caused by an indirect or a direct loss of FMRP. The loss of FMRP in neurons could result, indirectly, in aberrant astrocyte-mediated support functions as a result of impaired neuron-to-astrocyte signaling caused by the loss of neuronal FMRP. Alternatively, the astrocytes in an individual with FXS may also be deficient in FMRP and suffer abnormalities in normal functioning directly attributable to the loss of FMRP. Recently, our laboratory documented that FMRP is expressed in cells of the glial lineage and that FMRP was absent from putative astrocytes in the Fmr1 KO mouse (Pacey and Doering, 2007). Therefore, it is possible that astrocytes lack FMRP, specifically at a time during development when astrocyte support of neuron growth and synapse formation are vital, and this lack of FMRP could contribute to the abnormal neuron phenotype seen in FXS. Given that FMRP is a key regulator of translation of a number of mRNA targets in neurons (Brown et al., 2001; Miyashiro et al., 2003; Liao et al., 2008) and that the list is not yet complete, it is plausible that FMRP also regulates a subset of mRNAs in astrocytes. In this scenario, the loss of FMRP seen in FXS would result in aberrant protein translation in astrocytes in addition to that already documented in neurons. In turn, this could lead to altered astrocyte–neuron signaling and interfere with astrocyte-mediated neuronal growth and synaptic development. Although the present study provides strong evidence for a role of astrocytes in the development of the neurobiological abnormalities seen in FXS, the experiments do not identify the molecular basis of the alterations and cannot confirm whether they are a direct or indirect result of a lack of FMRP.
Future studies designed to investigate these possibilities would provide valuable information on the neurobiological processes that are altered in FXS. For example, in vitro studies evaluating the effect of FMRP transfection in FMR1 KO astrocytes, and the short interfering RNA-mediated specific downregulation of FMR1 in WT astrocytes, on WT neurons could provide more direct evidence for a role of astrocyte FMRP in the neurobiology of FXS. Such studies could yield insight into whether there are any astrocyte-secreted factors that are altered consequent to a deficiency of FMRP and would therefore provide novel avenues for therapeutic intervention. Additionally, in vivo studies targeting FMRP expression in astrocytes using the FMR1 conditional knock-out and FMR1 conditional expression mice would provide an invaluable in vivo correlate and offer insight into the functional consequences of alterations in astrocyte FMRP.
Footnotes
This work was supported by the Natural Sciences and Engineering Research Council of Canada and the Fragile X Research Foundation of Canada. We thank Momal Mazhar for her help in the quantification of the neurons and astrocytes.
References
- Bakker C, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen A, Oostra B, Reyniers E, De Boulle K, D'Hooge R, Cras P, Van Velzen D, Nagels G, Martin J, De Deyn PP, Darby JK, Willems PJ. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker GA. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science. 1980;209:809–810. doi: 10.1126/science.7403847. [DOI] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Beckel-Mitchener A, Greenough WT. Correlates across the structural, functional, and molecular phenotypes of fragile X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10:53–59. doi: 10.1002/mrdd.20009. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Castrén M, Tervonen T, Kärkkäinen V, Heinonen S, Castrén E, Larsson K, Bakker CE, Oostra BA, Akerman K. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:17834–17839. doi: 10.1073/pnas.0508995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry. 2004;9:417–425. doi: 10.1038/sj.mp.4001432. [DOI] [PubMed] [Google Scholar]
- Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, Weiler IJ. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci U S A. 2001;98:7101–7106. doi: 10.1073/pnas.141145998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizzetti M, Moore NH, Giordano G, Costa LG. Modulation of neuritogenesis by astrocyte muscarinic receptors. J Biol Chem. 2008;283:31884–31897. doi: 10.1074/jbc.M801316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ. The fragile X prevalence paradox. J Med Genet. 2008;45:498–499. doi: 10.1136/jmg.2008.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob S, Doering LC. Primary dissociated astrocyte and neuronal co-culture. In: Doering LC, editor. Protocols for neural cell culture. Ed 4. New York: Humana; 2009. pp. 269–284. [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- Lee A, Li W, Xu K, Bogert BA, Su K, Gao FB. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development. 2003;130:5543–5552. doi: 10.1242/dev.00792. [DOI] [PubMed] [Google Scholar]
- Liao L, Park SK, Xu T, Vanderklish P, Yates JR., 3rd Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc Natl Acad Sci U S A. 2008;105:15281–15286. doi: 10.1073/pnas.0804678105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci. 2003;6:153–160. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- Narro ML, Yang F, Kraft R, Wenk C, Efrat A, Restifo LL. NeuronMetrics: software for semi-automated processing of cultured neuron images. Brain Res. 2007;1138:57–75. doi: 10.1016/j.brainres.2006.10.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DM, Derber WJ, McClellan DA, Crnic LS. Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 2002;927:8–17. doi: 10.1016/s0006-8993(01)03309-1. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida H, Okabe S. Direct astrocytic contacts regulate local maturation of dendritic spines. J Neurosci. 2007;27:331–340. doi: 10.1523/JNEUROSCI.4466-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia. 2007;55:1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Powell EM, Geller HM. Dissection of astrocyte-mediated cues in neuronal guidance and process extension. Glia. 1999;26:73–83. doi: 10.1002/(sici)1098-1136(199903)26:1<73::aid-glia8>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Smith CB. Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:15758–15763. doi: 10.1073/pnas.242377399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KS. Cytogenetic abnormalities and fragile-X syndrome in autism spectrum disorder. BMC Med Genet. 2005;6:3. doi: 10.1186/1471-2350-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Wang H, Ku L, Osterhout DJ, Li W, Ahmadian A, Liang Z, Feng Y. Developmentally-programmed FMRP expression in oligodendrocytes: a potential role of FMRP in regulating translation in oligodendroglia progenitors. Hum Mol Genet. 2004;13:79–89. doi: 10.1093/hmg/ddh009. [DOI] [PubMed] [Google Scholar]