

Abstract
A variety of chromatin remodeling complexes are thought to orchestrate transcriptional programs that lead neuronal precursors from earliest commitment to terminal differentiation. Here we show that mammalian neurons have a specialized chromatin remodeling enzyme arising from a neurospecific splice variant of LSD1/KDM1, histone lysine specific demethylase 1, whose demethylase activity on Lys4 of histone H3 has been related to gene repression. We found that alternative splicing of LSD1 transcript generates four full-length isoforms from combinatorial retention of two identified exons: the 4 aa exon E8a is internal to the amine oxidase domain, and its inclusion is restricted to the nervous system. Remarkably, the expression of LSD1 splice variants is dynamically regulated throughout cortical development, particularly during perinatal stages, with a progressive increase of LSD1 neurospecific isoforms over the ubiquitous ones. Notably, the same LSD1 splice dynamics can be fairly recapitulated in cultured cortical neurons. Functionally, LSD1 isoforms display in vitro a comparable demethylase activity, yet the inclusion of the sole exon E8a reduces LSD1 repressor activity on a reporter gene. Additional distinction among isoforms is supported by the knockdown of neurospecific variants in cortical neurons resulting in the inhibition of neurite maturation, whereas overexpression of the same variants enhances it. Instead, perturbation of LSD1 isoforms that are devoid of the neurospecific exon elicits no morphogenic effect. Collectively, results demonstrate that the arousal of neuronal LSD1 isoforms pacemakes early neurite morphogenesis, conferring a neurospecific function to LSD1 epigenetic activity.
Introduction
Chromatin represents the physiological substrate of epigenetic regulation, underlying several biological processes, including replication, transcriptional activity, and cell differentiation (Kouzarides, 2007; Wu et al., 2009). In the CNS, chromatin integrates a plethora of converging signaling pathways, leading to short- and long-term changes in gene expression that are crucial for neuronal commitment and terminal maturation, as well as for neuroplasticity throughout life (Walton et al., 1999; Tartaglia et al., 2001; Borrelli et al., 2008). Although a variety of ubiquitously expressed chromatin-remodeling complexes assist tissue-specific transcription factors, neurorestricted chromatin-remodeling factors have been identified only recently (Olave et al., 2002; Barak et al., 2004; Lessard et al., 2007).
Lysine specific demethylase 1 [LSD1 (now named KDM1)] (Allis et al., 2007) is a flavin-dependent histone demethylase that specifically removes methyl groups from Lys4 of histone H3 (Shi et al., 2004; Forneris et al., 2005a), relating this function to a wide range of biological processes, such as development, differentiation, cancer, and neurological disorders (Shi, 2007; Forneris et al., 2008; Nottke et al., 2009). Its structure and function is conserved from yeast to human (Dallman et al., 2004; Lakowski et al., 2006), and it is typically associated to CoREST, a corepressor protein, and histone deacetylases HDAC1 and HDAC2 (Ballas et al., 2001; Humphrey et al., 2001; You et al., 2001; Hakimi et al., 2002; Shi et al., 2003). Human LSD1 consists of 852 amino acids and comprises an N-terminal SWIRM domain, involved in protein interactions, and a C-terminal amine oxidase domain, which contains an insertion that forms the CoREST interacting site (the so-called “tower domain”) (Chen et al., 2006; Stavropoulos et al., 2006). The three-dimensional structure of LSD1 in a ternary complex with its histone peptide substrate and CoREST has been solved, providing an explanation for the biological relevance of CoREST in mediating substrate binding and recognition (Yang et al., 2006; Forneris et al., 2007). Mechanisms that tune LSD1 activity have been extensively investigated, including interactions with protein partners (Lee et al., 2005; Shi et al., 2005) and concomitant modifications on histone tail residues surrounding the demethylation site (Forneris et al., 2005b; Forneris et al., 2006).
Given the central role of LSD1 in chromatin remodeling, we investigated the process of alternative splicing on LSD1 as additional regulatory mechanism. Alternative splicing occurs at higher frequency in singleton genes than multigene families (Kopelman et al., 2005; Xing et al., 2006), and LSD1 displays a unique paralog in the human genome AOF1 (now named KDM1B) (Shi et al., 2004; Karytinos et al., 2009). Here, we report the identification and functional characterization of four mammal-specific LSD1 isoforms generated by either single or double inclusion of two alternatively spliced exons, E2a and E8a. Interestingly, although the inclusion of E2a is an event occurring in all tissues, the isoforms retaining E8a display a neurospecific pattern of expression, dynamically regulated during mammalian brain development and synaptic maturation: in such critical phases, a fine balance of LSD1 isoforms allows differentiating neurons to acquire a proper morphology.
Materials and Methods
Total RNA extraction, reverse transcription-PCR, and quantitative reverse transcription-PCR analysis.
Total RNA was isolated using the RNeasy Mini Kit (Qiagen), and the purified RNA was treated with RNase-free DNase set (Qiagen) to remove any residual DNA. Quantitative reverse transcription (qRT)-PCR analysis was performed on an iQ5 Real-Time PCR Detection System (Bio-Rad) using the iScriptTM two-Step RT-PCR Kit with SYBR Green (Bio-Rad). The relative expression of the investigated genes was quantified after normalization against β-actin. RT-PCR was performed with BioTaq DNA Polymerase (Boline) with primer pairs reported in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Evaluation of splicing inclusion events by relative quantity fluorescent-PCR.
To quantitatively measure the relative amount of each isoform, we used a PCR-based method in which a fluorochrome-conjugated (FAM) forward primer (Sigma-Aldrich) and a reverse unmodified one were used to amplify all the expected isoforms in a single reaction. PCR products, mixed together with a suitable internal lane ROX-conjugated size standard (500ROX Standard; Applied Biosystems), were separated by capillary electrophoresis under denaturing conditions, and the amount of each amplified product was measured as microsatellite sample, based on related fluorescence unit levels using the software GeneMapper. For a more detailed protocol, see supplemental Material and Methods (available at www.jneurosci.org as supplemental material).
Cell culture, transient transfections, and reporter gene assays.
HeLa cells were cultured and transfected as described previously (Andres et al., 1999); the experiments were performed 48 h after transfection. 5xUAS–TK–LUC reporter plasmid (Chen et al., 1998) was used at 1:1, 1:0.125, 1:0.06, and 1:0.03 molar ratio relative to the expression plasmids pGal4–LSD1 and pGal4–LSD1-8a. Control experiments were performed by using equivalent molar amounts of empty vectors. DNA was kept constant by adding pBSIIKS (Stratagene) in every experiment; 0.05 μg of pRL–TK–vector (Promega) reporter vector was cotransfected to normalize for transfection efficiency. The luciferase reporter activity was determined with the Dual-Luciferase reporter assay system (Promega) according to the instructions of the manufacturer. For each construct, the values of Firefly luciferase were normalized over Renilla luciferase (both expressed as relative luminescent units). The activity of each construct was expressed as a percentage of the promoter-less plasmid pGal4–vector.
Coimmunoprecipitation assay.
Coimmunoprecipitation (IP) experiments were performed as reported previously (Battaglioli et al., 2002). HeLa cells were harvested 48 h after transfection in IP buffer (10% glycerol, 0.5 mm EDTA 0.5% Triton X-100, 1 mm PMSF, and 1× protease inhibitor cocktail (Sigma-Aldrich). Cellular extract (0.5 mg was reacted with 1.25 μg of rabbit polyclonal anti-CoREST antibody (Millipore), mouse monoclonal anti-hemagglutinin (HA) (Santa Cruz Biotechnology), or 1.25 μg of mouse monoclonal anti-green fluorescent protein (GFP) (Roche Molecular Biochemicals) with overnight rocking at 4°C. The immunoprecipitates were collected with rProtein G Agarose (Invitrogen). After incubation, the beads were washed four times with the IP buffer containing 5% glycerol and 0.1% Triton X-100. The immunoprecipitates were then eluted with 2× SDS sample buffer and analyzed by Western blot.
Immunoblot assay and antibodies.
Electrophoretically separated proteins from lysates (input) or immunoprecipitates were transferred from denaturing polyacrylamide gels to pre-equilibrated nitrocellulose membrane (Schleicher & Schuell). SigmaMarker High range (Sigma-Aldrich), whose molecular weight ranged from 36,000 to 200,000 kDa, was used to characterize the migration of LSD1 doublet.
The membranes were blocked in Tris-buffered saline and 0.1% Tween 20 (TBST) containing 5% nonfat dry milk and reacted at 4°C overnight with appropriate primary antibody in TBST with 5% dry milk, rinsed, and incubated with appropriate secondary antibody horseradish peroxidase conjugate, developed with ECL Western Blotting detection reagent (Pierce Thermo Fisher Scientific), and revealed with x-ray films (Eastman Kodak).
Antibodies used included the following: CoREST (Millipore), HA (Santa Cruz Biotechnology), HDAC2 (Millipore), panLSD1 antibody (Diagenode), Shank (NeuroMab), and C-terminus-binding protein (Cell Signaling Technology). Guanylate kinase domain-associated protein (GKAP), glutamate receptor-interacting protein (GRIP), and calcium/calmodulin-dependent serine kinase (CASK) (rabbit; gifts from M. Sheng), neuronal nitric oxide synthase (nNOS) (Santa Cruz Biotechnology), GluR1 (Oncogene), and GluR2/3 (Millipore Bioscience Research Reagents). Antibody against E8a-containing isoforms was generated as follows: a peptide containing exon E8a GQADTVKVPKE was synthesized, injected into rabbits, and used to prepare peptide affinity columns for antibody purification from antisera by Primm.
Expression and purification of recombinant LSD1 splice variants.
Recombinant human LSD1 splice variants were expressed as truncated proteins lacking the N-terminal 122 residues, following the same protocols used for the conventional isoform (Forneris et al., 2005a). Proteins were purified by affinity chromatography, taking advantage of the N-terminal His6 purification tag. The tag was then removed by cleavage with tobacco etch virus protease. During protein purification, SDS-PAGE analysis showed that the LSD1 proteins were subject to rapid precipitation and unrecoverable degradation. The problem was solved by copurification with a C-terminal fragment (residues 305-482) of recombinant CoREST by tandem-affinity chromatography as described previously (Forneris et al., 2007). The resulting LSD1–CoREST complex is stable at 4°C for several weeks as judged by SDS-PAGE and activity assays (see next section).
Biochemical assays.
The enzymatic activities of LSD1 splice variants were measured at 25°C using a Cary 100 UV/Vis spectrophotometer (Varian) following published protocols (Forneris et al., 2005a) (Table 1). Methylated synthetic peptides corresponding to the 21 aa N-terminal tail of histone H3 were used as substrates for biochemical analysis (purchased from Thermo Fisher Scientific).
Table 1.
Kinetic parameters for LSD1 and the splicing variants
| kcat (min−1)a | Km (μm)a | |
|---|---|---|
| LSD1 + CoREST | 7.35 ± 0.28b | 5.12 ± 1.04b |
| LSD1-2a + CoREST | 9.38 ± 0.65 | 6.31 ± 1.44 |
| LSD1-8a + CoREST | 5.19 ± 0.48 | 4.55 ± 1.65 |
| LSD1-2a/8a + CoREST | 5.47 ± 0.20 | 7.52 ± 1.00 |
aApparent steady-state kinetic parameters were determined as described by using a 21 aa H3 peptide monomethylated at Lys4 (Forneris et al., 2005).
bData were taken from Forneris et al. (2007).
Crystallization and structure determination.
Samples of purified human recombinant LSD1 splice variants in complex with CoREST in 25 mm potassium phosphate, pH 7.2, and 5% (w/v) glycerol were concentrated using an Amicon Ultra concentration device (Millipore) to a final concentration of 8 mg/ml. Crystals were grown at 20°C by hanging-drop vapor diffusion method by mixing equal volumes of protein samples with reservoir solutions containing 1.2 m sodium/potassium tartrate and 100 mm N-(2-acetamido)iminodiacetic acid, pH 6.5 (Forneris et al., 2007). Crystals were transferred in a solution containing 1.6 m sodium/potassium tartrate, 100 mm N-(2-acetamido)-2-iminodiacetic acid, pH 6.5, 10% glycerol, and 2 mm Lys4Met H3 peptide (i.e., the 21 N-terminal residues of H3 with Lys4 mutated to Met) and flash cooled in liquid nitrogen. Data collections were performed at the beam-lines ID14-EH1 and ID23-EH2 of the European Synchrotron Radiation Facility. Data processing was performed using the program MOSFLM (Leslie, 1999) and CCP4 (Collaborative Computational Project, Number 4, 1994). The structures of the LSD1–CoREST complexes were solved by molecular replacement using the program AMORE of the CCP4 suite. The initial search model was generated from the Protein Data Bank entry 2IW5, deprived of cofactors, substrates, and ligands. Refinement was performed out using Refmac5 (Murshudov et al., 1997). Data collection and refinement statistics are reported in Table 2. Structure analysis, validation, and modeling were performed using the program COOT (Emsley and Cowtan, 2004). Figures were generated with PyMOL (www.pymol.org).
Table 2.
Data collection and refinement statistics
| LSD1-8a + CoREST complex with H3 inhibitor peptidea | |
|---|---|
| Space group | I222 |
| Unit cell (Å) | a = 119.7 |
| b = 181.2 | |
| c = 233.4 | |
| Resolution (Å) | 3.0 |
| Rsymbc (%) | 11.3 (49.7) |
| Completenessc (%) | 98.9 (99.8) |
| Unique reflections | 50,357 |
| Redundancy | 4.1 (4.1) |
| I/σc | 10.2 (2.1) |
| Rcrystd (%) | 20.7 |
| Rfreed (%) | 24.9 |
| RMS bond length (Å) | 0.017 |
| RMS bond angles (°) | 1.81 |
aThe final model consists of residues 171-836 of LSD1-8a (including residues Asp-Thr-Val-Lys corresponding to exon 8a, which are inserted after Ala369 and are named 369A-369B-369C-369D), a FAD molecule, residues 308-440 of CoREST, and residues 1-16 of the Lys4Met peptide.
bRsym = Σ|Ii − <I>|/ΣIi, where Ii is the intensity of ith observation and <I> is the mean intensity of the reflection.
cValues in parentheses are for reflections in the highest resolution shell.
dRcryst = Σ| Fobs − Fcalc|/Σ|Fobs|, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. The set of reflections used for Rfree calculations and excluded from refinement was extracted from the structure factor file relative to the Protein Data Bank entry 2IW5.
Cortical neuron cultures and immunostaining.
The cortical neuron cultures were prepared from embryonic day 18 (E18) to E19 rat brain (Charles River) as described previously (Romorini et al., 2004), plated on 18-mm-diameter coverslips, and grown on 12-well plastic tissue culture plates (Iwaki; Bibby Sterilin). The neurons were transfected using calcium phosphate precipitation (transfection efficiency of 1%). Cells were fixed with a PBS solution containing 4% paraformaldehyde for 10 min. Cells were incubated with anti-HA (1:100; sc80; Santa Cruz Biotechnology) for 3 h in GDB buffer (30 mm phosphate buffer, pH 7.4, containing 0.2% gelatin, 0.5% Triton X-100, and 0.8 m NaCl), followed by 1 h incubation with cyanine 3-conjugated secondary antibody (The Jackson Laboratory) and mounted in VectaDAPI medium (Vector Laboratories). Images were acquired on a Carl Zeiss LSM5 510 laser-scanning confocal microscope (generously donated by Fondazione Monzino) at 63× magnification. Morphometric analysis was performed using NIH ImageJ software.
Results
Identification of novel LSD1 isoforms evolutionarily conserved in mammals
The annotated human LSD1 gene (also known as AOF2) sequence (GenBank accession number NM_015013) encodes for an mRNA that results from the fusion of 19 exons and gives rise to a protein of 852 aa (Fig. 1A). To further investigate the structure of the LSD1 gene, we performed an analysis of its genomic region using GenomeVISTA tools, comparing human and mouse LSD1 genes (Fig. 1B). We found a very high conservation degree (>95%) in correspondence of the known 19 exons and a lower conservation in all the flanking intronic sequences. However, two regions located inside introns 2–3 and 8–9 emerged for the highest homology (Fig. 1B,C), indicating the possible existence of two additional exons subject to alternative splicing within these conserved intronic sequences. On this basis, we performed a search in the University of California, Santa Cruz database (version March 2006) and found an additional known gene (GenBank accession number BC040194; IMAGE identification number 4815528) related to AOF2 gene transcript. Such sequence, derived from an mRNA expressed in human hippocampus, contained two yet undescribed exons that we named exons 2a and 8a (E2a and E8a). E2a is 60 bp long and encodes for 20 aa, whereas the E8a is 12 bp long and is translated into 4 aa with sequence Asp-Thr-Val-Lys. The inclusion of the two exons does not alter the reading frame and results in a protein of 876 aa that we named LSD1-2a/8a. In this LSD1 isoform, the amino acids coded by E2a localize between the N-terminal disordered region and the SWIRM domain, whereas the four residues of E8a immediately precede the CoREST-binding tower domain, which is inserted within the amine oxidase domain (Fig. 1A). The alternatively spliced introns present canonical donor/acceptor splice sites (supplemental Table 1, available at www.jneurosci.org as supplemental material) and display a very high conservation degree between human and mouse (Fig. 1C), a typical feature of alternatively spliced exons (Sorek and Ast, 2003).
Figure 1.

Genomic organization of human LSD1 gene. A, Schematic representation of the human LSD1 protein domains together with its exons ranging from 1 to 19; asterisks indicate the location of annotated alternative exons (E2a and E8a). Different colors in grayscale indicate functional domains. N-terminal unstructured region (white) coded by exons 1–2, SWIRM domain (black) coded by exons 2–4, the SWIRM-oxidase connector (black striped) coded by exon 5, the amine oxidase domain (gray), coded by exons 6–9 and exons 13–19, and the tower domain (gray striped) coded by exons 10–12. Residue Met1 of this sequence corresponds to the first amino acid of the protein characterized (Shi et al., 2004). B, Human AOF2 alignment across vertebrates by GenomeVista browser (http://pipeline.lbl.gov/cgi-bin/GenomeVista). The “peaks and valleys” graphs represent percentage conservation at a given genomic coordinate between aligned sequences and the human sequence. Human exons are numbered. The top and bottom percentage bounds are shown to the right of every row. Regions of high conservation are colored as exons (dark gray) or noncoding (light gray). Conserved regions are defined as regions with identity of 70% or higher that are wider than or equal to “minimal conservation width” (100 bp). C, Enlarged view of 650 bp of the alignment between human and mouse intronic regions containing the two alternatively spliced exons (E2a and E8a) and one constitutively included exon (E13). Highlighted bars above the conservation area correspond to annotated E2a, E8a, and E13 of human AOF2. Dark gray areas within the conservation graph mark exons; light gray areas mark conserved (above 70%) nonexonic sequences.
An extended search for partial and complete transcripts containing one or both identified LSD1 exons was performed in the genomic sequences of different mammalian species, based on human LSD1 mRNA (GenBank accession number BC040194) (http://www.ncbi.nlm.nih.gov/BLAST/): all investigated species displayed at least one alternative exon, and, most importantly, all E8a-containing expressed sequence tags (ESTs) turned out to derive from the nervous system, as shown in supplemental Table 2 (available at www.jneurosci.org as supplemental material). We extended our analysis to other vertebrates to compare AOF2 gene structure throughout evolution. Whereas E2a was present and highly conserved in lizard (data not shown), chicken, and mammals, E8a is fully preserved in mammals only.
Exon E8a-retaining LSD1 splice variant is restricted to neuronal lineage
The existence of an annotated human mRNA containing both E2a and E8a in an open reading frame prompted us to further analyze alternatively spliced isoforms containing the identified exons and to characterize their transcriptional profile on several samples derived from human, mouse, and rat tissues. The presence of LSD1 splicing isoforms was first investigated in a panel of total RNA samples from adult human tissues and cell lines (Fig. 2A,B) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Through selective amplification of isoforms containing E8a (hPCR1), we found that LSD1-2a/8a was present in brain and testis, whereas the LSD1 with the sole E8a (LSD1-8a) was detected exclusively in brain tissues. Conversely, the amplification of isoforms excluding E8a (Fig. 2B, hPCR2) gave two products in all analyzed tissues, indicating that native LSD1 and LSD1-2a isoforms are ubiquitously expressed (see scheme in Fig. 2C). The identification of four mRNA species transcribed from the human AOF2 gene indicates that (1) retention of E2a and E8a are two independent events, and (2) the inclusion of E8a is a tissue-specific event that occurs only in neuronal tissues and testis.
Figure 2.

Mammalian LSD1 transcript undergoes alternative splicing and produces four isoforms with different tissue distribution. A, Exon structure of the mammalian LSD1 gene and position of the primers used to identify full-length, polyadenylated transcripts characterized by the presence or absence of E2a and E8a. Primers are indicated as F for forward or R for reverse, and the numbers indicate the exons where each primer anneals. Total RNA from human adult tissues (B) and total RNA from mouse adult brain tissues (D) were tested for LSD1 splicing variant expression by RT-PCR. The cDNA were amplified with primers covering the entire coding sequence (primers 1F and 19R) and reamplified with two different nested PCR, one including E8a (PCR1) and one excluding E8a (PCR2). β-Actin was used as control. C, Structure of the four LSD1 variants. E, F, Isoform-relative quantification of LSD1 splicing variants in mouse brain areas, primary neuronal cultures, and astroglia. rqf-PCR on cDNA obtained from total RNA of the indicated samples. Amplicons were quantified by related fluorescence units (RFU) by GeneMapper software and graphed as percentage relative to the sum of all the isoforms. Neurospecific LSD1 isoforms are shown in black and dark gray, whereas ubiquitous ones are shown in light gray and white. G, Splicing generates four different LSD1 proteins. Western blots of total protein extracts from mouse brain and heart probed with a panLSD1 antibody and migration of recombinant LSD1 isoforms transfected in HeLa cells probed with anti-HA antibody. All the indicated samples were run on the same polyacrylamide gel. H, Anti-LSD1-8a antibody specificity was assessed by Western blot on total protein extracts from heart and brain rat tissues and HeLa cells transfected with pCGN vector (mock), HA-tagged LSD1, or HA-tagged LSD1-8a cDNAs.
We extended the characterization of the tissue distribution of the four LSD1 splicing isoforms in adult mouse brain using the same approach performed on human tissues. As shown in Figure 2D, all the analyzed areas coexpress the four LSD1 isoforms. The overall amount of LSD1 measured by real-time qPCR showed comparable amounts of LSD1 transcripts in the different areas (data not shown). To further refine our analysis, we set up a method, namely rqf-PCR (for detailed protocol, see supplemental data, available at www.jneurosci.org as supplemental material) in which coamplified LSD1 isoforms are analyzed at high resolution by capillary electrophoresis (for an example of two typical electropherograms, see supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This method allowed us to finely monitor differences in the expression levels of LSD1 isoforms among brain areas. This analysis (Fig. 2D,E) revealed that the four isoforms are similarly expressed in all investigated brain regions, suggesting a controlled balance between the inclusion and exclusion events within adult murine CNS. Furthermore, because neuronal tissues contain variable percentage of glial components, we asked whether the presence of the neurospecific LSD1 (nLSD1) doublet detected in all evaluated nervous tissues might be attributable to neuronal rather than glial histotype or both. Therefore, we performed a fluorescent rqf-PCR on selected rat primary cultures, that is hippocampal neurons, cortical neurons, and cortical astroglia. The analysis revealed that glia is completely devoid of nLSD1 isoforms but retained the ubiquitous LSD1 (uLSD1) ones (Fig. 2F), whereas both hippocampal and cortical neurons retain the full pattern of the four identified isoforms, confirming that E8a inclusion within nLSD1 variants strictly relates to the neuronal histotype. Because glial components (identified as GFAP-positive cells) do not exceed 2–3% in the evaluated neuronal cultures and the overall LSD1 protein levels did not appear to differ between neurons and glia (data not shown), we conclude that rqf-PCR quantification of coexisting LSD1 variants is reliably attributable to neurons.
Alternative splicing generates four functional proteins
Rat brain and heart tissues were assayed for the expression of LSD1 by SDS-PAGE. Immunoblotting by a panLSD1 antibody revealed a discrete doublet of ∼110 kDa in both tissues (Diagenode). The doublet was further characterized by comparing the migration of endogenous LSD1 with tagged isoforms that were individually transfected in HeLa cells. As shown in Figure 2G, the isoforms that contain exon E2a (LSD1-2a and LSD1-2a/8a) are upper shifted from the isoforms without it (LSD1 and LSD1-8a). The electrophoresis mobility displayed by the tagged LSD1 isoforms resumes the migration pattern of endogenous LSD1 proteins, hence suggesting that the doublet shared by and observed in neuronal and non-neuronal tissues is most probably attributable to the presence and absence of exon E2a (20 aa long) within LSD1. Nonetheless, we do not exclude that posttranslationally modified LSD1 isoforms may reside underneath the bands of the identified doublet.
Furthermore, because inclusion of the sole exon E8a (4 aa only) produces no detectable variation in electrophoretic mobility of the 110 kDa protein, the LSD1 and LSD1-2a cannot be distinguished from LSD1-8a and LSD1-2a/8a, respectively. To demonstrate the existence of endogenous proteins containing the exon E8a, we produced an antibody specifically raised against a short peptide containing the amino acids Asp-Thr-Val-Lys (exon E8a sequence). The anti-E8a antibody specificity was tested by ELISA against the recombinant proteins (data not shown) as well as against the transfected cDNAs in HeLa cells (Fig. 2H). This antibody detected the presence of E8a-containing isoforms in rat brain tissues but not in heart, confirming that the four mRNA generated by alternative splicing are indeed translated in four proteins.
Effect of E2a and E8a inclusion on LSD1 enzymatic activity in vitro and three-dimensional structure
To analyze the enzymatic activity of the four different LSD1 splice variants, we expressed three human recombinant LSD1 isoforms (LSD1-2a/8a, LSD1-2a, LSD1-8a) and we performed comparative biochemical assays using histone H3 peptides. Both E2a and E8a had a strong destabilizing effect on the purified recombinant LSD1, which prevented their further biochemical investigation. Fortunately, in vitro reconstitution of the complex formed by LSD1 with the corepressor protein CoREST by tandem-affinity purification strategies (Forneris et al., 2007) provided far more stable protein samples, enabling their structural and biochemical characterization. This finding indicated that the splicing variants retain the ability to form a stable complex with CoREST. Biochemical assays using histone H3 peptides revealed that all three LSD1 isoforms bound to CoREST can demethylate Lys4 of histone H3 with a catalytic efficiency virtually identical to that displayed by conventional LSD1 (Table 1). Moreover, they are totally inactive on peptides monomethylated on Lys9, demonstrating that alternative splicing does not affect substrate specificity. We further investigated the recombinant LSD1-2a/8a, LSD1-2a, and LSD1-8a proteins by x-ray crystallography. Crystals of their complex with the C-terminal region of CoREST (residues 305-482) and a 21 aa H3 peptide were obtained under identical conditions to those used for LSD1 protein (Forneris et al., 2007) (Fig. 3A). We found that, as for the native enzyme, also in the splicing variants, the N-terminal residues preceding Pro171 are disordered and not visible in the electron density map. This finding implies that, even in the presence of exon E2a (inserted between residues 170 and 171), the N-terminal region of LSD1 remains unstructured, at least in the crystalline state. Therefore, the structures containing E2a were indistinguishable from those lacking this insertion, and we shall focus our structural analysis to LSD1-8a in complex with CoREST and the histone peptide.
Figure 3.
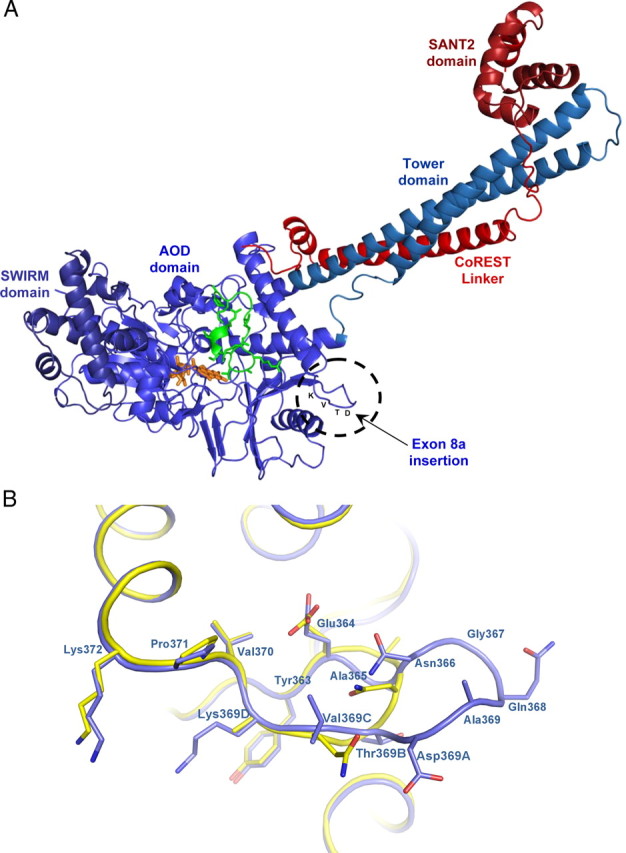
Comparative structural analysis of LSD1 and the LSD1-8a splice variant. A, Overall crystal structure of LSD1-8a–CoREST in complex with a histone peptide. LSD1-8a (residues 171-840) is in light blue, CoREST (residues 308-440) in red, and the histone H3 peptide (residues 1-16) in green. The FAD cofactor is in the orange ball-and-stick representation. The insertion site of E8a (residues Asp369A-Thr369B-Val369C-Lys369D) is highlighted. B, Close-up view of LSD1-8a structure at the site of exon E8a insertion. LSD1-8a structure is in blue, and it is superimposed onto native LSD1 (yellow; Protein Data Bank entry 2v1d) (Forneris et al., 2007). The orientation of the proteins is the same as in Figure 3A. The side chains of exon E8a residues are labeled in bold. Exon E8a insertion protrudes from the main body of the protein.
The overall conformation of LSD1-8a is very similar to that of the native protein with a root-mean-square deviation of 0.30 Å for 666 Cα atoms. The E8a residues Asp-Thr-Val-Lys inserted between Ala369 and Asp370 of the conventional isoform generate an antiparallel β-turn located in proximity of one of the two helices that define the tower domain (Fig. 3A). Structural superpositions show that the presence of E8a does not cause any local conformational change (Fig. 3B). Likewise, the conformations of CoREST and of the bound histone peptide are identical to those observed in the structure of LSD1–CoREST–peptide complex. The exon residues are located on the rim of the open cleft that forms the substrate-binding site, but they are not in direct contact with either the histone peptide or CoREST (Fig. 3A). This observation is in agreement with the biochemical evidence that the enzymatic activity and substrate specificity of LSD1 splice variants are very similar to those of conventional LSD1 (Table 1). It is noteworthy that the E8a residues form a sort of protrusion that emerges from the main body of the protein. Such a feature indicates that these residues could easily form a docking site for other protein partners, which remain to be identified.
Inclusion of the neurospecific E8a exon modulates LSD1 repressive activity on a reporter gene
Because LSD1 repressive function on target genes has been assessed previously (Shi et al., 2004), we wanted to determine whether the inclusion of either exon E2a or E8a in LSD1 isoforms might alter its activity. Therefore, we transfected LSD1 splice isoforms in different cellular systems and compared their effect on luciferase reporter gene. As shown in Figure 4A, in HeLa cells, the inclusion of exon E2a does not cause any change in luciferase expression (LSD1-2a vs LSD1, 1.31 ± 0.48 vs 1.12 ± 0.24; p = 0.34, t test), whereas the presence of exon E8a results in a significantly reduced repression of luciferase reporter, as evaluated in the HeLa cell line (LSD1-8a vs LSD1, 3.11 ± 1.16 vs 1.12 ± 0.24, p = 0.008; LSD1-2a/8a vs LSD1, 4.17 ± 0.78 vs 1.12 ± 0.24, p = 5.3e−16, t test). Differences in repressive activity were further evaluated on a scale of reporter: repressor molar ratios in HeLa and SH-SY5Y cell lines (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Notably, also in rat cortical neurons, in which the neuronal LSD1 isoforms are physiologically expressed, LSD1-8a and LSD1 exhibit different repressor properties (LSD1-8a vs LSD1 in Fig. 4B at a molar ratio 1:0.5, 89.41 ± 2.82 vs 69.99 ± 5.68, p = 0.018; molar ratio 1:1, 64.16 ± 4.01 vs 43.85 ± 3.22, p = 0.029, t test).
Figure 4.
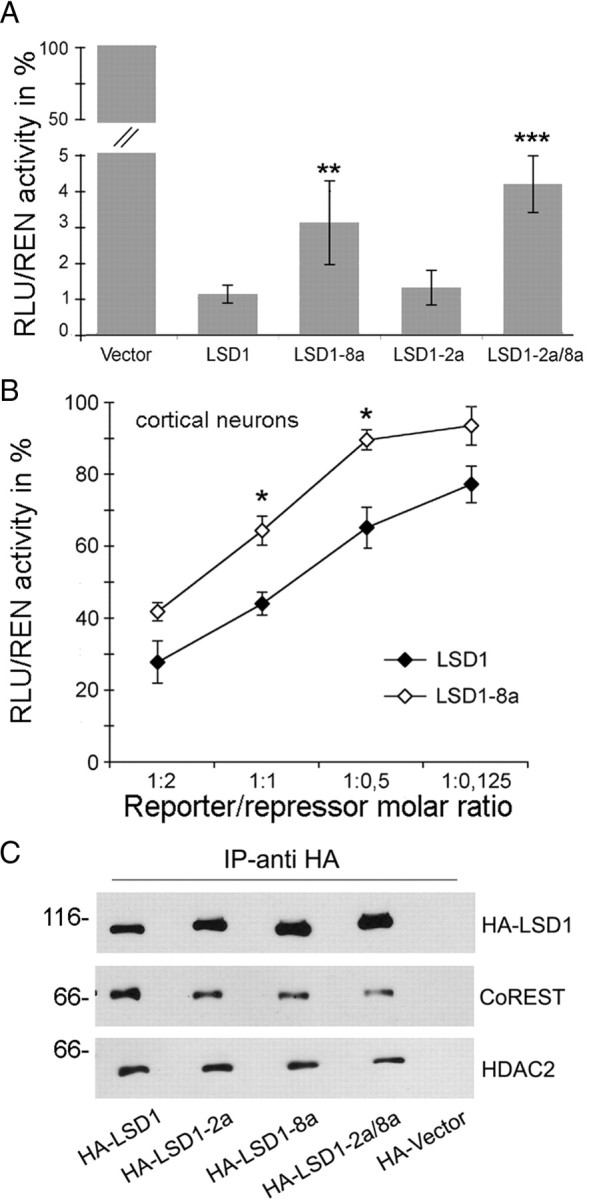
Effect of the included exon E2a and/or E8a on LSD1 repressor activity and affinity for corepressor partners. A, The four indicated LSD1 splice variants fused to Gal4–DBD were assayed for their ability to repress a reporter gene on HeLa cells at a constant reporter/repressor molar ratio. B, In rat cortical neurons, LSD1 and LSD1-8a were compared at different reporter/repressor molar ratios. The luciferase activity normalized over the activity of a cotransfected renilla reporter is expressed as percentage of the activity of the Gal4–DBD vector at each molar ratio. Values are derived from at least three independent experiments. In A and B, a Student's t test (l Stat t I ≥ T α/2) was applied to percentage values by comparing splicing isoforms with LSD1. *p < 0.05; **p < 0.01; ***p < 0.001. C, Whole-cell immunocomplexes from HeLa cells overexpressing the indicated HA–LSD1 isoforms were obtained by HA antibodies and separated by SDS-PAGE. The Western blots were probed with antibodies to HA, CoREST, and HDAC2.
All recombinant LSD1 splice isoforms can be assembled into a CoREST complex
The corepressor activity of LSD1 depends in part on the multiprotein complex to which it belongs. Therefore, we tested whether the presence of E2a, E8a, or both might interfere with the formation of the complex with CoREST and associated proteins. Using anti-HA-antibody, we isolated LSD1 immunocomplexes from HeLa cells transfected with the four HA-tagged LSD1 isoforms. In all isolated immunocomplexes, we were able to detect the presence of CoREST and HDAC2 (Fig. 4C). Furthermore, in brain extracts, LSD1 and CoREST take part to the same corepressor complex (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). This finding, together with the biochemical and structural data (Fig. 3), demonstrate that inclusion of either exon does not preclude LSD1 interaction with known molecular partners and that LSD1 splice variants can function as corepressor factors similarly to the conventional isoform.
A mechanism to explain the functional implication of the LSD1 isoforms diversity might arise from the combinatorial incorporation of different isoforms. Indeed, assembly of higher-order HDAC1/2 complexes requires two heterotrimers each formed by CoREST, LSD1, and either HDAC1 or HDAC2 (Humphrey et al., 2001). Therefore, we tested whether different isoforms might be incorporated into the same higher-order HDAC1/2 complex. HeLa cells were cotransfected with GFP-tagged LSD1 along with different HA-tagged LSD1 isoforms. Supplemental Figure 4B (available at www.jneurosci.org as supplemental material) shows that it is possible to form hetero-oligomeric complexes in which different LSD1 isoforms are incorporated.
Expression of neurospecific LSD1 isoforms is regulated throughout brain development
Because inclusion of E8a is a neurorestricted event, we inferred its function within CNS by relating its expression profile to peculiar stages of neuronal differentiation. Time course analysis of the relative amount of each LSD1 splice isoform was performed by rqf-PCR. Rat cortical tissues were collected at several developmental stages and analyzed (Fig. 5A,B). At early embryonic stages, all of the four LSD1 isoforms are detectable, with preponderant expression of the LSD1 and LSD1-2a (42 and 38%, respectively; data not shown) over neurospecific isoforms (LSD1-8a and LSD1-2a/8a, 12 and 8%, respectively; data not shown). Later on, within the perinatal window between E18.5 and postnatal day 1 (P1), a rapid inversion of the proportions occurs: E8a-containing isoforms undergo a threefold increase shifting from 12 to 50% and from 8 to 25%, respectively, with a concomitant threefold decrease of LSD1 and LSD1-2a isoforms (Fig. 5A,B), and preponderance of E8a-containing isoforms is maintained until P7. Thereafter, LSD1 isoforms reach comparable levels and stabilize to the values measured in adult cortical cortex (compare with Fig. 2E). Time course analysis performed on cerebellar tissues revealed a similar expression pattern of LSD1 splice isoforms (data not shown).
Figure 5.

Inclusion of neurospecific E8a in LSD1 transcripts is dynamically modulated during neuronal development, whereas inclusion of E2a is a steady event. The relative amount of each LSD1 isoform was measured by rqf-PCR; cDNA were obtained from total RNA from rat embryonic cortex (E18.5), postnatal rat cortex (PN), and adult rat cortex. Graphs represent the relative percentage of each isoform with respect to the sum of the four. Only two isoforms are shown per graph; A compares LSD1 isoform with LSD1-8a, whereas B compares LSD1-2a isoform with LSD1-2a/8a. Values shown are mean ± SD. C, Exon inclusion frequency of exons E2a and E8a. Each represented series relates to the overall inclusion of either E8a (black squares) calculated as the sum of LSD1-2a/8a and LSD1-8a relative percentage and E2a (white squares), calculated as sum of LSD1-2a and LSD1-2a/8a relative percentage, at each indicated developmental stage. D, Total LSD1 transcript quantification by qRT-PCR on total RNA extract from the indicated rat cortex samples normalized on β-actin. Samples are expressed as fold increase relative to the LSD1 value at E18.5. Western blot on total protein samples from the indicated development cortical stages with a panLSD1 antibody or neurospecific LSD1 antibody (E) and with the indicated synaptic markers (F).
From these data, we calculated the inclusion frequency of either exon during development. Inclusion frequency of E2a, derived from the sum of LSD1-2a and LSD1-2a/8a (Fig. 5C, white squares), is rather constant, whereas the inclusion of E8a, calculated as the sum of LSD1-8a and LSD1-2a/8a (Fig. 5C, black squares), appears to be developmentally regulated. Overall LSD1 transcription and protein levels decrease between E18.5 and P1 and remain stable along subsequent stages of development (Fig. 5D,E). Because astroglia express only the ubiquitous isoforms (LSD1 and LSD1-2a) (Fig. 2F), a decrease in tissue glial composition may misleadingly indicate an increase in neurospecific isoforms (LSD1-8a and LSD1-2a/8a). Therefore, we also analyzed GFAP expression inferring glial relative contribution to isoform quantification. GFAP real-time qPCR analysis was performed from E18.5 to postnatal stages (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Consistent with previous reports (Qian et al., 2000; Fox et al., 2004), GFAP is expressed at low levels in the perinatal window and constantly increases during postnatal development. This demonstrates that the detected increase of E8a is indeed caused by a neurospecific splice event, with no regard to glial composition. A parallel analysis showed that several synaptic markers arise during the perinatal window (E18.5 to P1) with a progressive increase over developmental stages (Fig. 5F), indicating that the inclusion frequency of E8a increases concomitantly with early stages of synaptogenesis.
Neurospecific LSD1 contributes to neurite morphogenesis during early phases of cortical development
LSD1 expression profile suggests a possible implication of neurospecific E8a-containing isoforms in neuronal development. To test this hypothesis, we perturbed the expression of LSD1 isoforms within rat cortical neurons, which represent a suitable model of neuronal maturation, as assessed through the expression of synaptic markers that mirror postnatal development (Fig. 6A) (Lee and Sheng, 2000; Sala et al., 2000). This model also recapitulates the physiological expression pattern of LSD1 isoforms that we initially observed in vivo: the four LSD1 isoforms are all detectable at day in vitro 0 (DIV0) (corresponding to rat E18.5), with LSD1-2a and LSD1 being most abundant; as neuronal maturation proceeds, neurospecific E8a-containing LSD1 isoforms progressively increase (DIV2) and become preponderant (DIV6) (Fig. 6B,C). The overall inclusion frequency of either exon confirmed that E8a inclusion is developmentally regulated, whereas E2a inclusion does not change (Fig. 6D). Furthermore, LSD1 protein level analyzed by a panLSD1 antibody showed the same decrease during neuronal maturation that was observed in vivo (Fig. 6E). Also in this case, we verified whether any detectable variation of LSD1 isoforms might be influenced by a change in the proportion between neurons and glia during maturation in vitro. As indicator of astroglial contribution, we measured GFAP transcript, confirming (Fox et al., 2004) a persistently low expression from embryonal stage DIV0 (E18.5) to DIV4 and a robust increase detectable not sooner than DIV8 (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material) that is after the inversion of LSD1 isoforms proportion has occurred. Again, we can reliably assume that the increase of E8a inclusion in our cellular model is attributable to the neuronal differentiation, with no regard to glial composition.
Figure 6.

LSD1 splicing analysis in a rat cortical neuron maturation system. A, In vitro maturation of cortical neurons prepared from E18.5 embryos (DIV0) was assessed by Western blot analysis of the indicated synaptic markers on total protein samples from the indicated DIV. Graphs represent the relative percentage of each isoform with respect to the sum of the four. Only two isoforms are shown per graph; B compares LSD1 isoform with LSD1-8a, whereas C compares LSD1-2a isoform with LSD1-2a/8a. Values shown are mean ± SD. D, Exon inclusion frequency of exons E2a and E8a. Each represented series relates to the overall inclusion of either E8a (black squares), calculated as the sum of LSD1-2a/8a and LSD1-8a relative percentage, and E2a (white squares), calculated as the sum of LSD1-2a and LSD1-2a/8a relative percentage, at each indicated developmental stage. E, Western blot on total protein samples from the indicated DIV with a panLSD1 antibody.
To infer the function of LSD1 isoforms within neurons, we knocked them down differentially by generating short hairpin RNAs (shRNAs) specific for either neurospecific exon E8a or the splice junction between exon E8 and E9, which is shared among ubiquitous LSD1 isoforms. Hairpins containing vectors were generated and tested for isoform specificity and efficacy (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Cortical neurons were transfected at DIV4 with hairpins containing vectors and analyzed at DIV8 (Fig. 7A–C). We evaluated phenotypic traits that describe neuronal morphogenesis during in vitro maturation, including cumulative neurite length, the number of branches, and neurite width by Sholl's analysis performed on increasing radii from centered soma (Fig. 7D–F) (Sholl, 1953). The knockdown of ubiquitous isoforms ensued little or no effect when compared with control according to cumulative neurite arborization, branch count, and neurite width. Conversely, the silencing of neurospecific isoforms altered neurite morphogenesis by eliciting a significant decrease of the cumulative neurite arborization (Fig. 7D), a reduced number of secondary branches (Fig. 7E), and a reduced average neurite width (Fig. 7F). [The following series are scrambled compared with shRNAvs8a or shRNAvs8/9 conditions with related p values: neurite length (in μm) in Figure 7D, 897.21 ± 225.9, 664.42 ± 170.9, p = 4.99e−4; 1063.44 ± 229.5, p = 0.06; neurite branches in Figure 7E, 11.08 ± 1.54, 8.88 ± 1.59, p = 0.048; 11.16 ± 2.13, p = 0.95; neurite width (in μm) in Figure 7F at inner radius, 1.42 ± 0.12, 0.95 ± 0.078, p = 4.95e−9; 1.29 ± 0.14, p = 0.16; intermediate radius, 1.27 ± 0.11, 0.85 ± 0.074, p = 7.21e−9; 1.12 ± 0.12, p = 0.08; outer radius, 1.13 ± 0.1, 0.79 ± 0.064, p = 1.01e−6; 0.98 ± 0.098, p = 0.05; two tailed t test.]
Figure 7.
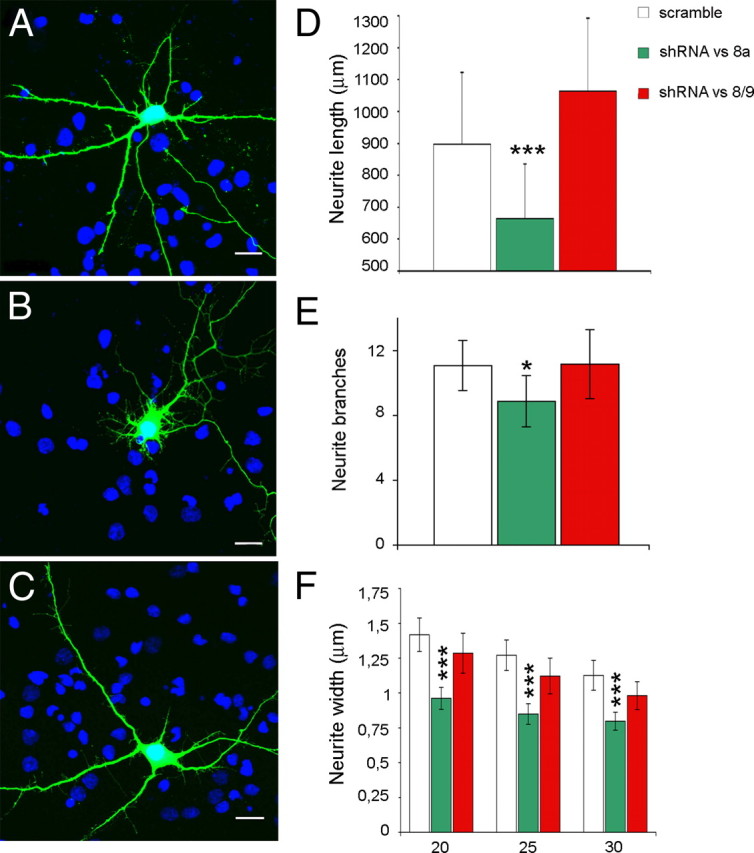
Effect of neurospecific or ubiquitous LSD1 knockdown by shRNAs on neurite morphology in rat cortical neurons. Cultured cortical neurons were transiently transfected with pSuper GFP Neo control (scramble) (A), pSuper engineered with shRNA against exon E8a (shRNA vs 8a) (B), and pSuper engineered with shRNA against the splice junction between exons E8 and E9 (shRNA vs 8/9) (C). Morphology was analyzed for EGFP-positive neurons with DAPI (4′,6′-diamidino-2-phenylindole) counterstain. D, Cumulative neurite length in differentially LSD1 knocked down neurons is indicated as average ± SEM in micrometers. E, Secondary branches count is indicated ± SEM. F, Average neurite width by Sholl analysis calculated on inner, intermediate, and outer Sholl's circles corresponding to 20, 25, and 30 μm radii, respectively. Values shown are mean ± SEM width in micrometers. Student's t test (l Stat t I ≥ T α/2) was applied to values by comparing each condition with control scramble. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars, 20 μm.
Furthermore, we evaluated neurite morphogenesis after overexpressing LSD1 neurospecific isoforms, because this was expected to exert opposite effects to those observed during knockdown experiments. Neurons were transfected at DIV4 with enhanced GFP (EGFP) vector alone (mock condition) (Fig. 8A) and cotransfected with HA-tagged LSD1 isoforms, both neuronal (pCGN–LSD1-8a plus pCGN–LSD1-2a/8a indicated as nLSD1 condition in Fig. 8B) and ubiquitous ones (pCGN–LSD1 plus pCGN–LSD1-2a indicated as uLSD1 condition in Fig. 8C). As expected, overexpression of neurospecific isoforms induced an increase in the morphometric parameters compared with controls (Fig. 8D–F), whereas overexpression of ubiquitous isoforms did not result in any significant effect. [The following series are mock compared with nLSD1 or uLSD1 conditions with related p values: neurite length (in μm) in Figure 8D, 783.7 ± 116.9, 943.8 ± 132.1, p = 0.03; 799.9 ± 101.5, p = 0.41; neurite branches in Figure 8E, 17.24 ± 5.67, 23.92 ± 4.64, p = 0.03; 12.56 ± 2.63, p = 0.07; neurite width (in μm) in Figure 8F at inner radius, 0.835 ± 0.09, 1.239 ± 0.11, p = 2.12e−8; 0.855 ± 0.07, p = 0.36; intermediate radius, 0.7339 ± 0.09, 1.1313 ± 0.11, p = 2.27e−8; 0.7419 ± 0.06, p = 0.44; outer radius, 0.5995 ± 0.08, 0.8876 ± 0.07, p = 2.01e−7; 0.6225 ± 0.05, p = 0.32; one-tailed t test]. To discern the relative contribution of each E8a-containing isoform in mediating the morphogenic effect on neurons, we performed a parallel experiment, transfecting either LSD1-8a or LSD1-2a/8a. As shown in supplemental Figure 7 (available at www.jneurosci.org as supplemental material), LSD1-8a is responsible for the morphogenic effect with LSD1-2a/8a partially recapitulating the phenotype that was observed under the nLSD1 condition (Fig. 8) in which both isoforms were present.
Figure 8.
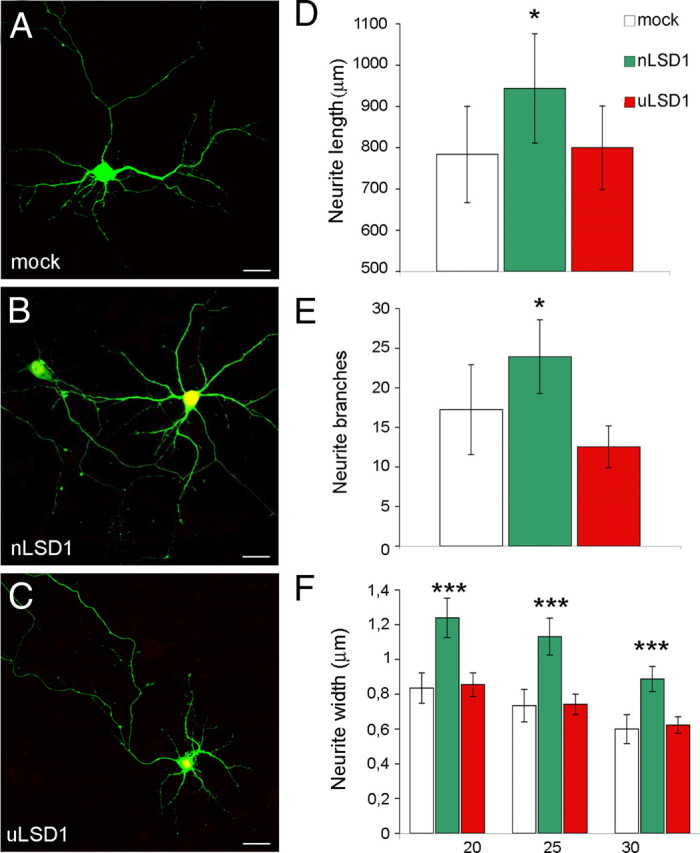
Effect of overexpression of neurospecific or ubiquitous LSD1 isoforms on neurite morphology in rat cortical neurons. Primary rat cortical neurons were transiently cotransfected with pCGN vector (mock) (A), EGFP together with HA–LSD1-8a plus LSD1-2a/8a (nLSD1) (B), HA–LSD1 plus LSD1-2a (uLSD1) (C). Morphology was analyzed in EGFP-positive neurons (mock) or double-labeled EGFP- and HA-positive neurons. D, Cumulative neurite length in differentially LSD1 transfected neurons is indicated as average ± SEM in micrometers. E, Secondary branches count is indicated ± SEM. F, Average neurite width by Sholl analysis calculated on inner, intermediate, and outer Sholl's circles corresponding to 20, 25, and 30 μm radii, respectively. Values shown are mean ± SEM width in micrometers. Student's t test (l Stat t I ≥ T α) was applied to values by comparing each condition with mock. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars, 20 μm.
In conclusion, although the knockdown of neurospecific LSD1 isoforms delays neurite morphogenesis, overexpression of the same seems to anticipate features that normally arise at later stages under physiological conditions. Conversely, perturbation of ubiquitous LSD1 variants, either by knock down or transfection, sorted no statistically significant effect when compared with controls. Because our conditions only differed for E8a retention in both experiments, the morphogenic effects likely rely on exon E8a presence.
Discussion
Alternative splicing is considered one of the most powerful biological mechanisms that convey diversification of gene function without a corresponding increase in gene number (Kopelman et al., 2005; Parmley et al., 2007), giving rise to evolutionary complexity, especially for mammalian species. Notably, most of splicing events occur on CNS-related transcripts and participate in fundamental nervous processes, from cell-related ones, such as axon guidance and synapse formation (Lipscombe, 2005; Licatalosi and Darnell 2006; Ule and Darnell, 2006), to higher cognitive functions, including learning and memory (Grabowski and Black, 2001). Although LSD1 function has been extensively investigated, its epigenetic role in the nervous system has not been elucidated yet. Nonetheless, LSD1 involvement in neuronal processes has been inferred through the transcriptional silencer REST of which LSD1 is a known corepressor (Shi et al., 2004): REST prevents the expression of neuron-specific genes outside CNS, regulating it throughout neurogenesis (Ballas et al., 2005).
REST is also expressed in the adult nervous system (Palm et al., 1998; Garriga-Canut et al., 2006), in which its repressor function can be modulated by neurospecific alternative splicing (Palm et al., 1998; Shimojo et al., 1999; Zuccato et al., 2003).
Here, we report that mammalian neurons have specialized chromatin remodeling complexes as a result of the presence of neurospecific splice variants of LSD1. A comparative analysis of LSD1 genomic sequence across vertebrates and exon retrieval from EST databases led us to identify four mammal LSD1 variants, generated by either single or double inclusion of two alternatively spliced exons, namely E2a and E8a. Thus, there are four LSD1 isoforms: LSD1 (i.e., the conventional protein), LSD1-2a, LSD1-8a, and LSD1-2a/8a. The former two are ubiquitous, whereas the latter ones are restricted to the neuronal histotype. Biochemical studies on the recombinant enzymes showed that all three splicing isoforms retain the histone H3 Lys4 demethylase activity displayed by the conventional LSD1. Most interestingly, E8a results in a small protruding loop proximal to the catalytic site as shown by the three-dimensional structure (Fig. 3).
The processing of LSD1 transcript undergoes a dynamic modulation within the perinatal window, when fundamental processes take place, such as phylopodia formation, neurite sprouting (Lee and Sheng, 2000; McAllister, 2000), and the expression of the first synaptogenic markers. In this phase, exon E8a inclusion frequency greatly increases, shifting the ratio toward LSD1 neurospecific isoform production. Transcriptional profiling of the developing rat brain revealed that the most dramatic changes in gene expression occur postpartum (Stead et al., 2006), underscoring the relevance of early postnatal life as a critical phase of neural organization and differentiation. The dynamics of LSD1 isoforms that we observed in vivo is fairly reproduced by cultured cortical neurons, in which a perturbation of exon E8a-containing isoforms results in phenotypic changes: early-stage silencing of neurospecific isoforms delays neurite maturation, whereas overexpression of the same enhances it, as revealed by morphometric analysis. Notably, isoforms that are devoid of E8a elicited no phenotypical changes relative to control, and this suggests that the 4 aa peptide coded by E8a is responsible for LSD1 morphogenic effect on differentiating neurons, possibly regulating the proper timing of neurite maturation. These results relate the “wave-like” perinatal expression of exon E8a to the acquisition of neuronal morphology, setting the basis for LSD1 role in regulating transcription of development-related genes. Remarkably, an enhanced neurite complexity has already been described in a similar cellular system by the overexpression of the methyl binding protein MeCP2 (Jugloff et al., 2005), whose function has already been related to the regulation of a particular class of REST target genes in differentiating neurons (Ballas et al., 2005).
Hints on LSD1 functional tunability come from trancriptional assays performed in rat cortical neurons in which neurospecific exon E8a reduces LSD1 repressor activity on a reporter gene. Several mechanisms may be proposed to explain how exon E8a can modulate the repressor activity of LSD1: the simplest hypothesis is that neurospecific LSD1-containing complexes regulate specific subsets of target genes that differ from those regulated by ubiquitous LSD1. A second hypothesis is that the inclusion of E8a provides a tool to tune LSD1 epigenetic activity on shared targets. A mechanistic distinction between neurospecific and ubiquitous isoforms may arise from posttranslational modifications on E8a induced by external stimuli. The structural analysis demonstrated that the presence of exon E8a creates a loop protruding from the surface of the protein. This observation suggests that exon E8a may endow LSD1 with a posttranslational modification site, possibly having a regulatory role. This may result in several functional alterations, including neurospecific LSD1 affinity for CoREST or other transcriptional factors, as well as a shift toward an activatory role (Garcia-Bassets et al., 2007; Wang et al., 2007; Gatta and Mantovani 2008). Moreover, because the four LSD1 isoforms can be expressed within the same neuronal type and they can mutually interact, multiple combinations may occur to widen the repertoire of corepressor complexes. Combinatorial assembly of neurospecific factors has already been described (Olave et al., 2002), increasing the complexity of epigenetic regulation within CNS (Lessard et al., 2007; Wu et al., 2007). In this context, it is feasible that the recruitment of LSD1 isoforms into CoREST complexes might be regulated by different environmental cues, and the frequency at which the splicing process occurs would determine the availability of such epigenetic components.
To conclude, our work suggests that the regulated expression of neurospecific LSD1 splice variants pacemakes early neurite morphogenesis, prospecting important implications for LSD1 splicing in the acquisition and dynamic maintenance of the neuronal phenotype by a mechanism in which different splice isoforms can coordinate the regulation of target genes.
Footnotes
This work was supported by the Italian Ministry of University and Scientific Research (Progetti di Rilevante Interesse Nazionale PRIN07 and PRIN08 and Fondo per gli Investimenti della Ricerca di Base programs to A.M. and C.S.), Fondazione Telethon-Italy Grants GGP07078A (E.B.), GGP06208 (C.S.), and GGP07078 (E.G.), Fondazione Cariplo Projects 2006-0779 (C.S.) and 2007-5561 (A.M.), Associazione Italiana per la Ricerca sul Cancro Grant 5410 (A.M.), and Compagnia di San Paolo Project 2005-1964 (C.S.). We are very grateful to Julia E. Dallman, Beatrice Bodega, and Emma Paola Sturani for stimulating discussions.
Coordinates of the LSD1-8a–CoREST–histone peptide complex have been deposited in the Protein Data Bank with accession number 2x0l.
References
- Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Andrés ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci U S A. 1999;96:9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Barak O, Lazzaro MA, Cooch NS, Picketts DJ, Shiekhattar R. A tissue-specific, naturally occurring human SNF2L variant inactivates chromatin remodeling. J Biol Chem. 2004;279:45130–45138. doi: 10.1074/jbc.M406212200. [DOI] [PubMed] [Google Scholar]
- Battaglioli E, Andrés ME, Rose DW, Chenoweth JG, Rosenfeld MG, Anderson ME, Mandel G. REST repression of neuronal genes requires components of the hSWI.SNF complex. J Biol Chem. 2002;277:41038–41045. doi: 10.1074/jbc.M205691200. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Nguyen PH, Courey AJ. A role for Groucho tetramerization in transcriptional repression. Mol Cell Biol. 1998;18:7259–7268. doi: 10.1128/mcb.18.12.7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc Natl Acad Sci U S A. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Dallman JE, Allopenna J, Bassett A, Travers A, Mandel G. A conserved role but different partners for the transcriptional corepressor CoREST in fly and mammalian nervous system formation. J Neurosci. 2004;24:7186–7193. doi: 10.1523/JNEUROSCI.0238-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005a;579:2203–2207. doi: 10.1016/j.febslet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005b;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Dall'Aglio A, Fraaije MW, Battaglioli E, Mattevi A. A highly specific mechanism of histone H3–K4 recognition by histone demethylase LSD1. J Biol Chem. 2006;281:35289–35295. doi: 10.1074/jbc.M607411200. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Battaglioli E, Mattevi A. LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci. 2008;33:181–189. doi: 10.1016/j.tibs.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Fox IJ, Paucar AA, Nakano I, Mottahedeh J, Dougherty JD, Kornblum HI. Developmental expression of glial fibrillary acidic protein mRNA in mouse forebrain germinal zones: implications for stem cell biology. Brain Res Dev Brain Res. 2004;153:121–125. doi: 10.1016/j.devbrainres.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, Ju BG, Ohgi KA, Wang J, Escoubet-Lozach L, Rose DW, Glass CK, Fu XD, Rosenfeld MG. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–518. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, Roopra A. 2-Deoxy-d-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9:1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- Gatta R, Mantovani R. NF-Y substitutes H2A-H2B on active cell-cycle promoters: recruitment of CoREST-KDM1 and fine-tuning of H3 methylations. Nucleic Acids Res. 2008;36:6592–6607. doi: 10.1093/nar/gkn699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Grabowski PJ, Black DL. Alternative RNA splicing in the nervous system. Prog Neurobiol. 2001;65:289–308. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci U S A. 2002;99:7420–7425. doi: 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, Howard BH. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem. 2001;276:6817–6824. doi: 10.1074/jbc.M007372200. [DOI] [PubMed] [Google Scholar]
- Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005;19:18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284:17775–17782. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman NM, Lancet D, Yanai I. Alternative splicing and gene duplication are inversely correlated evolutionary mechanisms. Nat Genet. 2005;37:588–589. doi: 10.1038/ng1575. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Roelens I, Jacob S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J Mol Neurosci. 2006;29:227–239. doi: 10.1385/JMN:29:3:227. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Lee SH, Sheng M. Development of neuron-neuron synapses. Curr Opin Neurobiol. 2000;10:125–131. doi: 10.1016/s0959-4388(99)00046-x. [DOI] [PubMed] [Google Scholar]
- Leslie AG. Integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Lipscombe D. Neuronal proteins custom designed by alternative splicing. Curr Opin Neurobiol. 2005;15:358–363. doi: 10.1016/j.conb.2005.04.002. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb Cortex. 2000;10:963–973. doi: 10.1093/cercor/10.10.963. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nottke A, Colaiácovo MP, Shi Y. Developmental roles of the histone lysine demethylases. Development. 2009;136:879–889. doi: 10.1242/dev.020966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olave I, Wang W, Xue Y, Kuo A, Crabtree GR. Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev. 2002;16:2509–2517. doi: 10.1101/gad.992102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmley JL, Urrutia AO, Potrzebowski L, Kaessmann H, Hurst LD. Splicing and the evolution of proteins in mammals. PLoS Biol. 2007;5:e14. doi: 10.1371/journal.pbio.0050014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, Temple S. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron. 2000;28:69–80. doi: 10.1016/s0896-6273(00)00086-6. [DOI] [PubMed] [Google Scholar]
- Romorini S, Piccoli G, Jiang M, Grossano P, Tonna N, Passafaro M, Zhang M, Sala C. A functional role of postsynaptic density-95-guanylate kinase-associated protein complex in regulating Shank assembly and stability to synapses. J Neurosci. 2004;24:9391–9404. doi: 10.1523/JNEUROSCI.3314-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M. Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci. 2000;20:3529–3536. doi: 10.1523/JNEUROSCI.20-10-03529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M, Paquette AJ, Anderson DJ, Hersh LB. Protein kinase A regulates cholinergic gene expression in PC12 cells: REST4 silences the silencing activity of neuron-restrictive silencer factor/REST. Mol Cell Biol. 1999;19:6788–6795. doi: 10.1128/mcb.19.10.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurone of the visual motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Ast G. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 2003;13:1631–1637. doi: 10.1101/gr.1208803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat Struct Mol Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- Stead JD, Neal C, Meng F, Wang Y, Evans S, Vazquez DM, Watson SJ, Akil H. Transcriptional profiling of the developing rat brain reveals that the most dramatic regional differentiation in gene expression occurs postpartum. J Neurosci. 2006;26:345–353. doi: 10.1523/JNEUROSCI.2755-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B. Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem. 2001;276:37585–37593. doi: 10.1074/jbc.M101683200. [DOI] [PubMed] [Google Scholar]
- Ule J, Darnell RB. RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr Opin Neurobiol. 2006;16:102–110. doi: 10.1016/j.conb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Walton M, Henderson C, Mason-Parker S, Lawlor P, Abraham WC, Bilkey D, Dragunow M. Immediate early gene transcription and synaptic modulation. J Neurosci Res. 1999;58:96–106. [PubMed] [Google Scholar]
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron. 2007;56:94–108. doi: 10.1016/j.neuron.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Wang Q, Lee C. Evolutionary divergence of exon flanks: a dissection of mutability and selection. Genetics. 2006;173:1787–1791. doi: 10.1534/genetics.106.057919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc Natl Acad Sci U S A. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]