

Abstract
Oligomers of the β-amyloid (Aβ) peptide have been indicated in early neuropathologic changes in Alzheimer's disease. Here, we present a synthetic Aβ20-42 oligomer (named globulomer) with a different conformation to monomeric and fibrillar Aβ peptide, enabling the generation of highly Aβ oligomer-specific monoclonal antibodies. The globulomer-derived antibodies specifically detect oligomeric but not monomeric or fibrillar Aβ in various Aβ preparations. The globulomer-specific antibody A-887755 was able to prevent Aβ oligomer binding and dynamin cleavage in primary hippocampal neurons and to reverse globulomer-induced reduced synaptic transmission. In amyloid precursor protein (APP) transgenic mice, vaccination with Aβ globulomer and treatment with A-887755 improved novel object recognition. The cognitive improvement is likely attributable to reversing a deficit in hippocampal synaptic spine density in APP transgenic mice as observed after treatment with A-887755. Our findings demonstrate that selective reduction of Aβ oligomers by immunotherapy is sufficient to normalize cognitive behavior and synaptic deficits in APP transgenic mice.
Introduction
Although being an abundant peptide with a high turnover rate (Bateman et al., 2006), β-amyloid (Aβ) is considered a critical contributor to the generation and progression of Alzheimer's disease (AD). Aβ is a major component of amyloid plaque deposits in the brain, one of the defining pathological hallmarks of this type of dementia (Hardy and Selkoe, 2002). Although production and metabolism of high amounts of Aβ are associated with normal brain function, several hypotheses have been developed how this peptide may become pathogenic for neurons and brain function.
Based on the high prevalence of the 42-aa-long version of Aβ in amyloid plaques, the amyloid cascade hypothesis of Hardy and Higgins (1992) suggested that an overproduction of the more hydrophobic Aβ1-42 compared with the more abundant Aβ1-40 triggers aggregation of Aβ into fibrils and plaques, which initiates AD pathology. However, this hypothesis is not able to explain an obvious discrepancy between amyloid plaque load and degree of dementia in AD (Katzman et al., 1988). In 1999, several groups reported a much better correlation of the soluble Aβ fraction in postmortem AD brain extracts with disease symptoms (Lue et al., 1999; McLean et al., 1999; Wang et al., 1999). These finding supported data that connected the pathological nature of Aβ to its oligomeric state (Lambert et al., 1998). Thus, Aβ oligomers became the focus of AD research and have subsequently been generated synthetically (Barghorn et al., 2005), harvested from medium after release by amyloid precursor protein (APP)-transfected CHO cell cultures (Walsh et al., 2002), or isolated from the brain of APP transgenic mice (Lesné et al., 2006). These preparations were useful to demonstrate Aβ oligomer pathology in animal models (Walsh et al., 2002; Lesné et al., 2006) and indicated in primary hippocampal neuronal culture that their major pathogenic mechanisms in AD pathology is impairment of synaptic activity (Lacor et al., 2007; Shankar et al., 2007). However, molecular characteristics of the pathological Aβ oligomers are still not fully elucidated, mainly because the preparations are either containing different Aβ species with undefined stability [e.g., so-called amyloid-derived diffusible ligands (ADDLs)] (Lambert et al., 1998) or because they were resistant to purification when extracted from biological sources (Walsh et al., 2002; Lesné et al., 2006). Thus, it remains under discussion whether the pathology of Aβ oligomers is related to its size, e.g., trimer (Walsh et al., 2002) or dodecamer (Lesné et al., 2006), or to its conformation. Likewise, the available preparations did not allow generation of Aβ oligomer-selective monoclonal antibodies that do not bind Aβ monomers and fibrils and thus would have been able to prove that neutralization of Aβ oligomers is sufficient to reduce AD pathology.
Here, we show that a synthetic Aβ oligomer preparation can be used to generate monoclonal antibodies that selectively detect Aβ oligomers in APP transgenic mouse and AD brain tissue. Importantly, this type of antibody is able to prevent pathological effects of Aβ oligomers in vitro and in vivo, indicating that neutralization of Aβ oligomers by specific antibodies is sufficient for efficacy in a preclinical AD model and should be therefore tested for therapeutic efficacy in AD.
Materials and Methods
Aβ20-42 globulomer and antibody generation.
The Aβ20-42 globulomer was generated from the Aβ1-42 globulomer by limited proteolysis with thermolysine (1:50). This synthetic Aβ oligomer proved extremely resistant to physical and chemical treatment over time (for details, see Barghorn et al., 2005). Monoclonal antibodies were generated from mice immunized with Aβ20-42 globulomer according to standard procedures and tested for their selectivity to Aβ20-42 globulomer versus other Aβ conformers using a standard dot blot assay.
Biacore analysis.
The Aβ20-42 globulomer antigen was immobilized on Biacore sensor chips CM5, and kon and koff for A-887755 binding was determined according to standard techniques.
Surface-enhanced laser desorption/ionization–mass spectrometry analysis.
Aβ peptide mass was determined by surface-enhanced laser desorption/ionization–mass spectrometry (SELDI-MS) using H4 protein chips (Bio-Rad) and α-cyano-4-hydroxy-cinnamic acid in 50% acetonitril/0.5% trifluor acetic acid as matrix.
Tissue extracts.
Aβ peptides were extracted from brain samples using two different buffer conditions. Clinically characterized human brain neocortical samples (supplemental Table 1, available at www.jneurosci.org as supplemental material) were homogenized in either 3% SDS extraction buffer [0.1 g/ml in 50 mm Tris, 150 mm NaCl, 0.5% Triton X-100, 1 mm EGTA, 3% SDS, and 1% Na-desoxycholate, pH 7.4, with complete protease inhibitor cocktail (Roche Diagnostics) and 1 mm PMSF] or PBS extraction buffer (0.1 g/ml in 20 mm NaH2PO4, and 140 mm NaCl, pH 7.4, with complete protease inhibitor cocktail and 1 mm PMSF) with ∼20 strokes in a glass potter and followed by an ultra turrax homogenizer for 30 s. The 3% SDS samples were incubated for 30 min at room temperature and centrifuged for 10 min at 16,000 × g at 20°C. The PBS samples were incubated for 30 min on ice and centrifuged for 30 min at 16,000 × g at 20°C, and the supernatants were analyzed for Aβ peptides using quantitative immunoprecipitation. Extraction of Aβ peptides from Tg2576 mice (Taconic Farms) brain (3% SDS only) followed the same protocol. Clinically characterized human plasma and CSF samples (supplemental Tables 2, 3, available at www.jneurosci.org as supplemental material) were also used for immunoprecipitation.
Immunoprecipitation.
For immunoprecipitation, mouse anti-Aβ antibodies were coupled to anti-mouse antibody magnetic beads (Dynabeads). The samples were incubated with the magnetic beads overnight at 4°C, washed three times with PBS, eluted with 50% acetonitrile/0.5% trifluoroacetic acid, and subjected to standard quantitative Western blot.
Cell culture and Aβ binding assay.
Hippocampal rat brain neurons (embryonic day 18) were cultured for ∼21 d in vitro in 24-well plates on poly-d-lysine-coated coverslips in Neurobasal medium with B27 supplement at 37°C, 5% CO2. For antibody-mediated Aβ globulomer binding inhibition, 8.3 nm Aβ20-42 globulomer (calculated from the initial 100 nm Aβ monomer) was incubated for 1 h at 37°C with 16.7 nm A-887755 or a non-Aβ-binding isotype control antibody [IgG2a, directed against key limpet hemocyanine (Abbott Bioresearch Center)] and subsequently applied for 15 min at 37°C to the hippocampal neurons. After intensive washing, cells were fixed and stained with immunofluorescence-labeled antibodies to Aβ globulomer and the neuron-specific marker MAP2 and with DAPI (4′,6′-diamidino-2-phenylindole) for cell-body localization.
Aβ-induced dynamin cleavage.
Aβ was aggregated according to the literature (Kelly et al., 2005; Kelly and Ferreira, 2006) with slight modifications. Briefly, Aβ1-40 (American Peptide) was dissolved in serum-free culture medium at 0.1 mg/ml and incubated at 37°C for 4 d. Anti-Aβ globulomer antibody A-887755, the non-Aβ-binding IgG2a isotype control antibody, or PBS was incubated with the aggregated Aβ at 25°C for 1 h under constant agitation in a final volume of 0.225–1 ml. The mixtures were added to the culture medium resulting in a final concentration of 5 μm Aβ (calculated from initial Aβ monomer) and 2 μm antibody, respectively. Every treatment was performed in triplicate, and wells without addition of Aβ or antibodies were included as additional controls. Cells were cultivated for another 24 h and briefly inspected by light microscopy before being processed for Western blot. Treatment with Aβ did not induce overt neuronal death during the incubation period.
Slice culture experiment.
Hippocampal slice cultures were prepared from 9-d-old Wistar rats (Janvier). In brief, 400 μm hemisphere slices were prepared using a tissue chopper (Mickle Laboratory Engineering Co. Ltd.) and cultured on Millicell-CM membranes (Millipore Corporation) in high-K medium [40% basal medium Eagle with Earle's salts, 25% horse serum, 25% Earle's balanced salt solution, 1 mm Glutamax I, 28 mm glucose (40%), and 10% 250 mm NaHEPES (Sigma-Aldrich; all other ingredients were from Invitrogen)] at 34°C, 5% CO2 for 3 d and then in Neurobasal A medium [96.4% Neurobasal A medium, 2% B-27 supplement, 1 mm l-glutamine (Invitrogen), and 25 mm d-glucose (Sigma-Aldrich)]. At 24 h before recording, 160 nm Aβ20-42 globulomer, globulomer plus 100 nm A-887755, or globulomer ultrafiltrate plus 830 nm SDS (vehicle control) was applied to the slice culture medium. Shortly before recording, slice cultures were placed in a Haas-interface recording chamber (Harvard Apparatus) and allowed to equilibrate for at least 60 min at 32°C. The Schaffer collateral was then stimulated with bipolar pulses (0.1 ms/phase) using a 0.5 MΩ bipolar Tungsten electrode (WPI), and field EPSP amplitudes were recorded with glass electrodes (0.7–1.1 MΩ, GC150F-15; Harvard Apparatus). Signals were digitized using a power CED 1401 (Cambridge Electronic Design) and analyzed using Signal 2.14 software (Cambridge Electronic Design).
Immunohistology.
Brain material of 19-month-old Tg2576 mice (Taconic Farms) and autopsy material of two Alzheimer's disease patients (RZ16 and RZ55; obtained from BrainNet) was used. The animals were deeply anesthetized and transcardially perfused with 0.1 m PBS to flush the blood. Then the brain was removed from the cranium and divided longitudinally. One hemisphere of the brain was shock frozen and used for Aβ oligomer extraction (see above), and the other was fixated by immersion into 4% paraformaldehyde. The immersion-fixated hemisphere was cryoprotected by soaking in 30% sucrose in PBS and mounted on a freezing microtome. The entire forebrain was cut into 40 μm sections that were collected in PBS and used for the subsequent staining procedure. The human brain material was an ∼1 cm3 deep-frozen block of the neocortex. A small part of the block was immersion fixated in 4% paraformaldehyde and further treated like the mouse brain material. Antibody staining was performed by incubating the sections with a solution containing 0.07–7.0 μg/ml of the conformation-independent antibodies 6E10 (Signet Labs) and 6G1 (generated by Abbott) (Barghorn et al., 2005) or the globulomer-specific antibody A-887755 in accordance with the following protocol (at room temperature if not indicated otherwise). Floating sections were transferred into ice-cold 0.3% H2O2 for 30 min, subsequently washed in Tris-buffered saline with Tween 20 (TBST) (10× concentrate; S3306, 1:10 in Aqua bidest; Dako Cytomation), and incubated with 5% donkey serum/TBST for 20 min. Then, sections were directly transferred to the primary antibody solution and incubated for 24 h. After thorough washing in TBST, nonspecific staining was blocked by blocking serum (Vectastain Elite ABC peroxidase kit) for 20 min, washed again in TBST, and incubated with a biotinylated donkey anti-mouse antibody (1:500 in TBST; Jackson ImmunoResearch) for 60 min. Before and after incubation with StreptABComplex (Dako Cytomation), for 60 min, the sections were washed in TBST. Finally, the sections were stained with diaminobenzidine (Vectastain Elite ABC peroxidase kit) for 20 min, mounted on glass slides, air dried, dehydrated, and coverslipped in xylol-free embedding medium (X-tra kitt; Medite).
Staining was photographed using a Carl Zeiss Axioplan microscope, and amyloid staining was additionally quantified by graphically excising 10 randomly selected plaques from the histological images using the ImagePro 5.0 image analysis system and determining their average grayscale value. Optical density values were calculated from the grayscale values by subtracting the mean background density of the stained material from the density of amyloid plaques (0%, no plaque staining above surrounding background; 100%, no transmission/maximal staining). All analysis was performed blinded to the used antibody.
For analysis of tissue cross-reactivity, FITC-labeled A-887755 was prepared and incubated at 0.1 and 10 μg/ml with standard tissues of cynomolgus monkey and man (Charles River).
Behavioral experiments.
For the cognition testing, male and female APP transgenic mice carrying the London mutation (APP/L) (Moechars et al., 1999) and age-matched littermate wild-type control mice were bred in-house and were individually housed under standard laboratory conditions with access to food and water ad libitum. Experimental procedures were approved by the Animal Welfare Office of reMYND or Abbott and were performed in accordance with the European and German national guidelines as well as recommendations and policies of the National Institutes of Health Principles of Laboratory Animal Care (1996 edition). All experiments were performed during the light phase of a 12 h light/dark cycle.
The novel object recognition task was performed in 4.5-month-old mice. For this test, the mice were individually placed and accustomed to an arena and then exposed to two identical elements (blue pyramid, green cube, yellow cylinder of similar size, ∼4 cm) for 10 min (acquisition phase). The duration and frequency with which the mouse explored the objects were recorded. During the retention phase, 2.5 h later, mice were returned to the arena that now contained, in addition to the known object, an unknown object randomly selected from the other objects. Recognition of the new object was recorded as the time during which the mouse was exploring the old object relative to total time (exploration of old and new object), the recognition index. A mouse that does not remember the known object will consider it as equally interesting as the new object and spend an equal amount of time on exploring it (recognition index of 50%), whereas a mouse that remembers the known object will consider it as not interesting and therefore show a significantly higher recognition index. APP/L mice are known to have memory deficits in this test at 4.5 months of age, i.e., a recognition index of ∼50% (Dewachter et al., 2002)
In a first study (active immunization), 1.5-month-old APP/L mice received 100 μg of Aβ1-42 monomer (in 0.1% NH4OH), Aβ1-42 globulomer, or Aβ20-42 globulomer in PBS mixed with an equal amount of complete Freund's adjuvant intraperitoneally, followed by booster injections with the same amount of antigen in incomplete Freund's adjuvant every third week for 3 months before being subjected to the object recognition task. In a second study (passive immunotherapy), the task was performed in 4- to 5-month-old APP/L mice 1 d after receiving the last of three injections of 500 μg of antibody (6G1 or A-887755) or 250 μl of saline (vehicle) separated by 1 week.
For the Morris water maze task, a custom-made pool (100 cm diameter) with white, nontransparent walls was located in a well-lit room with a number of distal extramaze cues. The daily filled and drained water (25°C) was made opaque with coffee creamer, and water surface was ∼10 cm from the top of the pool edge. A clear Plexiglas escape platform (9 cm diameter) was located 0.5 cm below the water surface in the center of one of eight equally sized pool sectors (∼20 cm from the edge of the platform to the pool wall). The platform location was selected randomly for each mouse but was kept constant for each individual mouse throughout training. For each trial, a mouse was released from any of eight start boxes (filled with water and separated from the pool by the manually operated sliding door) and had 60 s to locate the escape platform, where it was allowed to remain for 10 s. The mouse was then returned to its holding cage, which was located under an incandescent read heat lamp. If the platform was not found in the allotted time, the mouse was gently placed on the platform using an appropriately sized plastic spoon. The mice were given two trials per day for 5 d, followed by a final training after the weekend. Four mice were trained successively in a group enabling intertrial intervals to be 5 min. The mice were tracked in the pool by a Ethovision video tracking system (Noldus). APP/L mice or transgenic littermates of 5.5 months of age were divided into four groups: transgenic mice treated with PBS or with A-887755 (500 μg, i.p.) and wild-type mice treated with PBS or with A-887755. Antibody or PBS were injected on days 1, 6, 11, and 16 of the experiment. Water maze training took place on days 12–16 and day 19. Measures of latency to find the platform, path length, and swimming speed were obtained for each trial, and then averaged daily values were subjected to three-way ANOVA with repeated measures on time. Whenever applicable for repeated measures analysis, Mauchly's test of sphericity was applied and the degrees of freedom were corrected to more conservative values using the Huynh–Feldt's ε for any terms involving factors in which the sphericity assumption was violated.
Spine density quantification.
Female Tg2576 mice (5.5 months of age; Taconic Farms) were treated with A-887755 (500 μg in PBS, i.p.) or vehicle once weekly for 3 weeks. Then, the mice were deeply anesthetized and perfused with 0.9% saline (10 min). The right hemisphere was dissected out and processed for a modified Golgi–Cox staining as described by the manufacturer (Rapid Golgi; FD NeuroTechnologies). Coronal sections (240 μm) were obtained using a freezing-sliding microtome, mounted on 2% gelatin-coated glass slides, and stored in Golgi solution in the dark at room temperature. After 2 weeks, sections were rinsed, dehydrated, and cleared of xylenes, and slides were coverslipped and allowed to dry before quantitative analysis. Several pyramidal neurons impregnated with the Golgi solution were readily identified in the dorsal hippocampal region by their characteristic triangular soma shape and numerous dendritic spines. At least four neurons per animal were three-dimensionally reconstructed by NeuroLucida Software (MicroBrightField). For spine quantification, a 100× oil-immersion objective was used to identify mushroom-like spines in dendrites longer than 10 μm. Straight branches were preferred to have a clear presentation of mushroom-like spines. At least three proximal (proximal dendrites, 30–120 μm from soma) and three distal dendritic segments (distal dendrites, 220–340 μm from soma) were quantified in each neuron. Spines located on proximal dendrites and spines located on distal dendrites were analyzed separately. Statistical analysis of the spine density was performed with the average of the total number of spines per micrometers per neuron of each mouse/group.
Statistical analysis.
Averaged data are calculated and shown as means ± SEM. For evaluation of statistical differences, data were checked for normal distribution (Prism; GraphPad Software) and compared by parametric or nonparametric ANOVA. Thus, a nonparametric Kruskal–Wallis test followed by Dunn's test was performed for the electrophysiological study. Most other studies were analyzed by ANOVA, followed by post hoc Bonferroni's t test. For the dynamin cleavage study, a two-way ANOVA followed by post hoc Holm–Sidak test was used, and, for the water maze test, a three-way-ANOVA was used (see above). A level of p < 0.05 was considered significant.
Results
Oligomer-selective antibodies by immunization with N-terminally truncated Aβ20-42 globulomer
We had described previously a synthetic Aβ1-42 oligomer preparation that contains exclusively one biochemically defined oligomeric species and is not contaminated with Aβ1-42 monomers or Aβ1-42 fibrils (Barghorn et al., 2005). We used this preparation, named Aβ1-42 globulomer (Fig. 1a), to raise oligomer-specific mouse monoclonal antibodies. However, most of the resulting antibodies were directed against the hydrophilic flexible and less structure-defining N-terminal part of the Aβ peptide shared by all Aβ sequence-containing species. Antibody 6G1 is an example of this type of antibody and not able to discriminate between monomers, oligomers, or fibrils of Aβ (Fig. 1b). After cleaving the N terminus by limited proteolysis, a more condensed and hydrophobic oligomer, Aβ20-42 globulomer, was obtained (Fig. 1a). This truncated globulomer presented an epitope for raising antibodies that were now able to discriminate between Aβ oligomer and all other Aβ species, especially monomers and fibrils, and soluble APP in dot blot assays (Fig. 1b). Biacore binding analysis for the most specific antibody, A-887755, on Aβ20-42 globulomer immobilized sensor chips demonstrated a high-affinity binding for the antigen (KD of 1.56 nm; kon = 9.37 × 105 m−1/s−1and koff = 1.46 × 10−4 s−1).
Figure 1.

Generation of Aβ20-42 globulomer and selective antibodies. a, The Coomassie-stained SDS-PAGE shows the characteristic apparent band pattern of Aβ20-42 globulomer after limited proteolysis of Aβ1-42 globulomer with thermolysine. b, Dot blot of decreasing amounts of diverse Aβ conformers demonstrate the selectivity of mouse monoclonal anti-Aβ globulomer antibodies. Top row, Immunization of mice with Aβ1-42 globulomer generated 6G1-type antibodies that are Aβ conformer nonspecific and comparable with the commercially available Aβ conformer nonspecific 6E10 antibody. Bottom row, Aβ20-42 globulomer-immunized mice generated highly selective antibodies recognizing Aβ1-42 and Aβ20-42 globulomer (A-887756) or Aβ20-42 globulomer only (A-887755).
Monoclonal antibody A-887755 selectively binds soluble Aβ oligomers in brain tissue extracts from AD patients and APP transgenic mice
Although the specificity of the anti-globulomer antibodies is a very interesting in vitro property, the most relevant question was the ability of these antibodies to detect natural Aβ species in vivo. We used the most specific antibody, A-887755 (Fig. 1b), to immunoprecipitate soluble Aβ from the neocortex of an AD patient and analyzed the precipitated Aβ species using SELDI-MS. Although A-887755 was highly specific for the truncated Aβ20-42 globulomer in the dot blot assay in vitro, it was also able to bind to natural soluble Aβ1-42, Aβ1-40, and Aβ(x-40,42) species of various length in the 3% SDS AD brain extract (Fig. 2a). When we compared the amount of Aβ detected by the Aβ20-42 globulomer-derived antibodies in the extract with the amount of Aβ detected by the conformation-independent Aβ antibody 6E10 (Fig. 1b), for Aβ1-40 and Aβ1-42, a fraction of between 25 and 50% of total soluble Aβ was found to bind to A-887755 and A-887756 (Fig. 2b). The in vitro highly selective antibody A-887755 was considered most interesting and subsequently further characterized. In vivo oligomer specificity was tested by comparing A-887755-immunoprecipitated soluble Aβ from AD brain extracts with 6E10 immunoprecipitation, the latter binding to all soluble Aβ species. When the 3% SDS brain extract was pretreated with formic acid, the secondary structure of the Aβ oligomers was destroyed. As a consequence, A-887755 was no longer able to detect extracted soluble Aβ, although 6E10 did (Fig. 2c, first two bars). Because globulomer is generated by incubation with SDS in vitro, we checked the possibility that Aβ detected by A-887755 had been freshly generated during extraction. Therefore, synthetic Aβ monomer was spiked into and incubated with a 3% SDS extract of a non-AD brain before homogenization and immunoprecipitation. This procedure did not result in Aβ that could be precipitated by A-887755, although 6E10 detected spiked Aβ as expected (Fig. 2c, right bar), suggesting that the Aβ20-42 globulomer-derived antibody A-887755 specifically binds to oligomeric Aβ without detecting Aβ monomer even when present far in excess over oligomers. For quantification of the amount of Aβ oligomers in AD and control brains, formic acid extracts could not be used because this procedure destroys oligomers. Therefore, we only used both PBS and 3% SDS extracts to study the amount of soluble Aβ oligomers immunoprecipitated with 6E10, A-887755, and a non-Aβ-binding IgG2a control antibody in several AD and control brains. Although the concentration of total soluble Aβ, as detected by 6E10, was ∼75 ng/g in the PBS extract, approximately fivefold more Aβ was extracted with 3% SDS, probably because of dissolving additional lipid-bound Aβ. In comparison, the amount of soluble Aβ oligomers, as detected by A-887755, was a significant fraction of total Aβ in the PBS (12%) and 3% SDS (48%) extracts (Fig. 2d). Importantly, APP transgenic mice also generate A-887755-detectable Aβ oligomers and are therefore suitable for testing antibody efficacy by neutralization of these oligomers (Fig. 2e). The fraction of SDS-soluble Aβ oligomers in Tg2576 brain is lower (16%) as in the brain of AD patients.
Figure 2.

Binding of A-887755 in brain tissue of AD patients and APP transgenic mice ex vivo. a, Aβ20-42 globulomer-selective antibody A-887755 immunoprecipitates a variety of soluble N- and C-terminally truncated but also full-length Aβ1-40 and Aβ1-42 peptides as shown by SELDI-MS analysis of a 3% SDS AD brain extract. Longer APP fragments like the β stub C99 were not precipitated by A-887755. b, Quantification of Aβ1-40 and Aβ1-42 by Aβ globulomer-selective antibodies A-887756 and A-887755, expressed as the fraction of the total amount of the respective Aβ peptide in the 3% SDS extract of a representative AD brain determined by immunoprecipitation with the less selective antibody 6E10. c, The epitope recognized by A-887755 (black column) can be destroyed by pretreatment with formic acid (FA) (gray column). This epitope is not generated by adding synthetic Aβ monomer before the 3% SDS extraction to a control brain (white column), suggesting that A-887755 selectively recognizes an epitope present in endogenous Aβ oligomers. d, e, Soluble Aβ levels determined by immunoprecipitation of 3% SDS and PBS extracts of AD brain (filled symbols; n = 6) and control brain (open symbols; n = 3) (d), and a 3% SDS extract of the hippocampus of 10-month-old Tg2576 mice (n = 6) (e). Total Aβ levels and oligomeric Aβ levels were obtained with nonspecific 6E10 (circles) and globulomer-selective A-887755 (squares), respectively. Nonspecific background levels were determined by binding of a non-Aβ-reactive IgG2a isotype antibody (triangles) that was higher in AD brain because of higher total Aβ peptide levels. For clinical and pathophysiological characterization of the AD samples, see supplemental Table 1 (available at www.jneurosci.org as supplemental material).
A-887755 does not bind to Aβ monomer or amyloid ex vivo
We next evaluated whether A-887755 detects Aβ monomer or deposited Aβ in vivo. The ex vivo discrimination of A-887755 to Aβ monomer was obtained in CSF and plasma of several AD and age-matched control patients. Although 6E10 detected Aβ in both fluids, A-887755 was not able to detect more material than a non-Aβ-binding IgG isotype control antibody (Fig. 3a), suggesting that free soluble Aβ oligomers do not exist in CSF and plasma above a concentration of 5 pg/ml, which was the detection limit of our method. Following the idea that Aβ oligomers might be associated with lipids that might hinder A-887755 immunoprecipitation, we added 3% SDS to CSF in analogy to the extraction of Aβ oligomers from Alzheimer's disease brain. Nevertheless, this did not lead to any detectable immunoprecipitation of Aβ oligomers with A-887755, although the Aβ unselective antibody 6E10 was capable of immunoprecipitating monomeric Aβ peptide (data not shown; note that the 3% SDS had been suitably diluted before immunoprecipitation).
Figure 3.
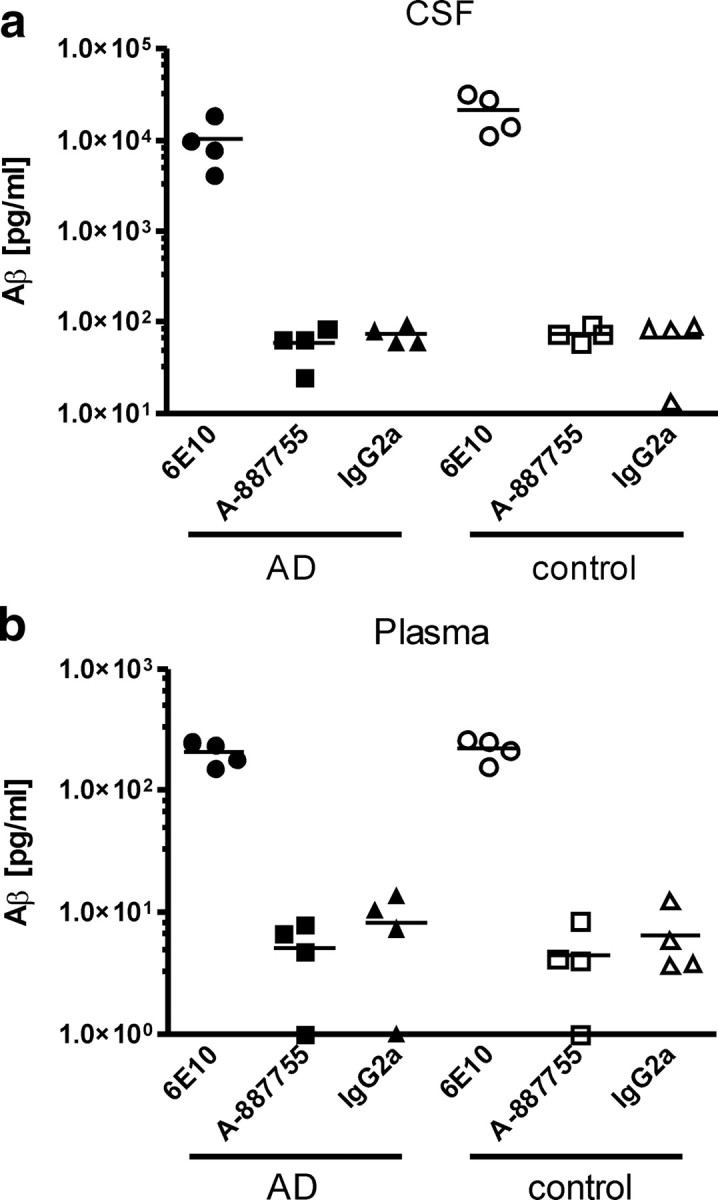
A-887755 does not bind to Aβ monomer in vivo. Soluble Aβ levels determined by immunoprecipitation of 3% SDS extracts of CSF (a) and plasma (b) of AD patients (filled symbols; n = 4) and age-matched controls (open symbols; n = 4). Total Aβ levels and oligomeric Aβ levels were obtained with nonspecific 6E10 (circles) and globulomer-selective A-887755 (squares), respectively. Nonspecific background levels were determined by binding of a non-Aβ-reactive IgG2a isotype antibody (triangles). The lack of A-887755 binding in CSF and plasma indicates absence of detectable levels of free Aβ oligomers in these fluids and high selectivity of A-887755 versus abundant Aβ monomer. For clinical and pathophysiological characterization of the AD samples, see supplemental Tables 2 and 3 (available at www.jneurosci.org as supplemental material).
Then, we investigated the selectivity of A-887755 toward Aβ deposits. Conformation-independent 6E10 and 6G1 but not A-887755 were able to bind to deposited Aβ in the parenchyma or vessels of AD patients (Fig. 4a,b). Binding of 6E10 and A-887755 was tested at various concentrations. Although 6E10 staining of Aβ deposits dose dependently increased, A-887755 did not bind to Aβ deposits up to 7 μg/ml (Fig. 4c). Staining of Tg2576 mouse brains revealed a similar picture as observed in AD brain. Although 6E10 heavily stained amyloid plaques and smaller Aβ deposits, A-887755 only showed background staining (Fig. 4d), again demonstrating that the antibody does not bind to deposited Aβ. A histological cross-reactivity analysis with several tissues of cynomolgus monkey and humans did not result in any specific binding of FITC-labeled A-887755.
Figure 4.

A-887755 does not bind to amyloid in vivo. a, b, Neocortical sections of two AD patients incubated with 0.7 μg/ml of different antibodies. Strong binding of the Aβ conformer-independent antibodies but no binding of A-887755 to Aβ deposits in the brain parenchym (a) and in the vessels (b; arrows) was observed. Scale bar: a, b, 100 μm. c, Quantitative analysis of plaque binding in AD brain sections by optical density of staining after different concentrations of 6E10 (black squares) and A-887755 (white circles). Data are means ± SEM of 10 plaques (background subtracted). d, Quantitative analysis of plaque binding in neocortical sections of Tg2576 mice at 0.7 μg/ml 6E10 (black bar) or A-887755 (white bar) reveals a similar strong difference as in AD patients. Data are means ± SEM of 10 plaques (background subtracted).
A-887755 neutralizes Aβ oligomer-derived neuropathogenic effects in hippocampal neurons and slice cultures
We were then interested in the pathological effects of Aβ20-42 globulomer and their neutralization by A-887755. In a first step, we demonstrated binding of Aβ20-42 globulomer to rat primary hippocampal neurons (Fig. 5a). The punctuated specific binding to neurons was prevented by A-887755 but not by a non-Aβ-binding IgG isotype control antibody. Because this binding pattern is reminiscent of synaptic binding, we also studied evoked synaptic transmission in rat hippocampal slice cultures. Incubation with Aβ20-42 globulomer resulted in a significant decrease in the input/output curve. Preincubation of Aβ20-42 globulomer with A-887755 fully prevented the reduced synaptic transmission (Fig. 5b).
Figure 5.
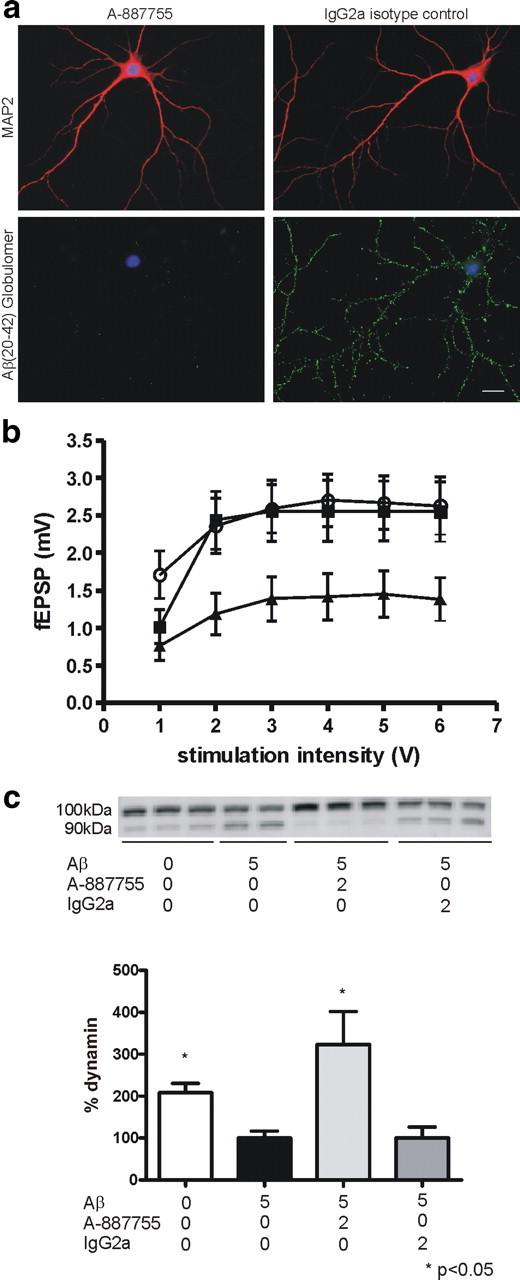
Aβ20-42 globulomer-derived antibodies neutralize Aβ oligomer-derived neuropathogenic effects in hippocampal neurons and slices. a, Incubation of A-887755 antibody with Aβ20-42 globulomer in a 2:1 molar ratio (8.3 nm Aβ20-42 globulomer/16.7 nm A-887755) prevents binding of the Aβ20-42 globulomer to hippocampal neurons, in contrast to the Aβ nonspecific isotype control IgG2a antibody. b, Antibody A-887755 prevents synaptic deficits induced by Aβ20-42 globulomer. Input/output relation was recorded in CA1 from slice cultures treated with 160 nm Aβ20-42 globulomer (filled triangles), 160 nm Aβ20-42 globulomer plus 100 nm A-887755 (filled squares), or vehicle (open circles) for 24 h. Globulomer-induced suppression was significant with p = 0.013, reversal with p = 0.027 (2-way repeated measures ANOVA with Holm–Sidak post hoc analysis). fEPSP, Field EPSPs. c, Antibody A-887755 protects hippocampal neurons against Aβ-induced dynamin cleavage. Top, Samples representing repeated experimental conditions from a single experiment are shown and treatment concentrations (in μm) are indicated below. Cells without addition of Aβ show mostly intact (∼100 kDa) dynamin I signals, which is decreased and reverted to an ∼90 kDa cleavage product in cells treated with 5 μm Aβ. Antibody A-887755 treatment reverts the cleavage, whereas an IgG2a control antibody is without a pronounced protective effect. Bottom, Quantification of the dynamin signal of three independent experiments (expressed as percentage dynamin ± SEM after normalization to 5 μm Aβ treatment) revealed a statistically significant protective effect of A-887755 (one-way ANOVA, Kruskal–Wallis test followed by Dunn's test; *p < 0.05).
Dynamin, a protein involved in synaptic vesicle release, may be involved in Aβ oligomer-induced synaptic pathology because aggregated Aβ1-40 has been shown recently to induce cleavage of dynamin I in hippocampal neurons (Kelly et al., 2005; Kelly and Ferreira, 2006). Using densitometric analysis of β-tubulin-normalized protein level, we also observed a marked decrease in the amount of uncleaved dynamin I and a concomitant increase of the inactive cleavage product after incubation of hippocampal neurons with aggregated Aβ1-40 for 24 h (Fig. 5c). Preincubation of aggregated Aβ1-40 with A-887755 completely prevented the dynamin I cleavage. In contrast, an IgG2a isotype control antibody did not provide any protection from cleavage (Fig. 5c).
Vaccination with Aβ20-42 globulomer mounts an Aβ oligomer-selective antibody response and normalizes cognition in APP/L mice
To address the therapeutic potential of our Aβ oligomer-specific antibody in a preclinical model, we immunized 1.5-month-old APP/L mice with Aβ20-42 globulomer. The selectivity profile of the antibody response in the serum of each individual mouse was determined 3 months later by dot blot against decreasing amounts of the secreted form of APP (sAPPα), Aβ1-40 monomer, Aβ1-42 monomer, Aβ1-42 fibrils, Aβ1-42 globulomer, and Aβ20-42 globulomer. The whole group of Aβ20-42 globulomer-immunized mice showed a very consistent antibody profile, indicating a very selective immune response toward the Aβ20-42 globulomer antigen without any cross-reaction to other Aβ species (Fig. 6a,b). Thus, Aβ20-42 globulomer vaccination of APP/L mice resulted in a monospecific immune response with a similar selectivity profile as shown for A-887755 (Fig. 1b). In contrast, immunization with full-length Aβ1-42 globulomer and Aβ1-42 monomer resulted in a broadband antiserum comprising antibodies not only reacting against Aβ oligomers but also against monomers and sAPPα in some mice (Fig. 6b), i.e., comparable with the selectivity profile of 6G1 (Fig. 1b). To assess the functional consequence of this endogenous antibody response, the mice were subjected to a novel object recognition task (Fig. 6c). The Aβ oligomer-specific immune response was able to normalize the inability of PBS-injected control APP/L mice to recognize a new object. The Aβ oligomer-specific immune response was associated with the improved ability of APP/L mice to recognize a new object, suggesting that antibodies to other Aβ species or sAPPα are not necessary or may even prevent this effect (see Aβ1-42 monomer vaccination in Fig. 6c).
Figure 6.

Oligomer-specific antibodies generated by vaccination with Aβ20-42 globulomer in APP/L mice improve cognition. a, Active immunization with Aβ20-42 globulomer reproducibly mounts an oligomer-specific antibody response. Representative dot blot of the antiserum from an APP/L mouse immunized with Aβ20-42 globulomer shows that the immune response is highly selective for Aβ globulomer and resembles that of A-887755. b, Specific titers of antibodies in APP/L mice from active immunization with Aβ1-42 monomer, Aβ1-42 globulomer, and Aβ20-42 globulomer. Immunization results in titers for Aβ1-42 monomer (light gray bars), Aβ1-42 globulomer (dark gray bars), Aβ20-42 globulomer (black bars), and sAPPα (white bars). Data are relative titers expressed as pEC20 (in picomoles) ± SEM. Detection limit for the monomer and globulomer of Aβ was 100 pmol (pEC20 = −2) and for sAPPα was 1 pmol (pEC20 = 0; therefore, remaining bar below 0 cross-hatched shaded). Although Aβ1-42 monomer and globulomer raised antibodies against all species, including sAPPα, Aβ20-42 globulomer mounted an antibody response specific for the globulomer (Aβ oligomer) epitope. c, Active immunization with Aβ20-42 globulomer improves performance in the novel object recognition task. Performance in novel object recognition of these mice [wild type (wt), white bars; APP/L, black and gray bars]. Recognition index was calculated as the time spent with exploring the new compared with both objects (e.g., 66% meaning double as long for the new object). Data are means ± SEM of the number of animals indicated in the bars. Statistical differences were calculated by ANOVA (p < 0.01), followed by post hoc Bonferroni's t test (*p < 0.05 vs PBS). Circles indicate significant recognition of new objects above chance level (i.e., 50%; °°°p < 0.001).
A-887755 normalizes cognitive performance in APP/L mice
To address the hypothesis that exogenous application of anti-Aβ oligomer antibodies has the same in vivo efficacy as vaccination, we treated 4.5- to 7-month-old APP/L mice with A-887755. The experiments were conducted at the age when cognitive differences between transgenic and nontransgenic mice had been found in previous studies but which was well before amyloid plaque deposition (Moechars et al., 1999; Dewachter et al., 2002). In the novel object recognition task, poor performance of the 4.5-month-old PBS-treated APP/L mice in the novel object recognition task was reversed by 3 week treatment with either the broadband Aβ antibody 6G1 or the Aβ oligomer-selective antibody A-887755 (Fig. 7a). Again, the oligomer-selective anti-Aβ immunotherapy was sufficient for reversing the Aβ-induced deficit. A second experiment evaluated effects of A-887755 on learning in the water maze task. Although 7-month-old APP/L mice performed significantly worse than their wild-type controls, treatment with A-887755 for 10 d before the starting the water maze training abolished the difference between transgenic and nontransgenic mice (Fig. 7b).
Figure 7.

Oligomer-specific A-887755 improves cognitive performance in APP/L mice. a, Treatment with the oligomer-specific A-887755, rather than with less Aβ oligomer-selective antibodies, is sufficient to improve performance in the novel object recognition task. In the novel object recognition test of APP/L mice [wild type (wt), white bars; APP/L, black and gray bars] recognition index was calculated as the time spent with exploring the new compared with both objects (e.g., 66% meaning double as long for the new object). Data are means ± SEM of the number of animals indicated in the bars. Statistical differences were calculated by ANOVA (p < 0.01), followed by post hoc Bonferroni's t test (*p < 0.05, **p < 0.01 vs PBS). Circles indicate significant recognition of new objects above chance level (i.e., 50%; °p < 0.05, °°°p < 0.001). Treatment with the A-887755 antibody eliminated the difference between transgenic and wild-type mice. b, Treatment with the oligomer-specific A-887755 improves learning in the water maze. In the Morris water maze over the days of training, mice learned to find the submerged platform, and this learning was reflected in the overall main effect of time revealed by ANOVA (F(5,287) = 3.4, p < 0.01). After several days of training, wild-type (WT) control animals substantially improved and swam shorter distances to the platform, and these traveled distances were shorter than those in transgenic (TG) mice treated with PBS. Treatment with the A-887755 antibody eliminated the difference between transgenic and wild-type mice as suggested by the genotype × treatment × time interaction (F(5,287) = 2.4, p < 0.05).
A-887755 normalizes synaptic spine density in Tg2576 mice
Finally, we were interested in the anatomical basis of Aβ oligomer pathology and its normalization by A-887755. As suggested by recent reports of pathogenic effects on synapses by Aβ oligomers in vitro (Lacor et al., 2007; Shankar et al., 2007), we focused on synaptic spine density in APP transgenic mice. Because we had identified a reduction of synaptic spines in 6-month-old Tg2576 mice, we injected A-887755 for 3 weeks in 5-month-old mice of this strain and quantified the synaptic spine density in hippocampal CA1 by Golgi staining. In the basal dendrites of pyramidal neurons, a reduced density of synaptic spines was observed that was normalized by A-887755 treatment (Fig. 8b). A quantitative analysis revealed a significantly reduced synaptic spine density in the basal but not the apical CA1 pyramidal dendrites of Tg2576 mice that was normalized in the proximal part of the dendrites by A-887755 (Fig. 8a).
Figure 8.

Oligomer-specific A-887755 improves synaptic spine density in Tg2576 mice. Five-month-old APP transgenic Tg2576 or nontransgenic control mice were intraperitoneally injected with 500 μg of A-887755 or PBS (vehicle) once weekly for 3 weeks. Three days after the last injection, the animals' brains were Golgi stained for reconstruction of neuronal morphology. The density of dendritic mushroom-type spines of CA1 pyramidal cells was quantitatively analyzed. a, Quantitative analysis of spine density in basal proximal dendrites (30–120 μm from soma) showed a significant reduction of mushroom-type spines in Tg2676 mice that was reversed by A-887755 (ANOVA, F(2,173) = 8.75, p < 0.05; post hoc Bonferroni's t test, *p < 0.05, **p < 0.01). Spine density was not changed in other parts of the CA1 pyramidal neuron dendrites. WT, Wild type. b, Golgi-stained basal dendrite of two Tg2576 mice treated with PBS or with A-887755. Scale bar, 10 μm.
Discussion
Our understanding of the neuropathology of Aβ has changed considerably with the accumulating evidence that Aβ oligomers are probably the most neuropathogenic Aβ species for synapses and cognition (Walsh and Selkoe, 2007). Toxic Aβ oligomers of different size have been studied, and the smallest apparent oligomeric species of Aβ in SDS-PAGE, a dimer, has been isolated recently from AD brain and its pathology demonstrated in preclinical AD models (Shankar et al., 2008). Toxicity of quite different Aβ oligomers raise the important question if size, i.e., Aβ dimer, dodecamer, or any other multimer size, is the main characteristic for its neuropathogenic nature or whether Aβ becomes toxic because of a conformational switch triggering subsequent formation of various misfolded Aβ oligomers structurally different from Aβ monomer or fibril. Aβ oligomer-containing brain extracts of AD patients (Shankar et al., 2008) or APP transgenic mice (Lesné et al., 2006) provided strong evidence for the toxic nature of Aβ oligomers but could not reveal their neuropathologic mechanism. The idea to study this mechanism by synthetic Aβ oligomers was first addressed with the ADDL preparation (Lambert et al., 1998). However, ADDLs turned out as a mixture of Aβ oligomers of different size and residual Aβ monomer, which could not answer the question of size or conformation of Aβ oligomer neuropathology. To test the conformation hypothesis, we have introduced Aβ1-42 globulomer as a homogenous and stable oligomer (Barghorn et al., 2005). Although the hypothesis of a different conformation to Aβ monomer and fibril was supported by the demonstration of separate aggregation pathway in vitro (Gellermann et al., 2008), Aβ1-42 globulomer did still produce antibodies such as 6G1 that detected all Aβ species, including sAPPα. We hypothesized that this was attributable to the N-terminal sequence of Aβ shared by all these species and therefore removed hydrophilic N-terminal amino acids by limited proteolysis. This procedure resulted in a poorly soluble Aβ20-42 globulomer, which, however, had the advantage of mounting a monospecific antibody response in APP transgenic mice yielding highly Aβ oligomer-selective monoclonal antibodies such as A-887755. This antibody bound only to Aβ20-42 globulomer but not to Aβ1-42 globulomer in dot blots, suggesting recognition of N-terminally truncated Aβ oligomers only. However, when using AD and APP transgenic mouse brain extracts, A-887755 was able to detect full-length Aβ, suggesting that synthetic Aβ1-42 globulomer is very tightly packed, leaving no access for antibodies that bind to regions beyond amino acid 20. In fact, antibodies generated with synthetic Aβ20-42 globulomer were able to exclusively bind to misfolded Aβ oligomers in AD brain tissue, both full-length and truncated, but not to Aβ monomer or fibrils, even when highly abundant as in CSF or amyloid plaques, respectively. Thus, Aβ20-42 globulomer and A-887755 were identified as excellent tools to address the following questions: (1) what is the structural basis of the special neuropathologic conformation of Aβ oligomers, (2) how abundant are the neuropathogenic Aβ oligomers in the brain of AD patients, and (3) does neutralization of this oligomeric Aβ conformer provide therapeutic efficacy in preclinical models of AD?
Assuming a conformational change as the basis of Aβ oligomer neuropathology, our data suggest that the relevant structural motif is located in the C-terminal part of the Aβ sequence between amino acids 20 and 42. Previous work by Teplow et al. (2006) hint to an Aβ21–30 loop region that may distinguish Aβ monomer, oligomers, and fibrils. We have recently analyzed the globulomer structure by nuclear magnetic resonance and found that the characteristic globulomer epitope is formed by a loop consisting of amino acids 20–30 and that A-887755 particularly binds to this 1oop (Yu et al., 2009). The change of Aβ conformation from the nonpathogenic monomer to the globulomer likely relies on additional factors such as lipids or lipoproteins (Barghorn et al., 2005), e.g., apolipoprotein E (Tokuda et al., 2000). Why and when the pathological event of Aβ misfolding takes place in Alzheimer's disease will be a most interesting question for additional studies with our conformation-dependent antibody. So far, we have observed highly specific binding of A-887755 only to Aβ oligomer but not to any other physiological or misfolded peptide, such as α-synuclein, which is in contrast to the binding of antibody A11 to several misfolded proteins (Kayed et al., 2003).
Another important question addressable by A-887755 is the amount and distribution of neuropathogenic Aβ oligomer in the brain. The monoclonal antibody does not detect the abundant Aβ monomers in CSF nor the accumulating deposited Aβ fibrils in brain tissue. Therefore, all peptides in brain extracts detected by the monoclonal antibody were misfolded Aβ peptides. Initially, this conformational switch of the loop comprising amino acids 20–30 may occur at the monomeric state, but our data suggest that this change from physiological to the pathological state is immediately stabilized by oligomerization. Compared with the total amount of Aβ peptides in brains of AD patients or aged APP transgenic mice, the soluble oligomer fraction is small. Our data demonstrate, however, that in SDS extractable material of disease brain, containing only soluble Aβ peptides, up to 50% of total Aβ was detected by A-887755. The remaining 50% of total Aβ may not have been detected because these were Aβ monomers, Aβ oligomers in a different conformation, or inaccessible for A-887755 because the antibody binding region was occupied by lipids or other binding partners. When comparing the binding of A-887755 with PBS and SDS extracts, extraction with 3% SDS increased the soluble fraction of the presumably neuropathogenic oligomers, suggesting that they are not freely diffusible but rather associated with membrane components or matrix proteins in vivo. This is supported by the lack of binding to biological fluids, i.e., no Aβ globulomer epitope detection above detection limit of 5 pg/ml for plasma and 70 pg/ml for CSF. If misfolded Aβ oligomers were freely diffusible, A-887755 would have been able to detect them in the CSF. The fact that a small amount of Aβ oligomers can be extracted by PBS and that they are enriched in the extracellular fraction (Lesné et al., 2006) suggests that they are weakly attached on the extracellular side. Specific binding of Aβ oligomers to neurons is suggested by the binding of the synthetic Aβ20-42 globulomer and ADDLs (Lacor et al., 2007) to hippocampal neurons. Recently, it has been shown that that presence of collagen VI prevents binding of Aβ oligomers to neurons, suggesting a high affinity to extracellular matrix protein (Cheng et al., 2009). We have also observed a strong binding of poorly soluble Aβ20-42 globulomer to extracellular matrix proteins such as laminin and collagen (our unpublished observation). It is therefore conceivable that not only formation of neuropathogenic Aβ oligomers is dependent on lipids but that oligomers also bind to lipids and extracellular matrix at the site of generation and are widely distributed in contrast to fibrillar Aβ in plaques. This, however, does not preclude that the concentration of Aβ oligomers can be highest around the plaques (Koffie et al., 2009) and that endogenous Aβ oligomers may serve as a seeding core for plaque formation (Meyer-Luehmann et al., 2006; Shankar et al., 2008). The comparison of the effects of A-887755 with that of conformation-independent anti-Aβ antibodies will help to better understand a potential interaction between plaque and oligomer-type amyloid deposits.
Toxicity of Aβ oligomers has been reported to be closely associated with synapses. Synthetic and natural Aβ oligomer preparations bind to synaptic sites on hippocampal neurons and interfere with synaptic transmission (Lacor et al., 2004, 2007; Shankar et al., 2007). This interaction is considered the pathological basis of dementia in AD and of morphological and functional deficits in APP transgenic mice (Walsh and Selkoe, 2007). Aβ immunotherapy has been used in the past to improve cognitive function in APP transgenic mice (Dodart et al., 2002; Hartman et al., 2005). Although the previously used conformation-independent antibodies, e.g., 266 or 10D5, should be able to neutralize Aβ oligomers, part of their efficacy may have been associated with binding to deposited fibrillar or soluble monomeric Aβ (Seubert et al., 2008). A-887755 or the oligomer-specific polyclonal antibodies after active vaccination were also efficacious in improving cognitive function in APP transgenic mice, demonstrating that selective neutralization of Aβ oligomers is sufficient to obtain this effect. The striking reduction of synaptic spines by Aβ oligomers in hippocampal neurons as a morphological substrate of reduced cognitive function was reversed by antibody 6E10, which recognizes all Aβ species with intact N terminus (Shankar et al., 2007). We have shown a similar reduction of synaptic spines in hippocampal CA1 dendrites in APP transgenic mice. A-887755 was able to partially reverse this deficit, confirming that an Aβ oligomer-selective treatment is sufficient to ameliorate neuropathology. Amyloid plaque removal seems unable to stop ongoing dementia in AD patients (Holmes et al., 2008). Thus, neutralization of Aβ oligomers appears to be a more attractive primary focus of Aβ immunotherapy. If effective immunotherapy is achieved by neutralization of misfolded Aβ oligomers, a conformation-independent anti-Aβ antibody might have some unwanted side effects. Binding to continuously generated Aβ monomers will consume these antibodies and reduce their ability to neutralize the less abundant Aβ oligomers. Binding to fibrillar Aβ will remove plaques but may also increase the risk of inflammatory responses, such as microhemorrhages in APP transgenic mice or vasogenic edema in AD patients (Pfeifer et al., 2002; Racke et al., 2005). A number of anti-Aβ oligomer-selective antibodies have been described, but they have limitations because they are polyclonal like the A11 antibody (Kayed et al., 2003) or they clearly recognize fibrillar Aβ as well and still bear the associated side-effect risk (Lee et al., 2006, Lambert et al., 2007). Strictly Aβ oligomer-specific monoclonal antibodies such as A-887755 that are devoid of any monomer or fibril binding will be extremely helpful tools to fully evaluate the therapeutic potential of Aβ oligomer-selective immunotherapy. Although efficacy of A-887755 by reducing soluble Aβ oligomers has been demonstrated in APP transgenic mice before any plaque deposition, the clinical situation is quite different. The antibody has to prove its potency also in the presence of a high concentration of deposited Aβ and it will be interesting and important to test this in the preclinical mouse model.
In summary, the use of Aβ20-42 globulomer allowed us to conclude that the neuropathogenic effect of Aβ oligomers is based on a specific conformation rather than on a specific molecular weight. Using the globulomer conformation, we were able to generate the specific monoclonal antibody A-887755, which was helpful in demonstrating the presence of Aβ oligomers in brain tissue of AD patients or APP transgenic mice but not in age-matched nondiseased individuals. Finally, the improved spine density and cognitive function in APP transgenic mice after A-887755 treatment raises the hope that selective Aβ oligomer immunotherapy is a promising therapeutic approach without side effects associated with general anti-Aβ therapy. In addition, the globulomer concept will assist in reaching a clearer understanding of the molecular mechanisms of Aβ oligomer pathology, like a recently described interference with the presynaptic P/Q-type calcium current (Nimmrich et al., 2008).
Footnotes
We are grateful to our colleagues from Abbott, in particular Gerry Fox, Gerhard Gross, and Alfred Hahn for helpful discussion, Nathan Rustay for providing Tg2576 mouse brains, and Bodo Janson and Tanja Georgi for excellent technical support. We thank Manuela Neumann (Brain-Net, Munich, Germany) for providing the AD and control brain samples.
The salaries of authors (except for I.v.d.A. and F.v.L.) and the study were funded as part of an ongoing drug discovery program at Abbott Laboratories.
References
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H. Globular amyloid β-peptide oligomer: a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JS, Dubal DB, Kim DH, Legleiter J, Cheng IH, Yu GQ, Tesseur I, Wyss-Coray T, Bonaldo P, Mucke L. Collagen VI protects neurons against Aβ toxicity. Nat Neurosci. 2009;12:119–121. doi: 10.1038/nn.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Reversé D, Caluwaerts N, Ris L, Kuipéri C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 Inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002;22:3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Gellermann GP, Byrnes H, Striebinger A, Ullrich K, Mueller R, Hillen H, Barghorn S. Aβ-globulomers are formed independently of the fibril pathway. Neurobiol Dis. 2008;30:212–220. doi: 10.1016/j.nbd.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an amyloid-β antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. β-amyloid-induced dynamin 1 degradation is mediated by N-methyl-d-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Vassar R, Ferreira A. β-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer's disease. J Biol Chem. 2005;280:31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Mononclonal antibodies that target pathological assemblies of Aβ. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-β peptide (Aβ) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Aβ precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral β-amyloidogenesis as governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C. Amyloid β oligomers (Aβ1-42 globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium channels. J Neurosci. 2008;28:788–797. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, Kling K, Schenk D, Johnson-Wood K, Schroeter S, Gill D, Jacobsen JS, Pangalos M, Basi G, Games D. Antibody capture of soluble Aβ does not reduce cortical Aβ amyloidosis in the PDAPP Mouse. Neurodegener Dis. 2008;5:65–71. doi: 10.1159/000112834. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brain impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, Baumketner A, Shea JE, Urbanc B, Cruz L, Borreguero J, Stanley HE. Elucidating amyloid β-protein folding and assembly: a multidisciplinary approach. Acc Chem Res. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Zlokovic B, Smith JD, Ladu MJ, Rostagno A, Frangione B, Ghiso J. Lipidation of apolipoprotein E influences its isoform-specific interactions with Alzheimer's amyloid beta peptides. Biochem J. 2000;348:359–365. [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Aβ Oligomers: a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]