

Abstract
Short-term habituation is a basic form of learning that is analyzed in different species and using different behavioral models. Previous studies on mechanisms of short-term habituation yielded evidence for a potential role of group III metabotropic glutamate receptors (mGluRIIIs). Here we tested the hypothesis that mGluRIII mediate short-term habituation of startle in rats, combining electrophysiological experiments in vitro with behavioral studies in vivo. We applied different mGluRIII agonists and antagonists on rat brainstem slices while recording from startle-mediating neurons in the caudal pontine reticular nucleus (PnC) and monitoring synaptic depression presumably underlying habituation. Furthermore, we injected the mGluRIII antagonist (RS)-α-phosphonophenylglycine (MPPG) and the agonist l-(+)-2-amino-4-phosphonobutyric acid (l-AP4) into the PnC of rats in vivo and measured its effect on startle habituation. Our results show that activation of mGluRIIIs in the PnC strongly inhibits startle-mediating giant neurons in vitro. Accordingly, l-AP4 reduced startle responses in vivo. However, synaptic depression in the slice was not disrupted by mGluRIII antagonists or agonists. Correspondingly, the in vivo application of the mGluRIII antagonist MPPG failed to show any effect on short-term habituation of startle responses. We therefore conclude that mGluRs are expressed within the primary startle pathway and that they inhibit startle responses upon activation; however, this inhibition does not play any role in synaptic depression and short-term habituation of startle. This is in contrast to the role of mGluRIIIs in other forms of habituation and supports the notion that there are different mechanisms involved in habituation of sensory-evoked behaviors.
Introduction
Habituation is a non-associative form of learning that is crucial to adjust behavioral responses to repeated sensory stimuli. It can be observed throughout the animal kingdom. Habituation mechanisms are probably well conserved, but there is also evidence that different mechanisms contribute to habituation, even within the same model (Rankin, 2000, 2009; Giles and Rankin, 2009).
One well studied rodent model system for habituation is short-term habituation to odors, as it can be measured as a decline in odor-evoked, heart-rate orienting responses, or as a decreased magnitude of odor-evoked responses in the piriform cortex (Wilson, 2000, 2009). It has been shown that the activation of group III metabotropic glutamate receptors (mGluRIIIs) within the olfactory sensory pathway mediates this decrease (Best et al., 2005). In a different behavioral rodent model, namely the short-term habituation of the startle response to acoustic stimuli, preliminary evidence for a potential role of mGluRIIIs has also been reported (Weber et al., 2002). Here we tested the hypothesis that activation of mGluRIIIs within the primary startle pathway underlies short-term habituation of acoustic startle responses, analogous to the mechanisms described for odor habituation.
The startle response is a protective response, elicited by a sudden and intense sensory stimulus that activates muscles within a few milliseconds (for review, see Davis, 1984; Koch, 1999; Yeomans et al., 2002). The primary startle pathway is very short: secondary sensory neurons within the startle pathway project onto giant neurons in the caudal pontine reticular formation (PnC), the sensorimotor interface of the primary startle pathway (Yeomans and Frankland, 1995; Lee et al., 1996; Schmid and Weber, 2002; Yeomans et al., 2002; Schmid et al., 2003). PnC giant neurons in turn project directly onto spinal, cranial, and facial motoneurons to elicit the startle response (Lingenhöhl and Friauf, 1992, 1994). Startle responses are modulated by emotions, learning, and sensorimotor gating, and presumably mediated by projections from different brain areas to the PnC (for review, see Koch and Schnitzler, 1997). In contrast, short-term habituation is considered to be caused by synaptic changes within the primary startle pathway itself, specifically by depression of synaptic input from sensory afferents to PnC giant neurons (Davis et al., 1982; Lingenhöhl and Friauf, 1992; Weber et al., 2002; Simons-Weidenmaier et al., 2006). There is preliminary evidence for a possible role of mGluRIIIs in depression of this synaptic input (Davis and Gendelman, 1977; Weber et al., 2002).
The fast synaptic transmission of startle signals in the PnC is mediated by AMPA and NMDA receptors (Krase et al., 1993; Weber et al., 2002). The action of metabotropic glutamate receptors is generally slower and rather modulatory. mGluRIIIs include mGluRs 4, 6, 7, and 8, and are coupled via Gi/Go to inhibition of adenylyl cyclase activity (Gerber et al., 2007). They are expressed predominantly in axonal terminals where they regulate transmitter release (Pinheiro and Mulle, 2008).
Here we tested the hypothesis that mGluRIIIs are functionally expressed in the PnC and that their activation induces synaptic depression of sensory afferents to the PnC neurons, which is assumed to underlie short-term habituation of startle. In parallel, we tested whether mGluRIII activation in vivo by stereotaxic injection of agonists and antagonists affects short-term habituation of startle responses.
Materials and Methods
Electrophysiology
Preparation of brain slices.
Details of the preparation have been reported previously (Simons-Weidenmaier et al., 2006). In brief, Sprague Dawley rats (10–15 days old; Charles River Laboratories) were deeply anesthetized with halothane, decapitated, and the brains were rapidly removed and transferred into ice-cold preparation solution containing the following (in mm): 210 sucrose, 26 NaHCO3, 1.3 MgSO4 1.2 KH2PO4, 2 MgCl2, 2 KCl, 2 CaCl2, 10 glucose, 3 myoinositol, 2 sodium pyruvate, and 0.4 ascorbic acid, equilibrated with 95% O2/5% CO2. Coronal slices (400 μm) were then cut with a vibratome (HM 650 V; Microm) in a submerged chamber filled with ice-cold preparation solution. Slices were transferred into a holding chamber filled with artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 26 NaHCO3, 1.2 KH2PO4, 1.3 MgSO4 2 KCl, and 10 glucose. CaCl2 (2 mm) was added a few minutes after the slices had been transferred. The holding chamber was heated for 1 h to 34°; slices were then kept at room temperature. All experiments were performed in accordance with German and Canadian animal protection laws.
Electrophysiological recording.
For recording, slices were transferred into a superfusion recording chamber mounted on an upright fixed stage microscope (Zeiss) with infrared differential interference optics. Superfusion rate was 2–3 ml of ACSF per minute. Patch-clamp recordings were made at room temperature under visual guidance by an infrared-sensitive camera (Kappa). Patch electrodes were pulled out of borosilicate capillaries (Science Products) and filled with a solution containing the following (in mm): 130 potassium gluconate, 5 KCl, 0.6 EGTA, 10 HEPES, 2 MgCl2, pH 7.2 (KOH), 270–290 mosm. Electrodes had a resistance of 2–5 MΩ. Giant neurons in the area of the PnC were identified by a soma diameter >35 μm. All measurements were made in the voltage-clamp configuration at −70 mV. Presynaptic stimuli were applied by bipolar concentric tungsten electrodes (SNEX; Science Products), connected to a stimulator (Isostim A320, WPI). The stimulation electrodes were positioned in the trapezoid body, ventrolateral to the lateral superior olive to stimulate auditory fibers arising from the cochlear nucleus and the cochlear root, and medial to the principle nucelus V (Pr5) to stimulate trigeminal fibers cruising from the Pr5 to the PnC (Schmid et al., 2003; Simons-Weidenmaier et al., 2006). Recordings were made using an Axopatch 200B amplifier and digitized by Digidata 1320 (both from Molecular Devices). The data were filtered with a 10 kHz low-pass filter with a sampling rate of 20 kHz. The pClamp 8.0.2 software (Molecular Devices) was used for data acquisition and analysis. Stimulus intensities were kept low to avoid generating spikes in the neurons. Stimulus duration was 150 μs. Paired pulses were applied with a 50 ms interstimulus interval (ISI). Measurements were repeated 10 times at 0.2 Hz for each cell before (control) and during synaptic depression induced by a sequence of 20 bursts. Ten traces were averaged for each condition and the paired-pulse ratio was determined for each cell from the average trace. Synaptic depression was induced by a sequence of 20 burst stimuli at 1 Hz. Each burst consisted of four stimuli given within 18.6 ms (spacing, 6 ms), which evoked one large compound EPSC (cEPSC) due to the large membrane time constant of giant neurons. At least 5 min recovery time was always allowed before applying a sequence of 20 bursts. The paired-pulse measurements under synaptic depression conditions followed immediately after the end of the 20 burst sequence. All EPSC amplitudes were quantified by setting the baseline current to zero and measuring the most negative current value of the evoked EPSC or cEPSC. Values are expressed as means ± SEM. Series resistance and seal quality was monitored at the beginning and several times during recordings to assure constant measurement conditions. Recordings were discarded when access resistance was larger than 30 MΩ or leak current more than −300 pA, or when one of these parameters was altered during recording.
Drugs were applied by adding them to the superfusing ACSF solution. l-(+)-2-amino-4-phosphonobutyric acid (l-AP4) was applied at a concentration of 50 μm, (RS)-α-phosphonophenylglycine (MPPG) at 200 μm, and RS-α-methylserine-O-phosphate (MSOP) at 100 μm. They were stored as 100× stock solutions in aqua bidest at 4°C.
Behavior
Animals.
Male Sprague Dawley rats (Charles River), weighing 250–350 g, were used in these studies. They were housed in groups of 5–6 animals under a continuous 12 h light-dark cycle (light on at 07:30) with 12 g/rat/d rat chow and water available ad libidum. The experiments were done in accordance with the ethical guidelines for the care and use of laboratory animals for experiments and were approved by the local animal care committee (Regierungspräsidium Tübingen, ZP 4/02, and University of Western Ontario).
Surgery and intracranial injections.
Rats were anesthetized with ketamine/xylazine (9:1; 100 mg/kg i.p.) and placed in a stereotaxic frame with blunt ear bars. Two stainless steel guide cannulae (diameter, 0.7 mm) were implanted bilaterally into the brain aiming at the PnC [−9.8 mm caudal, ±1.0 mm lateral, −9.6 mm ventral to bregma (Paxinos and Watson, 2005)]. The cannulae were fixed to the skull with dental cement and three anchoring screws. After surgery and between the experiments, the cannulae were fitted with stylets (diameter, 0.4 mm) to maintain patency. Rats were tested after full recovery of 5–7 d.
The rats received infusions of the mGluR group II/III-specific antagonist MPPG (Biotrend) or l-AP4 (Tocris Bioscience) dissolved in ACSF or saline. Stainless steel injectors were inserted into the implanted cannulae (diameter, 0.4 mm) and 0.5 μl of drug solution or ACSF was injected bilaterally with a velocity of 0.1 μl/min immediately before behavioral testing. Concentrations were as indicated in the text and figures. The injectors were left in place at least for additional 2 min to allow diffusion of the solution away from them before they were removed and replaced by the stylets.
Test on startle habituation.
The acoustic startle responses were measured in identical sound-attenuated test chambers (100 × 80 × 60 cm3). The rats were placed in holders resting on a piezoelectric accelerometer (custom-made at the University of Tübingen and Med Associates). Movements of the rats resulted in voltage output changes in the accelerometers. These changes were amplified, digitized, and then analyzed by a computer. A loudspeaker delivered acoustic startle stimuli and a continuous white background noise [52 dB sound pressure levels (SPL)]. Acoustic stimuli were generated by a computer using a digital signal processor board (Medav, SigGen, and Med Associates). The whole-body startle amplitude was calculated from the difference between the peak-to-peak voltage output of the accelerometer in time windows of 80 ms after and 80 ms before the startle stimulus onset.
Each animal received injections of ACSF and MPPG or saline/ACSF and l-AP4 into the PnC in a pseudorandomized order on 2 test days separated by a wash-out period of at least 5 d (unless otherwise noted). Immediately after the infusions, the rats were placed into the test cage and allowed to acclimatize for 10 min. For the MPPG/ACSF experiments, startle stimuli (10 kHz tone pulse, 20 ms duration including 0.4 ms rise and fall times, 30 s interstimulus interval) were presented with different SPL to evaluate the individual startle threshold for each animal. For threshold detection, the ascending method of limits was used, i.e., the first stimulus had an intensity of 60 dB SPL and the following stimuli were gradually increased by 2.5 dB SPL until the first startle response was detected. The animal was observed by the experimenter and startle response was defined as a fast flexor contraction of the neck, the shoulder, and finally the whole body. After detecting the first startle response, 20 startle stimuli were presented with an intensity of 20 dB SPL over the individual startle threshold. This protocol was used since it produces the most robust startle habituation curve in our laboratory (P. Pilz, personal communication). For later experiments (high dose of MPPG and l-AP4 injections), 20 startle stimuli with 105 dB SPL (which is ∼20 dB above threshold) were presented.
Histology.
After completion of the behavioral tests, the rats were killed by an overdose of Nembutal. Small amounts (∼0.1 μl) of thionin were injected through the cannulae in some cases to make it easier to determine the localization of the injector tips. The brains were then removed and immersion fixed with 8% paraformaldehyde in PBS with 20% sucrose. Coronal sections of 60 μm were taken on a freezing microtome and stained with hematoxylin and eosin (H&E) using a standard protocol. The injection sites were drawn onto plates taken from a rat brain atlas (Paxinos and Watson, 2005).
Statistical analysis
Paired t tests and ANOVAs were used for statistical analysis of the raw data as appropriate. In the electrophysiological experiments, n refers to the number of neurons; typically, only one neuron was recorded per animal. In behavioral experiments, n refers to the number of animals. A single-factor ANOVA was used to analyze the effect of a drug. A score for synaptic depression/habituation was determined by the following equation: 100 − [mean of the last five cEPSCs (or startle responses) × 100/mean of first two cEPSCs (or startle responses)]; therefore expressing the percentage of response decline. Paired t tests were used to compare scores. To determine the effect of a drug on synaptic depression/habituation, two-factor ANOVAs with repeated measurements with treatment and startle trial number as within-subject factors were performed. A p value of <0.05 was used as the criterion for statistical significance.
Results
Electrophysiology
Effect of the mGluR agonist l-AP4 on synaptic transmission
First, we tested whether functional mGluRIIIs are expressed in the PnC by applying the specific agonist l-AP4 (50 μm). All recorded neurons were located in the PnC and had a soma diameter larger than 35 μm, qualifying them as giant neurons (Lingenhöhl and Friauf, 1992). Trigeminal and auditory afferent fibers were stimulated and stimulation intensities were adjusted so that EPSCs in PnC giant neurons reached amplitudes between −300 and −400 pA, around two-thirds of the maximum amplitude. Figure 1, A and B, shows the effect of bath application of l-AP4 on evoked EPSCs. Trigeminally evoked EPSCs decreased by 78% (from 477 ± 60 pA control to 107 ± 30 pA in presence of l-AP4; ANOVA, F(2,21) = 18.19, p < 0.0001). Auditory EPSCs decreased by 72% (from 528 ± 78 pA to 150 ± 35 pA; ANOVA, F(2,18) = 11.48, p = 0.0011). This indicates that mGluRIIIs are expressed in the PnC and their pharmacological activation inhibits synaptic transmission from sensory afferents to the PnC giant neurons. Resting membrane potentials and input resistance of neurons remained unchanged during l-AP4 perfusion (data not shown). Analysis of the paired pulse ratio (PPR) (EPSC2 amplitude/EPSC1 amplitude) showed a trend to higher PPR during l-AP4 inhibition. The PPR of auditory-evoked EPSCs increased from 1.51 ± 0.04 to 1.85 ± 0.33 pA and of trigeminally evoked EPSCs from 1.34 ± 0.13 to 1.96 ± 0.31 pA, and both PPR declined upon washout to 1.42 ± 0.08 and 1.48 ± 0.22 pA, respectively (Fig. 1C). However, a paired t test revealed a p = 0.078 for trigeminal and p = 0.031 for auditory PPR alterations.
Figure 1.
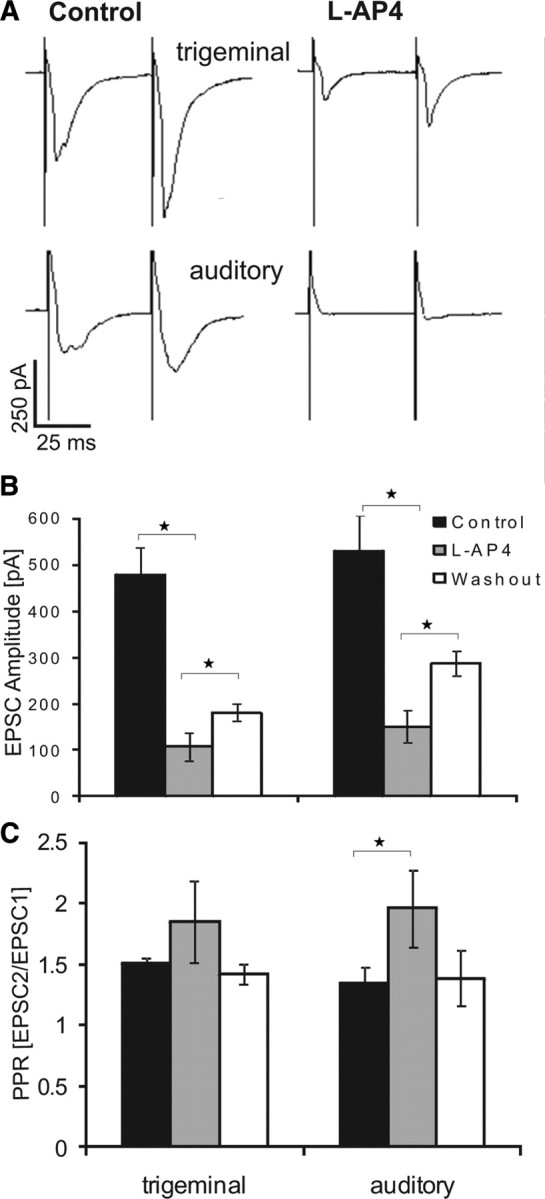
l-AP4 inhibits synaptic transmission in PnC giant neurons. A, Sample traces of EPSCs evoked by stimulation of trigeminal (top) and auditory (bottom) afferent fibers under control conditions and in presence of l-AP4. B, Average EPSC amplitudes under control conditions, in presence of 50 μm l-AP4, and after wash out. l-AP4 significantly decreased EPSC amplitudes. This was partly reversed by subsequent wash out. C, Paired pulse ratio (EPSC2/EPSC1 amplitude) under the different conditions. l-AP4 significantly increased the paired-pulse ratio of auditory EPSCs and there was a trend to an increased PPR with trigeminal stimulation, indicating a possibly presynaptic effect of l-AP4 (n = 8 cells for trigeminal and n = 7 cells for auditory stimulation, error bars indicate SEM, asterisks indicate statistically significant differences, p < 0.05).
Effect of mGluR antagonists on synaptic transmission
To test whether group III mGluRs are activated during synaptic transmission from sensory afferents to PnC giant neurons, the group II/III-specific antagonist MPPG (200 μm) or the group III-specific antagonist MSOP (100 μm) were applied by being added to the bath solution, and EPSC amplitudes were compared with control measurements in the same experimental setting as described above. Trigeminally evoked EPSC amplitudes averaged 365 ± 55 pA under control conditions, 334 ± 50 pA in presence of MPPG, and 300 ± 69 pA after a 10 min wash out of MPPG (n = 14). Auditory EPSC amplitudes were 335 ± 41 pA (control), 332 ± 41 pA (MPPG), and 300 ± 69 pA (after a 10 min wash out period) (n = 13) (Fig. 2A,B). We found no significant effect of MPPG on trigeminally or auditory-evoked synaptic current amplitudes in PnC giant neurons (ANOVA, df(2,38), F = 0.306, p = 0.7383; and ANOVA, df(2,36), F = 0.682, p = 0.513, respectively). The application of MSOP revealed similar results: trigeminally evoked EPSCs were 378 ± 45 pA under control conditions, 342 ± 46 pA in presence of MSOP, and 330 ± 29 pA after 10 min wash out; auditory EPSCs were 331 ± 69 pA under control, 334 ± 68 pA with MSOP, and 299 ± 59 pA after washout (n = 6) (Fig. 2C,D). Therefore, there was no significant effect of MSOP on trigeminally evoked synaptic currents (ANOVA, df(2,13), F = 0.368, p = 0.6979) nor on auditory EPSCs (ANOVA, df(2,13), F = 0.086, p = 0.9182). These results indicate that mGluRIIIs are not activated during baseline synaptic transmission in the PnC at the low stimulation frequencies used here (two stimuli every 5 s), because mGluRIII antagonists had no effect.
Figure 2.

Group III mGluR antagonists do not affect synaptic transmission. A, Afferent fibers within the startle pathway were presynaptically stimulated and EPSCs were measured in PnC giant neurons under control conditions and in presence of 200 μm MPPG. Sample traces of EPSCs evoked by stimulation of trigeminal (top) and auditory (bottom) afferent fibers under both conditions. B, Average EPSC amplitudes under control conditions, in the presence of MPPG, and after wash out. MPPG did not significantly change EPSC amplitudes (n = 14 cells for trigeminal and n = 13 cells for auditory stimulation). C, Sample traces of EPSCs evoked by stimulation of trigeminal (top) and auditory (bottom) afferent fibers under control conditions and in presence of 100 μm MSOP. D, Average EPSC amplitudes under control condition, in presence of MSOP, and after wash out. MSOP did not significantly change EPSC amplitudes (n = 6 cells, error bars indicate SEM).
How are mGluRIIIs in the PnC activated? Group III mGluRs have been shown to be located either in areas adjacent to synaptic zones or, if they are of low affinity, within synaptic zones (Pinheiro and Mulle, 2008). In both cases, they are activated by a high concentration of glutamate accumulating in the synaptic cleft upon stimulation at high frequencies. This turns mGluRIIIs into excellent candidates for mediating synaptic depression specifically induced by short, high-frequency trains of action potentials—a form of plasticity that has been proposed to underlie short-term habituation of startle responses (Weber et al., 2002; Simons-Weidenmaier et al., 2006). If this hypothesis is true, mGluRIII antagonists should block synaptic depression and an mGluRIII agonist should induce it. Moreover, short-term habituation of startle should be disrupted by injection of an mGluRIII antagonist into the PnC in vivo. We tested this using the same antagonists and agonist as in the experiments above.
Effect of mGluRIIIs on synaptic depression
As previously reported, short trains of four action potentials (interstimulus interval, 4–6 ms) applied at 1 or 0.1 Hz lead to synaptic depression that lasts a couple of minutes, after which the synapses fully recover (Weber et al., 2002; Simons-Weidenmaier et al., 2006). To test the role of mGluRIIIs in this form of synaptic depression, we induced synaptic depression under control conditions, subsequently applied mGluRIII antagonists or agonists by adding them to the bath solution, and then repeated the induction of synaptic depression after the synapses had fully recovered from depression during a 10 min recovery period. Figure 3A displays the course of synaptic depression under control conditions, in presence of MPPG and after wash out, always with a 10 min recovery period in between. MPPG had no influence on synaptic depression (repeated-measurements ANOVA, for interaction of drug × synaptic depression: trigeminal, df(1,64), F = 0.16, p = 0.69; auditory, df(1,56), F = 0.09, p = 0.76) (Fig. 3A). This was confirmed by calculating scores of synaptic depression: under both conditions, control and in presence of MPPG, the average amplitude of the last five cEPSCs was significantly depressed by ∼15–30% of the average amplitude of the first two cEPSCs (paired t test, p < 0.001 for both inputs). There was no difference in scores of synaptic depression between drugs (paired t test, trigeminal, p = 0.63, auditory, p = 0.85) (Fig. 3C). In summary, MPPG did not interfere with synaptic depression. A different antagonist, MSOP, also did not affect synaptic depression (ANOVA, F(1,28) = 0.02, p = 0.90 for interaction drug × synaptic depression with trigeminal stimulation; F(1,28) = 0.31, p = 0.58 for interaction of drug × synaptic depression with auditory stimulation) (Fig. 3B). Synaptic depression was always significant (p < 0.001) and scores of synaptic depression were unchanged by MSOP (paired t test, p = 0.27 for trigeminal and p = 0.49 for auditory stimulation) (Fig. 3C).
Figure 3.
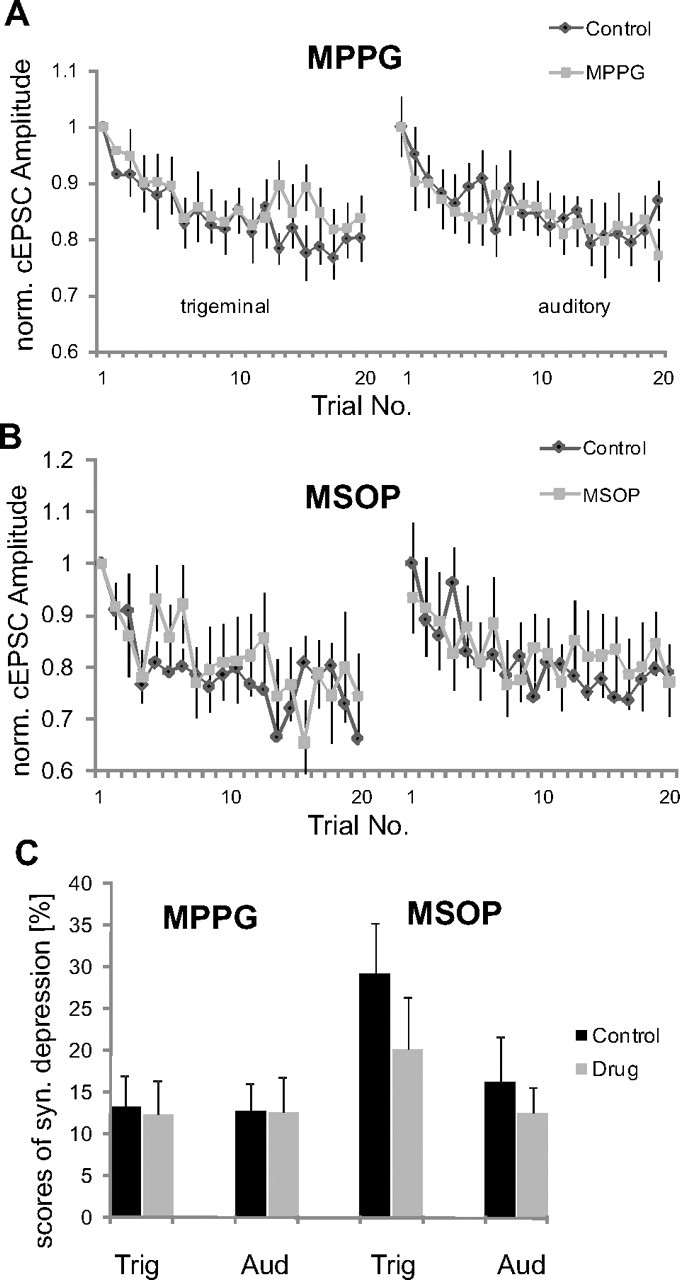
Group III mGluR antagonists do not interfere with synaptic depression. A, Synaptic depression induced by 20 bursts of four presynaptic action potentials under control conditions, and in the presence of MPPG. cEPSC amplitudes of each cell were normalized to the first EPSC amplitude in each condition to compare the time course of synaptic depression (n = 13 cells). B, Synaptic depression in the presence of MSOP (n = 8 cells). C, Percentage of synaptic depression before and during application of MPPG and MSOP. None of the group III mGluR antagonists interfered with synaptic depression (error bars indicate SEM). Trig, trigeminal; Aud, auditory; syn., synaptic; norm., normalized.
Next, we tested whether inhibition of synaptic signals by the mGluR agonist l-AP4 involves the same mechanisms as synaptic depression. Synaptic depression was induced under control conditions and in presence of l-AP4 after a 10 min recovery period. If l-AP4-induced synaptic inhibition is mediated by the same mechanisms as synaptic depression, in presence of l-AP4 no further synaptic depression should be induced by burst stimulation. Figure 4A shows synaptic current amplitudes of a typical cell under the respective conditions. As also displayed in Figure 1, l-AP4 significantly reduced baseline startle amplitudes. Figure 4B overlays average synaptic current amplitudes normalized to the first synaptic signal under each condition to compare the relative amount of synaptic depression. Although synaptic currents were significantly reduced by l-AP4 (see above; Fig. 4C), amplitudes still declined in an exponential way upon repeated burst stimulation by 20–30% (Fig. 4D). Thus, l-AP4 did not interact with synaptic depression induced by burst stimulation (trigeminal, ANOVA, F(1,24) = 0.09, p = 0.75; auditory, F(1,20) = 1.43, p = 0.24) (Fig. 4B,D). This is confirmed by paired t test on scores of synaptic depression that show an always-significant synaptic depression (p < 0.0001), and no change in scores by l-AP4 (p = 0.50 for trigeminal and p = 0.22 for auditory stimulation). These experiments show that mGluRs are able to inhibit PnC giant neurons but that they are not involved in synaptic depression in the PnC induced by burst stimulation, which is thought to underlie short-term habituation.
Figure 4.
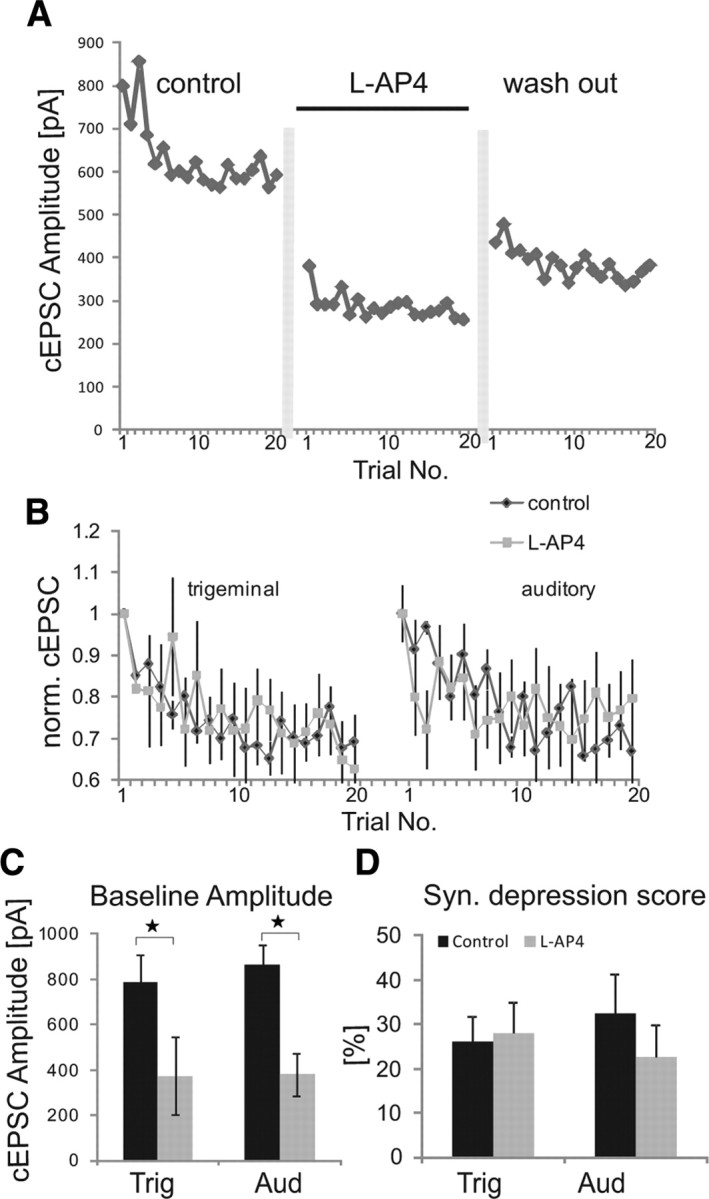
l-AP4 and synaptic depression. A, Auditory cEPSC amplitudes of a sample cell under control conditions, during application of l-AP4, and after wash out. Vertical gray bars indicate a >10 min recovery period. B, Effect of l-AP4 on synaptic depression induced by stimulation of trigeminal and auditory afferents. To compare synaptic depression, amplitudes of each cell were normalized to the first EPSC amplitude in each drug condition (n = 6 cells). C, Baseline amplitudes (mean of the first two cEPSC of each cell) of trigeminal- and auditory-evoked cEPSCs are significantly decreased (as indicated by asterisks) in the presence of l-AP4. D, Scores of synaptic depression under different conditions. Although l-AP4 reduced the amplitudes of synaptic signals, it did not interfere with synaptic depression (error bars indicate SEM). Trig, trigeminal; Aud, auditory; syn., synaptic.
Behavior
MPPG and short-term habituation
In the following, we tested the validity of our in vitro results for short-term habituation of startle in vivo. Specifically, we tested whether local injections of mGluRIII agonists and antagonists into the PnC interfere with startle and short-term habituation of startle. According to our in vitro results, neither activation nor inhibition of mGluRIIIs should influence short-term habituation. We first injected the MPPG into the PnC of rats and tested them for short-term habituation using startle stimuli 25 dB SPL over the individual startle threshold. Only animals with proper cannula placements (as assessed by histology later on) were included into final data analysis. Figure 5 shows an example of an H&E-stained brain where a small amount of dye was injected postmortem to mark the tips of the cannulae. Note that the amount of injected dye was approximately a fifth of the amount of injected drug, staining therefore does not reflect the spread of drugs. Animals received a saline injection and one of three MPPG concentrations in a pseudorandomized order on 2 separate testing days that were at least 3 d apart. Injection of 200 μm, 500 μm, or 2 mm MPPG (0.5 μl per side) did not affect habituation of startle (Fig. 6A–C). Habituation scores (last five/first two responses) were not significantly different between any of the MPPG groups and their respective saline controls (Fig. 6D; Table 1). A dependent two-way ANOVA comparing each drug concentration with its respective saline controls confirmed a significant effect of trial number (habituation) for all three MPPG concentrations, but no significant effect of drug treatment or interaction of drug treatment with trial number (Table 1). The baseline startle-response amplitudes (first two responses under each condition) were also not affected by MPPG injections when compared with their respective saline controls (Table 1).
Figure 5.
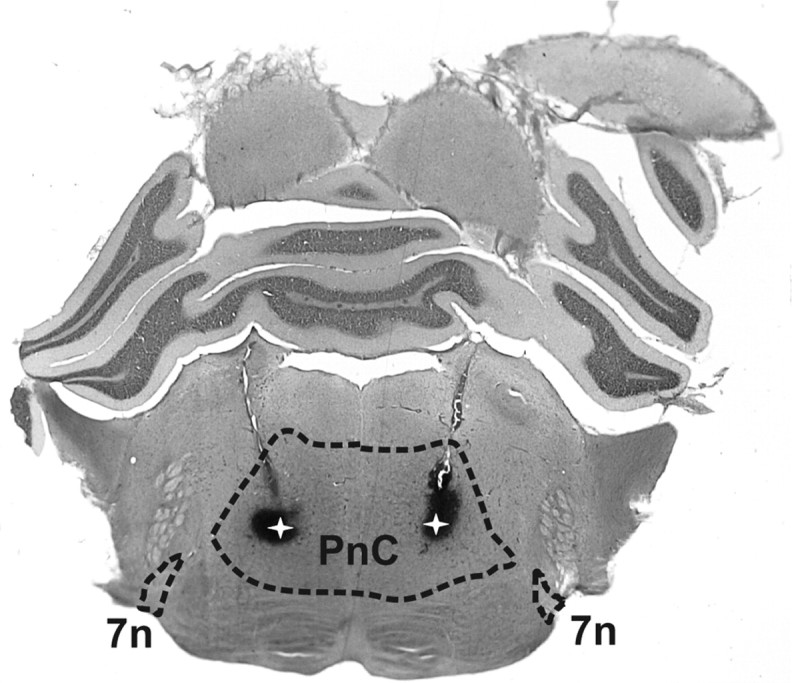
Sample brain slice showing the localization of the chronically implanted cannulae. Photograph of a 60-μm-thick brain slice from a rat that was tested for startle responses after saline injection plus injection of one drug (0.5 μl each), and which had received an injection of ∼0.1 μl of thionin postmortem. The slice was stained using a standard H&E staining protocol. The dashed lines show the boundaries of the PnC and the seventh nerve (7n). The latter was used (along with the lateral superior olive) as a landmark to identify the PnC. The white stars within the dark patches mark the localization of the cannula tips within the PnC. Note that the spread of thionin does not reflect the spread of injected drugs, since a considerably smaller amount of dye was injected to avoid lesions by this third injection (animals with clear lesions in the PnC were omitted from data analysis). Furthermore, thionin was injected postmortem. Its sole purpose was to mark the tips of the cannulae.
Figure 6.

MPPG does not interfere with short-term habituation of startle. A–C, MPPG (5 μl) was stereotaxically injected into each side of the PnC in the following concentrations: 200 μm (A; n = 13 animals), 500 μm (B; n = 8 animals), and 2 mm MPPG (C; n = 6 animals). None of the injections disrupted habituation. D, Habituation scores (first two responses × 100/last five responses) show a decrease to at least 60% of the initial startle response under each condition (error bars indicate SEM). sal, Saline.
Table 1.
Statistical analysis of behavioral experiments
| t test baseline response | t test habituation score | ANOVA factor drug | ANOVA factor trial (habituation) | ANOVA interaction drug × trial | N animals | |
|---|---|---|---|---|---|---|
| MPPG 200 μm | 0.556 | 0.67 | 0.93 | 0.031* | 0.56 | 12 |
| MPPG 500 μm | 0.315 | 0.87 | 0.96 | 0.002* | 0.63 | 8 |
| MPPG 2 mm | 0.13 | 0.44 | 0.09 | <0.0001* | 0.16 | 6 |
| l-AP4 1 mm | 0.025* | 0.77 | 0.96 | <0.0001* | 0.72 | 9 |
| MPPG + l-AP4 | 0.94 | 0.67 | 0.96 | 0.0004* | 0.72 | 6 |
Statistical analysis of the drug effects on baseline startle and habituation score, as assessed by a paired, two-tailed t test, and on habituation of startle, assessed by two-way dependent ANOVAs with “drug treatment” as first factor and “startle trial” as within-group factor. Each drug group was tested against its respective saline control, except the last group (MPPG + l-AP4), which was tested against the MPPG 2 mm-only injections in the same animals. All statistical analyses were performed with StatEL (AD Scientific).
*, p values indicating significant (p < 0.05) effects.
Electrophysiological experiments showed a strong decrease in synaptic transmission in the PnC upon activation of mGluRIIIs through l-AP4. According to the in vitro data, local application of l-AP4 in vivo should therefore generally suppress startle responses but should not disrupt short-term habituation. Indeed, local injection of l-AP4 into the PnC decreased the amplitude of startle responses when compared with injection of ACSF (paired t test, p = 0.025) (Fig. 7A). Both groups, however, showed significant short-term habituation, as shown by an unchanged habituation score (paired t test, p = 0.25) (Fig. 7B). A two-way dependent ANOVA confirmed a significant effect of the factor trial number and the factor drug (df(1,28), F = 7.69, p = 0.024), but no effect of l-AP4 on habituation (interaction treatment with trial number) (Table 1).
Figure 7.

Effect of local injection of l-AP4 on startle and short-term habituation. A, Local injection of 0.5 μl of l-AP4 (1 mm) into each side of the PnC significantly reduced baseline startle amplitudes (n = 9 animals). B, Local injections did not alter the habituation score when compared with saline injections. C, Time course of habituation after saline injection and l-AP4 injection (n = 8 animals, error bars indicate SEM, asterisk indicates statistically significant differences, p < 0.05).
As reported above, injections of MPPG had not shown any effects on habituation or baseline startle. As a positive control, we checked for the activity of 2 mm MPPG by combining it with 1 mm l-AP4. Coinjections of 2 mm MPPG should antagonize the l-AP4 effect on baseline startle. Rats shown in Fig. 6A received two injections (saline and 2 mm MPPG) on different test days, and all received a third injection with 2 mm MPPG and 1 mm l-AP4. Injected animals still habituated (Fig. 8A) and the habituation scores were not different between saline or MPPG only and MPPG plus l-AP4 (t test, p = 0.67 and 0.81, respectively) (Fig. 8B). Baseline startle responses were also not different between these two groups (t test, p = 0.06 and p = 0.94, respectively), indicating that 2 mm MPPG antagonized the l-AP4 effect on baseline startle (Fig. 8B; Table 1).
Figure 8.
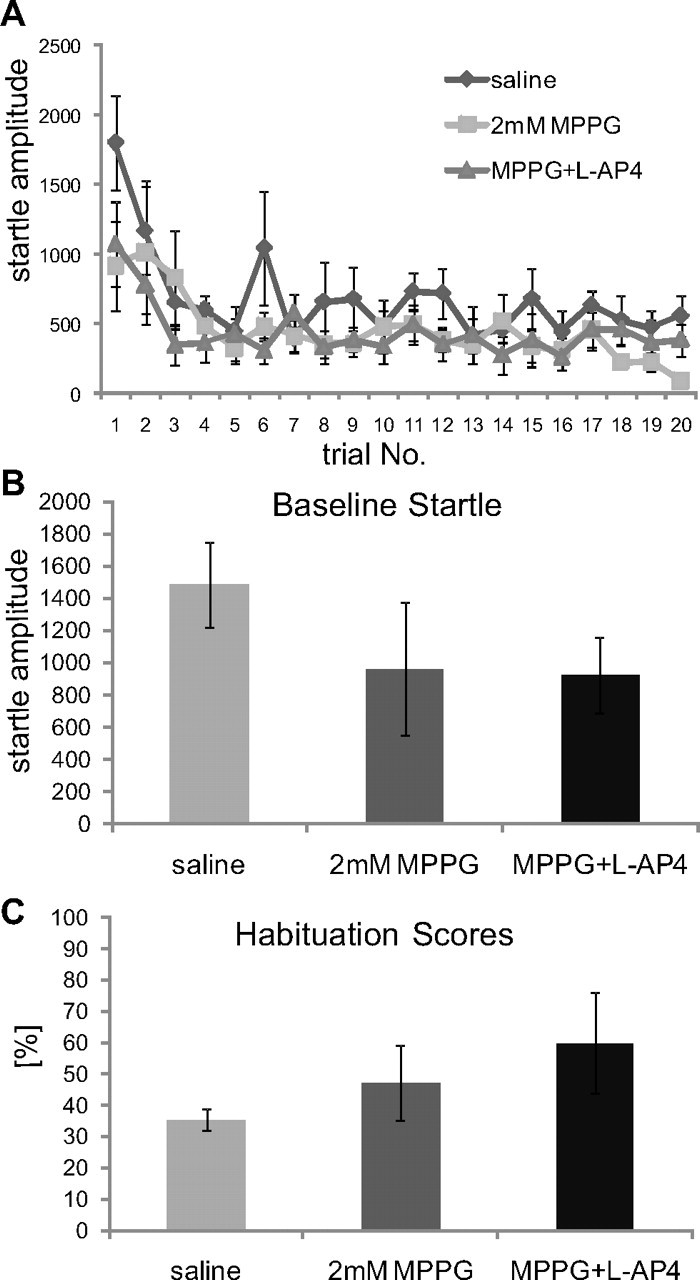
Combined injections of MPPG and l-AP4. Animals were injected with saline, MPPG 2 mm only, or with a combination of 2 mm MPPG and 1 mm l-AP4. A, Course of habituation. B, Effect on baseline startle amplitudes. MPPG (2 mm) had a trend to decrease baseline startle responses; however, this was not statistically significant. Most importantly, combined injections of l-AP4 and MPPG did not decrease baseline startle amplitudes compared with MPPG alone, showing that the l-AP4 effect that was shown in Figure 7 was antagonized by MPPG (n = 6 animals). C, Habituation scores remain unchanged.
In summary, the behavioral experiments confirm an inhibitory role for mGluRIIIs in the PnC on startle signaling, as predicted by the in vitro electrophysiological results that showed PnC giant neuron inhibition by mGluRIII agonists. Also, in accordance to the in vitro results that show no interference of this inhibition with synaptic depression by burst stimulation, mGluRIII activation does not interfere with short-term habituation of startle. Our results support the validity of the slice model for studying mechanisms underlying short-term habituation of startle, and they show that group III mGluRs do not mediate short-term habituation of startle under the given parameters.
Discussion
The primary startle pathway in rats has been extensively studied in the past, which makes it an excellent model for studying cellular and molecular mechanisms underlying behavior and behavioral plasticity, using both in vivo and in vitro studies.
In the present study, we tested the hypothesis that activation of group III mGluRs mediates short-term habituation of acoustic startle responses, analogous to their role in odor habituation (Best et al., 2005; Yadon and Wilson, 2005; Linster et al., 2009; Wilson, 2009). Specifically, we tested whether activation of mGluRIIIs inhibits synaptic transmission in the PnC and whether this inhibition is responsible for synaptic depression and short-term habituation of startle. The mGluR agonist l-AP4 applied by the bath solution had a strong inhibitory effect on PnC giant neurons. This clearly indicates that there are functional mGluRs expressed in the PnC and that activation of these receptors powerfully inhibits synaptic transmission from sensory afferent fibers to PnC giant neurons. In accordance with this synaptic effect, in vivo application of l-AP4 by stereotaxic injections into the PnC significantly reduced startle response amplitudes, as would be expected when inhibiting synaptic transmission to startle-mediating neurons in the PnC.
However, mGluRIII inhibition does not play a role in homosynaptic depression proposed to underlie short-term habituation of startle responses. Synaptic currents reduced by l-AP4 were still subject to synaptic depression. Furthermore, the mGluRIII antagonists MPPG and MSOP failed to show any effect on synaptic transmission in PnC giant neurons, even when afferent sensory fibers were repeatedly stimulated by bursts. Apparently, mGluRIIIs are not activated by glutamate released from these afferent sensory fiber synapses. In accordance with these electrophysiological results, mGluRIII agonists and antagonists injected into the PnC did not disrupt short-term habituation in vivo. The combined injection with l-AP4 showed that MPPG reversed the l-AP4 effect on baseline startle responses, therefore demonstrating that it was injected into the right place and in an effective dose.
The lack of mGluRIII effect on synaptic depression is in contrast to a former report, where the application of MPPG affected synaptic depression in PnC giant neurons (Weber et al., 2002). In this former study, synaptic depression was induced by trains of four stimuli with 4 ms ISI under control conditions and this stimulation protocol was then repeated after recovery and in the presence of MPPG. We have since observed, however, that a 4 ms ISI sometimes leads to action-potential failures due to the refractory state of the stimulated axons. These failures tend to increase during the course of the experiment, resulting in more trains that comprised only three action potentials (our unpublished data). It has also been shown that trains of three action potentials are not sufficient for inducing synaptic depression (Simons-Weidenmaier et al., 2006). The increase in action-potential failures might have been responsible for the observed effect in the former study. In the present study we have increased the ISI to 6 ms.
Alternatively, the change in stimulation protocol might have lead to an activation of a different mechanism. It has been shown in odor habituation in rats that different stimulus protocols can activate entirely different mechanisms (McNamara et al., 2008). Although the stimulus presentation time in mGluR-mediated odor habituation is longer than in startle experiments (4–20 s vs 20 ms), the interstimulus intervals were comparable between the two studies (10 s vs 15 s). The involvement of mGluRs in odor habituation might therefore be specific for the stimulus mode or the type of observed behavior. Nevertheless, different stimulus protocols will have to be used in both in vivo and in vitro future studies on acoustic startle to investigate the potential variability of the underlying mechanisms.
One drawback of the slice preparation used in this study lies in the age of animals of which slices were obtained (10–15 days old). Increasing myelinization of neurons impedes visual identification of PnC giant neurons in older animals. The acoustic startle response can be earliest measured in vivo with the onset of hearing at postnatal days 12–13, when the auditory meatus opens. Rats show acoustic startle responses from that moment on as well as habituation (S. Schmid, K. Shaik, and K. Carlton, unpublished data). The wiring of auditory pathways is already functional at postnatal day 8 (Tokimoto et al., 1977), thus we can assume that the auditory structures responsible for acoustic startle are also established at that time. Indeed, we measured auditory synaptic input from stimulated afferents for all PnC giant neurons investigated. Furthermore, studies addressing the tactile startle in developing rats revealed that these startle response can be measured as early as postnatal day 6 (Hickl et al., 1999). The tactile startle pathway converges with the auditory at the level of the PnC (Yeomans et al., 2002; Schmid et al., 2003); it can therefore be assumed that the motor part of the startle pathway is also established by that age.
Our experiments underline the validity of the synaptic depression model in the slice for short-term habituation of startle. There are several invertebrate models where synaptic depression as an underlying mechanism for habituation of an escape response was studied, such as Aplysia, Drosophila, and C. elegans. Whereas the depletion of releasable neurotransmitter as the underlying mechanisms for synaptic depression has been widely accepted at least in the Aplysia model (Gingrich and Byrne, 1985; for review, see Gover and Abrams, 2009), other mechanisms have also been proposed (for review, see Gover and Abrams, 2009). To date, cellular and molecular mechanisms underlying short-term habituation have not been completely resolved in any animal model. There is an increasing body of evidence for a common mechanism among species that is presynaptic and calcium dependent (Rose and Rankin, 2001; Weber et al., 2002; Simons-Weidenmaier et al., 2006; Gover and Abrams, 2009; Rankin et al., 2009). Our original hypothesis that activation of presynaptic mGluRIIIs induces synaptic depression would have been consistent with this idea, since group III mGluRs interfere with calcium-activated processes by inhibiting adenylyl cyclase. However, the present data clearly rejects any role for group III mGluRs in synaptic depression or short-term habituation, at least within the parameters used in this study.
In conclusion, this study excludes group III mGluRs as candidates for mediating synaptic depression underlying short-term habituation of startle under the present conditions, in contrast to their role in short-term habituation to odors. Short-term habituation occurs on the sensory part of the respective pathways; nevertheless, our work adds to an existing body of evidence that different habituation and synaptic depression mechanisms underlie habituation to different sensory stimuli (e.g., odor versus acoustic). While there might be different habituation mechanisms in different pathways on one hand, habituation to sensory stimuli that elicit a fast escape response might, on the other hand, be phylogenetically old, and probably highly conserved. Therefore, to unravel the molecular mechanisms underlying acoustic startle habituation in rats, it might be worth looking at studies of habituation deficits in invertebrate models such as Drosophila or C. elegans, where a large number of different mutants can be screened. In the future, it will be intriguing to see to what extent habituation mechanisms are conserved across species and to what extent they can vary within a single animal.
Footnotes
This work was funded by the German Research Council (Schm1710) and the Natural Science and Engineering Research Council of Canada.
References
- Best AR, Thompson JV, Fletcher ML, Wilson DA. Cortical metabotropic glutamate receptors contribute to habituation of a simple odor-evoked behavior. J Neurosci. 2005;25:2513–2517. doi: 10.1523/JNEUROSCI.5298-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. New York: Plenum; 1984. The mammalian startle response. [Google Scholar]
- Davis M, Gendelman PM. Plasticity of the acoustic startle response in the acutely decerebrate rat. J Comp Physiol Psychol. 1977;91:549–563. doi: 10.1037/h0077345. [DOI] [PubMed] [Google Scholar]
- Davis M, Parisi T, Gendelman DS, Tischler M, Kehne JH. Habituation and sensitization of startle reflexes elicited electrically from the brainstem. Science. 1982;218:688–690. doi: 10.1126/science.7134967. [DOI] [PubMed] [Google Scholar]
- Gerber U, Gee CE, Benquet P. Metabotropic glutamate receptors: intracellular signaling pathways. Curr Opin Pharmacol. 2007;7:56–61. doi: 10.1016/j.coph.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Giles AC, Rankin CH. Behavioral and genetic characterization of habituation using Caenorhabditis elegans. Neurobiol Learn Mem. 2009;92:139–146. doi: 10.1016/j.nlm.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Gingrich KJ, Byrne JH. Simulation of synaptic depression, posttetanic potentiation, and presynaptic facilitation of synaptic potentials from sensory neurons mediating gill-withdrawal reflex in Aplysia. J Neurophysiol. 1985;53:652–669. doi: 10.1152/jn.1985.53.3.652. [DOI] [PubMed] [Google Scholar]
- Gover TD, Abrams TW. Insights into a molecular switch that gates sensory neuron synapses during habituation in Aplysia. Neurobiol Learn Mem. 2009;92:155–165. doi: 10.1016/j.nlm.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickl A, Pilz PKD, Schnitzler HU. The sensorimotor interface mediating the startle response is functional before the onset of hearing. Proceeding of the 1st Göttingen Conference of the German Neuroscience Society; Stuttgart: Thieme; 1999. p. 809. [Google Scholar]
- Koch M. The neurobiology of startle. Prog Neurobiol. 1999;59:107–128. doi: 10.1016/s0301-0082(98)00098-7. [DOI] [PubMed] [Google Scholar]
- Koch M, Schnitzler HU. The acoustic startle response in rats: circuits mediating evocation, inhibition and potentiation. Behav Brain Res. 1997;89:35–49. doi: 10.1016/s0166-4328(97)02296-1. [DOI] [PubMed] [Google Scholar]
- Krase W, Koch M, Schnitzler HU. Glutamate antagonists in the reticular formation reduce the acoustic startle response. Neuroreport. 1993;4:13–16. doi: 10.1097/00001756-199301000-00003. [DOI] [PubMed] [Google Scholar]
- Lee Y, López DE, Meloni EG, Davis M. A primary acoustic startle pathway: obligatory role of cochlear root neurons and the nucleus reticularis pontis caudalis. J Neurosci. 1996;16:3775–3789. doi: 10.1523/JNEUROSCI.16-11-03775.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingenhöhl K, Friauf E. Giant neurons in the caudal pontine reticular formation receive short latency acoustic input: an intracellular recording and HRP-study in the rat. J Comp Neurol. 1992;325:473–492. doi: 10.1002/cne.903250403. [DOI] [PubMed] [Google Scholar]
- Lingenhöhl K, Friauf E. Giant neurons in the rat reticular formation: a sensorimotor interface in the elementary acoustic startle circuit? J Neurosci. 1994;14:1176–1194. doi: 10.1523/JNEUROSCI.14-03-01176.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linster C, Menon AV, Singh CY, Wilson DA. Odor-specific habituation arises from interaction of afferent synaptic adaptation and intrinsic synaptic potentiation in olfactory cortex. Learn Mem. 2009;16:452–459. doi: 10.1101/lm.1403509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara AM, Magidson PD, Linster C, Wilson DA, Cleland TA. Distinct neural mechanisms mediate olfactory memory formation at different timescales. Learn Mem. 2008;15:117–125. doi: 10.1101/lm.785608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. 5th Edition. Burlington, MA: Elsevier Academic; 2005. The rat brain in stereotaxic coordinates. [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Rankin CH. Context conditioning in habituation in the nematode Caenorhabditis elegans. Behav Neurosci. 2000;114:496–505. [PubMed] [Google Scholar]
- Rankin CH, Abrams T, Barry RJ, Bhatnagar S, Clayton DF, Colombo J, Coppola G, Geyer MA, Glanzman DL, Marsland S, McSweeney FK, Wilson DA, Wu CF, Thompson RF. Habituation revisited: an updated and revised description of the behavioral characteristics of habituation. Neurobiol Learn Mem. 2009;92:135–138. doi: 10.1016/j.nlm.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK, Rankin CH. Analyses of habituation in Caenorhabditis elegans. Learn Mem. 2001;8:63–69. doi: 10.1101/lm.37801. [DOI] [PubMed] [Google Scholar]
- Schmid S, Weber M. Neurons of the superior olivary complex do not excite startle-mediating neurons in the caudal pontine reticular formation. Neuroreport. 2002;13:2223–2227. doi: 10.1097/00001756-200212030-00012. [DOI] [PubMed] [Google Scholar]
- Schmid S, Simons NS, Schnitzler HU. Cellular mechanisms of the trigeminally evoked startle response. Eur J Neurosci. 2003;17:1438–1444. doi: 10.1046/j.1460-9568.2003.02565.x. [DOI] [PubMed] [Google Scholar]
- Simons-Weidenmaier NS, Weber M, Plappert CF, Pilz PK, Schmid S. Synaptic depression and short-term habituation are located in the sensory part of the mammalian startle pathway. BMC Neurosci. 2006;7:38. doi: 10.1186/1471-2202-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokimoto T, Osako S, Matsuura S. Development of auditory evoked cortical and brain stem responses during the early postnatal period in the rat. Osaka City Med J. 1977;23:141–153. [PubMed] [Google Scholar]
- Weber M, Schnitzler HU, Schmid S. Synaptic plasticity in the acoustic startle pathway: the neuronal basis for short-term habituation? Eur J Neurosci. 2002;16:1325–1332. doi: 10.1046/j.1460-9568.2002.02194.x. [DOI] [PubMed] [Google Scholar]
- Wilson DA. Odor specificity of habituation in the rat anterior piriform cortex. J Neurophysiol. 2000;83:139–145. doi: 10.1152/jn.2000.83.1.139. [DOI] [PubMed] [Google Scholar]
- Wilson DA. Olfaction as a model system for the neurobiology of mammalian short-term habituation. Neurobiol Learn Mem. 2009;92:199–205. doi: 10.1016/j.nlm.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadon CA, Wilson DA. The role of metabotropic glutamate receptors and cortical adaptation in habituation of odor-guided behavior. Learn Mem. 2005;12:601–605. doi: 10.1101/lm.41405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans JS, Frankland PW. The acoustic startle reflex: neurons and connections. Brain Res Brain Res Rev. 1995;21:301–314. doi: 10.1016/0165-0173(96)00004-5. [DOI] [PubMed] [Google Scholar]
- Yeomans JS, Li L, Scott BW, Frankland PW. Tactile, acoustic and vestibular systems sum to elicit the startle reflex. Neurosci Biobehav Rev. 2002;26:1–11. doi: 10.1016/s0149-7634(01)00057-4. [DOI] [PubMed] [Google Scholar]