

Abstract
Neuronal communication relies on rapid and discrete intercellular signaling but neither the molecular mechanisms of the exocytotic machinery that define the timing of the action potential-evoked response nor those controlling the kinetics of transmitter release from single synaptic vesicles are known. Here, we investigate how interference with the putative force transduction between the complex-forming SNARE (soluble N-ethylamide-sensitive factor attachment protein receptor) domain and the transmembrane anchor of synaptobrevin II (SybII) affects action potential-evoked currents and spontaneous, quantal transmitter release at mouse hippocampal synapses. The results indicate that SybII-generated membrane stress effectively determines the kinetics of the action potential-evoked response and show that SNARE force modulates the concentration profile of cleft glutamate by controlling the rate of transmitter release from the single synaptic vesicle. Thus, multiple SybII actions determine the exquisite temporal regulation of neuronal signaling.
Introduction
Presynaptic transmitter release is temporally locked to the occurrence of action potentials in the nerve terminal. Previous studies using the Ca2+-uncaging method have shown that the duration of the presynaptic Ca2+-signal determines strength and timing of evoked transmitter release (Bollmann et al., 2000; Schneggenburger and Neher, 2000; Bollmann and Sakmann, 2005). In contrast, changes in the Ca2+-affinity of synaptotagmin I, the classical Ca2+-sensor of neuronal exocytosis, have failed to alter the time course of the synchronous action potential-evoked response (Rhee et al., 2005). Thus, it is unclear whether the time course of the local, rapidly decaying [Ca2+] at the release site or intrinsic kinetics of the fusion apparatus control the timing of action potential-evoked release.
Strength and precision of quantal postsynaptic responses are similarly crucial to meet the speed requirements of neuronal signaling. At the presynaptic site the strength of quantal transmission can be modified by altering the efflux of neurotransmitter from small synaptic vesicles (SSVs). It became clear that neurotransmitter transporters determine the amount of transmitter stored within a SSV and by this can regulate the magnitude of quantal signaling (Zhou et al., 2000; Ishikawa et al., 2002; Yamashita et al., 2003; Fremeau et al., 2004; Wojcik et al., 2004; Wilson et al., 2005). These experiments suggested that the effective transmitter cleft concentration is insufficient to activate all receptors (Bekkers and Stevens, 1991; Liu and Tsien, 1995; Forti et al., 1997; Liu et al., 1999) and pointed to the possibility that cleft glutamate can serve as a regulation point for synaptic strength (Krupa and Liu, 2004). The membrane-bridging assembly of the neuronal SNARE (soluble N-ethylamide-sensitive factor attachment protein receptor) proteins Syntaxin, SNAP-25 (synaptosomal-associated protein of 25 kDa) and synaptobrevin II (SybII) is central in exocytosis of secretory cells (Jahn and Scheller, 2006). Using mouse chromaffin cells we have previously shown that a tight molecular coupling between the complex-forming SNARE domain and transmembrane domain (TMD) of SybII is crucial for priming of chromaffin granules, their exocytosis initiation and fusion pore expansion (Kesavan et al., 2007). Yet, molecular studies on the kinetics of transmitter release from single vesicles have relied preferentially on neuroendocrine cells like PC12 cells (Wang et al., 2001), chromaffin cells (Borisovska et al., 2005; Sorensen et al., 2006; Kesavan et al., 2007) and the neuromuscular junction of Drosophila (Pawlu et al., 2004). What presynaptic mechanisms shape transmitter discharge from SSVs at central synapses and to what degree they are able to determine amplitude and time course of fast miniature EPSCs (mEPSCs) has remained enigmatic.
Here we studied, whether the molecular force of SNARE proteins provides a rate-limiting step for action potential-evoked transmission and whether it can alter the fundamental unit of synaptic signaling, the quantal event. For this, we expressed SybII mutant proteins carrying an extended juxtamembrane region in hippocampal neurons that are genetically devoid of SybII (Schoch et al., 2001; Borisovska et al., 2005). Our results show that increasing the physical distance between the SNARE domain and the TMD of SybII determines priming of SSVs, governs their stimulus-secretion coupling in response to single action potentials and controls the speed of neurotransmitter release from single vesicles.
Materials and Methods
Hippocampal cultures for electrophysiology.
Autaptic cultures of hippocampal neurons were prepared at E18 from SybII knock-out mice, as described previously (Bekkers and Stevens, 1991; Schoch et al., 2001). Hippocampi were dissected from brain and digested for 20 min at 37°C with 10 units of papain (Worthington) followed by gentle mechanical trituration. Neurons diluted to a density of 1000 cells/ml were seeded onto a layer of glia microislands resulting in coculture of glia and nerve cells. Only islands containing single neurons were used for electrophysiology. For mass cultures, neuronal cell suspensions were plated at a density of 300 cells/mm2 on 25 mm coverslips coated with 0.5 mg/ml poly-d-lysine (Sigma). Cultures were maintained at 37°C in an incubator, humidified with 95% air and 5% CO2 in NBA (Invitrogen), supplemented with 2% B-27 (Sigma), 1% Glutamaxx (Invitrogen) and 2% penicillin/streptomycin (Invitrogen). To prevent astrocytic overgrowth, cultures were treated for 24 h with a mixture of mitotic inhibitors 40 μm/1 mm FUDR/Uridine (Sigma). Recordings were performed at room temperature on days 14–17 of culture.
Viral constructs.
cDNAs encoding for SybII and its mutants were subcloned into pRRL.sin.cPPT.CMV.WPRE lentiviral transfer vector (Follenzi et al., 2000), which contains a cPPT sequence of the pol gene and the posttranscriptional regulatory element of woodchuck hepatitis virus (Follenzi et al., 2002). To identify positive transfectants, SybII proteins were expressed as fusion constructs with the monomeric red fluorescent protein (mRFP) protein linked to the C-terminal domain of SybII via a 9 aa linker (GGSGGSGGT). According to the protocol from Kesavan et al., the following amino acids (underlined) were inserted into the juxtamembrane region of SybII: 4 aa, KNKLGGKL; 5 aa: KNKLGGSKL, 6 aa: KNKLGGSGKL, 7 aa: KNKLGGSGGKL, 8 aa: KNKLGGSGGSKL, 11 aa: KNKLGGSGGSGGSKL. The KL sites, flanking the GGS motif, encode for HindIII restriction sites and were generated to allow a primer-based elongation of the inserted amino acid-stretch to facilitate the cloning process. All constructs were verified by DNA sequence analysis.
Lentiviral production.
The transfer vector plasmid and the helper plasmids were transfected into human embryonic kidney HEK293T cell line using Lipofectamine 2000 according to a modified Invitrogen protocol. Briefly, after 14 h, the transfection medium was replaced with OptiMEM, 10% FCS and 100 mm sodium pyruvate and 48 h later the virus was harvested, filtered (0.4 μm PVDF membrane, Millipore) and concentrated using a centrifugal filter device (100 kDa molecular weight cutoff; Amicon Ultra-15; Millipore). The viral particles were immediately frozen and stored at −80°C. For each virus, a titer test on neuronal cultures was performed to adjust the number of infectious units to a transfection efficacy of >80%. Neurons were transfected at day 1 in vitro by adding 100–300 μl viral suspension to the culture medium.
Immunostaining.
Mass neuronal cultures were fixed for 30 min at room temperature in PBS containing 4% paraformaldehyde and 4% sucrose. Cultures were quenched for 10 min with 50 mm NH4Cl in PBS, blocked for 1 h in PBS containing 3% BSA and 0.1% Triton X100. Primary and secondary antibodies were diluted in 3% BSA, 0.1% Triton X-100 in PBS. The following primary antibodies were used: Anti-SybII (mouse monoclonal 69.1, antigen epitope amino acid position 1–14, kindly provided by R. Jahn, Göttingen, Germany), Anti-Synaptophysin (mouse monoclonal, Synaptic Systems), Anti-GluR4 (Millipore), Anti-vGlut1 (Synaptic Systems). Cells were incubated with primary antibodies overnight. After rinsing they were incubated once with PBS containing 0.5 m NaCl, washed briefly in PBS. Alexa-Fluor 488 (Invitrogen) was applied for 2 h. After washing in PBS, cells were imaged immediately or mounted on Glycerol or ProLong AntiFade (Invitrogen).
Epifluorescence pictures were digitized with an AxioCam MRm-CCD camera (Carl Zeiss) at the focal plane of the axonal network with a 25× and 100× objective (Zeiss). Images were acquired with the software Axiovison 4.5 (Zeiss) and analyzed with MetaMorph software (Universal Imaging Inc.). Immunopositive spots were determined using a threshold-based detection routine, with the threshold adjusted to the background signal of the dendrite. Immunosignals were quantified as mean fluorescent intensity. For the analysis of synaptic density, Synaptophysin-positive puncta were counted along 50 μm length of neuronal process. Statistics regarding the protein expression level and the synapse density are based on one-way ANOVA of wild-type cells (n = 25), SybII-ko cells (n = 25) and mutant cells (25 cells per mutant variant). Mean synaptic fluorescence intensity and synapse density was analyzed from 150 to 200 synapses per cell. Cells were collected from three cultures obtained from three different mice.
Electrophysiology.
Whole-cell voltage-clamp recordings of synaptic currents were obtained from isolated autaptic neurons. All experiments include measurements from >3 different culture preparations and were performed on age-matched neurons that derived from mice of the same litter. Patch pipettes (R Tip 3–4 MΩ) were filled with intracellular solution containing the following (in mm): 137.5 K-gluconate, 11 NaCl, 2 MgATP, 0.2 Na2GTP, 1.1 EGTA, 11 HEPES, 11 d-glucose, pH 7.3. The standard extracellular solution consisted of (in mm) 130 NaCl, 10 NaHCO3, 2.4 KCl, 4 Ca2+, 4 MgCl2, 10 HEPES, 10 d-glucose, pH 7.3 with NaOH, osmolarity, 310 mOsm. To minimize the potential contribution of GABAergic currents the reversal potential of chloride-mediated currents was adjusted to the holding potential. To verify the recording of AMPA receptor (AMPAR) currents neurons were superfused with DNQX (25 μm) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Neurons were voltage-clamped at −70 mV (without correction for the liquid junction potential, V LJ 9.8 mV) with an EPC10 amplifier (HEKA Electronic) under control of Pulse 8.5 program (HEKA Electronic) and stimulated by membrane depolarizations to +10 mV for 0.7 ms every 5 s (0.2 Hz). Cell with an average access resistance of 6–12 MΩ and with 70–80% resistance compensation were analyzed. The average access resistances/resistance compensation was 11 ± 1 MΩ/78 ± 2% for wild-type cells and 10 ± 0.5 MΩ/77 ± 1% for 11 aa mutant cells. Current signals were low-pass filtered at 2.9 kHz (four pole Bessel filter EPC10) and digitized at a rate of 10 or 50 kHz. The readily releasable pool (RRP) was determined by a 5 s application of hypertonic sucrose solution (500 mm sucrose) using a gravity-fed fast-flow system (Bruns, 1998). For recording spontaneous miniature EPSC (mEPSCs), mass cultures of hippocampal neurons were bathed in Ringer's solution containing 1 μm tetrodotoxin (TTX). To determine the mEPSC properties with reasonable fidelity and to prevent the detection of “false events” (due to random noise fluctuations), spontaneous mEPSCs with a peak amplitude >15 pA (∼5 times the SD of the background noise, SybII: 3.2 ± 0.08 pA, 11 aa: 3.05 ± 0.09 pA) and a charge criterion >25 fC were analyzed using a commercial software (Mini Analysis, Synaptosoft, Version 6.0.3) were analyzed. The false event rate (λf), given by λf = fce −θ2/2rms2 (Colquhoun and Sigworth, 1995), calculates to 0.01 Hz (threshold (θ), 15 pA; bandwith (fc), 2.9 kHz), representing less than one hundredth of the mEPSC frequencies in SybII or mutant neurons. Both the cumulative charge integral of the EPSC (see Fig. 1 F) and the mEPSC decay (see Fig. 4) were fitted with two exponentials setting t = 0 to ∼10% and ∼95% of the maximum amplitude, respectively.
Figure 1.
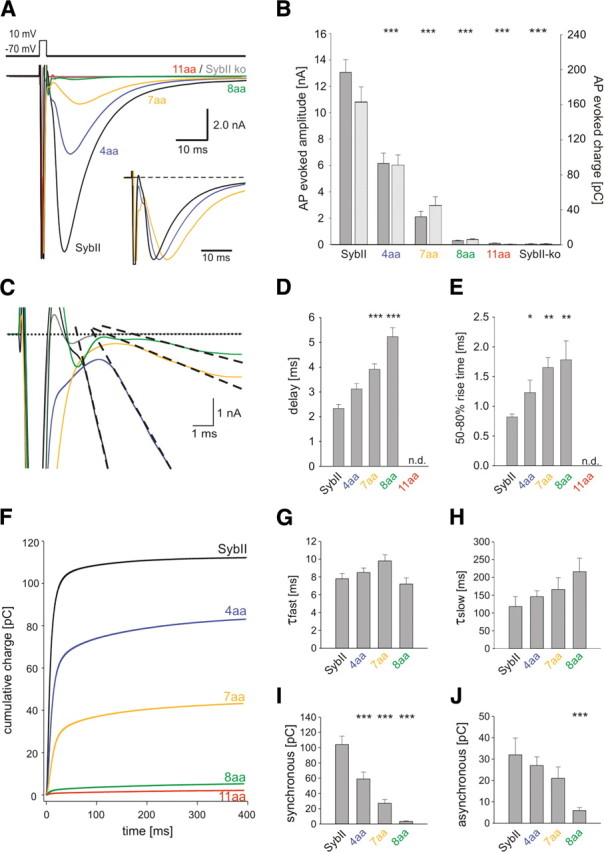
Extending SybII's juxtamembrane region decreases evoked neurotransmitter release in a linker-length-dependent fashion. A, Averaged EPSCs of hippocampal autaptic neurons mediated by SybII (SybII, n = 16) and its mutant variants (4 aa, n = 20; 7 aa, n = 19; 8 aa, n = 14; 11 aa, n = 10). Amino acid insertions of increasing length gradually decrease the evoked EPSC. The 11 aa insertion was unable to rescue secretion compared with SybII knock-out cells (SybII ko, gray trace). Inset, normalized repsonses. B, Amplitude (dark gray bars) and charge (light gray bars) of the EPSC strongly depend on the length of the linker. C, Representative EPSCs during the first 10 ms after an action potential. Compared with SybII (black), mutant proteins (4 aa, blue; 7 aa, yellow; 8 aa, green) delay the onset of the EPSC defined as the intercept between a linear fit of the EPSC rising phase (dashed lines) and the base line (dotted line). D, Mean synaptic delay measured between the end of the stimulus and the onset of the action potential-evoked EPSC. Note the increase in delay with longer linkers. E, The 50–80% rise time of the EPSC. F, Integral of the averaged EPSC for SybII and its mutant variants. Double exponential fit (data not shown) was used to approximate the data and to distinguish a fast (τfast = 6–10 ms) and a slow (τslow = 100–200 ms) phase of release. G, H, Average τfast and τslow. I, J, Magnitude of the fast and the slow phase of release with increasing linker length (SybII, n = 12; 4 aa, n = 17; 7 aa, n = 15; 8 aa, n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA.
Figure 4.
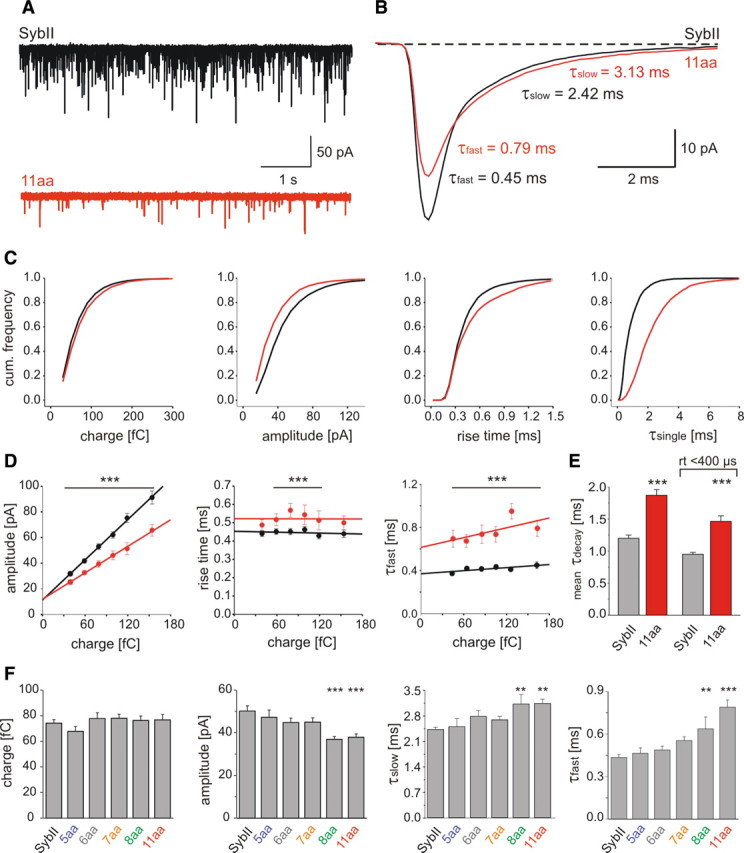
Extension of SybII's juxtamembrane region slows quantal signaling. A, Exemplary recordings of mEPSCs activity from ko neurons expressing SybII or the 11 aa mutant (11 aa). B, Ensemble average of mEPSCs from SybII (black, 19 cells, 300–400 events/cell) and the 11 aa mutant (red, 16 cells, 100–130 events/cell). The displayed SSV signals exhibit a similar charge but different amplitudes (SybII: 74 fC, 45 pA, 11 aa: 75 fC, 34 pA). Numbers indicate τdecays. C, Properties of SSV signals mediated by SybII (black, n = 15,293) or 11 aa mutant (red, n = 3829) displayed as cumulative frequency distributions for the indicated parameters. D, Independent of quantal charge, mutant mEPSCs (red) show smaller peak amplitudes, longer 10–90% rise times and τdecays than SybII signals (black). Continuous lines, Linear regressions. Events were grouped according to their charge. Bar indicates statistical differences between groups with identical charge. E, For events with an amplitude >15 pA and those selected with an additional 10–90% rise time restriction (<400 μs), the mean weighted τdecay of mutant signals is ∼1.56-fold and ∼1.54-fold slower than for SybII, respectively. F, Extension of SybII's juxtamembrane region reduces the mEPSC amplitude and progressively prolongs the mEPSC decay without affecting the quantal charge. Values are given as mean ± SEM and data were collected from the following number of cells: SybII (n = 33), 5 aa (n = 13), 6 aa (n = 13), 7 aa (n = 22), 8 aa (n = 31), 11 aa (n = 21). *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA versus SybII.
Statistical analyses.
Values are given as mean ± SEM, and n refers to the number of cells. To test for statistically significant differences, one-way ANOVA and a Tukey–Kramer posttest for comparing groups were used, if not indicated otherwise.
Results
Increasing the distance between the SNARE motif and the TMD of SybII gradually reduces evoked transmitter release
Patch-clamp recordings from hippocampal autaptic cultures (Bekkers et al., 1990; Bekkers and Stevens, 1991) were used to study how extending the juxtamembrane region of SybII affects neurotransmitter release. Synaptic responses (EPSCs) were evoked by brief somatic depolarizations (0.7 ms step from −70 to 10 mV) and measured as peak inward currents a few milliseconds after stimulation. SybII-ko neurons are nearly devoid of any evoked or spontaneous transmitter release (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). SybII ko neurons were infected with lentiviral expression constructs encoding for SybII or mutant variants that carry a flexible linker of different lengths in the juxtamembrane region of the protein (for details see Materials and Methods). Compared with the wild-type protein, the insertion of a 4 aa linker strongly reduces the amplitude of the action potential-evoked EPSCs. Increasing the length of the linker to 7, 8, and 11 aa further decreases the amplitude of the EPSC (Fig. 1 A,B). With the 11 aa insertion the evoked signals are nearly abolished and indistinguishable from those measured in knock-out neurons. The evoked EPSC charge exhibits a similar linker-length-dependent depression of release (Fig. 1 B). Thus, amino acid insertions immediately preceding the TMD gradually decrease the action potential-evoked EPSC.
v-SNARE linker mutants delay the synaptic response
The time course of vesicle exocytosis will, in principle, be determined by (1) the action potential-evoked Ca2+-transient (2) the affinity/kinetics of the Ca2+-sensor and (3) the intrinsic reaction kinetics of the Ca2+-triggered fusion process. Synaptotagmin I (Syt I) is required for synchronous release, but alterations in its apparent Ca2+-sensitivity do not change the time course of the synchronous component of the EPSC (Rhee et al., 2005). In contrast, SybII linker mutants significantly delay the onset of the release response (Fig. 1 C). On average, the synaptic delay progressively increases by extending the linker's length (SybII: 2.33 ± 0.17 ms; 4 aa: 3.12 ± 0.22 ms; 7 aa: 3.91 ± 0.22 ms; 8 aa: 5.22 ± 0.36 ms, Fig. 1 D). The EPSC rise time is a measure of the time window during which release probability is elevated. Lengthening the linker causes a significant increase in the 50–80% rise time (Fig. 1 E) leading to an overall increase in the time to peak (Fig. 1 A, inset). The 50–80% rise time of EPSCs recorded in wild-type cells (n = 43) shows no correlation with their amplitudes, ranging from 2 to 17 nA (r 2 = 0.05). Thus, size differences in the amplitude of the EPSCs, as observed for SybII and the mutant constructs, are unlikely to account for their kinetic differences. For further analyzing the kinetics of neurotransmitter release, we integrated the EPSC over time. The cumulative charge plot is well approximated with a sum of two exponential functions (Fig. 1 F). In neurons expressing the linker mutants, neither the time constant of the synchronous nor that of the asynchronous component is significantly altered when compared with the kinetics of SybII-mediated signals (Fig. 1 G,H). Still, both components of the synaptic response decrease in magnitude with increasing linker length, with the synchronous phase showing a higher sensitivity than the asynchronous phase (Fig. 1 I,J). Together, these data indicate that a tight molecular link between the complex-forming SNARE motif and the TMD of SybII controls the triggering of the synaptic response indicating a postpriming function of v-SNARE proteins during SSV exocytosis.
Linker mutations affect neither expression of SybII nor synaptogenesis
To analyze the expression level of SybII and its mutants, we used immunostainings with an antibody against SybII's N terminus. As shown in Figure 2, mutant proteins do not differ from the wild-type protein regarding the level or pattern of protein expression. The similar dotted appearance of the immunosignals indicates correct sorting of the proteins to synapses (Fig. 2 A, insets). Staining against the vesicular marker protein Synaptophysin shows that SybII-ko neurons exhibit the same synapse density as wild-type cells and further demonstrates that expression of mutant variants in SybII-ko neurons leaves the synapse number unchanged (Fig. 2 C,D). These results indicate that linker mutations do not interfere with synaptogenesis. Moreover, no differences in expression levels of the vesicular glutamate transporter vGLUT1 were found, ruling out the possibility of potential alterations in vesicular trafficking of proteins required for neurotransmitter filling (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Thus, neither variations in protein expression (or sorting) nor deficiencies in synapse formation or in vesicle filling can be held responsible for the phenotype of the mutant protein.
Figure 2.
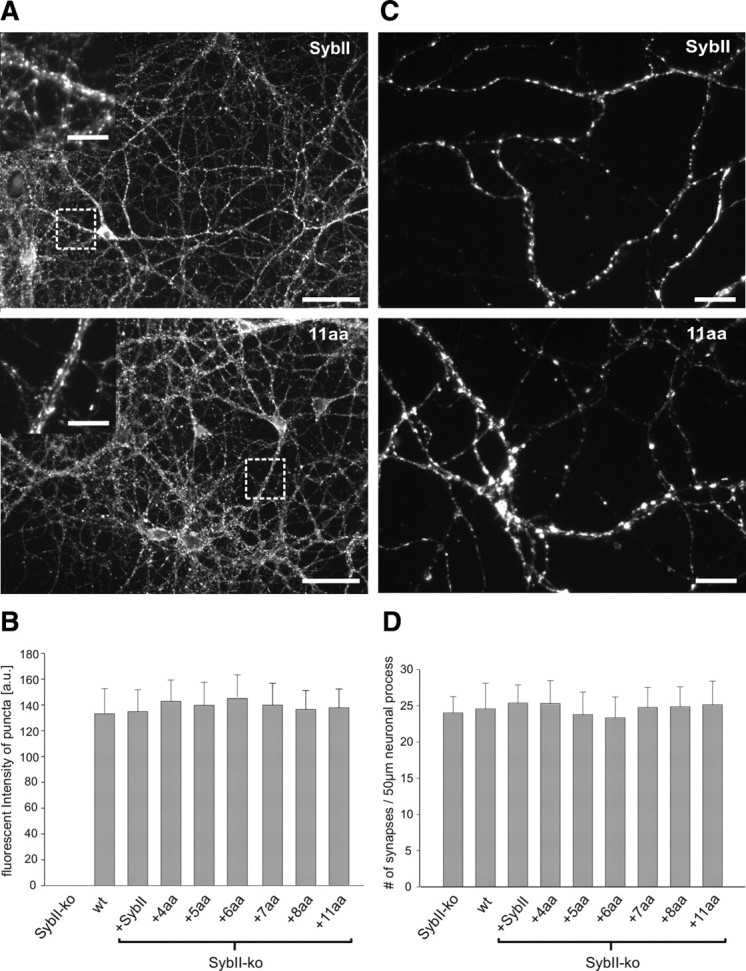
SybII-mutations do not alter either synapse density or protein expression. A, Representative epifluorescence images of SybII-ko neurons expressing SybII and the 11 aa mutant. Insets show the similar synaptic localization of SybII and the mutant protein at higher magnification. B, Quantification of immunosignals for SybII and its mutants. Mean fluorescence intensity of SybII labeling is nearly identical for the Syb variants and reaches the level of wild-type neurons (n = 25 cells/condition, p = 0.5, one-way ANOVA). No signal is detectable from synaptic varicosities of SybII-ko neurons. C, Synapse density is indistinguishable in SybII and 11 aa mutant cells, as judged from frequency and pattern of synaptophysin-positive puncta. D, Number of synaptophysin-positive puncta determined per 50 μm length of neuronal process is unchanged (n = 25 cells/condition, p = 0.6, one-way ANOVA). Scale bars: A, 50 μm, insets 25 μm; C, 5 μm.
Readily releasable pool and release probability decrease with longer linkers
Important physiological factors that might change for mutant secretion and contribute to the reduced EPSC amplitude are the size of the RRP of synaptic vesicles and the release probability (RP). Only a fraction of synaptic vesicles in close proximity to the active zone are primed and constitute the RRP (Rizzoli and Betz, 2005). The RRP can be quantified by application of hypertonic solution for several seconds. The integral of the transient inward current produced by concomitant release of glutamate provides a direct estimate on the number of vesicles within the RRP (Rosenmund and Stevens, 1996). As shown in Figure 3, B and C, the size of the RRP significantly and systematically decreases with lengthening of SybII's juxtamembrane region, indicating that linker mutations interfere with the establishment or maintenance of the vesicle's release-ready state.
Figure 3.

The pool of readily releasable vesicles decreases by lengthening the juxtamembrane region of SybII. A, Averaged evoked response of SybII ko autaptic neurons expressing SybII. After 5 s the same cell was stimulated by superfusion with hypertonic solution (see B, top trace). B, Exemplary secretory responses to stimulation with hypertonic solution. The RRP charge is defined by the time integral over the first 1.2 s after the onset of the sucrose response. C, D, Readily releasable pool charge and release probability show a stringent length requirement of SybII‘s juxtamembrane region (SybII, n = 12; 4 aa, n = 20; 7 aa, n = 17; 8 aa, n = 14; 11 aa, n = 10). *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA.
To further elucidate functional defects caused by the extended linker mutants, we analyzed the vesicular release probability in response to single action potential stimulation (Fig. 3 A). The ratio of charges evoked by an action potential and that during subsequent sucrose stimulation provides an estimate on the release probability. Indeed, the release probability decreases with longer amino acid insertions (Fig. 3 D), confirming the view that lengthening the intramolecular distance between the SNARE motif and the TMD of SybII impairs Ca2+-triggered exocytosis at the postpriming level. Together, our results show that force transmission from the SNARE complex to the vesicle membrane functions in multiple steps in exocytosis.
SybII action underlies the speeding of the mEPSC time course
To test whether the distance between the complex-forming SNARE domain and the TMD of Synaptobrevin II is also critical for quantal signaling, we recorded spontaneous EPSCs (mEPSCs) in the presence of 1 μm TTX using mass cultures of hippocampal neurons. Compared with the SybII protein expression of the 11 aa mutant in SybII ko neurons reduces the frequency of spontaneous events (Fig. 4 A, SybII: 7.3 ± 1.3 Hz, n = 33, 11 aa: 2.9 ± 0.6 Hz, n = 21, p = 0.014). Notably, the frequency of mEPSCs recorded with the 11 aa mutant is ∼10-fold higher than with SybII ko neurons (0.3 ± 0.1 Hz, n = 9, supplemental Fig. S1, available at www.jneurosci.org as supplemental material), emphasizing the gain-of-function phenotype even with long linker mutants. The mean amplitude of 11 aa-mediated mEPSCs (37.0 ± 1.6 pA, n = 21) is significantly smaller than that of SybII signals (50.4 ± 2.4 pA, n = 33, p = 0.001). The decay of averaged SybII mEPSCs, aligned to the midpoint of their rise time (Fig. 4 B), is best approximated by a sum of two exponentials revealing an initial fast (τfast) and a subsequent slower component (τslow) of decay (Afast −24.7 ± 2.1 pA, τfast 0.45 ± 0.02 ms, Aslow −17.4 ± 1.1 pA, τslow 2.42 ± 0.7 ms, n = 19). Compared with SybII, the initial phase of the 11 aa mEPSC is characterized by both a significantly lower amplitude (Af −17.2 ± 1.1 pA, n = 16, p = 0.006) and slower time course (τfast 0.79 ± 0.05 ms, p = 0.001). In contrast, the second component retains a nearly unchanged amplitude (Aslow −14.8 ± 1.3 pA), but also exhibits a slower decay (τslow 3.13 ± 0.12 ms, n = 16). Together, the linker mutant protein diminishes the peak amplitude and slows the decay of the mEPSC. A comparison of the corresponding frequency distributions shows that the amplitude distribution of 11 aa-mediated mEPSCs is shifted to lower values without changing their quantal charge (Fig. 4 C). This result is similar to linker effects on catecholamine release from chromaffin granules (Kesavan et al., 2007). τ values from single exponential fits (τsingle) to individual responses confirm the significant difference between the decays of SybII and 11 aa signals (SybII 1.2 ± 0.06 ms, 11 aa 1.87 ± 0.09, p < 0.001). The 10–90% rise time of quantal signals differs for rise times longer than 300 μs, but is unchanged for more rapidly rising signals, most likely due to the limiting response time of the cell-electrode system. The relative changes in τsingle between the mutant and the SybII signals are preserved, when we restrict our analysis to signals with rise times faster than 400 μs, suggesting that experimental inconsistencies like variable dendritic filtering are unlikely to account for the observed differences in mEPSC decay (events >15 pA, τsinglesybII/τsinglemut: 0.64; events rt <400 μs, τsinglesybII/τsinglemut: 0.65, Fig. 4 E) or peak amplitude (events >15 pA, AsybII/Amut: 1.36, events rt <400 μs, AsybII/Amut: 1.40). Quantal signals vary greatly with respect to their magnitude. As shown in Figure 4 D, the event's amplitude scales proportionally to the event's charge (slope 0.52 pA/fC, r 2 = 0.99) and is significantly reduced over the entire range of charges for 11 aa signals (slope 0.34 pA/fC, r 2 = 0.99). In the same line, the kinetic parameters like 10–90% rise time and τfast are shifted to longer times for the mutant protein when compared with SybII-signals (Fig. 4 D). Thus, alterations in magnitude and time course of the mutant signals are independent of quantal charge and affect small and large events to the same degree. The combined set of data for the different mutants with 5, 6, 7, 8 and 11 aa insertion reveals a linker-length-dependent attenuation of the mEPSC amplitude that is accompanied with a strong and significant prolongation of the event's decay(Fig. 4 F). Thus, SybII action determines magnitude and time course of the mEPSC.
SybII action governs the time course of cleft glutamate
Motivated by the observations above, we studied whether alterations of the mutant-mediated mEPSCs could arise from a change in the temporal profile of glutamate experienced by the AMPA receptors. The rapidly dissociating competitive antagonist γ-d-glutamylglycine (γ-DGG), which competes with glutamate on the time-scale of AMPA-mEPSCs, generates a block that is inversely related to the transmitter concentration (Liu et al., 1999). We used γ-DGG (200 μm) to probe v-SNARE-dependent changes in the peak glutamate concentration. If the cleft concentration of glutamate is significantly higher for SybII- than for 11 aa-mediated events, one would expect that SybII mEPSCs are less affected than 11 aa mEPSCs when challenged with the rapidly equilibrating blocker. To minimize the loss of events below the amplitude threshold (15 pA), we restricted our analysis to recordings with an average minimum event charge of 100 fC. Application of γ-DGG results in a reversible reduction of the mEPSC peak amplitude (Fig. 5 A,B) that is on average ∼1.7-fold stronger for the mutant than for the wt protein (15.0 ± 2.2% for SybII and 25.3 ± 5.2% for 11 aa insertion, p = 0.05, Fig. 5). These experiments corroborate the view that the linker mutant produces lower effective glutamate concentrations than SybII. The stronger reduction in the frequency of the mutant signals caused by the antagonist (due to the loss of events below the detection threshold) is consistent with their lower amplitude (Fig. 5 E, top). Interestingly, the first component of the SybII mEPSC is less attenuated by the antagonist than its second component (Fig. 5 F), suggesting that the kinetically distinct phases of the mEPSC are governed by different transmitter concentrations. This effect is less pronounced for the mutant protein (Fig. 5 F), a notion that is compatible with the view that the mutant protein slows transmitter release, producing lower peak glutamate concentrations during the initial phase of the mEPSC. Neither the decay of the first nor of the second phase is kinetically changed by γ-DGG (Fig. 5 G). This is possible, because the expected acceleration of the mEPSC time course might in part be compensated by reduced desensitization of the postsynaptic receptors in the presence of the competitive antagonist (Wong et al., 2003).
Figure 5.
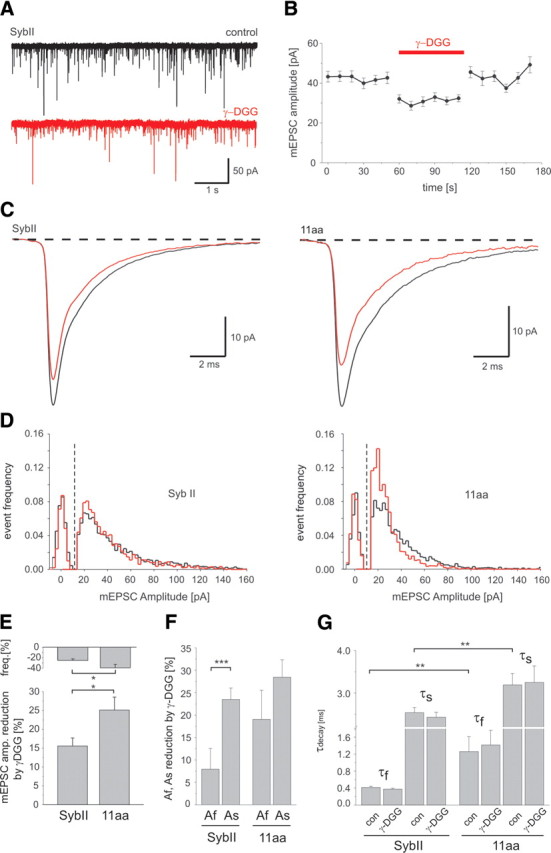
v-SNARE actions controls the glutamate concentration profile in the synaptic cleft. A, Exemplary traces of spontaneous mEPSC mediated by SybII in the absence (black traces; control) and in the presence of 200 μm γ-DGG (red traces). B, γ-DGG causes a reversible attenuation of the mEPSC amplitude. Application of γ-DGG (red bar) is bracketed by control runs with superfusion of Ringer's solution. C, Ensemble average of mEPSCs of cells expressing SybII (left, n = 17) or the 11 aa mutant (right, n = 16) recorded in the absence (black trace) and in the presence of γ-DGG (red trace). Note the different scaling for the left and the right panel. The relative inhibition is stronger for the 11 aa mutant (control: 109 fC, 33 pA, γ-DGG: 75 fC, 25 pA) than for SybII (control: 102 fC, 42 pA, γ-DGG: 82 fC, 35 pA). D, Peak amplitude distributions of mEPSCs in the absence (black) and during exposure of γ-DGG (red) for SybII (left) and the 11 aa insertion (right). Background noise distributions (peak centered at 0 pA) were obtained from 10 to 20 ms of recordings in which no mEPSCs were evident. E, The mEPSC amplitude of 11 aa events is more sensitive to γ-DGG than that of SybII events (SybII, n = 17; 11 aa, n = 16). Top illustrates the reduction in mEPSC frequency caused by γ-DGG. F, Differential effect of γ-DGG on the magnitude of the fast (Af) and slow (As) component of the mEPSC decays measured for SybII and 11 aa signals. G, Mean mEPSC τdecays in the presence and in the absence of γ-DGG for both SybII and 11 aa. The decay time constants, τfast (τf) and τslow (τs), are significantly slower for mutants signals compared with SybII. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA.
To verify that the effects of γ-DGG are due to a competitive interaction with glutamate, we examined the action of 6,7-dinitroquinoxaline-2,3-dione (DNQX), a high affinity antagonist, which slowly dissociates from the receptors and therefore blocks synaptic signals at the same level regardless of the effective glutamate concentration (Liu et al., 1999). With concentrations of DNQX (80–150 nm), that caused a peak amplitude reduction similar to that produced by γ-DGG, we observed no differences between the SybII and the 11 aa signals (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Moreover, DNQX produces a similar fractional block of the mEPSC's first and second component for both proteins.
AMPARs composed of the GluR2, -3 and -4 flop splice variants show faster desensitization than those containing flip variants (Mosbacher et al., 1994; Geiger et al., 1995; Koike et al., 2000; Koike-Tani et al., 2005). Therefore, we considered whether the altered mEPSC time course in 11 aa mutant cells could arise from a larger contribution of flip isoforms. To test this, we took advantage of splice-dependent effects of low concentrations of cyclothiazide (CTZ, 30–40 μm) affecting preferentially flip over flop variants (Partin et al., 1994). As shown in supplemental Figure S4 (available at www.jneurosci.org as supplemental material), CTZ neither affects the amplitude nor the 10–90% rise time, but uniformly prolongs the time constant of the mEPSC decay by ∼80%, regardless of the protein expressed. Therefore, an altered contribution of flip/flop subunits to synaptic AMPARs is unlikely to account for the slowing of the 11 aa mEPSC.
Together, the experiments indicate that mutant-mediated changes of the mEPSC are due to lower glutamate concentrations in the synaptic cleft rather than changes in the postsynaptic receptor composition. They suggest that v-SNARE force governs the time course of transmitter release from SSVs and thereby controls the temporal profile of cleft glutamate and the quantal signal.
Discussion
Molecular mechanisms that determine the timing of the action potential-evoked response or shape the kinetics of elementary SSV signals are fundamental to all forms of neurotransmission. Our experiments show how key properties of evoked and spontaneous postsynaptic signals in hippocampal neurons gradually change by increasing the intramolecular distance between the SNARE domain and the TMD of SybII. They provide the first evidence that SybII-generated membrane stress is a rate-limiting step for timing of the EPSC and show that SNARE force changes the concentration profile of cleft glutamate by controlling the rate of transmitter release from single synaptic vesicles. Thus, multiple SybII actions determine the exquisite temporal regulation of neuronal signaling.
Linkers impair priming and stimulus secretion coupling of SSVs
Extending the juxtamembrane region of SybII gradually reduces the EPSC. Neither differences in expression levels of the protein variants nor changes in synapse number were detected. In addition, we observe a linker-length-dependent reduction in the pool of primed vesicles, as judged by hypertonic sucrose application. This agrees with previous observations, showing that insertions of 12 or 24 aa between the SNARE domain and the TMD abolishes SybII's ability to support vesicle priming (Deák et al., 2006). Yet, it contrasts with observations for null mutants of Complexin or Synaptotagmin at hippocampal synapses, indicating that linker mutations do not interfere with binding of these proteins to the SNARE complex (Geppert et al., 1994; Xue et al., 2008). Since linker extensions do not perturb SNARE complex assembly (McNew et al., 1999; Deák et al., 2006), it is likely that efficient priming of synaptic vesicles demands, beside trans-SNARE complex formation, a tight molecular link between the SNARE domain and the TMD.
Priming of secretory vesicles involves a large variety of synaptic proteins (Sudhof, 2004), whereas only a few molecular steps should be required to initiate fusion of SSVs at the millisecond time scale in response to the rapid release-site Ca2+-transient. Localization of SSVs relative to the site of Ca2+-influx is crucial for exocytosis timing (Adler et al., 1991; Llinás et al., 1992; Wadel et al., 2007; Young and Neher, 2009), but changes in the Ca2+-sensitivity of synaptotagmin failed to alter the EPSC kinetics (Rhee et al., 2005). Furthermore, experiments with Ca2+-uncaging suggested that reaction steps of the fusion machinery may become rate-limiting for EPSCs in response to local, ultrarapid Ca2+-transients evoked by single action potentials (Bollmann and Sakmann, 2005). Indeed, our experiments show that extending the juxtamembrane region of SybII systematically increases the time lag between stimulus and onset of the postsynaptic response. Since linker mutations do not change Ca2+-sensitivity of secretion (Kesavan et al., 2007), we conclude that a tight intramolecular coupling between SybII's SNARE domain and the TMD represents a rate-limiting step for SSV exocytosis. Thus, SNARE complex formation exerts mechanical force that is transmitted to membranes to initiate fusion and delays the moment by which exocytosis follows the Ca2+-signal. Given the short duration of the presynaptic Ca2+-signal, the altered release probability observed for the mutant proteins may be due to delayed stimulus-secretion coupling. Together, both reductions in RRP size and release probability contribute to the decreased mutant response.
While both evoked and spontaneous activity crucially depend on the presence of SybII, they obviously differ regarding their sensitivity to extensions of the juxtamembrane region. This notion is reminiscent of the differential sensitivity of synchronous and asynchronous exocytosis observed in chromaffin cells (Kesavan et al., 2007). The most straightforward interpretation is that spontaneous exocytosis requires less force from the SNARE machinery than evoked release, because other proteins like Complexin, which are required for rapid stimulus secretion coupling (Reim et al., 2001; Tang et al., 2006) may increase the energy barrier for synchronized exocytosis.
SybII action drives rapid transmitter release from SSVs
Flexible insertions into the juxtamembrane region of SybII attenuate the amplitude and slow the decay of mEPSCs in a linker-length-dependent fashion. This suggests that SNARE protein-generated membrane stress provides an essential force not only to initiate fusion but also to hasten membrane merger. This result is compatible with a recent study, showing that a continuous helix forms throughout the linker from the SNARE complex into the membrane leaving no flexible intermittent domain (Stein et al., 2009). Thus, force of SNARE-complex formation can be directly transmitted into membranes in form of bending stress promoting fusion. Such a scenario may cause changes in the structure of the fusion pore, as has been suggested for a SNAP-25 construct lacking the last 9 amino acids at its C terminus (Fang et al., 2008).
Given that SNAREs like SNAP-25 or SNAP-23 have been implicated in trafficking of glutamate receptors (Lan et al., 2001; Washbourne et al., 2004; Lau et al., 2010), one might alternatively speculate that the linker effects are due to changes in the number, spatial organization or subunit composition of postsynaptic receptors. Several lines of evidence render these possibilities unlikely. First, detailed analyses reveal that linker mutations selectively change amplitude and time course of the quantal signal without altering its charge. This counters the possibility that mutant-mediated events engage a lower number of postsynaptic receptors than SybII events. Importantly, this phenotype can be clearly distinguished from that of SNAP-25-null mutants, which show a reduction in mEPSC amplitude and a similarly strong reduction in charge (Delgado-Martínez et al., 2007). Second, misalignment of postsynaptic receptors relative to the release site should cause a reduction in amplitude together with an increase in the event's rise time, but cannot account for the strong changes in the decay time course (Fig. 4 E). Furthermore, “off-center” release should be again accompanied with a profound decrease in charge transfer per SSV signal (Franks et al., 2003; Wu et al., 2007). Third, potential alterations in the receptor properties are unlikely to be responsible for the mutant phenotype, because the subunit composition of the glutamate receptors (GluR1-3) did not change in hippocampal synapses that were chronically treated with tetanus toxin to specifically inactivate SybII (Harms et al., 2005). In addition, we found no difference in GluR4 expression (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Moreover, the similar efficacy of CTZ in prolonging the decay of wild-type and mutant signals excludes the possibility that varying contributions of flip/flop isoforms are responsible for the different time course of wild-type and 11 aa signals (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Together, these results indicate a presynaptic origin of the mutant phenotype.
SSVs release their content on a time scale of 100–200 μs, as suggested by Monte-Carlo simulations in hippocampal synapses (Wahl et al., 1996) or judged from amperometric measurements of SSV release (Bruns and Jahn, 1995; Bruns, 2004; Staal et al., 2004). Therefore, the difference in τdecay (Δτfast ∼400 μs, Fig. 4 B) of wild-type and mutant mEPSCs suggests a 2- to 3-fold slowing of transmitter release, agreeing well with the mutant's effects on transmitter release from chromaffin granules (Kesavan et al., 2007). Considering the overall differences in the speed of transmitter release from SSVs and chromaffin granules (∼20-fold), it demonstrates that SybII action dominates the transmitter discharge behavior over a wide range of release kinetics, regardless of vesicle size and membrane curvature. Together, v-SNARE action provides an essential force for rapid transmitter release from SSVs determining amplitude and kinetic properties of fast quantal transmission.
In close correlation, the low-affinity receptor antagonist γ-DGG attenuates mutant events more strongly than SybII events. Moreover, the differential sensitivity of the fast and slow phase to γ-DGG indicates that receptors experience during the mEPSC peak phase higher glutamate concentrations than during its slow tail, a scenario that is compatible with results on modeling the mEPSC time course (Silver et al., 1996; Wahl et al., 1996; Diamond and Jahr, 1997). The high-affinity antagonist DNQX, instead, produces a similar attenuation of SybII and mutant mEPSCs. Furthermore, it uniformly reduces the first and second component of the mEPSC decay, as expected for a slow-off-rate antagonist. Together, these observations predict a biphasic concentration waveform of cleft glutamate underlying the SSV event, where the first phase is determined in magnitude and kinetics by the rate of SybII-mediated transmitter release and the second phase is dominated by slow clearance of transmitter from the synaptic cleft.
Heterogeneity in the speed of glutamate release from SSVs has been implicated for a number of important mechanisms central to development and plasticity of the CNS (Zakharenko et al., 2002; Choi et al., 2003; Chen et al., 2004). Likewise, perturbing synaptic vesicle fusion with tetanus toxin, silences AMPA transmission and slows NMDA activation (Renger et al., 2001), agreeing with our observation that SNARE-mediated membrane stress is a rate-limiting factor for transmitter release from single vesicles. In vitro studies indicated that the energy of several SNARE complexes is required to make a vesicle fuse (Li et al., 2007; Domanska et al., 2009). Thus, SNARE-mediated membrane fusion should be determined by the efficacy of force transfer between SNARE domain and TMD as well as by the number of productive SNARE complexes engaged per fusion event. Intriguingly, quantal signaling changes during development of brain synapses in a remarkably similar fashion as observed for events mediated by SybII and its mutants (Yamashita et al., 2003; Cathala et al., 2005). This points to the possibility that alterations in SNARE force contribute to speeding of mEPSCs at mature synapses. Together, our experiments delineate a backbone of multiple SybII actions that guide the synaptic vesicle through the consecutive stages of exocytosis, controlling strength/timing of the action potential-evoked response and regulating the speed of glutamate release from single synaptic vesicles.
Footnotes
The work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB530 and GK1326) to D.B. and by the Homburger Forschungsförderung (HOMFOR). We are grateful to Drs. R. Mohrman, C. Grabner, and D. Stevens, and S. Bruns-Engers for valuable discussions, and to T. Südhof for providing the SybII-null mice. We thank K. Klingler, R. Dufke, S. Rumsey, and M. Wirth for excellent technical assistance.
References
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci U S A. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B. Control of synaptic strength and timing by the release-site Ca2+ signal. Nat Neurosci. 2005;8:426–434. doi: 10.1038/nn1417. [DOI] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Südhof T, Bruns D. v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 2005;24:2114–2126. doi: 10.1038/sj.emboj.7600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D. Serotonin transport in cultured leech neurons. Methods Enzymol. 1998;296:593–607. doi: 10.1016/s0076-6879(98)96042-6. [DOI] [PubMed] [Google Scholar]
- Bruns D. Detection of transmitter release with carbon fiber electrodes. Methods. 2004;33:312–321. doi: 10.1016/j.ymeth.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Bruns D, Jahn R. Real-time measurement of transmitter release from single synaptic vesicles. Nature. 1995;377:62–65. doi: 10.1038/377062a0. [DOI] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci. 2005;8:1310–1318. doi: 10.1038/nn1534. [DOI] [PubMed] [Google Scholar]
- Chen G, Harata NC, Tsien RW. Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc Natl Acad Sci U S A. 2004;101:1063–1068. doi: 10.1073/pnas.0307149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Fusion pore modulation as a presynaptic mechanism contributing to expression of long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:695–705. doi: 10.1098/rstb.2002.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E, editors. Single-channel recording. New York: Plenum; 1995. pp. 483–585. [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Martínez I, Nehring RB, Sørensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanska MK, Kiessling V, Stein A, Fasshauer D, Tamm LK. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 2009;284:32158–32166. doi: 10.1074/jbc.M109.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Q, Berberian K, Gong LW, Hafez I, Sørensen JB, Lindau M. The role of the C terminus of the SNARE protein SNAP-25 in fusion pore opening and a model for fusion pore mechanics. Proc Natl Acad Sci U S A. 2008;105:15388–15392. doi: 10.1073/pnas.0805377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A, Ailles LE, Bakovic S, Geuna M, Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet. 2000;25:217–222. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- Follenzi A, Sabatino G, Lombardo A, Boccaccio C, Naldini L. Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2002;13:243–260. doi: 10.1089/10430340252769770. [DOI] [PubMed] [Google Scholar]
- Forti L, Bossi M, Bergamaschi A, Villa A, Malgaroli A. Loose-patch recordings of single quanta at individual hippocampal synapses. Nature. 1997;388:874–878. doi: 10.1038/42251. [DOI] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ. Independent sources of quantal variability at single glutamatergic synapses. J Neurosci. 2003;23:3186–3195. doi: 10.1523/JNEUROSCI.23-08-03186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Harms KJ, Tovar KR, Craig AM. Synapse-specific regulation of AMPA receptor subunit composition by activity. J Neurosci. 2005;25:6379–6388. doi: 10.1523/JNEUROSCI.0302-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y, Takahashi T. A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron. 2002;34:613–621. doi: 10.1016/s0896-6273(02)00692-x. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Kesavan J, Borisovska M, Bruns D. v-SNARE actions during Ca(2+)-triggered exocytosis. Cell. 2007;131:351–363. doi: 10.1016/j.cell.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Koike M, Tsukada S, Tsuzuki K, Kijima H, Ozawa S. Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J Neurosci. 2000;20:2166–2174. doi: 10.1523/JNEUROSCI.20-06-02166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Tani M, Saitoh N, Takahashi T. Mechanisms underlying developmental speeding in AMPA-EPSC decay time at the calyx of Held. J Neurosci. 2005;25:199–207. doi: 10.1523/JNEUROSCI.3861-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa B, Liu G. Does the fusion pore contribute to synaptic plasticity? Trends Neurosci. 2004;27:62–66. doi: 10.1016/j.tins.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lau CG, Takayasu Y, Rodenas-Ruano A, Paternain AV, Lerma J, Bennett MV, Zukin RS. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J Neurosci. 2010;30:242–254. doi: 10.1523/JNEUROSCI.4933-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- Liu G, Tsien RW. Properties of synaptic transmission at single hippocampal synaptic boutons. Nature. 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- McNew JA, Weber T, Engelman DM, Söllner TH, Rothman JE. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol Cell. 1999;4:415–421. doi: 10.1016/s1097-2765(00)80343-3. [DOI] [PubMed] [Google Scholar]
- Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- Pawlu C, DiAntonio A, Heckmann M. Postfusional control of quantal current shape. Neuron. 2004;42:607–618. doi: 10.1016/s0896-6273(04)00269-7. [DOI] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Egles C, Liu G. A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron. 2001;29:469–484. doi: 10.1016/s0896-6273(01)00219-7. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Li LY, Shin OH, Rah JC, Rizo J, Südhof TC, Rosenmund C. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc Natl Acad Sci U S A. 2005;102:18664–18669. doi: 10.1073/pnas.0509153102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Silver RA, Colquhoun D, Cull-Candy SG, Edmonds B. Deactivation and desensitization of non-NMDA receptors in patches and the time course of EPSCs in rat cerebellar granule cells. J Physiol. 1996;493:167–173. doi: 10.1113/jphysiol.1996.sp021372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal RG, Mosharov EV, Sulzer D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 2004;7:341–346. doi: 10.1038/nn1205. [DOI] [PubMed] [Google Scholar]
- Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Wahl LM, Pouzat C, Stratford KJ. Monte Carlo simulation of fast excitatory synaptic transmission at a hippocampal synapse. J Neurophysiol. 1996;75:597–608. doi: 10.1152/jn.1996.75.2.597. [DOI] [PubMed] [Google Scholar]
- Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER, Jackson MB. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 2001;294:1111–1115. doi: 10.1126/science.1064002. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Liu XB, Jones EG, McAllister AK. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci. 2004;24:8253–8264. doi: 10.1523/JNEUROSCI.2555-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Xue L, Mohan R, Paradiso K, Gillis KD, Wu LG. The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J Neurosci. 2007;27:3046–3056. doi: 10.1523/JNEUROSCI.4415-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Stradomska A, Chen H, Brose N, Zhang W, Rosenmund C, Reim K. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci U S A. 2008;105:7875–7880. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Ishikawa T, Takahashi T. Developmental increase in vesicular glutamate content does not cause saturation of AMPA receptors at the calyx of held synapse. J Neurosci. 2003;23:3633–3638. doi: 10.1523/JNEUROSCI.23-09-03633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron. 2009;63:482–496. doi: 10.1016/j.neuron.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Petersen CC, Nicoll RA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol. 2000;525:195–206. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]