

Abstract
To examine the role of small RNAs in peripheral pain pathways, we deleted the enzyme Dicer in mouse postmitotic damage-sensing neurons. We used a Nav1.8-Cre mouse to target those nociceptors important for inflammatory pain. The conditional null mice were healthy with a normal number of sensory neurons and normal acute pain thresholds. Behavioral studies showed that inflammatory pain was attenuated or abolished. Inflammatory mediators failed to enhance excitability of Nav1.8+ sensory neurons from null mutant mice. Acute noxious input into the dorsal horn of the spinal cord was apparently normal, but the increased input associated with inflammatory pain measured using c-Fos staining was diminished. Microarray and quantitative real-time reverse-transcription PCR (qRT-PCR) analysis showed that Dicer deletion lead to the upregulation of many broadly expressed mRNA transcripts in dorsal root ganglia. By contrast, nociceptor-associated mRNA transcripts (e.g., Nav1.8, P2xr3, and Runx-1) were downregulated, resulting in lower levels of protein and functional expression. qRT-PCR analysis also showed lowered levels of expression of nociceptor-specific pre-mRNA transcripts. MicroRNA microarray and deep sequencing identified known and novel nociceptor microRNAs in mouse Nav1.8+ sensory neurons that may regulate nociceptor gene expression.
Introduction
Sensory neurons are essential for pain perception, and alterations in neuronal excitability contribute to inflammatory pain. Fast axonal transport would require hours or days to influence protein composition at nerve terminals, but local protein synthesis occurs in neuromas, considered a model for nerve terminals (Murashov et al., 2007; Huang et al., 2008). We wondered whether local protein synthesis regulated by microRNAs (miRNAs) could play a role in altered responses to noxious stimuli, either in terms of peripheral transduction or altered input into the CNS. As a first step to addressing such a mechanism, we examined the role of Dicer products in controlling acute and inflammatory pain.
The cytoplasmic ribonuclease III Dicer produces miRNAs from hairpin loop precursors (He and Hannon, 2004). miRNAs bind to 3′ untranslated region (3′UTR) sites of mRNAs where they recruit RNA-induced silencing complexes (RISCs), resulting in the repression of gene expression. Individual miRNAs may coordinately regulate dozens or hundreds of different mRNA transcripts (Lim et al., 2005). The Dcr-1 gene encoding Dicer has been shown to give rise to multiple transcripts (Singh et al., 2005). The coding sequence is conserved in all instances. Posttranscriptionally dicer transcripts are predicted to be heavily regulated by miRNAs based on bioinformatic analysis of its 3′UTR (Asirvatham et al., 2008). Cellular stresses, especially interferon induction have been shown to regulate Dicer activity (Wiesen and Tomasi, 2009). At the protein level the HIV transactivating response RNA binding protein (TRBP) is required for Dicer recruitment to argonaute proteins, serving as a platform for RISC assembly (Chendrimada et al., 2005).
In the nervous system, noncoding RNAs and miRNAs are implicated in a variety of processes including neuronal survival, dendrite outgrowth, transcriptional regulation, and trophic factor regulation (Giraldez et al., 2005; Vo et al., 2005; Schratt et al., 2006). Complex disease states, for example a familial Tourette syndrome case, have been ascribed to miRNA dysfunction resulting in aberrant trophic factor expression (Abelson et al., 2005). The coordinated expression of distinct repertoires of genes in neuronal or non-neuronal tissue has been linked to the actions of a functional repressor element 1(RE1)-silencing transcription factor (REST), which inhibits neuronal gene expression in non-neuronal cells, and to the miRNA-124a, which inhibits the expression of non-neuronal genes posttranscriptionally in neurons (Conaco et al., 2006). In addition, a double-stranded RNA corresponding to the neuron-restrictive silencing factor/REST consensus binding sequence for NRSE/RE1 has been shown to increase the expression of neuronal genes (Kuwabara et al., 2004). Thus, many aspects of neuronal function are regulated by small regulatory RNAs. Dendritically localized miRNA-134-negatively regulates dendritic spine size through translational inhibition (Schratt et al., 2006), and loss of Dicer in cortical neurons leads to significant increases in dendritic spine length (Davis et al., 2008). This evidence supports a role for miRNAs in local regulation of protein synthesis.
We therefore deleted Dicer in damage-sensing neurons that express Nav1.8 (a sensory neuron-specific voltage-gated sodium channel) using the Cre-loxP system and examined the functional consequences. Nav1.8+ neurons are known to play a critical role in inflammatory pain, as well as in responses to cold and noxious mechanosensation (Abrahamsen et al., 2008). We provide evidence that small RNAs are essential for the maintenance of normal levels of expression of transcripts in these neurons, which are essential for altering peripheral pain thresholds associated with inflammation.
Materials and Methods
Characterization of mutant mice.
Floxed dicer mice containing loxP sites flanking dicer exon 20 and 21 (Cobb et al., 2005) were crossed with the Nav1.8-Cre strain (Stirling et al., 2005) to affect dicer gene ablation in a defined subset of sensory neurons. The study population contained the homozygous floxed dicer gene and one copy of the Nav1.8-Cre allele [conditional Dicer knock-out (KO)], while homozygous floxed dicer littermates [Dicer wild type (WT)] were used as controls.
Genotyping analysis with PCR.
Genomic DNA was isolated from ear or dorsal root ganglia (DRGs). The dicer wild-type fragment (259 bp), floxed fragment (390 bp), and mutant fragment (309 bp) were detected by PCR with the primers 5′-AGTGTAGCCTTAGCCATTTGC-3′ (forward), 5′-CTGGTGGCTTGAGGACAAGAC-3′, and 5′-AGTAATGTGAGCAATAGTCCCAG-3′. The Nav1.8-Cre fragment (460 bp) was detected by PCR with primers 5′-TGTAGATGGACTGCAGAGGATGGA-3′ (forward) and 5′-AAATGTTGCTGGATAGTTTTTACTGCC-3′ (reverse). PCR primer positions are show in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
Neural marker labeling.
After deep anaesthetization with Euthatal (150 mg/kg, i.p., Rhone Merieux), animals were transcardially perfused with 10 ml of heparinized saline [0.9% (w/v) NaCl] followed by 25 ml of fresh prepared 4% (w/v) paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. DRGs were postfixed in the same fixative solution for 2 h at 4°C and then cryoprotected overnight in 30% (w/v) sucrose containing 0.02% sodium azide in 0.1 m PB at 4°C. These perfused DRGs were embedded in OCT embedding compound (BDH Chemicals) on dry ice and cut serially (11 μm thickness) on a cryostat and collected onto SuperFrost Plus slides (VWR International, 631-0108). After drying the slides at room temperature for 2 h, the perfused DRG sections were stained with anti-calcitonin gene-related peptide (CGRP), anti-N52, biotin-labeled isolectin B4 (IB4), and anti-c-RET antibodies. Fresh unperfused DRG sections were prepared as described previously (Zhao et al., 2006b) and stained with anti-peripherin and anti-neurofilament antibodies (N200). The immunostaining procedure followed as described previously (Zhao et al., 2006a). Briefly, After three washes in 1× PBS containing 0.3% Triton X-100 (PBST), slides were incubated with 10% goat serum in PBST at room temperature for 1 h and then incubated with primary antibodies overnight at 4°C in mouse anti-peripherin (1:1000; Sigma, catalog #P5117), rabbit anti-N200 (1:200; Sigma, catalog #N4142), rabbit anti-CGRP (1:50000; Sigma, catalog #C8198), mouse anti-N52 (1:1000; Sigma, catalog #N0142), biotin-labeled IB4 (1:600, Sigma, catalog #L2140), rabbit anti-c-RET (1:100; IBL International, catalog #18121). After three washes in PBST, sections were incubated in goat anti-rabbit Alexa Fluor 488 antibody (1:1000, Invitrogen, catalog #A-11017), anti-mouse IgG Alexa Fluor 594 (1:1000; Invitrogen, catalog #A-11037), or Cy3-conjugated streptavidin (1:1000; Jackson ImmunoResearch Laboratories, catalog #016-160-084) for 2 h. After 3 washes in PBST, the sections were mounted with Gel Mount Aqueous (Sigma, catalog #G0918). The sections were visualized using a fluorescent Leica microscope.
Microarray analysis.
In three experiments, total RNA was extracted from DRGs of Dicer conditional null (n = 2, all males) and floxed littermates (n = 2, all males) with RNeasy Mini Kit (Qiagen, catalog #74104). Five micrograms of total RNA was transcribed to cDNA with One-Cycle cDNA synthesis kit (Affymetrix, catalog #P/N 900431). Biotin-labeled cRNAs were prepared with IVT labeling kit (Affymetrix, catalog #P/N 900449) and fragmented with GeneChip sample cleanup module (Affymetrix P/N 900371). Six chips (GeneChip Mouse Genome 430 2.0 array, Affymetrix, with catalog #P/N 900449) were hybridized with fragmented biotin-labeled cRNA (20 μg for one chip). Hybridization and data analysis were performed by Microarray Services, Wolfson Institute for Biomedical Research, University College London, London, UK. After differential expression analysis, the lists of differentially expressed genes were compared. Genes showing significant negative expression in both models when compared with control were selected [fold change ≥2; false discovery rate (FDR) ≤0.05]. An evaluation was made of the linear relationship between fold change in Nav1.8 neuron-depleted mice (Abrahamsen et al., 2008) and fold change in the conditional Dicer-null mutant using Pearson's correlation:
 |
Quantitative real-time reverse-transcription PCR analysis of mRNA and pre-mRNA.
Total RNA was isolated with RNeasy Mini Kit (Qiagen, catalog #74104) from DRGs in accordance with the manufacturer's instructions. The DNA in the samples was removed with RNase-Free DNase set (Qiagen, catalog #79254). Reverse transcription was performed with 1 μg of RNA by using iScript Select cDNA synthesis kit (Bio-Rad, catalog #170-8896). Quantitative real-time reverse-transcription PCR (qRT-PCR) was then performed with iQ SYBR Green Supermix (Bio-Rad, catalog #170-8882). The following primer pairs were used: Edg7: 5′-TCCCATGAAGCTAATGAAGACAG-3′ (forward, for mRNA and pre-mRNA), 5′-TTCATGACGGAGTTGAGCAG-3′ (reverse, for mRNA), and 5′-CCTGGCACATAGTGGACATTT-3′ (reverse, for pre-mRNA); TrpC6: 5′-ATTGATAAGGAGAGCGATGAGG-3′ (forward, for mRNA), 5′-GGAATCATTGGTTCACACTTCAT-3′ (forward, for pre-mRNA), and 5′-AGGTCTTCTGTGTTCTGTGATTTC-3′ (reverse, for mRNA and pre-mRNA); Scn11a: 5′-AGCCCAACGAAGTGAAGAAA-3′ (forward, for mRNA and pre-mRNA), 5′-TCTCCAAGCCAGAAACCAAG-3′ (reverse, for mRNA), and 5′-GGTCCTGGCTACTGGGTTTAC-3′ (reverse, for pre-mRNA); Scn10a: 5′-AATCAGAGCGAGGAGAAGACG-3′ (forward, for mRNA), 5′-TGCAGATGCCTACACTGAGAAA-3′ (forward, for pre-mRNA), and 5′-CTAGTGAGCTAAGGATCGCAGA-3′ (reverse, for mRNA and pre-mRNA); Scn9a: 5′-ATTTCAGCAGATGTTAGACCGA-3′ (forward, for mRNA) and 5′-TCCCATAATCCTTTGTCCCA-3′ (reverse, for mRNA); Runx1: 5′-GGTGGACAGAGGAAGAGGTG-3′ (forward, for mRNA) and 5′-TTGCCACCTACCATAGAGCC-3′ (reverse, for mRNA); calcium/calmodulin-dependent protein kinase type II α (CaMKIIa): 5′-GAGTCCTACACGAAGATGTGC-3′ (forward, for mRNA), 5′-GGAAGCTGAGAGAACAATGGA-3′ (forward, for pre-mRNA), and 5′-GGTACTGAGTGATGCGGATA-3′ (reverse, for mRNA and pre-mRNA); apolipoprotein D (ApoD): 5′-GTCTCTTCACCACAGCCAAA-3′ (forward, for mRNA), 5′-TCTGCTTGTTGTTGTCTGTGC-3′ (forward, for pre-mRNA), and 5′-TTCTCCATCAGCGAGTAGTTG-3′ (reverse, for mRNA and pre-mRNA); β-actin: 5′-TTCTTTGCAGCTCCTTCGTT-3′ (forward, for mRNA and pre-mRNA), 5′-ATGGAGGGGAATACAGCCC-3′ (reverse, for mRNA), and 5′-CTAGGCGTAAAGTTGGCTGTG-3′ (reverse, for pre-mRNA). qRT-PCR analysis was performed using an iCycler (Bio-Rad).
Western blot analysis.
Proteins were isolated from freshly excised DRGs taken from Dicer-null and control mice. All steps were performed at 4°C. DRGs were homogenized in 100 μl of homogenization buffer (50 mm Tris-Cl, pH 8.0, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 0.5% nadeoxycholate, 0.1% SDS, 1 mm PMSF, 1 U/ml aprotinin, 10 μg/ml antipain, 1 μg/ml leupeptin, and 1 μg/ml pepstatin A) by using 15 strokes of a glass-Teflon homogenizer. Centrifugation for 15 min at 14,000 rpm removed the nuclear fraction and cell debris. Protein concentrations were determined with BCA protein assay kit (Pierce, catalog #23227), and then samples of 40 μg were separated on SDS-PAGE gel and blotted to nitrocellulose membrane (GE Healthcare) in transfer buffer (25 mm Tris-HCl, pH 8.3, 192 mm glycine, 0.1% SDS, and 20% methanol) for 1 h at 100 V with a Bio-Rad transfer cell system. The membrane was blocked with 5% nonfat milk in PBS-Tween (0.1% Tween 20) for 1 h at room temperature and then incubated with primary antibody anti-Camk2a (1:200; Santa Cruz Biotechnology catalog #sc-32288) and anti-ApoD (1:200; Santa Cruz Biotechnology, catalog #sc-34760) in blocking buffer overnight at 4°C. The membrane was washed three times with TBS-Tween and then incubated with secondary antibody goat anti-mouse or goat anti-rabbit IgG-HRP (Jackson ImmunoResearch Laboratories), or donkey anti-goat IgG-HRP (Santa Cruz Biotechnology, catalog #sc-2020) was applied at 1:1000 in TBS-Tween for 2 h at room temperature. Detection was performed by Western Lightning Chemiluminescence Reagent Plus (PerkinElmer, catalog #NEL104) and exposed to BioMax film (Kodak). After detection, the membranes were stripped of antibody by using high concentrations of glycine, SDS, and β-mercaptoethanol at 50°C. The membranes were then reprobed with anti β-actin primary antibody (1:1000, Santa Cruz Biotechnology, catalog #sc-47778) and subsequently with anti-mouse secondary antibody.
Tetrodotoxin-resistant current detection.
DRGs were excised from culled mice after CO2 anesthesia (Dicer conditional null or floxed littermate control), DRG neurons were cultured in DMEM, and the tetrodotoxin (TTX)-resistant currents in small DRG neurons were recorded as described previously (Stirling et al., 2005).
Behavioral test.
All tests were approved under the United Kingdom Home Office Animals (Scientific Procedures) Act 1986 and performed on animals aged between 8 and 16 weeks. Behavioral analysis was performed as described by Stirling et al. (2005) (rotarod, Hargreaves' apparatus, hotplate, von Frey, Randal–Selitto tests), Zimmermann et al. (2007) (cold plate), Foulkes et al. (2006) (formalin test), and Zhao et al. (2006a) (carrageenan). The complete Freund's adjuvant (CFA) inflammatory model was performed by injecting 20 μl of CFA into the plantar surface of left paw. Following the CFA injection, thermal hyperalgesia and mechanical allodynia were analyzed with Hargreaves and von Frey tests, respectively, at days 1, 2, 3, 4, and 10. The spontaneous pain assessment was performed in a modified version (Djouhri et al., 2006). Briefly, 20 μl of CFA was injected into the side of the right knee and the plantar surface of the right paw. After injection spontaneous foot lifting (SFL) were observed individually two times (5 min each time) on days 1, 2, and 4. The neuropathic pain assessment was performed with the Seltzer model (Seltzer et al., 1990). Briefly, animals were anesthetized using halothane. A 0.5 cm incision was made in the skin of the upper left leg and blunt scissors were used to part the muscle layers to access the sciatic nerve. A tight ligation of between one-half and one-third of the nerve was made using 6-0 mersilk suture (Ethicon). The skin was closed using 4-0 mersilk sutures (Ethicon). After at least 3 d of recovery, the mice were tested and analyzed. Responses to mechanical stimulation were assessed with von Frey filaments on days 3, 5, 8, 10, 15, and 30 after surgery. Baseline responses for mechanical stimulation were obtained with von Frey tests before surgery. Results from the responses to mechanical stimulation were expressed as the relative change in the 50% threshold at each time point after surgery (test/baseline) for each group ± SEM. The results for the two groups were compared using a two-way repeated-measures ANOVA test.
In vivo mouse spinal cord electrophysiology.
Intact anesthetized male and female mice (8–10 weeks) were used in all experiments in accordance with the requirements of the United Kingdom Animals (Scientific Procedures) Act 1986. After briefly inducing anesthesia with 3% halothane (66% N2O and 33% O2), mice were injected with urethane (240 mg/kg). Animals were placed in a stereotaxic frame and a laminectomy was performed to expose the L3–L6 spinal segments. Extracellular recordings were made from single wide dynamic range neurons using Parylene-coated tungsten electrodes (A-M Systems). Neurons were visualized on an oscilloscope and were isolated and discriminated by spike amplitude and waveform. All neurons had receptive fields over the hindpaw. Depth of the neuron was measured from the surface of the spinal cord. Electrical stimulation was administered by inserting two needles into the receptive field. A train of 16 stimuli was given (2 ms pulse duration, 0.5 Hz at three times the C-fiber threshold). The evoked neuronal responses were superimposed and separated on the basis of fiber conduction velocity and latency into total A-fiber-evoked (0–50 ms), and C-fiber-evoked (50–250 ms) action potentials. Neuronal responses occurring after the C-fiber latency band (250–800 ms) were classed as postdischarge. The “input” (nonpotentiated response) and the “wind-up” (potentiated response, shown by increased neuronal excitability to repeated stimulation) were calculated as follows: input = (action potentials (50–800 ms) evoked by first pulse at 3× C-fiber threshold) × total number of pulses (16). Wind-up = (total action potentials (90–800 ms) after 16 train stimulus at 3× C-fiber threshold) − input. A wide range of natural stimuli, including brush, von Frey filaments (Scientific Marketing Associates) in ascending order (1, 6, 8, 15, 26, and 60 g), and heat (35, 40, 45, and 50°C water jet), was applied to the receptive field for 10 s per stimulus. Data were captured and analyzed by a CED 1401 interface (Cambridge Electronic Design) coupled to a Pentium computer with Spike 2 software (poststimulus time histogram and rate functions).Data were analyzed from a total of 30 wide dynamic range neurons in Nav1.8-Cre+/− mice (n = 14) and Dicer KO mice (n = 16) mice. All neurons selected responded to innocuous and noxious stimuli. Statistical analysis was conducted using unpaired t tests to compare evoked responses of heterozygous Nav1.8-Cre control (Nav1.8-Cre+/−) and Dicer KO mice except for graded mechanical and thermal testing, where two-way ANOVA with Bonferroni's post hoc tests were used.
c-Fos immunohistochemistry.
Two hours after intraplantar injections of 20 μl of 5% formalin in right hindpaw, the Dicer conditional null mice (n = 5) and floxed littermates (n = 4) were deeply anesthetized with urethane (1.25 g/kg, intraperitoneal injection) and then animals were transcardially perfused with 10 ml of heparinized saline [0.9% (w/v) NaCl] followed by 25 ml of fresh prepared 4% (w/v) PFA (paraformaldehyde) in 0.1 m PB, pH 7.4. The lumbar segments (L2–L5) of the spinal cord were removed from the vertebral column. DRG were postfixed in the same fixative for 2 h at 4°C and then cryoprotected overnight in 30% (w/v) sucrose containing 0.02% sodium azide in 0.1 m PB at 4°C. The lumbar segments were then cut serially (40 μm thickness) on a freezing microtome. Every sixth section was collected into a Petri dish containing 0.1 m PB. The sections were blocked in 0.1 m PB containing 3% goat serum and 0.3% Triton X-100 at room temperature for 1 h, then stained with anti c-Fos antibody (1:2000, Abcam catalog #ab7963) in 50 mm Tris-Cl, pH 7.4, containing 0.3% Triton X-100 (TTBS) at room temperature for 24 h. After three washes with 0.1 m PB, the sections were incubated with biotinylated anti-rabbit IgG (1:500; Vector Laboratories, catalog #BA-1000) in TTBS at room temperature for 2 h. Then, after three washes as before, the sections were incubated with fluorescein avidin D (1:200, Vector Laboratories A-2001) in dark at room temperature for 1 h. After three washes with TTBS, the sections were put on the SuperFrost Plus slides (VWR International, catalog #631-0108) and mounted with Gel Mount Aqueous (Sigma, catalog # G0918) and coverslipped. The sections were visualized using a fluorescent Leica microscope, and the images were taken using the Xillix MicroImager system and analyzed with OpenLab software. The same setup of acquisition was used to acquire the pictures of both groups of mice. In each animal, the number of c-Fos-positive neurons was counted blind in four sections in laminae I and II of lumbar segment L4–L5.
Current-clamp recordings.
Current-clamp experiments were performed using an Axopatch 200B patch-clamp amplifier (MDS Analytical Technologies) in the I-clamp normal mode. Pipettes with resistances of 1–3 MΩ were pulled from borosilicate glass capillaries (Harvard Apparatus) using Flaming/Brown P-97 horizontal micropipette pullers (Sutter Instruments) and fire polished to improve sealing. All experiments were begun in voltage-clamp mode. Pipette capacitance transients were cancelled while in cell-attached mode, and whole-cell capacitance (10–40 pF) and series resistance (40–70%) were compensated once whole-cell mode had been attained. Whole-cell membrane currents were filtered at 1 kHz and sampled at 10 kHz. Data were acquired and analyzed using pCLAMP 9.2 software (MDS Analytical Technologies). All recordings were made at 22–24°C. In current-clamp experiments, action potential firing in small DRG neurons (<25 μm) was elicited by a series of 7 depolarizing current pulses (160 ms duration) from 0 to 140 pA in 20 pA step increments. Extracellular solution contained the following: 140 mm NaCl, 4 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, adjusted to pH 7.4 with NaOH, osmolarity 300–310 mosmol l−1. Pipettes were filled with an intracellular solution containing the following: 140 mm KCl, 1.6 mm MgCl2, 2 mm EGTA, 2.5 mm NaATP, 0.5 mm NaGTP, 10 mm HEPES and adjusted to pH 7.3 with KOH, osmolarity 300–310 mosmol l−1. Cells were continuously perfused with extracellular solution. The inflammatory soup was adapted from that used by Maingret et al. (2008) and contained the following: 10 μm bradykinin, 10 μm prostaglandin E2 (PGE2), 10 μm histamine, 10 μm ATP, and 10 μm 5-hydroxytryptamine (5-HT). To determine TTX sensitivity of action potentials, 300 nm TTX was applied at the end of experiments.
miRNA sequencing and bioinformatical analysis.
DRGs were dissected from either wild-type C57BL/6 or Dicer KO mice and suspended in RNAlater (Ambion) solution. DRG complements from several mice were pooled and homogenized in lysis buffer (Ambion) using a borosilicate glass pestle and mortar (Fisher Scientific). Small RNA was purified from homogenate using the mirVana miRNA isolation kit (Applied Biosystems), and the quantity and quality were checked using a NanoDrop spectrometer (Thermo Scientific). Samples were prepared for sequencing following the Illumina small RNA sample preparation kit protocol. Briefly, 10 μg of small RNA for each sample was subjected to electrophoresis on a 15% Urea Tris-borate-EDTA polyacrylamide gel. Gel fragments corresponding to oligonucleotides 16–26 bp were excised and purified. Retained small RNAs were then ligated sequentially with 5′ and 3′ Illumina adapters and subsequently purified. Using DNA primers complementary to the adapters, the samples were reverse transcribed to produce a cDNA library. For library validation, 1 μl of each cDNA sample was subcloned into TOPO vector (Invitrogen) and sequenced via conventional Sanger methodology. The remaining sample was subjected to Solexa sequencing using the Genome Analyzer II DNA sequencing platform (Illumina) (Hafner et al., 2008). Counts (frequency of each sequence) from the Dicer KO sample were normalized to those from wild-type sequencing by adjusting the raw counts by the ratio of control:Dicer KO. The miRDeep (Friedländer et al., 2008) algorithm package was used to identify known and potentially novel miRNAs from the sequences. The Mapping and Assembly with Qualities (Maq) algorithm (Li et al., 2008) was also used for sequence analysis by aligning Solexa sequences (Illumina) with murine miRNA hairpin sequences to estimate absolute copy number in both samples (given as a maximum coverage value). The miRNA database (miRBase revision 11) (Griffiths-Jones, 2004, 2006, 2008) was used for determining whether identified sequences are novel candidates.
Statistical analysis.
All values are presented as means ± SEM. Data were analyzed with two-way ANOVA and Student's t test. Differences were considered significant at p < 0.05.
Results
Dicer deletion does not result in sensory neuron loss
Cerebellar Purkinje cells and some other cell types die in the absence of Dicer (Schaefer et al., 2007). We therefore checked DRGs for any potential neuronal cell loss in adult animals, using markers for large and small diameter sensory neurons. We found no evidence for loss of neurons in Dicer conditional null mutants (Fig. 1A,C). The relative numbers and percentage of large (neurofilament positive) and small diameter neurons (peripherin positive) were the same as those of controls (∼62% small diameter peripherin-positive neurons) (Zhao et al., 2006a). However, when we subdivided the small diameter peripherin-positive neurons into the peptidergic (NGF dependent, TrkA positive) and IB4 binding nonpeptidergic populations, we found that there was a loss of IB4 staining, although peptidergic neurons expressing CGRP were present at normal levels (Fig. 1A). IB4 binding is a marker associated with the expression of c-RET, a tyrosine kinase receptor for GDNF neurotrophic family members (Luo et al., 2007). Staining with antibodies to c-RET showed that the neurons that respond to GDNF and normally express IB4 binding were still present, which is consistent with no loss of DRG neurons in the null mutant mice (Fig. 1A). The staining of primary afferent terminals in the dorsal horn was consistent with the data obtained from DRG cell bodies (Fig. 1B). The IB4 binding epitope is a terminal galactose residue found on a number of glycolipids and/or glycoproteins, e.g., V2-like variant of versican (Vcan) (Bogen et al., 2005) and laminin β2 (Lama2) (Fullmer et al., 2004) in small and medium diameter DRG neurons. The enzyme α(1, 3)galactosyltransferase catalyzes the addition of galactose residues to proteins (Fullmer et al., 2007). Both Vcan and Lama2 appear to be expressed at normal levels (microarray) in Dicer KO mice; however, glycoprotein galactosyltransferase α 1,3 (Ggta1) was downregulated by ∼2-fold in DRG in Dicer mutant mice, suggesting that a lower level of Ggta1 may underlie the loss of IB4 binding.
Figure 1.

Neural marker expression in DRGs of Dicer conditional null mice. A, DRG sections of Dicer conditional null mice and Dicer floxed littermate controls were labeled with anti-neurofilament (N200 and N52), anti-peripherin, anti-CGRP, anti-IB4, and anti-c-RET antibodies. The panels in the columns marked “Merged” are merged images of the panels in the two columns to the left. B, Cross sections of lumbar spinal cord were stained with anti-CGRP and IB4. In lamina I-II, CGRP-positive terminals were identified in both the Dicer knock-out mice and floxed littermate controls, whereas all detectable IB4-positive terminals were deleted compared with littermate controls. C, The proportions of neurofilament (N200) and peripherin-expressing neurons were normal in Dicer conditional null animals compared with floxed littermate controls. WT, White symbols; KO, black symbols. Scale bars, 50 μm.
Dicer deletion leads to deficits in inflammatory but not acute pain
The subsets of neurons that we targeted are known to be essential for inflammatory and acute cold and mechanical, but not neuropathic, pain (Abrahamsen et al., 2008). We therefore examined the consequences of deleting Dicer on pain behavior in the null mutants. Motor function measured on a rotarod was normal (Fig. 2A), allowing us to rule out the possibility of major motor deficits that would compromise behavioral tests. Responses to light (von Frey hairs) and noxious (Randall–Selitto apparatus) levels of mechanical stimulation, as well as noxious heat (Hargreaves apparatus and hot plate), were also normal (Fig. 2B–E). Thus there were no behavioral deficits in peripheral acute pain responses in the absence of miRNAs. However, cold pain behavior was attenuated (Fig. 2F), and this may be related to the lower levels of expression of the voltage-gated sodium channel Nav1.8, which is known to be essential for the transmission of information from damaged cold tissue (Zimmermann et al., 2007).
Figure 2.

Acute pain behavior and in vivo electrophysiology of wide dynamic range (WDR) in Dicer mutant mice. A, Rotarod studies showed no motor deficits in conditional Dicer-null animals (n = 12, WT; n = 20, KO). B, Acute mechanical pressure applied with the Randall–Selitto apparatus demonstrated identical behavior in KO and WT mice (n = 12, WT; n = 20, KO). C, Responses to low-threshold mechanical stimulation by Von Frey filaments are normal in the null mutants (n = 8, WT; n = 15, KO). D, Hargreaves' apparatus demonstrates identical latencies of response to thermal stimulation (n = 8, WT: n = 16, KO). E, Supra-spinal reflexes to heat (50 and 55°C) using the hot plate apparatus demonstrates identical latencies in KO and WT mice (n = 9, WT; n = 14, KO). F, Cold behavior (number of liftings) is reduced in the Dicer-null mutant mice (0°C) using the cold plate apparatus (n = 16, WT; n = 22, KO). G, A-fiber and C-fiber firing thresholds of WDR neurons in control Nav1.8Cre+/− mice (black) (n = 14) and Dicer KO mice (white) (n = 16). H, Responses to transcutaneous electrical stimulation of the receptive field (a train of 16 2-ms-wide electrical pulses at 0.5 Hz at 3× C-fiber threshold). Spikes evoked between 0 and 50 ms were classified as evoked by A-fiber input, and those evoked between 50 and 250 ms were classified as evoked by C-fiber input. Postdischarge (PD) describes the spikes that are evoked between 250 and 800 ms and correlate with C-fiber activity. Input is determined by the first stimulus of the train × 16 and reflects nonpotentiated C-fiber-evoked WDR neuron responses. Wind-up (WU) is determined by calculating additional spikes above the predicted constant baseline response and describes the increased neuronal excitability evoked by C-fibers to repeated constant stimulation. C-fiber activity, wind-up, and postdischarge were all significantly decreased in diphtheria toxin A chain (DTA) mice. I, Evoked responses from innocuous dynamic brush, noxious pinch, and a cold (1°C) water jet stimulus. J, Evoked responses from Nav1.8 Cre+/− mice (solid line) and Dicer−/− mice (dashed line) to graded von Frey filaments. K, Thermally evoked responses using water jets at graded temperatures (Temp). There was no significant difference between the two groups.
We next examined electrical input into the CNS, recording polysynaptic input into wide dynamic range neurons in anesthetized mice (Abrahamsen et al., 2008). The mean depths from the surface of the spinal cord of the neurons in heterozygous Nav1.8-Cre control and Dicer conditional null mice were 530 ± 62 and 432 ± 39 μm respectively, corresponding to the deep laminae. Evoked neuronal responses to electrical, mechanical, and thermal stimuli revealed no significant differences between the null and heterozygous Nav1.8-Cre control (Fig. 2G–K), confirming that acute pain signaling to the CNS is normal in Dicer conditional null mice. These data confirm that the Dicer null mutant contains a complete complement of healthy functional A-fiber- and C-fiber-associated sensory neurons. When we examined the altered pain states associated with inflammation however, there were substantial deficits in the Dicer-null mice. The second phase of the formalin response was attenuated in the null mutants (Fig. 3A,B), although heterozygous nulls were indistinguishable from wild-type controls in their behavior (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). This suggests that there is no haploinsufficency associated with Dicer heterozygotes. Carrageenan-evoked inflammation had little effect on pain thresholds in the Dicer-null mutants compared with controls (Fig. 3C). Carrageenan effects are mainly mediated through prostanoid signaling (Tanaka et al., 1998; Chao et al., 2006), while many aspects of formalin-evoked pain behavior that occur are transduced by the TRPA1 channel (McNamara et al., 2007). However, both inflammatory mechanisms are deficient in the Dicer null mice. We also tested inflammatory pain evoked by CFA. Thermal and mechanical responses were recorded on days 1, 2, 3, 4, and 10 after CFA injection. WT controls developed both thermal hyperalgesia and mechanical allodynia. However, the conditional KO mice displayed neither thermal hyperalgesia nor mechanical allodynia (Fig. 3D,E). Spontaneous pain evoked by inflammation was much reduced (Fig. 3F). Neuropathic pain, which does not require Nav1.8+ nociceptors (Abrahamsen et al., 2008), was unaffected (Fig. 3G).
Figure 3.

Inflammatory, neuropathic pain behavior, c-Fos expression in spinal cord, and excitability of DRG neurons. A, Time course of pain behavior in Dicer conditional null mutants after intraplantar injection of formalin. Time spent licking/biting the injected hindpaw was recorded (n = 8, WT; n = 10, KO). B, Two phases (0–10 min, 10–60 min) of licking behavior after formalin injection were analyzed, the second phase of which is attenuated in the Dicer-null mice (p < 0.01, t test). C, Intraplantar injection of carrageenan (n = 9, WT; n = 11, KO) caused a long-term thermal hyperalgesia that is attenuated in Dicer conditional mutant mice (p < 0.05, two-way repeated measured ANOVA). D, CFA-induced mechanical hyperalgesia was measured using von Frey filaments (n = 5, WT; n = 7, KO). A strong difference was observed. (p < 0.01, two-way repeated measured ANOVA). E, CFA-induced thermal hyperalgesia was measured using the Hargreaves instrument (n = 5, WT; n = 7, KO). A significant difference was observed (p < 0.05, two-way repeated measured ANOVA). F, Behavioral signs of spontaneous pain behavior were tested on CFA-treated mice. The number of spontaneous foot liftings (SFLs) was measured on days 1, 2, and 4 after intradermal injections of CFA in the right hind-paw and right knee (n = 11, WT; n = 10, KO). SFL on day 1 was much less in Dicer-null mutant mice than in floxed control mice (p < 0.05, t test). G, Sciatic nerve ligation following the Seltzer procedure caused a long-lasting mechanical hyperalgesia. Both Dicer conditional null mutants and floxed Dicer littermate controls showed an identical time course and level of allodynia after this injury. All data presented as means ± SEM; *p < 0.05, **p < 0.01. WT, White symbols, KO, black symbols.
We next used c-Fos staining to examine nociceptive input into the spinal cord in response to formalin injections (Fig. 4A,B) (Hunt et al., 1987). Two hours after intraplantar injection of formalin, the number of dorsal horn c-Fos-immunoreactive neurons was reduced by >50% compared with controls, consistent with diminished noxious input into the spinal cord in inflamed states in the Dicer null mutant (Fig. 4B). We examined whether the loss of inflammatory pain reflected an inability of null mutant Nav1.8+ sensory neurons to alter their excitability on treatment with inflammatory mediators. All wild-type Nav1.8+ neurons showed a consistent increase in excitability on treatment with inflammatory “soup” containing bradykinin, prostaglandin E2, histamine, ATP, and 5-HT. However, the majority of Dicer-null, mutant-derived Nav1.8+ sensory neurons showed normal thresholds of activation. Nineteen of twenty one (19 of 21) showed no increase in excitability on the addition of inflammatory mediators (Fig. 4C).
Figure 4.

c-Fos response in the spinal cord and sensitization of nociceptors by inflammatory mediator(s). A, Representative immunostaining for c-Fos in superficial laminae I and II of spinal cord. Contra, Contralateral side to formalin injection; Ispi, ipsilateral side to formalin injection. Scale bar, 100 μm. B, Quantification of c-Fos-positive neurons. The number of c-Fos-positive nuclei in superficial (laminae I, II) cord in L4 to L5 ipsilateral to the injection of formalin was counted (n = 4, WT; n = 5, KO). The number of c-Fos-positive neurons is reduced in the ipsilateral side in Dicer-null mutants (p < 0.05, t test). C, Sensitization of WT and KO nociceptors by inflammatory mediators. Top, Representative traces showing action potential firing produced by a 100 pA depolarizing current in control conditions (left) and in the presence of inflammatory soup (IS, right) in a WT DRG neuron. Bottom, Same as top, but in a Dicer KO DRG neuron.
Dicer deletion has differential effects on broadly expressed and nociceptor-enriched transcripts
Because we expected that some mRNA species would be upregulated in the absence of miRNAs, we examined the levels of mRNA transcripts present in Dicer-null mice and wild-type controls using microarray analysis (Fig. 5). We then used qRT-PCR to confirm altered levels of transcripts (Fig. 6A,B). Most transcripts (>39,000) were unaffected by Dicer deletion, although hundreds were expressed at higher levels than those in normal sensory neurons (Fig. 6B). Surprisingly, many sensory neuron-associated transcripts are downregulated rather than upregulated in the absence of Dicer. For example, qRT-PCR data (Fig. 6A) showed that the mRNA for the nociceptor-associated transcription factor Runx1 (Zhong et al., 2006) was downregulated, as were the peripheral neuron-specific TTX-resistant sodium channels Nav1.8 and 1.9 (Wood et al., 2004). A variety of other receptors and channel transcripts, such as the lysophosphatidic acid G-protein-coupled receptor (GPCR) Edg7 and the MAS-related GPCR Mrgpra3 found in subsets of sensory neurons, were dramatically downregulated, while other transcripts (for example ApoD, which is broadly expressed in a variety of tissues) were present at more than double their normal levels (Fig. 6B). The complete microarray data have been deposited at ArrayExpress (accession no. E-MEXP-1461). Small RNAs are required for normal expression of many sensory neuron-specific genes. The downregulation of nociceptor mRNA transcripts is surprising given the role of miRNAs in recruiting RNA silencing complexes, leading to the view that Dicer deletion should cause an upregulation of mRNA expression (He and Hannon, 2004). We examined the levels of pre-mRNA unspliced transcripts in DRG neurons using qRT-PCR with primers designed across intron–exon boundaries. We found that the lowered levels of mRNA expression seemed to be a consequence of lower levels of unspliced precursors (Fig. 6C). Thus, Dicer products are required for normal levels of transcription. Because a number of nociceptor-specific genes were downregulated in Dicer-null mutants, we compared the microarray data for transcripts regulated by Dicer deletion with previously obtained microarray data on the transcripts that are selectively expressed by Nav1.8+ neurons (Abrahamsen et al., 2008). An analysis using Pearson's correlation coefficient indicates a statistically significant linear relationship between the genes negatively expressed by a significant amount (fold change ≥2; FDR ≤0.05) in both Nav1.8+ neuron-depleted and Dicer knock-out mice, r = 0.73, p < 0.001 (Fig. 6D and supplemental Table 1, available at www.jneurosci.org as supplemental material). This suggests that there is a requirement for Dicer products to upregulate the levels of many sensory neuron-associated transcripts, but not other more globally expressed transcripts. One possible explanation for these observations is that deleting all miRNAs allows a repressor of sensory neuron gene transcription to be expressed at high levels in Dicer-null neurons. We therefore used brief pulses of protein synthesis inhibitors to see whether we could relieve potential inhibition of sensory neuron-specific pre-mRNA transcripts. The levels of Nav1.8 pre-mRNA measured with qRT-PCR were similarly increased in control and Dicer-null neurons by cycloheximide, a protein synthesis inhibitor, which is consistent with the loss of some inhibitory proteins that act independently of miRNAs in control and null mutant mice (Fig. 7E). ApoD mRNA and pre-mRNA were completely unaffected by cycloheximide (Fig. 7F). This does not support the existence of short-lived protein repressors of sensory neuron gene expression. However, this experiment does not rule out upregulation of stable transcriptional repressor proteins. Nonetheless, whether direct or indirect in their action, small RNAs are clearly required for normal expression of many sensory neuron genes.
Figure 5.
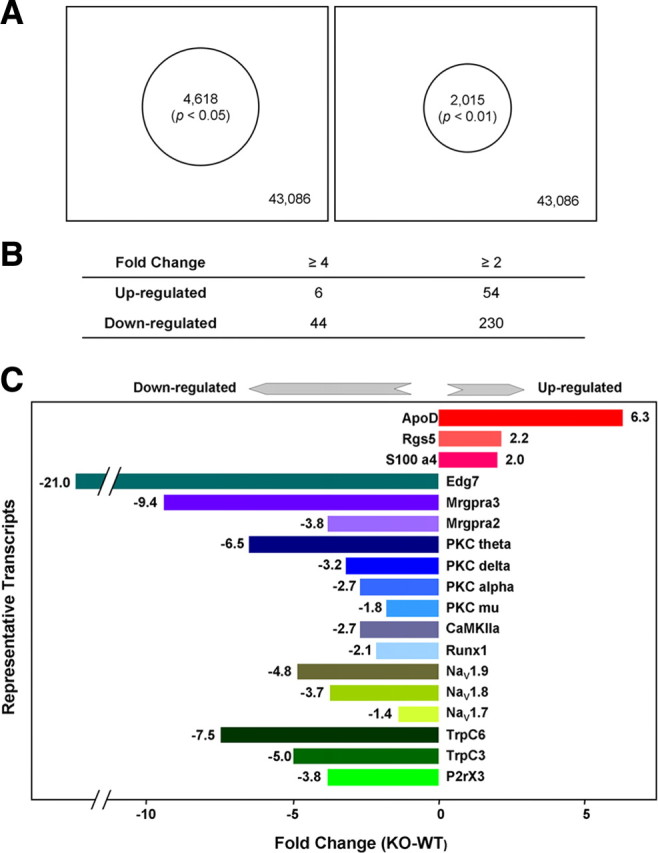
Microarray analysis. A, Summary of changed transcripts in Dicer conditional null mutant. After isolation of total RNA from DRGs of Dicer conditional null mice (n = 6, males) and floxed littermate control (n = 6, males), the expression of mRNA was detected with GeneChip (mouse genome 430 2.0 array). Four thousand six hundred and eighteen (4618) transcripts (p < 0.05) (left) and 2015 transcripts (p < 0.01) (right), changed from 43,086 transcripts in the DRGs of Dicer conditional null mice compared with floxed littermate controls. B, The number of transcripts with fold change is ≥4 and ≥2, respectively, in Dicer conditional null mutant versus floxed littermate controls (p < 0.01). C, The fold change of representative genes in Dicer conditional null mutant, p < 0.0001.
Figure 6.
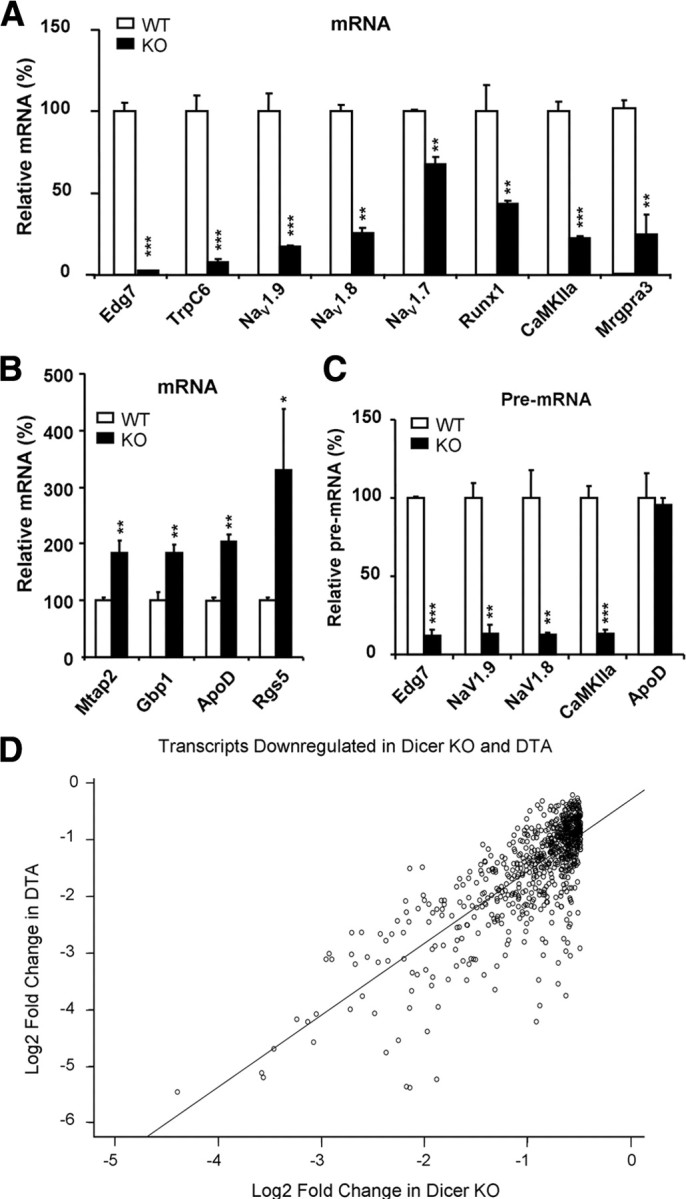
Representative qRT-PCR analysis of selected transcripts and correlation between fold decrease in Nav1.8+ neuron-depleted mice [diphtheria toxin A chain (DTA)] (Abrahamsen et al., 2008) and fold decrease in Dicer conditional null mutants. A, After isolation of DRG, total RNA was extracted from DRG. Representative mRNA and pre-mRNAs were subjected to analysis by qRT-PCR. Some of the DRG-specific gene (Edg7, TRPC6, Nav1.9, Nav1.8, Nav1.7, Runx1, CaMKIIa, and Mrgpra3) transcripts were significantly downregulated in Dicer knock-out mice. B, Representative DRG nonspecific gene (Mtap2, Gbp1, ApoD, and Rgs5) transcripts show significant upregulation in Dicer knock-out mice. C, The pre-mRNAs of DRG-specific genes (Nav1.8, Nav1.9) and Edg7 and CaMKIIa were downregulated in Dicer knock-out mice. No alteration of pre-mRNA of ApoD was detected in Dicer knock-out mice. The data are normalized with β-actin. Each bar represents n = 3 mice. All data are presented as means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, using t test. WT, White symbols, KO, black symbols. D, The Pearson product-moment correlation (r) measures the strength of the association between Log2 fold change of gene expression in DTA mice and Log2 fold change of gene expression in Dicer KO. The correlation coefficient is r = 0.937. If there was a perfect correlation (r = 1), all points would fall along the straight diagonal line.
Figure 7.

Expression and function of representative genes in DRGs of Dicer conditional null mice. A, Protein was extracted from DRGs and CaMKIIa and ApoD were analyzed with Western blots. B, Western blot relative density. The experiments were repeated at least three separate times. The data are normalized with β-actin. Each bar represents n = 3 mice. C, DRG sections were labeled with anti-CaMKIIa antibody and anti-neurofilament (N200) antibody. CaMKIIa is mainly expressed in small diameter DRG neurons of floxed dicer littermate controls, but not in Dicer conditional null mice. D, TTX-resistant sodium currents were measured in small diameter (≤25 μm) DRG neurons (n = 23) and presented as maximum current density. E, Effect of cycloheximide on expression of Nav1.8. The DRGs from Dicer conditional null mice and floxed littermate controls were cultured in DMEM at 37°C, 5% CO2. Three hours after treatment with or without 10 μg/ml cycloheximide, mRNA and pre-mRNA of Nav1.8 from DRG were subjected to analysis by qRT-PCR. The data are normalized with β-actin. Each bar represents n = 3 mice. F, Effect of cycloheximide on expression of ApoD. The data are normalized with β-actin. Each bar represents n = 3 mice. All data presented as means ± SEM; *p < 0.05, **p < 0.01, using t test. WT, White symbols, KO, black symbols. Scale bars, 50 μm.
Functional protein expression reflects altered transcript levels
We examined the effect of altered mRNA expression on protein levels detected with Western blots (Fig. 7A,B). There was a significant lowering of protein expression for CaMKIIa, an important modulator of glutamatergic signaling and synaptic plasticity (Ninan and Arancio, 2004), while the broadly expressed ApoD protein showed levels that were higher, reflecting an increase in mRNA (Figs. 6B, 7B). Interestingly, differences in mRNA and pre-mRNA levels were not always linearly correlated with altered protein levels in DRG cell bodies. For example, CaMKIIa protein was substantially downregulated concomitant with lower mRNA levels, but Nav1.8 protein levels were less dramatically lowered despite a similar substantial reduction in mRNA levels. This may reflect differential rates of protein degradation. We stained DRG sections with anti-CaMKIIa antibody. The result showed that CaMKIIa is mainly expressed in small and medium DRG neurons, and protein levels are strongly reduced in Dicer-null mutant mice (Fig. 7C). We also examined the functional levels of expression of the nociceptor-specific Nav1.8 sodium channels electrophysiologically (Baker et al., 2003). The peak tetrodotoxin-resistant Nav1.8 sodium current density was significantly reduced in the Dicer KO neurons (Fig. 7D). Thus, deficits at the level of pre-mRNA are reflected in lowered levels of mRNA, protein, and functional activity.
Known and novel miRNA candidates are enriched in nociceptive neurons
We examined the repertoire of miRNAs that are expressed in Nav1.8+ nociceptors. Known miRNAs were cataloged in Dicer-null and control mouse DRG extracts using microarrays (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and their distribution is summarized in the supplemental information (supplemental Table 2, available at www.jneurosci.org as supplemental material). More than 60 miRNA species were lost or downregulated in the Dicer knock-out (supplemental Table 2, available at www.jneurosci.org as supplemental material), implying that they are selectively expressed in Nav1.8+ nociceptors within DRGs. Because there is the possibility that nociceptor-specific miRNAs are involved in regulating the expression of nociceptor-specific genes, we used Solexa sequencing to identify known and novel miRNAs, comparing the miRNA sequences in control DRG with the miRNAs present in the Nav1.8+ neurons of Dicer-null mice. To elicit which known miRNAs are present in control and Dicer KO DRG, the miRDeep (Friedländer et al., 2008) bioinformatics pipeline and the MAQ algorithm (Li et al., 2008) was used. The most significantly enriched nociceptor miRNAs were identified as having 50% or greater loss in the Dicer-null versus control mice with counts of 20 or greater following normalization (Table 1). The datasets from microarray analysis and deep sequencing are reasonably consistent (supplemental Tables 2 and 3, available at www.jneurosci.org as supplemental material). We examined the expression levels of the broadly expressed miRNA-137 using Northern blots. In accordance with the >22% reduction detected by Solexa sequencing, we found a diminished signal in the dicer mouse DRG (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), thus helping to validate the miRNA measurements using microarrays and deep sequencing. Full MAQ results of miRNAs which are reduced are shown in supplemental Table 3, available at www.jneurosci.org as supplemental material. Solexa sequencing followed by miRDeep analysis allowed us to identify a cohort of novel candidate miRNAs associated with sensory neurons. The novel miRNAs identified are shown in Table 1.
Table 1.
Nociceptor-enriched known murine miRNAs and novel miRNA candidates identified through deep sequencing
| No. | Identifier | Mature sequence (5′-3′) | Fold change |
|---|---|---|---|
| 1 | mmu-miR-19b | UGUGCAAAUCCAUGCAAAACUGA | −14.60 |
| 2 | mmu-miR-365 | UAAUGCCCCUAAAAAUCCUUAU | −11.00 |
| 3 | mmu-miR-193 | AACUGGCCUACAAAGUCCCAGU | −10.80 |
| 4 | mmu-miR-377 | AUCACACAAAGGCAACUUUUGU | −5.71 |
| 5 | mmu-miR-181d | AACAUUCAUUGUUGUCGGUGGGU | −4.93 |
| 6 | mmu-miR-7a | UGGAAGACUAGUGAUUUUGUUGU | −4.03 |
| 7 | mmu-miR-181b | AACAUUCAUUGCUGUCGGUGGGU | −3.88 |
| 8 | mmu-miR-181a | AACAUUCAACGCUGUCGGUGAGU | −3.18 |
| 9 | mmu-miR-218 | UUGUGCUUGAUCUAACCAUGU | −3.13 |
| 10 | mmu-miR-551b | GCGACCCAUACUUGGUUUCAG | −2.86 |
| 11 | mmu-miR-145 | GUCCAGUUUUCCCAGGAAUCCCU | −2.61 |
| 12 | mmu-miR-20a | UAAAGUGCUUAUAGUGCAGGUAG | −2.50 |
| 13 | mmu-miR-7b | UGGAAGACUUGUGAUUUUGUUGU | −2.49 |
| 14 | mmu-miR-204 | UUCCCUUUGUCAUCCUAUGCCU | −2.44 |
| 15 | mmu-miR-23b | AUCACAUUGCCAGGGAUUACC | −2.39 |
| 16 | mmu-miR-497 | CAGCAGCACACUGUGGUUUGUA | −2.35 |
| 17 | mmu-miR-182 | UUUGGCAAUGGUAGAACUCACACCG | −2.34 |
| 18 | mmu-miR-34a | UGGCAGUGUCUUAGCUGGUUGU | −2.33 |
| 19 | mmu-miR-672 | UGAGGUUGGUGUACUGUGUGUGA | −2.32 |
| 20 | mmu-miR-122 | UGGAGUGUGACAAUGGUGUUUG | −2.27 |
| 21 | mmu-miR-139-5p | UCUACAGUGCACGUGUCUCCAG | −2.27 |
| 22 | mmu-miR-23a | AUCACAUUGCCAGGGAUUUCC | −2.25 |
| 23 | mmu-miR-196a | UAGGUAGUUUCAUGUUGUUGGG | −2.21 |
| 24 | mmu-miR-133a | UUUGGUCCCCUUCAACCAGCUG | −2.15 |
| 25 | mmu-miR-196b | UAGGUAGUUUCCUGUUGUUGGG | −2.09 |
| 26 | mmu-miR-138 | AGCUGGUGUUGUGAAUCAGGCCG | −2.08 |
| 27 | mmu-miR-487b | AAUCGUACAGGGUCAUCCACUU | −2.02 |
| 28 | DRG-1 | UGGACACUGGAGAGAGAGCUUUU | −4.88 |
| 29 | DRG-2 | AAGAUGGAGACUUUAACAUGGGU | −4.50 |
| 30 | DRG-3 | CAUCUUAGCAGUAUCUCCCAU | −4.00 |
| 31 | DRG-4 | CUCACUGAACAAUGAAUGCAA | −3.50 |
| 32 | DRG-5 | UUGGAGUUCAUGCAAGUUCUAACC | −2.13 |
| 33 | DRG-6 | GAUAUAACCACUGCCAGACUGA | −2.00 |
| 34 | DRG-7 | GUAAAGGCUGGGCUUAGACGUGG | −1.91 |
| 35 | DRG-8 | CUAAGGCAGGCAGACUUCAGUG | −1.81 |
| 36 | DRG-9 | AUGGAGAGACUUUGACAGCUCA | −1.50 |
| 37 | DRG-10 | CCCUGGGAGGAGACGUGGAUUC | −1.50 |
| 38 | DRG-11 | AGGACGAGCUAGCUGAGUGCU | 1.10 |
| 39 | DRG-12 | CCGACUUCUGGGCUCCGGCUUU | 1.12 |
| 40 | DRG-13 | AUGGUAGACCGGUGACGUACA | 1.13 |
| 41 | DRG-14 | GCUCUGACUUUAUUGCACUACU | 2.00 |
| 42 | DRG-15 | AGGGGAGCUAGGUAGAAAGCCA | 4.00 |
| 43 | DRG-16 | UAGAUUGUAGGCCCAUUGGAAGGGU | 5.00 |
| 44 | DRG-17 | CAGGUGCCAGCUCCUCCCUUC | (1, 0) |
| 45 | DRG-18 | GUGCAUGGGUUUGGAUUUAUGGCA | (2, 0) |
| 46 | DRG-19 | UGGGGUGGUGGACUCAGGCUUC | (2, 0) |
| 47 | DRG-20 | CUGUGGGAUCAGAUAGCCUGU | (2, 0) |
| 48 | DRG-21 | UGAUUGGAAGACACUCUGCAA | (3, 0) |
| 49 | DRG-22 | CGGUGCUGGUGGAGCAGUGAGCACG | (4, 0) |
| 50 | DRG-23 | GGGAGCCAGGAAGUAUUGAUGUU | −4.14 |
| 51 | DRG-24 | AGAGGUUUUCUGGGUCUCUGUUU | −1.81 |
| 52 | DRG-25 | AUGGAGGUCUCUGUCUGACUU | −1.57 |
| 53 | DRG-26 | ACCGUGGCUUUCGAUUGUUACU | −1.56 |
| 54 | DRG-27 | CAUGGAGGUCUCUGUCUGACU | −1.40 |
| 55 | DRG-28 | AAGGUAGAUAGAACAGGUCUUG | −1.08 |
| 56 | DRG-29 | AGAAUUGUGGCUGGACAUCUGU | 1.04 |
| 57 | DRG-30 | UGUAGGGAUGGAAGCCAUGAAA | 1.55 |
| 58 | DRG-31 | CAGGUGGAACACGAUGCAAUUU | 1.66 |
| 59 | DRG-32 | UUCAUUCGGCUGUCCAGAUGUA | 1.67 |
| 60 | DRG-33 | GGAUAUCAUCAUAUACUGUAAGU | 1.81 |
| 61 | DRG-34 | AUGGCUCUUACUGAAGACUAGCAGU | (4, 0) |
| 62 | DRG-35 | GAGGGAGGGACGGGGGCGGUGC | (3, 0) |
Numbers 1–27 show the top murine miRNAS that have the greatest loss between control and Dicer KO DRGs. All miRNAs show a >2-fold loss in the Dicer KO and have counts of >20 in control DRG tissue. Numbers 28–49 show novel miRNA candidates identified using the miRDeep algorithm, which predicts miRNAs based o a probabilistic model of Dicer processing combined with secondary structure precursor analysis. Numbers 50–62 show novel murine miRNA sequences predicted by miRDeep that have orthologs in the Sanger miRNA registry. The first and second numbers in parentheses indicate control and normalized Dicer KO counts, respectively.
Discussion
The cytoplasmic ribonuclease III Dicer generates small double-stranded RNAs (miRNAs and siRNAs) that have been implicated in a range of functions (Bartel, 2004). Interestingly, aberrant miRNA function has been correlated with Alzheimer's disease and schizophrenia, as well as a familial form of Tourette syndrome (Abelson et al., 2005; Beveridge et al., 2008; Hébert et al., 2008). The basis of these diseases may relate to the ability of miRNAs to regulate translation both negatively and positively (Bartel, 2004; Vasudevan et al., 2008). The present study demonstrates that small RNAs in peripheral damage-sensing neurons are required for altered inflammatory pain thresholds. In other neuronal conditional Dicer knock-outs, substantial neuronal cell loss has been observed (Schaefer et al., 2007). With the postmitotic deletion of Dicer in Nav1.8+ sensory neurons, we found no cell loss and normal expression of the vast majority of mRNA transcripts. A-fiber- and C-fiber-mediated electrophysiological input into the dorsal horn in response to tissue damage was also identical to that in heterozygous Nav1.8-Cre control mice.
Behavioral studies of motor function and responses to acute painful stimuli showed that Dicer nulls were apparently normal apart from a deficit in noxious cold sensing. Nav1.8 has been shown to be essential for this function, and the level of expression of this sodium channel is lowered in Dicer-null neurons reflecting lowered levels of mRNA and pre-mRNA transcripts. The response to inflammatory stimuli showed a much more dramatic phenotype, however. Dicer-null mice failed to alter pain thresholds in response to carrageenan, formalin, or complete Freund's adjuvant. This behavioral insensitivity to inflammatory stimuli was reflected at the cellular level in the inability of sensory neurons in culture to respond to bradykinin, ATP, 5HT, and PGE2 with the increased levels of electrical excitability that are shown by wild-type neurons. As a consequence, formalin-evoked induction of c-Fos in the dorsal horn of the spinal cord was greatly diminished in the null mutant mice.
There is an interesting correlation between the expression of tissue-specific genes that have important roles in nociceptor function, particularly in inflammatory pain (e.g., P2xr3, Nav1.8, Nav1.9, Runx-1, etc.) (Abrahamsen et al., 2008) and lowered levels of expression in the absence of Dicer. We found that predominantly nociceptor-specific transcripts were transcriptionally downregulated, while other genes were either expressed at normal levels or had higher levels of mRNA transcripts. Nav1.7, Nav1.8, and Nav1.9 are all intimately linked with altered pain thresholds in inflammation (Akopian et al., 1999; Nassar et al., 2004; Priest et al., 2005; Zimmermann et al., 2007) The cell types in which we deleted Dicer, Nav1.8+ nociceptors, are known to be critically important for inflammatory pain, but not for neuropathic pain, which, as we confirmed, develops normally with conditional Dicer deletion. Pain associated with chronic inflammatory diseases such as rheumatoid arthritis is a major clinical burden, so understanding and exploiting the role of miRNAs in altering inflammatory pain thresholds is of considerable interest.
What are the mechanisms by which miRNAs alter pain thresholds? The most obvious explanation is that the transcriptional deficits leading to lowered levels of functional protein expression compromise the mechanisms that regulate damage sensing and the transmission of noxious information. For example, mRNA that encodes TRPA1 mRNA, the channel that transduces formalin responses, is expressed at lower levels in the absence of Dicer (55%). We also found low levels of CaMKIIa, which may play an important presynaptic role in regulating input into the CNS. The lowered levels of expression of Nav1.7, Nav1.8, and Nav1.9, channels whose activity is regulated by inflammatory mediators, is likely to reduce the impact of inflammatory mediators on peripheral neuron excitability (Wood et al., 2004). In addition, the protein kinase cascades associated with prostanoid-induced altered peripheral pain thresholds may be compromised by the lowered levels of kinase transcripts present in the null mice. For example PKCι, η, α, and μ transcripts are all >4-fold downregulated in Dicer null neurons (Fig. 5C). Inflammatory mediators that enhance pain through effects on the thresholds of activation of sensory neurons had little or no effect on Dicer-null neurons. Thus, the observed deficits in pain plasticity are likely to reflect lowered levels of expression of previously described essential modulators (Khasar et al., 1998; Priest et al., 2005; Joseph et al., 2007) of sensory neuron excitability. We used both microarrays of miRNAs as well as Solexa sequencing to identify known and novel miRNAs that were lost from the total repertoire found in DRGs through ablation of Dicer in Nav1.8+ neurons. Because no miRNAs were present in the Nav1.8+ neurons required for inflammatory pain, we could not identify miRNAs that contribute to such pain. We found a number of new miRNA candidates that are specifically associated with damage-sensing neurons. We also found known miRNAs that have been associated with other tissues and functions. The exact mechanism by which small RNAs regulate normal expression within sensory neurons is currently under investigation. Recent attention has focused on effects on transcription (Janowski et al., 2007). A model invoking effects on the stability of antisense transcripts that act as repressors has been used to explain how miRNAs may activate transcription. P21 induction seems to depend upon the actions of the nuclease argonaute 2, which in conjunction with miRNAs degrades an inhibitory antisense transcript (Morris et al., 2008; Place et al., 2008). Antisense transcripts present at RNA polymerase II promoter regions are a common feature of expressed genes (Core et al., 2008; He et al., 2008; Seila et al., 2008).
In summary, we have identified a critical role for Dicer products in altering pain thresholds. Given the fact that normalizing pain thresholds is the ideal solution to the treatment of the vast clinical problem of chronic pain, these experiments suggest that that identification of the small RNA species essential for altering pain thresholds could provide an attractive new target for pain treatment. It will be interesting to see whether small RNAs are also key players in neuropathic pain in other sensory neuron populations and to explain the mechanism by which small RNAs increase nociceptor-specific transcript expression.
Footnotes
This work was supported by the Medical Research Council (J.Z., M.M., J.N.W.), Biotechnology and Biological Sciences Research Council (J.Z., J.N.W.), and the Wellcome Trust (M.A.N., J.N.W.) for support. This work was partially supported by grant no. R31-2008-000-10103-0 from the World Class University project of the Korean Ministry of Education, Science, and Technology and the National Research Foundation of Korea, and C.M.C. was supported by the Research Program of the University of Granada. We thank Marc Friedländer, Ziheng Yang, Huiliang Li, and Ann Sheasby for technical advice and Ramin Raouf and the London Pain Consortium for helpful discussions.
References
- Abelson JF, Kwan KY, O'Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, Davis NR, Ercan-Sencicek AG, Guez DH, Spertus JA, Leckman JF, Dure LS, 4th, Kurlan R, Singer HS, Gilbert DL, Farhi A, et al. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB. MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components. Mol Immunol. 2008;45:1995–2006. doi: 10.1016/j.molimm.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MD, Chandra SY, Ding Y, Waxman SG, Wood JN. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurons. J Physiol. 2003;548:373–382. doi: 10.1113/jphysiol.2003.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Beveridge NJ, Tooney PA, Carroll AP, Gardiner E, Bowden N, Scott RJ, Tran N, Dedova I, Cairns MJ. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet. 2008;17:1156–1168. doi: 10.1093/hmg/ddn005. [DOI] [PubMed] [Google Scholar]
- Bogen O, Dreger M, Gillen C, Schröder W, Hucho F. Identification of versican as an isolectin B4-binding glycoprotein from mammalian spinal cord tissue. FEBS J. 2005;272:1090–1102. doi: 10.1111/j.1742-4658.2005.04543.x. [DOI] [PubMed] [Google Scholar]
- Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci (Lond) 2006;110:167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, Wilson CB, Brockdorff N, Fisher AG, Smale ST, Merkenschlager M. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, McManus MT, Ullian EM. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28:4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L, Koutsikou S, Fang X, McMullan S, Lawson SN. Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J Neurosci. 2006;26:1281–1292. doi: 10.1523/JNEUROSCI.3388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes T, Nassar MA, Lane T, Matthews EA, Baker MD, Gerke V, Okuse K, Dickenson AH, Wood JN. Deletion of annexin 2 light chain p11 in nociceptors causes deficits in somatosensory coding and pain behavior. J Neurosci. 2006;26:10499–10507. doi: 10.1523/JNEUROSCI.1997-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- Fullmer JM, Riedl MS, Higgins L, Elde R. Identification of some lectin IB4 binding proteins in rat dorsal root ganglia. Neuroreport. 2004;15:1705–1709. doi: 10.1097/01.wnr.0000136037.54095.64. [DOI] [PubMed] [Google Scholar]
- Fullmer JM, Riedl M, Williams FG, Sandrin M, Elde R. Enzymes that synthesize the IB4 epitope are not sufficient to impart IB4 binding in dorsal root ganglia of rat. J Comp Neurol. 2007;501:70–82. doi: 10.1002/cne.21233. [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–838. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S. The microRNA registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, Holoch D, Lim C, Tuschl T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44:3–12. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW. The antisense transcriptomes of human cells. Science. 2008;322:1855–1857. doi: 10.1126/science.1163853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HL, Cendan CM, Roza C, Okuse K, Cramer R, Timms JF, Wood JN. Proteomic profiling of neuromas reveals alterations in protein composition and local protein synthesis in hyper-excitable nerves. Mol Pain. 2008;4:33. doi: 10.1186/1744-8069-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SP, Pini A, Evan G. Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature. 1987;328:632–634. doi: 10.1038/328632a0. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Bogen O, Alessandri-Haber N, Levine JD. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132:67–73. doi: 10.1016/j.pain.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell. 2004;116:779–793. doi: 10.1016/s0092-8674(04)00248-x. [DOI] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Luo W, Wickramasinghe SR, Savitt JM, Griffin JW, Dawson TM, Ginty DD. A hierarchical NGF signaling cascade controls Ret-dependent and Ret-independent events during development of nonpeptidergic DRG neurons. Neuron. 2007;54:739–754. doi: 10.1016/j.neuron.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Maingret F, Coste B, Padilla F, Clerc N, Crest M, Korogod SM, Delmas P. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J Gen Physiol. 2008;131:211–225. doi: 10.1085/jgp.200709935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV, Santoso S, Turner AM, Pastori C, Hawkins PG. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008;4:e1000258. doi: 10.1371/journal.pgen.1000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashov AK, Chintalgattu V, Islamov RR, Lever TE, Pak ES, Sierpinski PL, Katwa LC, Van Scott MR. RNAi pathway is functional in peripheral nerve axons. FASEB J. 2007;21:656–670. doi: 10.1096/fj.06-6155com. [DOI] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101:12706–12711. doi: 10.1073/pnas.0404915101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan I, Arancio O. Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron. 2004;42:129–141. doi: 10.1016/s0896-6273(04)00143-6. [DOI] [PubMed] [Google Scholar]
- Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A. 2008;105:1608–1613. doi: 10.1073/pnas.0707594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM, Liberator P, Iyer LM, Kash SF, Kohler MG, Kaczorowski GJ, MacIntyre DE, Martin WJ. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A. 2005;102:9382–9387. doi: 10.1073/pnas.0501549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, O'Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, Greengard P. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med. 2007;204:1553–1558. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Singh S, Bevan SC, Patil K, Newton DC, Marsden PA. Extensive variation in the 5′-UTR of Dicer mRNAs influences translational efficiency. Biochem Biophys Res Commun. 2005;335:643–650. doi: 10.1016/j.bbrc.2005.07.138. [DOI] [PubMed] [Google Scholar]
- Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH, Nassar MA. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain. 2005;113:27–36. doi: 10.1016/j.pain.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Cummins TR, Ishikawa K, Dib-Hajj SD, Black JA, Waxman SG. SNS Na+ channel expression increases in dorsal root ganglion neurons in the carrageenan inflammatory pain model. Neuroreport. 1998;9:967–972. doi: 10.1097/00001756-199804200-00003. [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Tong Y, Steitz JA. Cell-cycle control of microRNA-mediated translation regulation. Cell Cycle. 2008;7:1545–1549. doi: 10.4161/cc.7.11.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol. 2009;46:1222–1228. doi: 10.1016/j.molimm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JN, Boorman JP, Okuse K, Baker MD. Voltage-gated sodium channels and pain pathways. J Neurobiol. 2004;61:55–71. doi: 10.1002/neu.20094. [DOI] [PubMed] [Google Scholar]
- London Pain Consortium. Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S, Hathaway G, Morenilla-Palao C, Stirling C, Fitzgerald M, McMahon SB, Rios M, Wood JN. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006a;31:539–548. doi: 10.1016/j.mcn.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhao J, Nassar MA, Gavazzi I, Wood JN. Tamoxifen-inducible NaV1.8-CreERT2 recombinase activity in nociceptive neurons of dorsal root ganglia. Genesis. 2006b;44:364–371. doi: 10.1002/dvg.20224. [DOI] [PubMed] [Google Scholar]
- Zhong J, Pevny L, Snider WD. “Runx”ing toward sensory differentiation. Neuron. 2006;49:325–327. doi: 10.1016/j.neuron.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]