

Abstract
B-type natriuretic peptide (BNP) has been known to be secreted from cardiac myocytes and activate its receptor, natriuretic peptide receptor-A (NPR-A), to reduce ventricular fibrosis. However, the function of BNP/NPR-A pathway in the somatic sensory system has been unknown. In the present study, we report a novel function of BNP in pain modulation. Using microarray and immunoblot analyses, we found that BNP and NPR-A were expressed in the dorsal root ganglion (DRG) of rats and upregulated after intraplantar injection of complete Freund's adjuvant (CFA). Immunohistochemistry showed that BNP was expressed in calcitonin gene-related peptide (CGRP)-containing small neurons and IB4 (isolectin B4)-positive neurons, whereas NPR-A was present in CGRP-containing neurons. Application of BNP reduced the firing frequency of small DRG neurons in the presence of glutamate through opening large-conductance Ca2+-activated K+ channels (BKCa channels). Furthermore, intrathecal injection of BNP yielded inhibitory effects on formalin-induced flinching behavior and CFA-induced thermal hyperalgesia in rats. Blockade of BNP signaling by BNP antibodies or cGMP-dependent protein kinase (PKG) inhibitor KT5823 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester] impaired the recovery from CFA-induced thermal hyperalgesia. Thus, BNP negatively regulates nociceptive transmission through presynaptic receptor NPR-A, and activation of the BNP/NPR-A/PKG/BKCa channel pathway in nociceptive afferent neurons could be a potential strategy for inflammatory pain therapy.
Introduction
Neuropeptides and neuropeptide receptors are known to play important roles in pain transmission and modulation. Substance P and calcitonin gene-related peptide (CGRP) are expressed in small-diameter neurons in the dorsal root ganglion (DRG) and secreted from their afferent C- and Aδ-fibers in lamina I–II of the spinal cord in response to nociceptive stimulation, enhancing excitatory synaptic transmission (Schaible, 1996). Somatostatin and its receptors are expressed in a subset of small DRG neurons and activation of peripheral somatostatin receptors reduces inflammatory nociceptive behavior (Hökfelt et al., 1976; Carlton et al., 2001; Bar et al., 2004). Moreover, low levels of galanin, neurotensin, and neuropeptide Y are expressed in DRG neurons under normal circumstance, in addition to their expression in the spinal dorsal horn neurons (Todd et al., 1994; Simmons et al., 1995; X. Zhang et al., 1995, 1997, 1998; Polgar et al., 1999). These neuropeptides are also involved in the modulation of DRG neuron activity (X. Zhang et al., 1995; Xu et al., 1997). Importantly, the expression of both these neuropeptides and their receptors are strongly altered after peripheral nerve injury and/or inflammation (Ji et al., 1994, 1995; X. Zhang et al., 1995, 1998; Xiao et al., 2002), suggesting their potential roles in modulating pathological pain. Therefore, we are interested to further identify neuropeptides and their receptors that are expressed in DRG neurons and markedly regulated after peripheral tissue inflammation.
Natriuretic peptides (NPs) are a family of three structurally related paracrine factors, namely atrial NP (ANP), B-type NP (BNP), and C-type NP (Potter et al., 2009). The BNP, also known as brain natriuretic peptide, is expressed and secreted from the cardiac ventricles. This peptide binds to the natriuretic peptide receptor-A (NPR-A), a guanylyl cyclase receptor that is also sensitive to ANP, and increases intracellular cGMP level (Misono, 2002) to further modify biological functions through cGMP-dependent protein kinase (PKG), ion channels, and other effectors (Hofmann et al., 2006). Moreover, NPR-A is present in the vasculature, kidneys, and adrenal gland, and regulates vasorelaxant, natriuretic functions, and aldosterone synthesis (Hofmann et al., 2006). Although most studies on BNP expression and action have been focused on the cardiovascular system (Woodard and Rosado, 2007; Potter et al., 2009), recent studies show that BNP and NPR-A are also expressed at low levels in the CNS and may contribute to functions ranging from modulating synaptic transmission to neuroprotection (Cao and Yang, 2008). However, their expression and function in the peripheral nervous system remains mostly unknown.
In the present study, we found BNP and NPR-A are expressed in small DRG neurons and their expression is upregulated after peripheral tissue inflammation. Furthermore, BNP application reduces both the excitability of small DRG neurons and the hyperalgesic response in rat models of inflammatory pain. Thus, activation of the BNP/NPR-A pathway in nociceptive afferent neurons inhibits inflammatory pain, suggesting a therapeutic potential of BNP and other NPR-A agonists in pain treatment.
Materials and Methods
Animals and drugs.
All experiments were approved by the Committee of Use of Laboratory Animals and Common Facility, Institute of Neuroscience, Chinese Academy of Sciences. Male Sprague Dawley rats were provided by Shanghai Laboratory Animal Center (Shanghai, China) and housed under a 12 h light/dark cycle at 22–26°C, with ad libitum access to tap water and rat chow. BNP peptide was purchased from Phoenix Pharmaceuticals. PKG inhibitor (9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2, 3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester (KT5823), BKCa channel blocker iberiotoxin (IBTX), glibenclamide, glutamate, and complete Freund's adjuvant (CFA) were purchased from Sigma-Aldrich. BNP, IBTX, and glutamate were dissolved in distilled water. KT5823 and glibenclamide were prepared as stock solutions (1000×) in dimethyl sulfoxide as the datasheet suggested.
Microarray analysis.
Adult male rats (n = 100) received bilateral hindpaw intraplantar injection with 200 μl of CFA. They were killed at 0.5, 2, 4, 7, and 14 d (20 rats each group) after the injection. Lumbar 4 (L4) and L5 DRGs of these rats and 20 normal rats were dissected out. Total RNA was isolated from DRG using the Agilent total RNA isolation mini kit (Agilent Technologies) and used as templates for cDNA synthesis. In vitro transcription was performed using Agilent low RNA input fluorescent linear amplification kit (Agilent Technologies) in the presence of Cy3- and Cy5-CTP. Synthesized fluorescence-labeled cRNA was used for oligo microarray hybridization. Hybridization solution was prepared according to in situ hybridization kit plus (Agilent Technologies). Hybridization was performed using Agilent 22k rat oligo microarray (G4130B; Agilent Technologies) in hybridization oven at 60°C for 17 h in a dye-swap replication experimental design. Microarray scanner system (Agilent Technologies) was used for data analysis. After feature extraction by Feature Extraction software, log2 ratio values, which are equal to the ratio of Cy5-processed signal to Cy3-processed signal, were calculated and converted to fold change. Genes with a log2 ratio >1 (greater than twofold increase) were considered to be upregulated, and less than −1 (greater than twofold decrease) were considered to be downregulated. After filtration and normalization of each qualified gene, hierarchical clustering and visualization were performed using Cluster 3.0 and TreeView software (M. B. Eisen Laboratory, Stanford University, Stanford, CA).
Immunoblot analysis.
Adult male rats received bilateral intraplantar injection of CFA (200 μl) into hindpaws. The rats were allowed to survive for 1, 2, 4, and 7 d. Protein was extracted from L4 and L5 DRGs or L4–5 spinal segments by using RIPA buffer and concentration was determined by BCA kit (Pierce). The samples was loaded on SDS-PAGE, transferred, probed with antibodies, and visualized with enhanced chemiluminescence (ECL) (Thermo Fisher Scientific). Antibodies against BNP (1:1000; Santa Cruz), NPR-A (1:1000; Abcam), and GAPDH (1:50,000; Abcam) were used. Quantification was based on at least three independent experiments from different animal groups. ECL signal intensity of BNP or NPR-A versus GAPDH was quantified with Scion Image program (Scion Corporation).
Immunohistochemistry.
Adult male rats were fixed with 4% paraformaldehyde and 0.02% picric acid. Cryostat sections of L4 and L5 DRGs and spinal cord segments were processed for double-immunofluorescence staining with rabbit antibodies against NPR-A (1:500) or rabbit antibodies against BKCa channels (1:1000; Alomone Labs) and goat antibodies against CGRP (1:500; Biogenesis) or 10 μg/ml fluorescein-conjugated isolectin B4 (IB4) (Vector Laboratories). Cryostat sections of L4 and L5 DRGs without any fixation were used for double-immunofluorescence staining goat antibodies against BNP (1:500) with rabbit antibodies against NPR-A (1:500) or rabbit antibodies against tyrosine kinase receptor A (TrkA) (1:500; Santa Cruz) or rabbit antibodies against CGRP (1:500; DiaSorin) or 10 μg/ml fluorescein-conjugated IB4. Specificity of BNP, NPR-A, and BKCa channels immunostaining was tested by preabsorption of antibodies with 10−6 m corresponding immunogenic peptides.
Electrophysiology.
Neurons were acutely dissociated from L4 and L5 DRGs of 5-week-old rats with 1 mg/ml collagenase type 1A, 0.4 mg/ml trypsin type I, and 0.1 mg/ml DNase I in DMEM at 37°C for 40 min. Cells were plated on coverslips, and patch clamp was performed within 2∼12 h after plating. All of the recordings were made from small-diameter (<30 μm; 15–35 pF) DRG neurons. Normal extracellular solution (ECS) contained the following (in mm): 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4. To prepare ECS containing 0, 0.1, 0.25, and 0.5 mm Ca2+, 2.5, 2.4, 2.25, and 2.0 mm CaCl2 was replaced by the same concentration of MgCl2. The pipette solution contained the following (in mm): 140 KCl, 1 MgCl2, 2.5 CaCl2, 5 EGTA, 10 HEPES, 2 Na-ATP, and 0.3 Na-GTP, pH 7.3. Whole-cell recordings were performed if they matched all of the following criteria: >1 GΩ seal resistance before “breakthrough” with additional suction; a resting potential of −55 mV or more negative; and >100 MΩ input resistance after whole-cell mode was established. Data were collected using an EPC-9 patch-clamp amplifier and the Pulse software (version 8.31; HEKA Elektronik).
Transverse lumbar spinal cord slices (600 μm thick) with an attached dorsal root from adult rats were prepared with a vibrating microslicer and perfused in preoxygenated Krebs' solution for blind whole-cell patch-clamp recording. The Krebs' solution contained the following (in mm): 117 NaCl, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose, pH 7.4. The resistance of the patch-pipette electrodes was typically 4–10 MΩ. The pipette solution contained the following (in mm): 135 K-gluconate, 0.5 CaCl2, 2 MgCl2, 5 KCl, 5 EGTA, 5 HEPES, and 5 glucose, pH 7.3. Currents were filtered at 2 kHz and digitized at 5 kHz using an Axopatch 200B microelectrode amplifier and pCLAMP8.5 software (Molecular Devices). The holding potential was −70 mV. After blockade of GABAA receptor- and glycine receptor-mediated IPSCs by 20 μm bicuculline and 2 μm strychnine, monosynaptic glutamate-mediated EPSCs were evoked by orthodromic stimulation (100 μs duration, 0.2 Hz frequency) of the dorsal root via a suction electrode (Nakatsuka et al., 2000). Stimulus intensity was adjusted to 1.5 times the threshold for evoking detectable EPSCs in the C-afferent fibers.
Behavioral tests.
Injections and behavioral responses of animals were performed in a double-blind way. For the formalin test in adult male rats, 5% formalin (50 μl) was injected subcutaneously into the dorsal surface of the left hindpaw with a 30 gauge needle. The animals were placed into the chambers and nociceptive behavior was observed immediately after formalin injection. Nociceptive behavior was quantified as the numbers of flinches of the injected paw every 5 min, up to 60 min after injection. Formalin-induced flinching behavior was biphasic. The initial acute phase (0∼10 min, phase 1) was followed by a prolonged tonic response (10∼60 min, phase 2). Animals were used only once, and at the end of the experiment they were killed in a CO2 chamber. To induce chronic inflammation, CFA (50 μl) was injected subcutaneously into the ventral surface of the left hindpaw with a 30 gauge needle. To measure the thermal latency, rats were placed in plastic chambers on the surface of a 2-mm-thick glass plate, and the sensitivity to heat stimuli with a radiant heat stimulator (BME-410C; CAMS) was measured. The latency was determined as the duration from the beginning of heat stimuli to the occurrence of a hindpaw withdrawal reflex, with a cutoff time of 20 s. To measure mechanical threshold, rats were placed in plastic chambers on a mesh floor. Mechanical threshold was determined by assessing paw withdrawal to von Frey hairs of graded tensile strength. A positive response was defined as rapid withdrawal on application of the stimulus, which was then followed by application of the next finer von Frey filament. After a negative response, the next higher von Frey filament was applied.
Statistical analysis.
Data are presented as mean ± SEM. Statistical analysis for Western blot analysis, immunofluorescence, and electrophysiology was performed by Student's t test. Statistical analysis for behavioral tests was performed by two-way ANOVA, followed by Student's t test, to compare differences between treatments. Differences were considered to reach statistical significance when p < 0.05.
Results
BNP and NPR-A are expressed in DRGs and upregulated by inflammation
Total RNA from L4 and L5 DRGs of normal rats and that of rats 0.5, 2, 4, 7, and 14 d after CFA injection were analyzed with Agilent 22k rat oligo microarray. Three independent replicate experiments were carried out, and the gene expression data were filtered and subjected to statistical analyses. A total of 1257 gene probes, which represented 6.1% of total probes (20,500), was identified to be markedly regulated (greater than twofold; p < 0.05) in DRGs during at least one time interval after CFA treatment. Additional Unigene analysis revealed that these identified probes represented 1123 genes (509 increased and 614 decreased). Among them, 18 neuropeptides, 6 G-protein-coupled receptors for neuropeptides, and NPR-A were found (Burbach, 2010) (Fig. 1A).
Figure 1.

Expression of BNP and NPR-A is upregulated in DRGs after peripheral inflammation. A, Hierarchical clustering analysis of neuropeptides and neuropeptide receptors with marked changes in the microarray signal. Expression levels of 18 neuropeptides and 7 neuropeptide receptors in L4 and L5 DRGs are markedly increased after intraplantar CFA injection, including BNP and its receptor NPR-A. B, Western blot analysis shows that the levels of BNP and NPR-A are increased in L4 and L5 DRGs of rats after intraplantar injection of CFA, compared with that in control rats (Con). The protein level of BNP in DRGs is gradually increased and reaches the peak level on day 4 after CFA (n = 4). The amount of NPR-A protein is increased on day 1 after CFA and reaches the peak level on day 2 (n = 5). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is used as a loading control. *p < 0.05 versus control rats. Error bars indicate SEM.
These neuropeptides and receptors were subjected to hierarchical clustering analysis (Eisen et al., 1998) (Fig. 1A). Notably, correlated changes in the expression of four neuropeptides and their receptors were found in DRGs after peripheral tissue inflammation, including tachykinin, neurotensin, somatostatin, and BNP (Fig. 1A). All of these neuropeptides and their receptors were markedly upregulated. Interestingly, the mRNA levels of tachykinin and neurotensin were transiently increased on day 2 after CFA, whereas somatostatin and BNP were upregulated in a long-lasting manner from 0.5 to 14 d (Fig. 1A).
Since the expression and function of tachykinin, neurotensin, and somatostatin as well as their receptors in DRGs have been previously reported (X. Zhang et al., 1995; Malcangio and Bowery, 1999; Thermos et al., 2006), our study was then focused on the expression and potential function of BNP and its receptor in pain modulation. We next examined whether the protein levels of BNP and NPR-A were changed in DRGs after peripheral inflammation. Consistent with our microarray result, immunoblotting showed that the level of BNP in the DRG was gradually increased after CFA-induced inflammation and reached its peak level (2.61 ± 0.35 vs control; p = 0.002) 4 d after intraplantar CFA injection (Fig. 1B). The amount of NPR-A protein was also increased on day 1 (1.44 ± 0.19 vs control; p = 0.104) and reached the peak level on day 2 (1.58 ± 0.20 vs control; p = 0.045) after intraplantar injection of CFA. The expression of NPR-A returned to the baseline level 7 d after inflammation (Fig. 1B). These results suggest that the expression of both BNP and NPR-A in rat DRGs are increased after peripheral tissue inflammation.
BNP and NPR-A are increased in small DRG neurons after inflammation
The distribution of BNP and NPR-A in rat DRGs was further examined by immunochemistry. BNP and NPR-A were mainly distributed in small DRG neurons (cross-sectional area, <900 μm2) (Fig. 2A,B). Fifty percent of total DRG neurons were immunoreactive for BNP and 26% were positive for NPR-A (Fig. 2A). Immunostaining intensity for BNP and NPR-A in small DRG neurons was increased 2 d after intraplantar CFA injection (Fig. 2A–C), whereas the number of immunoreactive neurons was not apparently changed (Fig. 2A) (50 and 27% of total DRG neurons were immunoreactive for BNP and NPR-A, respectively). Therefore, the inflammation-induced upregulation of BNP and NPR-A mainly occurs in small DRG neurons.
Figure 2.

Expression of BNP and NPR-A in small DRG neurons is increased after peripheral inflammation. A, Immunostaining showed that BNP immunoreactivity and NPR-A immunoreactivity are mainly distributed in small DRG neurons in L4 and L5 DRGs. Expression of BNP and NPR-A in small DRG neurons are increased 2 d after intraplantar injection of CFA. The number of neurons immunostained for BNP is greater than that stained for NPR-A. All NPR-A-positive neurons are also stained for BNP. Scale bars, 50 μm. B, Size distribution of BNP- and NPR-A-positive DRG neuron profiles in L4 and L5 DRGs of control rats and the rats 2 d after intraplantar CFA injection. Sizes of BNP- and NPR-A-positive neuron profiles are located in the size range of small DRG neurons (neuronal cross-sectional area, <900 μm2) of rats. C, The intensity of BNP immunostaining is increased from 63.56 ± 0.69 (n = 608) in small DRG neurons of normal rats to 81.64 ± 0.87 (n = 606) 2 d after intraplantar injection of CFA. The immunofluorescence intensity of NPR-A in small DRG neurons is also increased from 51.14 ± 0.67 (n = 317) in control rats to 58.02 ± 0.83 (n = 324) 2 d after inflammation. ***p < 0.001 versus control rats. D, Approximately 52 and 53% of BNP-positive neurons are also stained for NPR-A in L4 and L5 DRGs of control rats and the rats 2 d after intraplantar CFA injection, respectively. However, almost all NPR-A-positive neurons contain BNP.
Using double-immunofluorescent staining, we further examined which subset of small DRG neurons expressed BNP and its receptor. Small DRG neurons are often classified as peptidergic neurons (CGRP-positive) and IB4-positive neurons. TrkA is expressed in most peptidergic small DRG neurons and some IB4-positive neurons (Silos-Santiago et al., 1995; Fang et al., 2006). Interestingly, we observed that almost all NPR-A-positive neurons also expressed BNP, whereas only one-half of BNP-immunoreactive neurons were labeled by NPR-A (Fig. 2A,D). BNP immunoreactivity was further found to be present in TrkA-positive small DRG neurons (Fig. 3A). Meanwhile, the BNP immunostaining was found in 95% of CGRP-containing small DRG neurons and in 89% of IB4-positive neurons (Fig. 3B). However, NPR-A immunoreactivity was predominantly present in CGRP-positive subset of small DRG neurons (Fig. 4A) and their afferent fibers in the spinal lamina I–II (Fig. 4B). The NPR-A immunoreactivity was strongly reduced in the ipsilateral lamina I–II of the spinal cord 4 d after unilateral dorsal root section, whereas that on the contralateral side remained intact (Fig. 4C), indicating that these NPR-A-immunoreactive fibers are sensory afferents. Additionally, NPR-A immunoreactivity was also observed along the blood vessels in the spinal cord (Fig. 4B), consistent with previous reports that NPR-A is widely distributed in cardiovascular tissues (Nakayama, 2005). Thus, BNP was expressed in most peptidergic small DRG neurons and IB4-positive neurons, whereas NPR-A was almost all present in peptidergic small DRG neurons.
Figure 3.
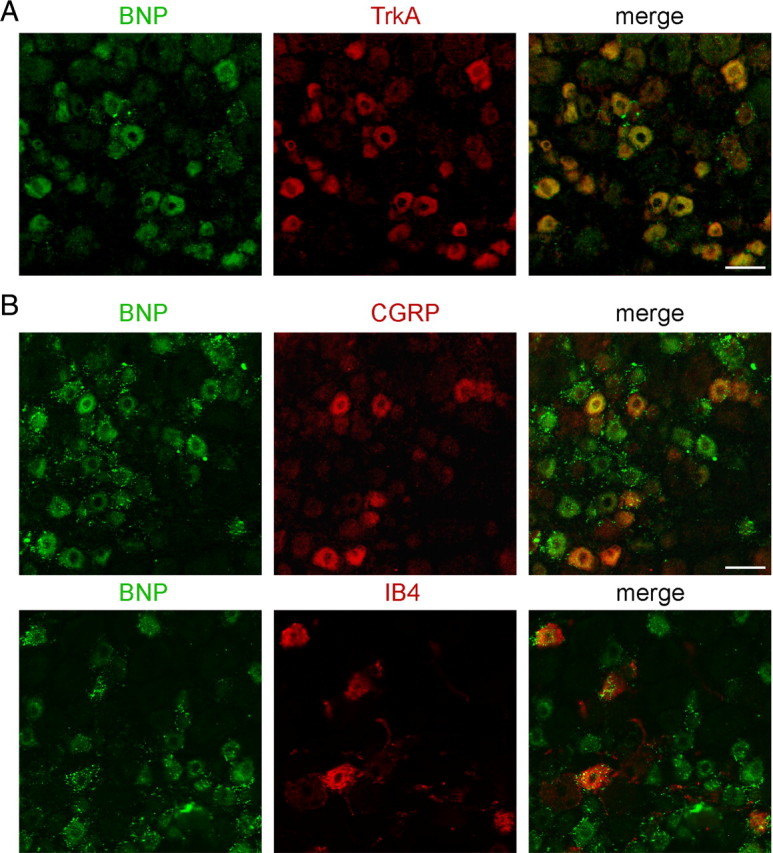
BNP is expressed in peptidergic and IB4-positive subsets of small DRG neurons. A, Double immunostaining shows that BNP is present in TrkA-immunoreactive small neurons in the rat DRG. B, Immunostaining of BNP is found in CGRP-containing subset and IB4-positive subset of small DRG neurons in the rat DRG. Scale bar, 50 μm.
Figure 4.

NPR-A is expressed in peptidergic subset of small DRG neurons. A, Double immunostaining shows that NPR-A is expressed in CGRP-containing subset of small neurons in L4 DRGs of adult rats. B, In lamina II of L4 spinal cord segment of the rat, most NPR-A-immunoreactive fibers contain CGRP. The boxed area is shown at high magnification. Moreover, NPR-A immunoreactivity is also found along blood vessel (arrows). Scale bar, 50 μm. C, The NPR-A immunoreactivity is markedly reduced in the ipsilateral dorsal horn of L5 spinal cord segment 4 d after unilateral section of L1–L6 dorsal roots, whereas that on the contralateral side remains intact. Scale bar, 50 μm.
BNP suppresses neuronal firing in the presence of glutamate
We then asked whether BNP could regulate the activity of small DRG neurons. Previous studies have shown that activation of NPR-A induces cGMP synthesis, and activates downstream PKG, which phosphorylates large-conductance Ca2+-activated K+ channels (BKCa channels) and increases their open probability, leading to the hyperpolarization of vascular smooth muscle cells and glomerular mesangial cells (Williams et al., 1988; Sansom and Stockand, 1996; Tanaka et al., 1998). Importantly, BKCa current is present in the vast majority of small DRG neurons and contributes to the repolarization of the action potential (X. L. Zhang et al., 2010). Moreover, BKCa channel opener NS1619 reduces firing frequency of small DRG neurons (X. F. Zhang et al., 2003). Thus, we proposed that BNP might exert inhibitory effects on small DRG neurons through modulating downstream BKCa channels. However, to our surprise, despite the significant reduction of the action potential amplitudes (from 44.39 ± 2.60 to 31.48 ± 3.17 mV; p < 0.001, paired Student's t test) (Fig. 5A), the action potential frequency under a current ramp (1 s duration, peak ranging from 100 to 500 pA) was not altered by puffing 100 ng/ml BNP to small DRG neurons. The ratio of the action potential frequency (after BNP treatment vs before treatment) was 1.13 ± 0.09 (n = 14) (Fig. 5A,B), which was same to the ratio under ECS puffing (1.13 ± 0.06; n = 22) (Fig. 5B).
Figure 5.
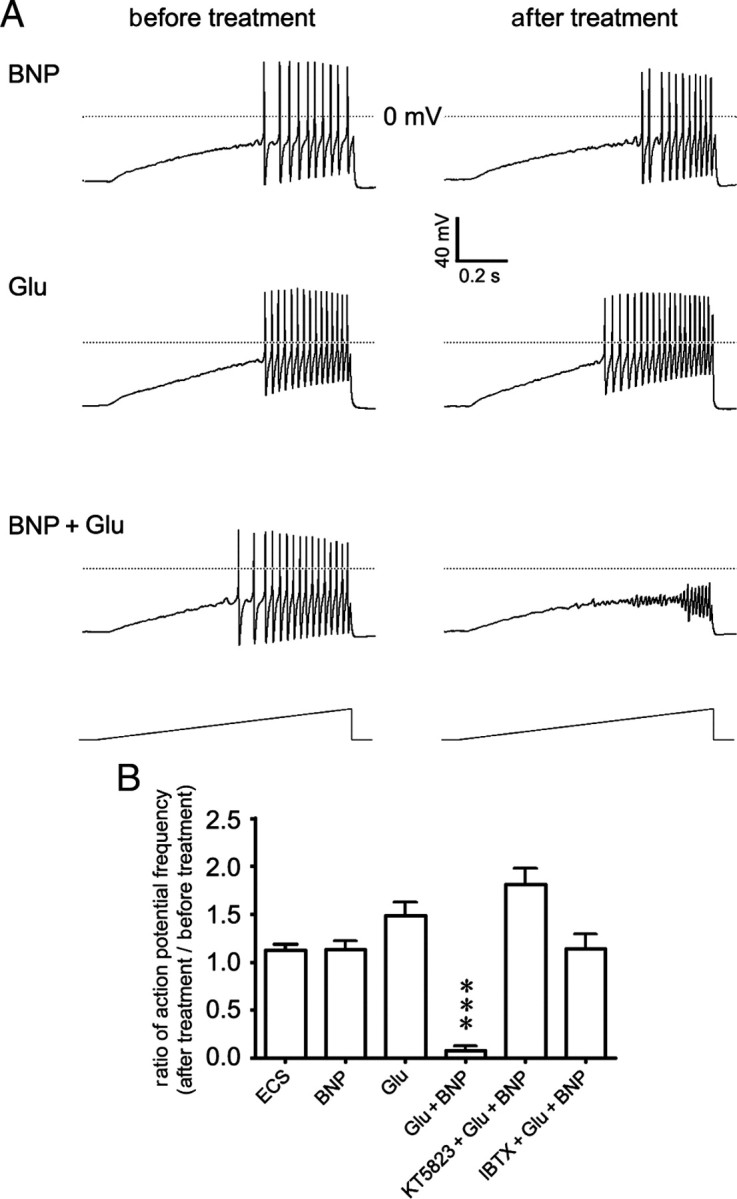
BNP reduces the excitability of small DRG neurons in the presence of glutamate. A, Whole-cell patch-clamp recording in a small neuron freshly dissociated from the rat DRG shows that the firing frequency of action potentials induced by depolarizing current ramp stimulation (1 s duration; peak ranging from 100 to 500 pA) is not apparently altered by puffing BNP (100 ng/ml) for 10 min (top) and is slightly increased by puffing glutamate (Glu) (10 μm) for 10 min. However, the firing of action potentials is strongly inhibited by puffing BNP (100 ng/ml) and glutamate (10 μm) for 10 min. B, The ratio of action potential frequency in small DRG neurons (after treatment vs before treatment) is not apparently altered by puffing ECS (1.13 ± 0.06; n = 22) or BNP (100 ng/ml; 1.13 ± 0.09, n = 14, p = 0.971, compared with puffing ECS), but is slightly increased by puffing 10 μm glutamate (Glu) (1.49 ± 0.14; n = 10; p = 0.035). Evoked action potentials are mostly inhibited by coapplied BNP and glutamate (0.08 ± 0.05; n = 21; ***p < 0.001). The BNP/glutamate-induced inhibition can be attenuated by preincubating neurons with either PKG inhibitor KT5823 (1.81 ± 0.17; n = 11) or BKCa channel blocker IBTX (1.14 ± 0.15; n = 10). Error bars indicate SEM.
We noticed that opening of BKCa channels relies on the elevation of intracellular Ca2+ and membrane depolarization (Fukao et al., 1999). Therefore, it might be possible that BNP alone was insufficient to suppress neuronal firing when the level of intracellular free Ca2+ was not elevated, though the open probability of BKCa channels was increased and the amplitude of action potentials was reduced. On activation of nociceptors, the excitatory transmitter glutamate is released (Carozzi et al., 2008; D'Mello and Dickenson, 2008) to elevate intracellular Ca2+ level through presynaptic glutamate receptors (Cochilla and Alford, 1999; Kerchner et al., 2001; Lee et al., 2002; Bardoni et al., 2004). We further hypothesized that BNP might regulate the opening of BKCa channels more efficiently in the presence of extracellular glutamate. To test this hypothesis, we puffed BNP (100 ng/ml) together with glutamate (10 μm) to small DRG neurons and examined whether the firing frequency could be affected. There was a 92.7 ± 7.2% reduction in the frequency of evoked action potentials (in 21 of 28 neurons examined) within 10 min after bath application of BNP, whereas the resting potential of tested neurons was not apparently affected (Fig. 5A,B). In contrast, puffing glutamate alone only slightly increased the firing frequency (1.49 ± 0.14; n = 10) (Fig. 5A,B). Since there is no commercially available specific antagonist of NPR-A, we used PKG inhibitor KT5823 and BKCa channel blocker IBTX to further evaluate the functional role of NPR-A-mediated signaling in the modulation of neuronal activity. The BNP-induced inhibitory effect on small DRG neurons in the presence of glutamate could be attenuated by pretreatment of 1 μm KT5823 (1.81 ± 0.17; n = 11) or 100 nm IBTX (1.14 ± 0.15; n = 10) (Fig. 5B). Together, BNP reduces the excitability of small DRG neurons through PKG/BKCa channel pathway in the presence of extracellular glutamate.
BNP increases open probability of BKCa channels
As activation of BKCa channels requires both intracellular Ca2+ concentration and membrane voltage to reach their thresholds, we next examined how BKCa channels reacted under different membrane potentials and Ca2+ concentrations in the presence of BNP. To obtain BKCa current, we used a voltage ramp from −60 to 100 mV (0.4 s duration) to trigger an outward current in small DRG neurons in whole-cell voltage-clamp configuration. Under normal ECS containing 2.5 mm Ca2+, this current was suppressed by BKCa channel inhibitor IBTX (100 nm) in all examined small DRG neurons (53.2 ± 5.0% decrease at 100 mV; p < 0.001; n = 21) (Fig. 6A). BKCa current was isolated by taking the difference between total current and IBTX-resistant current (X. L. Zhang et al., 2010). Bath application of BNP (100 ng/ml) for 10 min before IBTX treatment resulted in an enlarged current with 13.7 ± 2.4% increase, compared with the current before BNP treatment (at 100 mV; p < 0.001; 12 of 21 neurons) (Fig. 6A). BKCa current was also induced in ECS with 0.5 mm Ca2+, and this current was enhanced by BNP too (p = 0.006; 8 of 12 neurons) (Fig. 6B). Thus, BNP increases the open probability of BKCa channels by increasing their sensitivity to membrane voltage.
Figure 6.

BNP increases the open probability of BKCa channels. A, A voltage ramp from −60 to 100 mV (0.4 s duration) triggers an outward current in small DRG neurons in whole-cell voltage-clamp configuration. In small DRG neurons incubated in ECS with 2.5 mm Ca2+, the current is increased by bath-applied BNP (100 ng/ml) and suppressed by BKCa channel inhibitor IBTX (100 nm). In the absence of extracellular Ca2+, neither BNP nor IBTX affects the current. B, BKCa current is induced in small DRG neurons incubated in ECS with 0.5 or 2.5 mm Ca2+, and this current is increased by BNP (p = 0.006, n = 8 for 0.5 mm Ca2+ at 100 mV; p < 0.001, n = 12 for 2.5 mm Ca2+ at 100 mV). In the presence of 0, 0.1, or 0.25 mm extracellular Ca2+, BKCa current cannot be induced and BNP has no effect (p > 0.05, before treatment vs BNP treatment; n = 10 neurons/group). BKCa current is isolated by taking the difference between total current and IBTX-resistant current, and normalized to the cell membrane capacitance. **p < 0.01 and ***p < 0.001 versus control, paired Student's t test. Error bars indicate SEM. C, At −15 mV (dashed line), the voltage threshold for small DRG neurons to arise an all-or-none action potential, BNP plus glutamate caused a strong increase in BKCa current density (33.27 ± 6.06 pA/pF; n = 9), whereas smaller increase in BKCa current density is induced by BNP alone (9.09 ± 3.48 pA/pF; p = 0.004; n = 12) or glutamate (4.24 ± 2.32 pA/pF; p = 0.004; n = 8).
However, neither BNP nor IBTX affected the triggered current in Ca2+-free ECS (Fig. 6A), indicating BKCa channels may not open under this condition, consistent with a previous report that BKCa current in DRG neurons is dependent on Ca2+ influx via voltage-gated Ca2+ channels (X. L. Zhang et al., 2010). This result further suggests that it is BKCa channels to mediate BNP action, because BNP could not regulate the triggered current when BKCa channels were not opened. Furthermore, BKCa current could not be induced in small DRG neurons incubated in ECS with 0, 0.1, or 0.25 mm Ca2+, and BNP did not induce any significant effects (p > 0.05, before treatment vs after treatment of BNP; n = 10 neurons/group) (Fig. 6B), suggesting BNP did not alter the Ca2+ threshold of BKCa channels. Thus, BNP increases BKCa current by increasing the open probability of BKCa channels under different membrane potentials.
We further compared the change of BKCa current after the treatment of BNP, glutamate, or BNP plus glutamate (after treatment minus before treatment). The voltage threshold for small DRG neurons to arise an all-or-none action potential is approximately −15 mV (Rush et al., 2006). We found that BNP plus glutamate induced a robust increase in BKCa current density (33.27 ± 6.06 pA/pF; n = 9 of 12 cells) at the −15 mV membrane potential, which was much greater than that induced by BNP alone (9.09 ± 3.48 pA/pF; p = 0.004; n = 12 of 21 cells) or glutamate alone (4.24 ± 2.32 pA/pF; p = 0.004; n = 8) (Fig. 6C). This means an addition of 832 pA (33.27 pA/pF × 25 pF) outward K+ current to neutralize the inward Na+ current at −15 mV for a typical small DRG neuron (Cm = 25 pF). The generated K+ current may be big enough to inhibit the neuronal firing. However, in the case of BNP alone, the additional outward current was only ∼227 pA (9.09 pA/pF × 25 pF), which may not be sufficient to suppress the generation of action potential. This may explain the BNP-induced suppression of neuronal firing in the presence of extracellular glutamate.
Intrathecally applied BNP reduces inflammatory pain
Electrophysiological recording showed that the triggered current in all examined small DRG neurons were suppressed by BKCa channel inhibitor IBTX, consistent with previous report that BKCa current are present in the vast majority of small DRG neurons (X. L. Zhang et al., 2010). Immunochemistry showed BKCa channels were present in almost all small DRG neurons of rats (cross-sectional area, <900 μm2), including CGRP- and IB4-positive neurons (Fig. 7A). Therefore, BKCa channels, BNP, NPR-A are expressed in peptidergic subset of small DRG neurons. Interestingly, both NPR-A and BKCa channels were found to be mainly transported in the dorsal roots to the spinal cord, whereas BNP was transported in both the dorsal roots and the sciatic nerve (Fig. 7B). Furthermore, immunoblot signal of BNP was present in the dorsal spinal cord and was apparently increased 2 d after intraplantar injection of CFA (Fig. 7C).
Figure 7.
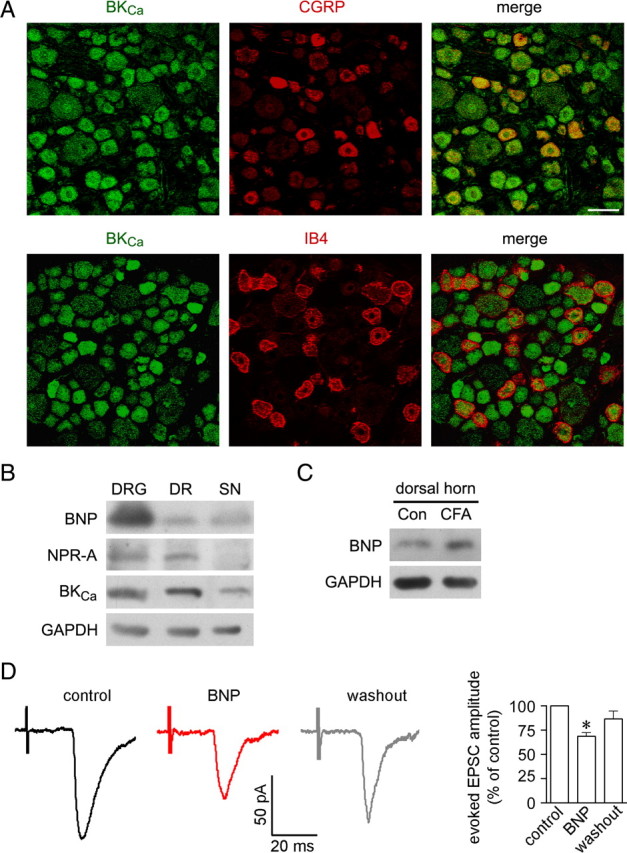
BKCa channels are expressed in small DRG neurons and centrifugally transported in correlation with BNP and NPR-A. A, Double immunostaining shows that all CGRP-containing small neurons express BKCa channels in the rat DRG. Scale bar, 50 μm. B, Immunoblotting shows that the relative amount of BNP, NPR-A, and BKCa channels in L4 and L5 DRGs of rats, L4 and L5 dorsal roots (DR), and the sciatic nerve (SN). C, Immunoblot signal of BNP is present in the dorsal horn of L4–L5 segments of the rat spinal cord and is elevated 2 d after intraplantar CFA injection. The image represents three independent experiments. D, The monosynaptic glutamate-mediated EPSCs of C-fibers are evoked by stimulating the dorsal roots at a frequency of 0.2 Hz (100 μs duration; 350–380 μA strength). Such evoked EPSCs are inhibited by following bath application of BNP (200 ng/ml) (n = 5 lamina II neurons of rat spinal cord). *p < 0.05 versus control. Error bars indicate SEM.
The effect of BNP on the synaptic transmission between the axonal terminals of afferent fibers and lamina II neurons was examined in the dorsal root-attached spinal cord slices. After blockade of GABAA receptor- and glycine receptor-mediated IPSCs with 20 μm bicuculline and 2 μm strychnine, the monosynaptic glutamate-mediated EPSCs of afferent C-fibers were evoked by 0.2 Hz stimulation (100 μs duration; 350–380 μA strength) of the dorsal root (Nakatsuka et al., 2000). Such evoked EPSCs could be inhibited by following perfusion of BNP (200 ng/ml) (p < 0.05; n = 5 of 8 recorded lamina II neurons) (Fig. 7D). This result, combined with the selective transportation of DRG neuron expressed NPR-A and BKCa to the spinal cord, indicates that the BNP/NPR-A/PKG/BKCa channel mechanism may mainly function at sensory afferents in the dorsal spinal cord.
We then examined whether BNP could exert inhibitory effects in the rat models of inflammatory pain in which neurotransmitter glutamate is released from nociceptive afferents (D'Mello and Dickenson, 2008). Subcutaneous injection of formalin into the left hindpaw of adult rats produced a typical pattern of flinching behavior (Fig. 8A) characterized by a biphasic time course (1∼10 min for phase 1, 10∼60 min for phase 2) (Araiza-Saldana et al., 2005). Intrathecal injection of BNP (applied 10 min before intraplantar formalin injection) induced a dose-dependent inhibitory effect on both phases of the formalin test (p < 0.001 for 100 ng, 500 ng, and 2 μg of BNP vs vehicle, respectively, ANOVA; phase 1: p = 0.099, 0.026, and 0.034, and phase 2: p = 0.007, <0.001, and = 0.004, for 100 ng, 500 ng, and 2 μg of BNP vs vehicle, respectively; n = 7 rats/group) (Fig. 8A). However, intraplantar injection of BNP (2 μg) did not affect paw withdrawal latency in normal rats (p = 0.980, ANOVA; n = 10 rats/group). The antinociceptive effect induced by intrathecal BNP (2 μg) could be reversed by intrathecal pretreatment (10 min before BNP treatment) of either PKG inhibitor KT5823 (500 ng) or BKCa channel blocker IBTX (500 ng), but not by ATP-sensitive K+ channel (KATP) blocker glibenclamide (50 μg) (phase 1: p = 0.038, 0.048, and 0.800; and phase 2: p = 0.003, 0.037, and 0.907 for cotreatment of 2 μg of BNP with KT5823 or IBTX or glibenclamide vs 2 μg of BNP, respectively; n = 7 rats/group) (Fig. 8B). However, KT5823, IBTX, or glibenclamide alone could not alter the formalin-induced flinching behavior (phase 1: p = 0.549, 0.568, and 0.873; and phase 2: p = 0.743, 0.906, and 0.764 for KT5823 or IBTX or glibenclamide vs vehicle, respectively; n = 7 rats/group). Thus, BNP induces an inhibitory effect on the formalin-induced nociceptive response by activating PKG/BKCa channel pathway.
Figure 8.

BNP inhibits formalin-induced flinching behavior. A, Intrathecal injection of BNP (100 ng, 500 ng, and 2 μg, applied 10 min before intraplantar formalin injection) dose-dependently reduces flinching behavior in both phases of the formalin test [p < 0.001 for 100 ng, 500 ng, and 2 μg of BNP vs vehicle (Veh), respectively, ANOVA; n = 7 rats/group]. B, The BNP-induced antinociceptive effect can be attenuated by intrathecal pretreatment (10 min before intrathecal injection of BNP) of PKG inhibitor KT5823 (500 ng) or BKCa channel blocker IBTX (500 ng) (phase 1: p = 0.038 and 0.048; phase 2: p = 0.003 and 0.037, for cotreatment of BNP with KT5823 or IBTX vs BNP alone, respectively; n = 7 rats/group), but not ATP-sensitive K+ channels blocker glibenclamide (Glib) (50 μg). Formalin-induced flinching behavior cannot be reduced by applying KT5823, IBTX, or Glib alone (n = 7 rats/group). *p < 0.05 and **p < 0.01 versus Veh; #p < 0.05 and ##p < 0.01 versus BNP alone. Error bars indicate SEM.
Chronic inflammation induced by intraplantar injection of CFA results in pronounced thermal and mechanical hyperalgesia with a reduction of paw withdrawal latency during radiant heat or von Frey filament stimulation (Ren and Dubner, 1999). Thermal hyperalgesia reached the peak level on day 1 after CFA and slowly recovered during 7–14 d after intraplantar injection of CFA, whereas mechanical hyperalgesia may last for weeks (Gould et al., 2004). Since the expression level of BNP in small DRG neurons was gradually increased in the first week after CFA injection, we speculated that BNP signaling pathway might be related to the recovery from thermal hyperalgesia. Intrathecal injection of BNP (2 μg) significantly increased paw withdrawal latency (p < 0.001, ANOVA; p = 0.011, 0.023, and 0.032 for 90, 120, and 180 min after BNP injection; n = 7 rats/group) during radiant heat test on day 2 after CFA (Fig. 9A), but had no effect on mechanical hyperalgesia (p = 0.928, ANOVA; n = 6 rats/group) (Fig. 9B). BNP also facilitated the recovery from thermal hyperalgesia on day 4 after CFA (p = 0.039 for 120 min after BNP injection; n = 7 rats/group). However, intrathecal injection of BNP antibodies (2 μg) to block the activity of endogenous BNP in the dorsal spinal cord or KT5823 (500 ng) to inhibit PKG impaired the recovery from thermal hyperalgesia on day 7 after CFA (p < 0.001, ANOVA; n = 6–7 rats/group) (Fig. 9C,D). However, intrathecal injection of BNP, BNP antibodies, or KT5823 did not apparently affect basal thresholds of thermal and mechanical nociceptive responses in normal rats (data not shown). Thus, both acute and chronic inflammatory pain can be reduced by activation of the BNP signaling pathway in nociceptive DRG neurons, and this pathway is essential for the recovery from thermal hyperalgesia during chronic inflammation.
Figure 9.

BNP inhibits CFA-induced thermal hyperalgesia. A, Two days after intraplantar injection of CFA in left hindpaw of rats, intrathecal injection of BNP (2 μg) significantly increases the paw withdrawal latency during radiant heat test (p < 0.001, ANOVA; n = 7 rats/group) but has no effect on mechanical hyperalgesia (p = 0.928, ANOVA; n = 6 rats/group) (B). C, CFA-induced thermal hyperalgesia reaches the peak level on day 1 after CFA (3.82 ± 0.16 s; n = 6) and recovers on day 7 (9.58 ± 0.41 s). Intrathecal injection of BNP antibodies (2 μg) attenuates the recovery from thermal hyperalgesia on day 7 after CFA (p < 0.001, ANOVA). D, CFA-induced thermal hyperalgesia reaches the peak level on day 1 after CFA (3.86 ± 0.09 s; n = 7) and recovers on day 7 (8.37 ± 0.23 s). Intrathecal injection of PKG inhibitor KT5823 (500 ng) impairs the recovery on day 7 after CFA (p < 0.001, ANOVA). Error bars indicate SEM.
Discussion
The present study reveals that both BNP and NPR-A are expressed in small DRG neurons and their expression levels are elevated by peripheral inflammation. Furthermore, BNP reduces the excitability of small DRG neurons in the presence of extracellular glutamate through activating PKG/BKCa channel pathway. Intrathecal BNP attenuates formalin- and CFA-induced nociceptive responses. Thus, BNP secreted from nociceptive afferents negatively regulates the excitatory synaptic transmission via activating presynaptic NPR-A/PKG/BKCa channel pathway. These findings suggest that activation of the BNP/NPR-A/PKG/BKCa channel pathway in nociceptive afferents is a potential approach for pain therapy.
Presynaptic mechanism for BNP-induced inhibition
Although autoreceptor-mediated regulatory mechanisms for classical neurotransmitters, such as dopamine and serotonin, are known to contribute to a local feedback mechanism in neural circuits (Feuerstein, 2008), evidence for autoreceptors in peptidergic system is still limited (Hökfelt et al., 2000). The presence of both BNP and NPR-A in peptidergic small DRG neurons and the effect of BNP on the firing rate of small DRG neurons suggest that NPR-A serves as an autoreceptor on some nociceptive afferent neurons. Our immunoblot data showed that BNP and NPR-A were transported in the dorsal roots to the spinal cord. Importantly, NPR-A was mostly transported in the dorsal roots, indicating NPR-A-mediated action of BNP mainly occurs at central afferent terminals. This notion is supported by the recording of an inhibitory action of BNP on the monosynaptic EPSCs evoked by C-fiber stimulation. Although we failed to immunostain BNP in afferent fibers in the spinal dorsal horn, immunoblot signal of BNP was found in the spinal dorsal horn and was elevated after inflammation. Previous studies reported that BNP immunoreactivity is present in afferent fibers in the spinal lamina I–II of rats and pigs, but not in the spinal interneurons (Kawata et al., 1989; Saper et al., 1989). Therefore, BNP might be released from primary sensory afferents in the dorsal spinal cord. Importantly, NPR-A is localized in CGRP-containing afferent fibers in the spinal dorsal horn. Thus, coordinated expression of BNP and NPR-A in peptidergic small DRG neurons may represent a mechanism for autoregulation of afferent synaptic transmission. Moreover, BNP released from some IB4-positive neurons might act at NPR-A on peptidergic sensory afferents in a paracrine manner.
Presynaptic inhibition of glutamate release has been considered to be a mechanism for pain modulation. Several neuropeptides, such as opioid peptides, somatostatin, and neuropeptide Y, have been found to presynaptically inhibit glutamate release through their receptors localized on axonal terminals (Tallent, 2008). Importantly, activation of presynaptic glutamate receptors results in a reduction of glutamate release (Cochilla and Alford, 1999; Kerchner et al., 2001; Lee et al., 2002; Bardoni et al., 2004). Our electrophysiological recording showed that application of BNP together with glutamate reduced the excitability of small DRG neurons through opening BKCa channels. This action may consequently reduce Ca2+-dependent neurotransmitter release. The requirement of glutamate action for the BNP-induced inhibition suggests that BNP may act in an activity-dependent manner. This notion is consistent with findings that BNP induced antinociceptive effects in inflammatory conditions that cause glutamate release in vivo (Carozzi et al., 2008; D'Mello and Dickenson, 2008), but not in normal rats. Interestingly, most of neuropeptide-induced presynaptic inhibition are mediated by their G-protein-coupled receptors (Tallent, 2008), whereas BNP receptor is a guanylyl cyclase and does not bind G-protein. Together, BNP may inhibit neurotransmitter release through activating presynaptic NPR-A-mediated signal pathway. This may represent a novel mechanism for regulating nociceptive afferent transmission.
Inhibition of inflammatory pain by activating BNP/NPR-A/PKG/BKCa channel pathway
Activation of NPR-A results in elevated cGMP/PKG signaling (Hofmann et al., 2006). We find that the BNP-induced inhibitory effects on neuronal excitability and nociceptive behavior were attenuated by PKG inhibitor KT5823, suggesting a requirement of cGMP/PKG signaling for the BNP action. Coordinately, cGMP is detected in afferent fibers in the superficial dorsal horn of spinal cord (Vles et al., 2000; de Vente et al., 2006). Intracellular concentration of cGMP is determined by the ratio of synthesis and breakdown. The NPR-A-mediated cGMP generation can be enhanced in the presence of different phosphodiesterase inhibitors (de Vente et al., 2006). Accordingly, inhibition of cGMP degradation by intrathecal administration of the phosphodiesterase 5 inhibitor sildenafil reduces inflammatory pain induced by formalin (Torres-Lopez et al., 2002; Araiza-Saldana et al., 2005). Therefore, the nociceptive afferent transmission may be regulated by factors that alter intracellular level of cGMP.
Furthermore, neurotransmission could be regulated through cGMP signaling in a neuron-specific manner. A large body of evidence shows that the cGMP synthesis induced by nitric oxide (NO) through NO-sensitive guanylyl cyclase contributes to the processing of nociceptive signals, and inhibition of cGMP synthesis can considerably reduce both inflammatory and neuropathic pain (Schmidtko et al., 2009). However, the NO-sensitive guanylyl cyclase is present in both neurokinin 1 receptor-positive projection neurons in lamina I of the spinal cord and inhibitory interneurons in lamina II–III, but not in DRG neurons (Schmidtko et al., 2008b). Thus, opposite effects of BNP and NO on nociceptive responses may be attributable to cGMP regulation in different populations of neurons in the spinal sensory circuits, namely BNP/cGMP signaling in primary afferent neurons and NO/cGMP signaling in spinal dorsal horn neurons.
Similar to the expression pattern of NPR-A, the PKG type I (cGKI) is present in small DRG neurons and their afferent fibers in the dorsal spinal cord (Qian et al., 1996). However, cGKI-null mutants show a reduction in formalin-evoked nociception and zymosan-induced hyperalgesia (Tegeder et al., 2004). In fact, the spinal cord of cGKI-knock-out mice is significantly smaller and contains fewer neurons than that in control mice (Tegeder et al., 2004). It cannot be excluded that the spinal structural disorder, presumably because of defects in axon guidance during embryogenesis (Schmidt et al., 2002), may contribute to the reduced nociception in cGKI-null mutants (Tegeder et al., 2004). However, cysteine-rich protein 2 (CRP2), a downstream effector of the cGKI, is also present in cGKI- and CGRP-containing afferent fibers in the spinal dorsal horn. Increased nociceptive responses in CRP2-deficient mice in inflammatory pain models indicates that CRP2 contributes to a inhibitory regulation of pain transmission (Schmidtko et al., 2008a). This is consistent with the notion that the PKG signaling pathway could be involved in a negative regulation of nociceptive afferent transmission.
Previous studies show that activation of NPR-A/cGMP/PKG pathway increases the open probability of BKCa channels to hyperpolarize the cell membrane through phosphorylation of the channels (Williams et al., 1988; Sansom and Stockand, 1996; Tanaka et al., 1998). BKCa current is found in the vast majority of small DRG neurons, contributing to the repolarization of action potential and the reduction of firing frequency (X. F. Zhang et al., 2003; X. L. Zhang et al., 2010). We show that BNP increases BKCa channel sensitivity to membrane voltage but cannot inhibit the neuronal firing without extracellular glutamate to elevate intracellular Ca2+. This result implies that BNP may not reduce the firing rate of nociceptive afferent neurons under normal circumstance but could suppress the neuronal firing in response to intensive noxious stimulation such as inflammation, which activates peripheral nociceptors and causes their central terminals releasing glutamate (Carozzi et al., 2008; D'Mello and Dickenson, 2008). Several lines of evidence we have found in this study support this idea. First, the baseline of nociception is not altered by applied BNP or BNP antibodies in normal rats. Second, intrathecal BNP inhibits formalin-induced flinching behavior and CFA-induced thermal hyperalgesia. Third, blockade of endogenous BNP signal pathway by intrathecal injection of BNP antibodies or PKG inhibitor attenuates the recovery from CFA-induced thermal hyperalgesia. Therefore, we propose that, in response to peripheral inflammation, activation of the BNP/NPR-A/PKG/BKCa channel pathway in nociceptive afferent neurons would switch on an intrinsic antinociceptive mechanism to reduce inflammatory pain transmission.
Potential role of BNP/NPR-A/PKG/BKCa channel pathway in pain therapy
Activation of the BNP/NPR-A pathway in the cardiac ventricles induces an adaptive response to cardiovascular strain and reduces ventricular fibrosis (Woodard and Rosado, 2007; Potter et al., 2009). Synthetic analogs of BNP have been proved in some countries for the treatment of decompensated heart failure. We find that inflammatory pain can be reduced by both endogenous and applied BNP. However, the baseline of nociception is not altered by BNP, in agreement with the notion that BNP may suppress neuronal firing when glutamate is extensively released from nociceptive afferents in response to intensive noxious stimulation. No effect of BNP on mechanical hyperalgesia may be attributable to the NPR-A expression in peptidergic small DRG neurons, which mainly respond to noxious thermal but not mechanical stimuli (Cavanaugh et al., 2009). Furthermore, since NPR-A and BKCa channels synthesized in the cell bodies of DRG neurons are mostly transported to the spinal cord, BNP exerts the suppressive effects mainly at central afferent terminals. Although the level of BNP in the dorsal spinal cord is increased after chronic inflammation, intrathecal BNP further reduces inflammatory pain and facilitates the sensory recovery, suggesting that BNP-binding sites on central afferent fibers may be unsaturated by endogenous BNP and applied BNP can further activate the NPR-A/PKG/BKCa channel pathway in nociceptive afferents. Therefore, in addition to the application in heart failure and other diseases, BNP and other NPR-A agonists may serve as analgesic molecules/compounds in pain treatment.
Conclusion
The present study shows the expression of BNP and its receptor NPR-A in small DRG neurons and the upregulation of this signaling system after peripheral tissue inflammation. Furthermore, BNP is found to inhibit the excitability of small DRG neurons in the presence of glutamate and to reduce inflammatory pain through activating the NPR-A/PKG/BKCa channel pathway. These results suggest that selective activation of NPR-A-mediated signal pathway in nociceptive afferent neurons with appropriate agonists could be a potential strategy for pain therapy.
Footnotes
This work was supported by National Natural Science Foundation of China Grants 30630029 and 30621062, Ministry of Science and Technology of the People's Republic of China 973 Project Grants 2006CB806604 and 2009CB522005, and Chinese Academy of Sciences Grant KSCX1-YW-R-31.
References
- Araiza-Saldana CI, Reyes-Garcia G, Bermudez-Ocana DY, Perez-Severiano F, Granados-Soto V. Effect of diabetes on the mechanisms of intrathecal antinociception of sildenafil in rats. Eur J Pharmacol. 2005;527:60–70. doi: 10.1016/j.ejphar.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Bar KJ, Schurigt U, Scholze A, Segond Von Banchet G, Stopfel N, Brauer R, Halbhuber KJ, Schaible HG. The expression and localization of somatostatin receptors in dorsal root ganglion neurons of normal and monoarthritic rats. Neuroscience. 2004;127:197–206. doi: 10.1016/j.neuroscience.2004.04.051. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach JP. Neuropeptides from concept to online database www.neuropeptides.nl. Eur J Pharmacol. 2010;626:27–48. doi: 10.1016/j.ejphar.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Cao LH, Yang XL. Natriuretic peptides and their receptors in the central nervous system. Prog Neurobiol. 2008;84:234–248. doi: 10.1016/j.pneurobio.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Du J, Davidson E, Zhou S, Coggeshall RE. Somatostatin receptors on peripheral primary afferent terminals: inhibition of sensitized nociceptors. Pain. 2001;90:233–244. doi: 10.1016/S0304-3959(00)00407-3. [DOI] [PubMed] [Google Scholar]
- Carozzi V, Marmiroli P, Cavaletti G. Focus on the role of glutamate in the pathology of the peripheral nervous system. CNS Neurol Disord Drug Targets. 2008;7:348–360. doi: 10.2174/187152708786441876. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A. 2009;106:9075–9080. doi: 10.1073/pnas.0901507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S. NMDA receptor-mediated control of presynaptic calcium and neurotransmitter release. J Neurosci. 1999;19:193–205. doi: 10.1523/JNEUROSCI.19-01-00193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vente J, Markerink-van Ittersum M, Vles JS. ANP-mediated cGMP signaling and phosphodiesterase inhibition in the rat cervical spinal cord. J Chem Neuroanat. 2006;31:263–274. doi: 10.1016/j.jchemneu.2006.02.005. [DOI] [PubMed] [Google Scholar]
- D'Mello R, Dickenson AH. Spinal cord mechanisms of pain. Br J Anaesth. 2008;101:8–16. doi: 10.1093/bja/aen088. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Djouhri L, McMullan S, Berry C, Waxman SG, Okuse K, Lawson SN. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J Neurosci. 2006;26:7281–7292. doi: 10.1523/JNEUROSCI.1072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein TJ. Presynaptic receptors for dopamine, histamine, and serotonin. Handb Exp Pharmacol. 2008;2008:289–338. doi: 10.1007/978-3-540-74805-2_10. [DOI] [PubMed] [Google Scholar]
- Fukao M, Mason HS, Britton FC, Kenyon JL, Horowitz B, Keef KD. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J Biol Chem. 1999;274:10927–10935. doi: 10.1074/jbc.274.16.10927. [DOI] [PubMed] [Google Scholar]
- Gould HJ, 3rd, England JD, Soignier RD, Nolan P, Minor LD, Liu ZP, Levinson SR, Paul D. Ibuprofen blocks changes in Nav 1.7 and 1.8 sodium channels associated with complete Freund's adjuvant-induced inflammation in rat. J Pain. 2004;5:270–280. doi: 10.1016/j.jpain.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Elde R, Johansson O, Luft R, Nilsson G, Arimura A. Immunohistochemical evidence for separate populations of somatostatin-containing and substance P-containing primary afferent neurons in the rat. Neuroscience. 1976;1:131–136. doi: 10.1016/0306-4522(76)90008-7. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides—an overview. Neuropharmacology. 2000;39:1337–1356. doi: 10.1016/s0028-3908(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Ji RR, Zhang X, Wiesenfeld-Hallin Z, Hökfelt T. Expression of neuropeptide Y and neuropeptide Y (Y1) receptor mRNA in rat spinal cord and dorsal root ganglia following peripheral tissue inflammation. J Neurosci. 1994;14:6423–6434. doi: 10.1523/JNEUROSCI.14-11-06423.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Zhang X, Zhang Q, Dagerlind A, Nilsson S, Wiesenfeld-Hallin Z, Hökfelt T. Central and peripheral expression of galanin in response to inflammation. Neuroscience. 1995;68:563–576. doi: 10.1016/0306-4522(95)94333-t. [DOI] [PubMed] [Google Scholar]
- Kawata M, Hirakawa M, Kumamoto K, Minamino N, Kangawa K, Matsuo H, Sano Y. Brain natriuretic peptide in the porcine spinal cord: an immunohistochemical investigation of its localization and the comparison with atrial natriuretic peptide, substance P, calcitonin gene-related peptide, and enkephalin. Neuroscience. 1989;33:401–410. doi: 10.1016/0306-4522(89)90219-4. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Wilding TJ, Li P, Zhuo M, Huettner JE. Presynaptic kainate receptors regulate spinal sensory transmission. J Neurosci. 2001;21:59–66. doi: 10.1523/JNEUROSCI.21-01-00059.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Bardoni R, Tong CK, Engelman HS, Joseph DJ, Magherini PC, MacDermott AB. Functional expression of AMPA receptors on central terminals of rat dorsal root ganglion neurons and presynaptic inhibition of glutamate release. Neuron. 2002;35:135–146. doi: 10.1016/s0896-6273(02)00729-8. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Bowery NG. Peptide autoreceptors: does an autoreceptor for substance P exist? Trends Pharmacol Sci. 1999;20:405–407. doi: 10.1016/s0165-6147(99)01388-7. [DOI] [PubMed] [Google Scholar]
- Misono KS. Natriuretic peptide receptor: structure and signaling. Mol Cell Biochem. 2002;230:49–60. [PubMed] [Google Scholar]
- Nakatsuka T, Ataka T, Kumamoto E, Tamaki T, Yoshimura M. Alteration in synaptic inputs through C-afferent fibers to substantia gelatinosa neurons of the rat spinal dorsal horn during postnatal development. Neuroscience. 2000;99:549–556. doi: 10.1016/s0306-4522(00)00224-4. [DOI] [PubMed] [Google Scholar]
- Nakayama T. The genetic contribution of the natriuretic peptide system to cardiovascular diseases. Endocr J. 2005;52:11–21. doi: 10.1507/endocrj.52.11. [DOI] [PubMed] [Google Scholar]
- Polgar E, Shehab SA, Watt C, Todd AJ. GABAergic neurons that contain neuropeptide Y selectively target cells with the neurokinin 1 receptor in laminae III and IV of the rat spinal cord. J Neurosci. 1999;19:2637–2646. doi: 10.1523/JNEUROSCI.19-07-02637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009;2009:341–366. doi: 10.1007/978-3-540-68964-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Chao DS, Santillano DR, Cornwell TL, Nairn AC, Greengard P, Lincoln TM, Bredt DS. cGMP-dependent protein kinase in dorsal root ganglion: relationship with nitric oxide synthase and nociceptive neurons. J Neurosci. 1996;16:3130–3138. doi: 10.1523/JNEUROSCI.16-10-03130.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R. Inflammatory models of pain and hyperalgesia. ILAR J. 1999;40:111–118. doi: 10.1093/ilar.40.3.111. [DOI] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103:8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom SC, Stockand JD. Physiological role of large, Ca2+-activated K+ channels in human glomerular mesangial cells. Clin Exp Pharmacol Physiol. 1996;23:76–82. doi: 10.1111/j.1440-1681.1996.tb03066.x. [DOI] [PubMed] [Google Scholar]
- Saper CB, Hurley KM, Moga MM, Holmes HR, Adams SA, Leahy KM, Needleman P. Brain natriuretic peptides: differential localization of a new family of neuropeptides. Neurosci Lett. 1989;96:29–34. doi: 10.1016/0304-3940(89)90238-3. [DOI] [PubMed] [Google Scholar]
- Schaible HG. On the role of tachykinins and calcitonin gene-related peptide in the spinal mechanisms of nociception and in the induction and maintenance of inflammation-evoked hyperexcitability in spinal cord neurons (with special reference to nociception in joints) Prog Brain Res. 1996;113:423–441. doi: 10.1016/s0079-6123(08)61102-4. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Werner M, Heppenstall PA, Henning M, More MI, Kuhbandner S, Lewin GR, Hofmann F, Feil R, Rathjen FG. cGMP-mediated signaling via cGKIα is required for the guidance and connectivity of sensory axons. J Cell Biol. 2002;159:489–498. doi: 10.1083/jcb.200207058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, Sausbier M, Rauhmeier I, Sausbier U, Niederberger E, Scholich K, Huber A, Neuhuber W, Allescher HD, Hofmann F, Tegeder I, Ruth P, Geisslinger G. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J Neurosci. 2008a;28:1320–1330. doi: 10.1523/JNEUROSCI.5037-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, Konig P, Heine S, Motterlini R, Ruth P, Schlossmann J, Koesling D, Niederberger E, Tegeder I, Friebe A, Geisslinger G. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J Neurosci. 2008b;28:8568–8576. doi: 10.1523/JNEUROSCI.2128-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Tegeder I, Geisslinger G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009;32:339–346. doi: 10.1016/j.tins.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Silos-Santiago I, Molliver DC, Ozaki S, Smeyne RJ, Fagan AM, Barbacid M, Snider WD. Non-TrkA-expressing small DRG neurons are lost in TrkA-deficient mice. J Neurosci. 1995;15:5929–5942. doi: 10.1523/JNEUROSCI.15-09-05929.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DR, Spike RC, Todd AJ. Galanin is contained in GABAergic neurons in the rat spinal dorsal horn. Neurosci Lett. 1995;187:119–122. doi: 10.1016/0304-3940(95)11358-4. [DOI] [PubMed] [Google Scholar]
- Tallent MK. Presynaptic inhibition of glutamate release by neuropeptides: use-dependent synaptic modification. Results Probl Cell Differ. 2008;44:177–200. doi: 10.1007/400_2007_037. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Aida M, Tanaka H, Shigenobu K, Toro L. Involvement of maxi-KCa channel activation in atrial natriuretic peptide-induced vasorelaxation. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:705–708. doi: 10.1007/pl00005228. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Del Turco D, Schmidtko A, Sausbier M, Feil R, Hofmann F, Deller T, Ruth P, Geisslinger G. Reduced inflammatory hyperalgesia with preservation of acute thermal nociception in mice lacking cGMP-dependent protein kinase I. Proc Natl Acad Sci U S A. 2004;101:3253–3257. doi: 10.1073/pnas.0304076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermos K, Bagnoli P, Epelbaum J, Hoyer D. The somatostatin sst1 receptor: an autoreceptor for somatostatin in brain and retina? Pharmacol Ther. 2006;110:455–464. doi: 10.1016/j.pharmthera.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC, Price RF, Neilson M. Immunocytochemical evidence that neurotensin is present in glutamatergic neurons in the superficial dorsal horn of the rat. J Neurosci. 1994;14:774–784. doi: 10.1523/JNEUROSCI.14-02-00774.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Lopez JE, Arguelles CF, Granados-Soto V. Participation of peripheral and spinal phosphodiesterases 4 and 5 in inflammatory pain. Proc West Pharmacol Soc. 2002;45:141–143. [PubMed] [Google Scholar]
- Vles JS, de Louw AJ, Steinbusch H, Markerink-van Ittersum M, Steinbusch HW, Blanco CE, Axer H, Troost J, de Vente J. Localization and age-related changes of nitric oxide- and ANP-mediated cyclic-GMP synthesis in rat cervical spinal cord: an immunocytochemical study. Brain Res. 2000;857:219–234. doi: 10.1016/s0006-8993(99)02434-8. [DOI] [PubMed] [Google Scholar]
- Williams DL, Jr, Katz GM, Roy-Contancin L, Reuben JP. Guanosine 5′-monophosphate modulates gating of high-conductance Ca2+-activated K+ channels in vascular smooth muscle cells. Proc Natl Acad Sci U S A. 1988;85:9360–9364. doi: 10.1073/pnas.85.23.9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard GE, Rosado JA. Recent advances in natriuretic peptide research. J Cell Mol Med. 2007;11:1263–1271. doi: 10.1111/j.1582-4934.2007.00125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, Yang L, Huang WJ, Fu G, Xu SH, Cheng XP, Yan Q, Zhu ZD, Zhang X, Chen Z, Han ZG, Zhang X. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci U S A. 2002;99:8360–8365. doi: 10.1073/pnas.122231899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZQ, Zhang X, Grillner S, Hökfelt T. Electrophysiological studies on rat dorsal root ganglion neurons after peripheral axotomy: changes in responses to neuropeptides. Proc Natl Acad Sci U S A. 1997;94:13262–13266. doi: 10.1073/pnas.94.24.13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xu ZQ, Bao L, Dagerlind A, Hökfelt T. Complementary distribution of receptors for neurotensin and NPY in small neurons in rat lumbar DRGs and regulation of the receptors and peptides after peripheral axotomy. J Neurosci. 1995;15:2733–2747. doi: 10.1523/JNEUROSCI.15-04-02733.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi T, Holmberg K, Landry M, Huang W, Xiao H, Ju G, Hökfelt T. Expression and regulation of the neuropeptide Y Y2 receptor in sensory and autonomic ganglia. Proc Natl Acad Sci U S A. 1997;94:729–734. doi: 10.1073/pnas.94.2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xu ZO, Shi TJ, Landry M, Holmberg K, Ju G, Tong YG, Bao L, Cheng XP, Wiesenfeld-Hallin Z, Lozano A, Dostrovsky J, Hökfelt T. Regulation of expression of galanin and galanin receptors in dorsal root ganglia and spinal cord after axotomy and inflammation. Ann N Y Acad Sci. 1998;863:402–413. doi: 10.1111/j.1749-6632.1998.tb10710.x. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Gopalakrishnan M, Shieh CC. Modulation of action potential firing by iberiotoxin and NS1619 in rat dorsal root ganglion neurons. Neuroscience. 2003;122:1003–1011. doi: 10.1016/j.neuroscience.2003.08.035. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Mok LP, Katz EJ, Gold MS. BKCa currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci. 2010;31:450–462. doi: 10.1111/j.1460-9568.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]