

Abstract
Inflammatory mediators through the activation of the protein kinase A (PKA) pathway sensitize primary afferent nociceptors to mechanical, thermal, and osmotic stimuli. However, it is unclear which ion conductances are responsible for PKA-induced nociceptor hyperexcitability. We have previously shown the abundant expression of Slack sodium-activated potassium (KNa) channels in nociceptive dorsal root ganglion (DRG) neurons. Here we show using cultured DRG neurons, that of the total potassium current, I K, the KNa current is predominantly inhibited by PKA. We demonstrate that PKA modulation of KNa channels does not happen at the level of channel gating but arises from the internal trafficking of Slack channels from DRG membranes. Furthermore, we found that knocking down the Slack subunit by RNA interference causes a loss of firing accommodation analogous to that observed during PKA activation. Our data suggest that the change in nociceptive firing occurring during inflammation is the result of PKA-induced Slack channel trafficking.
Introduction
According to the Melzack–Wall gate control theory (Melzack and Wall, 1965), neurons exhibit a range of responses according to various conditions. For example, pain-responsive neurons in the dorsal root ganglion (DRG), commonly known as nociceptors, exhibit intrinsic plasticity. Normally, nociceptors relay information to the CNS, indicating the location, nature, and intensity of the ensuing pain. During inflammation, nociceptors are sensitized: they have a lowered threshold of activation and increased spontaneous activity, resulting in symptoms of hyperalgesia. Inflammatory mediators such as prostaglandin E2 (PGE2) (Hingtgen et al., 1995), produce hyperalgesia through the activation of PKA (Gold et al., 1998; Khasar et al., 1998, 1999; Schnizler et al., 2008). The importance of PKA during inflammatory pain has been clearly established (Malmberg et al., 1997). Ion channel modulation by PKA is, therefore, responsible for the intrinsic plasticity associated with nociceptor sensitization (Taiwo et al., 1989, 1992; Taiwo and Levine, 1991). The duration of hyperalgesia can last from minutes to days, suggesting that membrane trafficking of ion channels (Ji et al., 2002; Zhang et al., 2005; Fabbretti et al., 2006) is likely to play an important role in nociceptor sensitization (Schmidt et al., 2009). It is not known, however, whether K+ channel trafficking occurs during inflammation. K+ conductances progressively diminish in DRG neurons after PKA stimulation (Evans et al., 1999), and PKA stimulation of DRG neurons causes hyperexcitability consistent with a reduction of K+ current (Song et al., 2006; Zheng et al., 2007).
KNa channels are abundantly expressed in the cell bodies and axons of nociceptive DRG neurons (Bischoff et al., 1998; Tamsett et al., 2009), and suggested to regulate DRG neuronal resting membrane potential (RMP) (Bischoff et al., 1998). In neuronal simulations, KNa channels caused firing accommodation (Bhattacharjee et al., 2005; Brown et al., 2008). These findings are congruent with the fact that KNa channels are a prominent rectifying K+ current in neurons (Dale, 1993; Hess et al., 2007; Budelli et al., 2009; Lu et al., 2010). However, direct empirical evidence for KNa channel contribution to neuronal excitability has been lacking.
KNa channels are encoded by the Slack and Slick genes (Bhattacharjee and Kaczmarek, 2005). Both channel subunits contain large C termini consisting of tandem RCK domains (regulators of K+ conductance) (Jiang et al., 2002; Ye et al., 2006) with many consensus PKA and protein kinase C (PKC) phosphorylation sites (Joiner et al., 1998; Bhattacharjee et al., 2003). Although sensitive to PKC regulation (Santi et al., 2006), recombinant Slack channels are unresponsive to PKA modulation (Nuwer et al., 2009). Most of the PKA consensus in Slack are located outside the RCK domains, suggesting that PKA modulation of Slack KNa channels should not affect gating but could involve protein–protein interaction and consequently trafficking of channels. Here we demonstrate that PKA activation in cultured DRG neurons produces hyperexcitability, a reduction in the KNa component of I K and Slack channel internalization. We also demonstrate that Slack KNa channel knockdown by RNA interference produces a loss of firing accommodation.
Materials and Methods
DRG neuronal culture and immunocytochemistry.
Dorsal root ganglia were dissected from E15 embryos of Sprague Dawley rats. The ganglia were dissociated in trypsin (2.5 mg/ml) for 40 min. Cells were plated on poly-d-lysine (100 μg/ml)- and laminin (3 μg/ml)-coated coverslips, and maintained on serum-free C2 medium containing β-nerve growth factor (NGF) (100 ng/ml). Unlike neonatal and adult DRG neurons, embryonic DRG neurons require NGF for survival (Chalazonitis et al., 1987). The day after dissection, cells were treated with 1 μm cytosine-β-d-arabinofuranoside for 2 d. Cells were allowed to recover for 2 d before use. For gene delivery experiments, neurons were supplemented with 1 mm nicotinamide. For Slack immunocytochemistry, after 5 d in culture, neurons were fixed in 4% paraformaldehyde over 30 min followed by a 30 min wash with PBS. Neurons were permeabilized with 0.1% Triton X-100 for 4 min and then washed again in PBS. Neurons were incubated with an anti-Slack antibody (1:1000) (Bhattacharjee et al., 2002) at 4°C overnight followed by a secondary AlexaFluor 546 goat anti-chicken antibody (1:1000) for 2 h at room temperature. After three washes in PBS, coverslips were then mounted on slides and imaged.
Electrophysiology.
For current-clamp recordings, a Multiclamp 700B (Molecular Devices) was used and data were stored digitally using a Digidata interface. Only neurons that had a resting membrane potential more negative than −40 mV and input resistance >100 MΩ were used. Depolarizing steps in increments of 10 pA from −10 to 200 pA (20 ms duration) were used to measure the action potential (AP) threshold current. Then the repetitive discharge of each cell was measured by injection of 2.5× threshold stimulus for 1000 ms. For voltage-clamp, nystatin-perforated patch-clamp recordings and inside-out recordings were performed using the gigaseal patch-clamp technique (Hamill et al., 1981). Electrodes had resistances of 4–5 MΩ for nystatin-perforated patch recordings and 7–10 MΩ for inside-out recordings. Data were acquired using an Axopatch 200B (Molecular Devices) and digitized at and filtered at either 5 kHz for nystatin recordings or 1 kHz for inside-out recordings. Data analysis was performed using Clampfit (Molecular Devices) and Origin (OriginLab Software). For current-clamp whole-cell and nystatin (125 μg/μl)-perforated patch recordings, the pipette solution contained the following (in mm): 124 K-gluconate, 2 MgCl2, 13.2 NaCl, 1 EGTA, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP, pH 7.2 (Wu and Pan, 2007). The bath solution contained (in mm) 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 15.6 HEPES, and 10 glucose, pH 7.4. For the nystatin (125 μg/μl)-perforated patch voltage-clamp, the extracellular solution contained the following (in mm): 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl26H2O, 10 HEPES, and 10 glucose, with pH 7.3. For 0 mm Na+ bath solution, 140 mm N-methyl glucamine (NMG) was used for Na+ replacement in the above solution. The internal solution contained the following (in mm): 97.5 K-gluconate, 32.5 KCl, 10 HEPES, 1 EGTA, and 2 MgCl26H2O, with pH 7.3. For inside-out recordings, a symmetric K+ solution was used containing the following (in mm): 130 KCl, 10 NaCl, 10 HEPES, 5 EGTA, and 2 MgATP, with pH 7.3. One millimolar TEA-Cl was added to the pipette solution to block contaminating large-conductance Slo K+ channels. 8-Bromo-cAMP and Rp-cAMPS were obtained from Calbiochem and the PKA catalytic subunit from Promega.
Molecular biology.
The NtermGFP-Slack construct was made by TOPO cloning Slack without its original start methionine, in frame into the N-terminal GFP/pcDNA6.2 plasmid (Invitrogen). The negative control or scrambled siRNA was purchased from OligoEngine (ID #105205) and Slack-specific siRNAs were obtained from Ambion (ID numbers are 51466, 191981, and 191980 for Slack siRNA numbers 1, 2, and 3, respectively). siRNA subcloning procedures were performed as per the manufacturer's guidelines. siRNAs were cloned into the DNA plasmid pHSVsi (kindly provided to us by Y. Saeki, Ohio State University, Columbus, OH) for transfection experiments. For transduction experiments, siRNAs were cloned into an adenovirus intermediate vector for recombination into the Adeno-X/ZsGreen1 (Clonetech) virus. The recombined Adeno-X virus was transfected into HEK 293A cells and amplified, and titers were calculated as multiplicity of infection (MOI) from HEK cells. We used 125 MOI of negative control siRNA versus 62.5 MOI of Slack siRNA#1 + 62.5 Slack siRNA#3 for Slack knockdown experiments. For Western analysis, DRG neurons were collected in lysis buffer containing protease inhibitors and Laemmli sample buffer (Bio-Rad). Samples were boiled for 10 min and loaded onto a Ready Gel (Bio-Rad) (4–15% Tris-HCl). After electrophoresis, the gel was transferred to a PVDF membrane, then blocked with nonfat dry milk prior incubation with an anti-Slack antibody (NeuroMab). The signal was detected using horseradish peroxidase-conjugated secondary antibody and chemiluminescent substrate kit (KPL). For PCR, mRNA was harvested from DRG neurons and HEK cells stably expressing Slack, using the RNeasy Mini kit (Qiagen) following the manufacturer's instructions. After quantification, 0.5 μg of RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). One-tenth of that reaction volume was used in a PCR with gene-specific primers for Slack producing a cDNA fragment of 100 bp.
Live-cell imaging and analysis.
All imaging experiments were performed at 5% CO2 and 37°C. Neurons were plated onto 35 mm glass dishes coated with laminin and poly-d-lysine (MatTek). Neurons were transfected after 5 d in culture with 1 μg of NtermGFP-Slack/ pcDNA6.2 plasmid using Lipofectamine LTX (Invitrogen). After 48 h, cells were labeled with 50 μg/μl concanavalin A-AlexaFluor 647 (Invitrogen) for 5 min. After two brief washes in PBS, fluorescing neurons were used for imaging. Images were acquired using a Zeiss LSM Meta Confocal Microscope with a 63× oil-immersion objective and acquired using LSM ImageExaminer software. Sectioning from the z-stack is divided upon time points and allows for specific selection of slices based on individual time points. We quantified the colocalization signal on one slice before and after PKA activation using ImageJ colocalization software (National Institutes of Health). This software analyzes the overlap coefficient from two channels (red and green) in overlapping confocal images. A resultant grayscale image is produced, and the gray values representing the area of overlap can be measured and quantified.
Membrane protein biotinylation.
DRG neurons were plated on a poly-d-lysine- and laminin-coated six-well plate. DRG neurons were treated with 250 μm 8-bromo-cAMP for 30 min at 7% CO2 and 37°C. Then 160 μl of 10 mm Sulfo-NHS-SS-Biotin (Thermo Scientific) was added to each well and incubated at room temperature for 45 min. During this time, 100 μl of Pierce Streptavidin Magnetic Beads (Thermo Scientific) were washed with PBS + 0.1% Tween. After 45 min, the cells were rinsed with PBS and lysed with a buffer containing PBS, 150 mm NaCl, 1% Nonidet P-40, and 0.1% SDS, pH 8.0. PMSF, a protease inhibitor cocktail, and a phosphatase inhibitor cocktail (Sigma) were also added to the lysis buffer. Samples were then incubated with 50 μl of the avidin beads and rotated at room temperature for 2 h. The samples were then washed twice in PBS. After removing the supernatant, beads were resuspended in Laemmli sample buffer (Bio-Rad) and boiled for 5 min. Samples were loaded onto a Ready Gel (Bio-Rad) (4–15% Tris-HCl). After electrophoresis, the gel was then transferred onto a PVDF membrane for 1 h and incubated in 5% nonfat dry milk as a blocking agent for 2 h. The anti-Slack (NeuroMab) primary antibody was used for Western analysis. The signal was detected using horseradish peroxidase-conjugated secondary antibody and chemiluminescent substrate kit (KPL). Densitometry analyses obtained from Western blots were conducted using Quantity One (Bio-Rad) software.
Results
Cultured DRG neurons contain Slack KNa channels
To investigate the modulation of native KNa channels by PKA, we used cultured embryonic DRG neurons. Like their adult counterparts (Tamsett et al., 2009), embryonic cultured DRG neurons abundantly express the Slack subunit (Fig. 1 A). We have confirmed this immunohistochemistry data by both PCR and Western analysis (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Voltage-clamp whole-cell recordings were conducted in these DRG neurons (Fig. 1 B), and when external Na+ was substituted with NMG, I K of DRG neurons was greatly reduced, to ∼40% its control value (n = 6). This is very similar to KNa reported in Xenopus spinal neurons (Dale, 1993), lamprey spinal cord neurons (Hess et al., 2007), and rat olfactory neurons (Budelli et al., 2009). These cultured embryonic neurons are ideal because they are also homogenous in their firing properties (Chen et al., 1987; Grigaliunas et al., 2002; Toman et al., 2004). Neurons are cultured in serum-free medium containing NGF. NGF is critical for embryonic neuronal survival (Chalazonitis et al., 1987); however, this condition also produces a highly enriched population of peptidergic DRG neurons (Bucelli et al., 2008). AP properties generally resemble those of mature peptidergic nociceptors (Fig. 1 C) (Table 1) exhibiting broad AP durations and large AP overshoots (McCarthy and Lawson, 1997; Fang et al., 2005; Binshtok et al., 2007). Conversely, afterhyperpolarizations were smaller, and AP base durations longer, than previously reported (Fang et al., 2005) (Table 1), but this is probably due to differences in recording technique and recording conditions. Nonetheless, cultured neurons universally exhibit firing accommodation during a sustained stimulus (2.5× the stimulus threshold) (Fig. 1 C). We have recorded 152 untreated neurons in this manner and have never observed repetitive firing.
Figure 1.
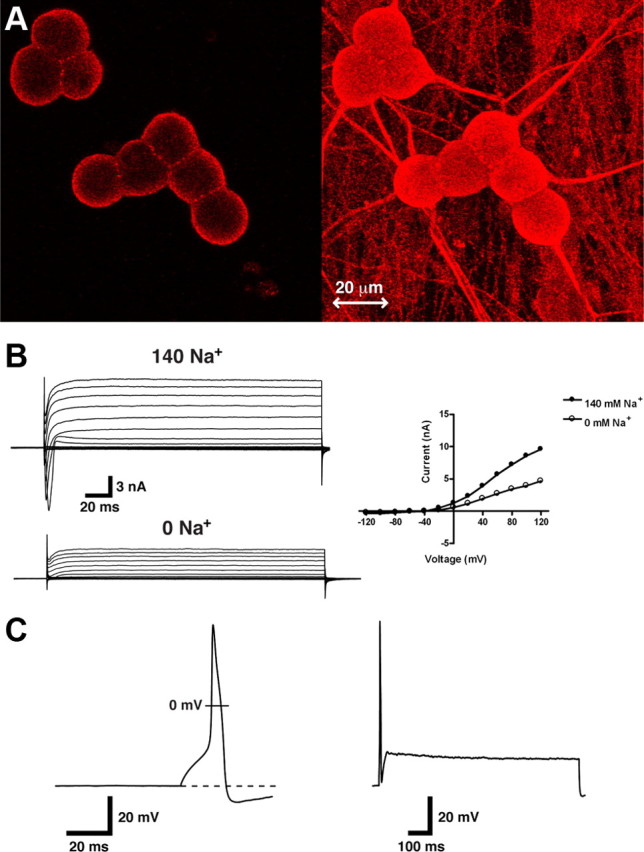
KNa channels in cultured DRG neurons. A. Slack immunolabeling in E15 cultured embryonic DRG neurons. Using confocal microscopy, we found robust staining localized at the outer membrane of all neurons (left panel). We also found immunoreactivity within the axonal projections of these neurons, which is more clearly seen with a full projection (right panel). B, Removal of extracellular Na+ reduces I K of DRG neurons. A DRG neuron was recorded using the voltage-clamp whole-cell technique. Protocol consisted of 200 ms steps from −120 mV to +120 mV. Voltage was clamped at −70 mV. Neuron was initially recorded in high external Na+ (top left panel) and then perfused with a solution in which Na+ was substituted with N-methyl-d-glucamine (bottom left panel). C, Representative action potential in an unstimulated neuron fired during a 10 ms pulse of 300 pA (left panel). Action potential properties resemble mature peptidergic nociceptors. During 2.5× threshold protocol maintained for 1000 ms, neurons exhibit firing accommodation, typically firing less than two action potentials (right column). This type of firing was observed in all unstimulated neurons recorded (n = 152).
Table 1.
Action potential properties of rat embryonic DRG neurons
| Total sample (n) | Peak (mV) | AHP (mV) | AP base duration (ms) | RMP (mV) | Repetitive firing (%) |
|---|---|---|---|---|---|
| 45 | 116 ± 0.5 | −5.8 ± 0.2 | 14.1 ± 0.4 | −53 ± 0.3 | 0 |
All recordings were performed using the whole-cell current-clamp technique. Repetitive firing is defined as firing greater than two action potentials during a suprathreshold stimulus. Values are means ± SEM. AHP, Afterhyperpolarization.
The KNa component of I K in DRG neurons is reduced after PKA activation
Also like their adult counterparts (Cui and Nicol, 1995), cultured embryonic DRG neurons exhibit hyperexcitability after PKA activation. Using nystatin-perforated patch whole-cell current-clamp on cultured DRG neurons, we typically see neurons fire less than two APs in response to 2.5× the stimulus threshold (Fig. 2 A) (n = 6) before PKA stimulation. After application of 250 μm 8-bromo-cAMP, the PKA activator, five of the six neurons exhibited a progressive loss of firing accommodation, and after 10 min no firing accommodation was observed (Fig. 2 A). Previous reports on the effects of PGE2/PKA on I K in DRG neurons are conflicting. When I K is recorded in neurons under Na+-free conditions, only a small inhibition of I K by PKA and PGE2 was observed (Akins and McCleskey, 1993; England et al., 1996). In contrast, when I K is recorded in normal Na+-containing Ringer's solution, both PGE2 and PKA activation by cpt-cAMP cause a progressive inhibition of I K (∼36% inhibition) over a period of 20 min. Using nystatin-perforated whole-cell voltage clamp, we assayed the effect of PKA activation on the I K in DRG neurons in the presence and absence of Na+ in the bath. In external solutions void of Na+ (0 mm), 15 min after application of 8-bromo cAMP we observed a significant decrease in I K, but the decrease was small; only a 12% decrease in I K was observed (Fig. 2 B) when normalized to control (Fig. 2 D). This level of inhibition is essentially identical to what was previously reported (Akins and McCleskey, 1993; England et al., 1996). In parallel experiments in which neurons were bathed in an extracellular bath containing physiological levels of Na+ (140 mm), application of 250 μm 8-bromo cAMP caused a much greater decrease in I K (Fig. 2 C) (n = 6). After application of 8-bromo cAMP, there was an ∼50% decrease in I K when normalized to control (Fig. 2 D), and this percentage decrease was significantly larger when compared to Na+-free conditions (p < 0.05, unpaired t test). These results strongly suggest that under physiological recording conditions, PKA strongly inhibits I K, particularly the KNa component of I K. The loss of firing accommodation caused by PKA then could likely be attributed to the inhibition of KNa.
Figure 2.

PKA inhibits the KNa component of I K. A, Representative action potential recorded in a neuron before (left panel) and after (right panel) application of 250 μm 8-bromo-cAMP (n = 6). Current-clamp recordings were performed using nystatin-perforated patches, and stimulation was 2.5× threshold for 1000 ms. Progressive loss of firing accommodation after 8-bromo-cAMP application was observed in five of the six neurons. B, C, Left, Representative traces of perforated-patch whole-cell recordings of I K recorded before and after application of 250 μm 8-bromo-cAMP in Na+-free external solution (B) and Na+-containing external solution (C). Holding potential was −50 mV, and currents were elicited with voltage steps from −120 mV to +120 mV in 20 mV increments. Current–voltage relationships before and after 8-bromo-cAMP are depicted in the right panels. Arrows indicate current level after 8-bromo-cAMP application. D, Peak current after 250 μm 8-bromo-cAMP addition at +80 mV normalized to control for both 140 and 0 mm Na+-containing extracellular solution (n = 6 for both; *p < 0.05). Error bars represent SEM.
KNa channel gating is not affected by PKA modulation
It was previously found that native KNa channels recorded in excised patches from olfactory neurons are insensitive to PKA modulation (Egan et al., 1992). Our group has recently shown that PKA does not directly affect Slack channels when expressed in HEK-293 cells (Nuwer et al., 2009). Still based on the observed inhibition of KNa by PKA described above, we sought to determine whether the gating of native KNa channels recorded from DRG neurons exhibited PKA sensitivity. Using inside-out patch-clamp recordings and after directly applying 100 U/ml of the PKA catalytic subunit directly to the patch, we found no statistically significant PKA-mediated inhibition of KNa channel gating (n = 6, p = 0.56) (Fig. 3).
Figure 3.

PKA did not affect KNa channel gating. A, Representative traces of excised inside-out patches of KNa channels from DRG neurons recorded before and after direct application of the PKA catalytic subunit (100 U/ml). Bath solution contained 1 mm TEA-Cl. Patch was held at 0 mV and stepped to −80 mV. Shown are three consecutive sweeps of 900 ms duration before and after catalytic subunit application. B, NPO of 10 s of channel recordings after PKA catalytic subunit addition normalized to control (n = 6). Error bars represent SEM. Results were not statistically significant (p = 0.562).
Slack channels are trafficked from the DRG neuronal membrane after PKA stimulation
Evidence indicates that different ion channels are trafficked in DRG neurons after PKA stimulation (Ji et al., 2002; Zhang et al., 2005; Fabbretti et al., 2006). We first investigated the membrane expression of Slack before and after PKA activation by labeling the N-terminal of the Slack protein with emerald green fluorescent protein (NtermGFP-Slack). We tagged the N-terminal instead of the C-terminal because the C-terminal portion of the protein contains all of the PKA consensus sites (Joiner et al., 1998; Nuwer et al., 2009) and a PDZ binding motif (Joiner et al., 1998). The NtermGFP-Slack produced Slack-like currents when expressed in HEK-293 cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), demonstrating that tagging the N-terminal with GFP did not affect functionality of the protein, nor did it prohibit the expression of the channel at the membrane. We then transfected DRG neurons with the NtermGFP-Slack construct and, after 48 h, performed live-cell imaging using confocal microscopy on successfully transfected neurons using an AlexaFluor-conjugated anti-concanavalin A antibody. Concanavalin A is localized to the membrane and thus served as a membrane marker. Fifteen minutes after the application of 8-bromo-cAMP (Fig. 4 A), there was a decrease in the amount of Slack (green) that colocalized with the concanavalin A (red), represented by a decrease in yellow punctate in overlapping images. We found a significant decrease in the colocalization signal (Fig. 4 B) in six independently transfected neurons (p < 0.05) after 8-bromo-cAMP application, indicating that NtermGFP-Slack channels are internalized after PKA activation. Although we did note some photobleaching in both the red and green signals over a period of 30 min (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), bleaching was minimal, and therefore, the decrease in overlap was due primarily to channel redistribution.
Figure 4.
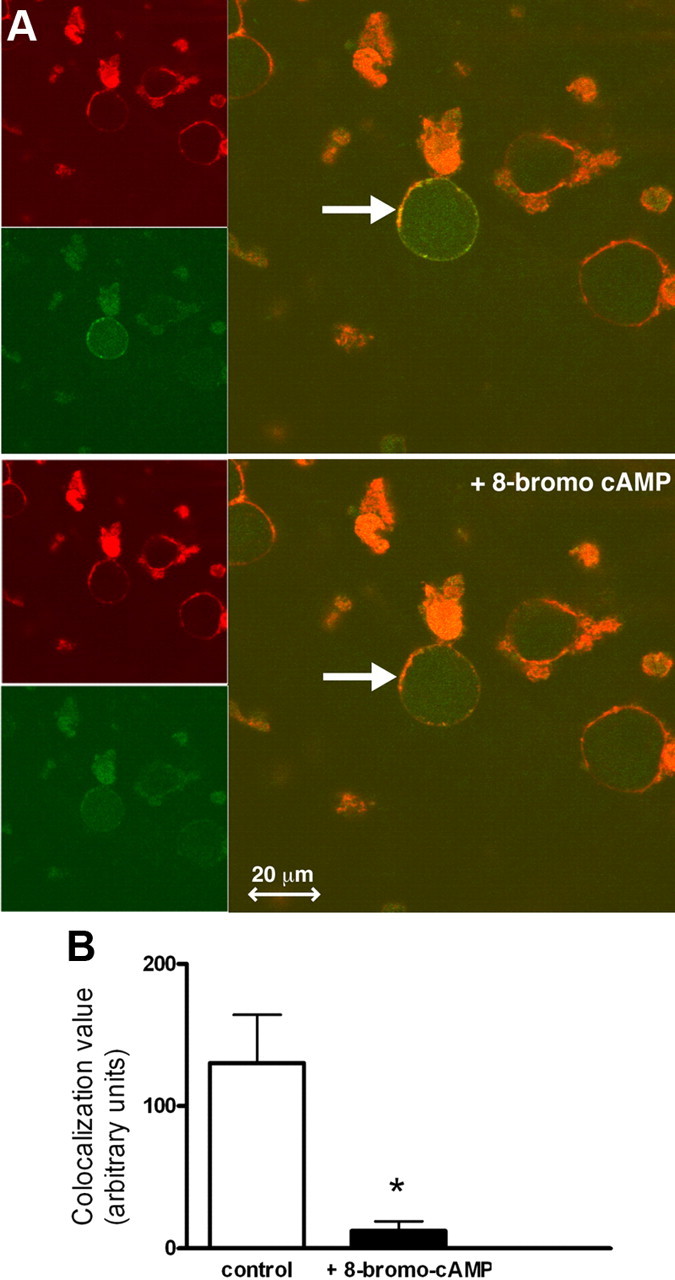
PKA activation decreases NtermGFP-Slack DRG neuronal membrane expression. A. Representative images a DRG neuron transfected with NtermGFP-Slack (green) colocalized with membrane-bound concanavalin A (red) as revealed by yellow punctate in overlapping images (marked by arrows), before (upper panel) and 15 min after (lower panel) 250 μm 8-bromo-cAMP application. Images were taken from a single slice of a z-stack using a 63× objective on a Zeiss confocal microscope. Chamber was kept at 5% CO2 and 37°C. B, PKA activation reduced the colocalization of NtermGFP-Slack with concanavalin A. Colocalization or gray values were determined using ImageJ colocalization software (n = 6; *p < 0.05). Error bars represent SEM.
We next investigated whether PKA-induced Slack channel internalization occurs with endogenous Slack channels. To do this, we conducted a surface-expression biotinylation assay. In unstimulated neurons, appreciable levels of biotinylated Slack protein could be pulled down with streptavidin beads and resolved by Western analysis (Fig. 5 A). In contrast, DRG neurons pretreated with 250 μm 8-bromo-cAMP for 30 min had reduced Slack protein present on the membrane. In all of the six independent experiments, we found a consistent reduction of Slack channels at the membrane after PKA stimulation. In half of those experiments, we conducted parallel studies in which PKA was inhibited by pretreating neurons with the cell-permeable inhibitor Rp-cAMPS (100 μm) for 30 min followed by 8-bromo-cAMP treatment (Fig. 5 B). In this case, surface Slack channel expression was comparable to unstimulated neurons (n = 3). These data suggest that the internalization of Slack channels occurs in a PKA-dependent manner. Our biotinylation assays thus confirm what was shown in the live-cell imaging experiments, that Slack membrane expression is reduced after PKA activation.
Figure 5.

PKA activation decreases membrane expression of endogenous Slack channels in DRG neurons. A. Representative blot of membrane Slack biotinylation assay. Membrane biotinylation and precipitation by streptavidin followed by Western analysis using a Slack-specific antibody were performed on untreated neurons, neurons treated with 250 μm 8-bromo-cAMP for 30 min, or neurons pretreated with Rp-cAMPS for 30 min followed by 250 μm 8-bromo-cAMP for 30 min. B, Densitometric analysis of Slack membrane expression when normalized to actin (n = 3; *p < 0.005). Error bars represent SEM.
Knockdown of Slack channels produces a loss of firing accommodation in DRG neurons
Because PKA activation produces DRG neuronal hyperexcitability, a substantial reduction in the KNa component of I K, and Slack channel internalization, we wanted to investigate whether Slack channels are indeed responsible for firing accommodation in DRG neurons. Because there are no Slack-specific inhibitors, we used RNA interference to knock down the Slack subunit. We used Slack-specific siRNAs sequences generated by Ambion and initially screened their ability to knock down Slack message in a Slack stable HEK cell line (Yang et al., 2006) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We then transfected neurons with plasmids that encode Slack-specific siRNAs and recorded whole-cell I K in Slack-specific siRNA and negative control siRNA-transfected neurons 72 h after transfection. We found that a combination of two different siRNAs at lower concentration produced the greatest amount of Slack subunit knockdown. Similar to the Na+ replacement experiments in Figure 1, we found an ∼40% reduction in I K in Slack siRNA-treated neurons versus negative control siRNA-treated neurons (Fig. 6 A,B). We confirmed knockdown of Slack unit by both Western analysis (Fig. 6 C) and immunocytochemistry (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). However, because transfection efficiency of cultured DRG neurons is only ∼5–8%, we used an adenoviral delivery method of siRNAs to neurons to confirm Slack protein knockdown. In our hands, we see >75% inoculation efficiency. After inoculating neurons with adenoviral Slack siRNAs, we saw appreciable knockdown of Slack protein after 72 h versus the negative control. Finally, we performed current-clamp recordings of untransduced, negative control siRNA- and Slack siRNA-inoculated neurons. Slack knockdown caused DRG neuronal depolarization [RMP = −49.8 ± 1 mV (n = 8), p < 0.01] vs the negative control [RMP = 52.7 mV ± 0.5 mV (n = 4)] and uninoculated neurons [RMP = 54.7 mV ± 0.8 mV (n = 3)]. Repetitive firing, defined as more than two APs in response to 2.5× the stimulus threshold, was observed in six of eight Slack siRNA-infected neurons, while no repetitive firing was observed in the negative control (n = 4) and uninfected (n = 3) neurons. These data strongly suggest that KNa channels contribute to DRG neuronal firing and that decreasing their activity produces DRG neuron hyperexcitability.
Figure 6.

Knockdown of Slack channels produces a loss of firing accommodation in DRG neurons. A, Representative whole-cell current I K recordings of DRG neurons transfected with negative control siRNA- (left panel) or Slack-specific siRNA- (right panel) encoding constructs. B, Averaged current–voltage relationship of I K measured from negative control siRNA and Slack siRNA (n = 8; *p < 0.05). C, Slack protein knockdown was confirmed by Western analysis on neurons inoculated with adenoviruses containing negative control or Slack-specific siRNA. D, Representative action potential recordings in untransfected and negative control siRNA- and Slack-specific siRNA-inoculated neurons. Current-clamp recordings were performed using a 2.5× threshold for 1000 ms. Loss of firing accommodation was observed only in Slack siRNA-treated neurons (6 of 8).
Discussion
When PGE2 is injected into the hindpaw of rats, hyperalgesia peaks at 1 h and continues for up to 3 h, and when the phosphodiesterase inhibitor rolipram is coadministered with PGE2, hyperalgesia can persist for many more hours (Ouseph et al., 1995). It is reasonable to conclude that ion channel trafficking in nociceptors is responsible for prolonged hyperalgesia. Indeed, nociceptive signals increase TRPA1 expression at the membrane through a PKA-dependent pathway (Schmidt et al., 2009), and an increased Nav1.8 surface expression after PKA activation has also been recently described (Liu et al., 2010). These studies suggest that Na+ entry increases in DRG neurons after PKA activation, and reducing the number of KNa channels at the plasma membrane will enhance the depolarizing effects of the increased Na+ currents.
It is debatable which ion channels are most important for PKA-induced DRG neuronal hyperexcitability. NaV1.7 is thought to be the primary Na+ channel responsible for nociception because of its ability to respond to ramp stimuli and thereby amplify generator potentials (Dib-Hajj et al., 2007). Surprisingly, NaV1.7 currents are not potentiated but in fact inhibited by PKA modulation (Vijayaragavan et al., 2004). Other studies have shown that NaV1.8 Na+ channel properties are modulated by PKA, NaV1.8 message is upregulated, and channels are trafficked to the membrane, but in NaV1.8 knock-out mice, inflammatory pain is reduced but not abolished (Akopian et al., 1999; Nassar et al., 2005). Moreover, thermal hyperalgesia after PGE2 administration to the hindpaw was equivalent in NaV1.8 +/+ and NaV1.8 −/− mice (Kerr et al., 2001). Therefore, it has been argued that the Na+ channels only play a minor role in increased DRG neuronal action potential firing following tissue damage (Momin and McNaughton, 2009). Alternatively, H-channels have been suggested to be more important for PGE2/PKA-induced hyperexcitability than Na+ channels (Momin and McNaughton, 2009). H-channels are not directly modulated by PKA in DRG neurons (Komagiri and Kitamura, 2007), so it is unclear how they can be essential to PKA-induced nociceptor sensitization. The case for K+ channels has been contentious. While some investigators have implicated a role for K+ channels in hyperexcitability during elevated cAMP/cGMP in nerve injury (Song et al., 2006; Zheng et al., 2007), the primary rationale for a lesser role if any for I K in PKA-mediated hyperexcitability is an apparent lack of modulation of I K by PKA. Prior studies have shown that cAMP cell-permeable analogs only caused a slight inhibition of I K (∼12–15%) (Akins and McCleskey, 1993; England et al., 1996). However, these studies were conducted in Na+-free external recording solutions, and when we record I K after PKA stimulation, we similarly see a small inhibition. On the other hand, when examining I K using whole-cell voltage clamp, over a longer period of time (e.g., 20 min), in Na+-containing external solutions, a larger decrease (36%) of I K by PKA in DRG neurons was observed (Evans et al., 1999). We found a near 50% PKA-mediated inhibition of I K using perforated patch-clamp experiments in solutions containing Na+ in physiological concentrations within 15 min. We would argue that our experimental approach is the most physiological and accurately reflects the inhibition of I K by PKA. Moreover, because KNa is a prominent rectifying current in different neurons (Dale, 1993; Hess et al., 2007; Budelli et al., 2009; Lu et al., 2010), we propose that any modulation studies of I K in neurons should always be conducted in Na+-containing external solutions.
Based on their intrinsic voltage dependence and gating properties (Joiner et al., 1998), computer simulations have predicted that Slack KNa channels cause firing accommodation in model neurons (Brown et al., 2008). In this report, using RNA interference, we have now shown that knockdown of Slack KNa channels produces a loss of firing accommodation. This marks the first direct demonstration of the contribution of Slack KNa channels to neuronal excitability. In addition to inflammatory pain, DRG neuronal hyperexcitability is also important to neuropathic pain pathophysiology (Chung and Chung, 2002). Curiously, in neuropathic pain, DRG neuronal hyperexcitability is also thought to arise as a direct result of Na+ channel upregulation (Novakovic et al., 1998; Roza et al., 2003; Devor, 2006). In conflict with this view is the normal development of neuropathic pain during nerve injury in various Na+ channel subtype knock-out mouse models (Porreca et al., 1999; Kerr et al., 2001; Nassar et al., 2005, 2006; Priest et al., 2005; Amaya et al., 2006; Krafte and Bannon, 2008). Alternatively, I K is reduced under neuropathic conditions (Kim et al., 2002; Tan et al., 2006), and therefore our findings strongly suggest that Slack KNa channels might be involved in multiple disease states associated with DRG neuronal hyperexcitability. Indeed, their ubiquitous and abundant expression patterns in DRG neuronal cell bodies and axons suggest that these channels act to prevent neuronal hyperexcitability. During inflammation and/or nerve injury, targeting these channels, either directly or at the proteins that regulate their trafficking, could offer a novel approach for analgesia.
Footnotes
This study was supported by a Junior Faculty Award from the American Diabetes Association and a John R. Oishei Foundation Grant to A.B. We thank L. Christen and W. Ruyechan for help with constructing the adenoviral siRNAs. We thank W. Sirgurdson for help with the confocal microscopy. We thank E. Daurignac for critical reading of this manuscript.
References
- Akins PT, McCleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience. 1993;56:759–769. doi: 10.1016/0306-4522(93)90372-m. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, Chessell IP, Tate S, Green PJ, Woolf CJ. The voltage-gated sodium channel Nav1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852–12860. doi: 10.1523/JNEUROSCI.4015-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+ Trends Neurosci. 2005;28:422–428. doi: 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol. 2002;454:241–254. doi: 10.1002/cne.10439. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci. 2003;23:11681–11691. doi: 10.1523/JNEUROSCI.23-37-11681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, von Hehn CA, Mei X, Kaczmarek LK. Localization of the Na+-activated K+ channel Slick in the rat central nervous system. J Comp Neurol. 2005;484:80–92. doi: 10.1002/cne.20462. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449:607–610. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- Bischoff U, Vogel W, Safronov BV. Na+-activated K+ channels in small dorsal root ganglion neurones of rat. J Physiol. 1998;510:743–754. doi: 10.1111/j.1469-7793.1998.743bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Spilianakis CG, Flavell RA, von Hehn CA, Bhattacharjee A, Kaczmarek LK. Amino-termini isoforms of the Slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J Physiol. 2008;586:5161–5179. doi: 10.1113/jphysiol.2008.160861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucelli RC, Gonsiorek EA, Kim WY, Bruun D, Rabin RA, Higgins D, Lein PJ. Statins decrease expression of the proinflammatory neuropeptides calcitonin gene-related peptide and substance P in sensory neurons. J Pharmacol Exp Ther. 2008;324:1172–1180. doi: 10.1124/jpet.107.132795. [DOI] [PubMed] [Google Scholar]
- Budelli G, Hage TA, Wei A, Rojas P, Jong YJ, O'Malley K, Salkoff L. Na+-activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat Neurosci. 2009;12:745–750. doi: 10.1038/nn.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalazonitis A, Peterson ER, Crain SM. Nerve growth factor regulates the action potential duration of mature sensory neurons. Proc Natl Acad Sci U S A. 1987;84:289–293. doi: 10.1073/pnas.84.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GG, Cole AE, MacDermott AB, Lange GD, Barker JL. The influence of skeletal muscle on the electrical excitability of dorsal root ganglion neurons in culture. J Neurosci. 1987;7:2412–2422. [PMC free article] [PubMed] [Google Scholar]
- Chung JM, Chung K. Importance of hyperexcitability of DRG neurons in neuropathic pain. Pain Pract. 2002;2:87–97. doi: 10.1046/j.1533-2500.2002.02011.x. [DOI] [PubMed] [Google Scholar]
- Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2-induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- Dale N. A large, sustained Na(+)- and voltage-dependent K+ current in spinal neurons of the frog embryo. J Physiol. 1993;462:349–372. doi: 10.1113/jphysiol.1993.sp019559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Egan TM, Dagan D, Kupper J, Levitan IB. Properties and rundown of sodium-activated potassium channels in rat olfactory bulb neurons. J Neurosci. 1992;12:1964–1976. doi: 10.1523/JNEUROSCI.12-05-01964.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Vasko MR, Nicol GD. The cAMP transduction cascade mediates the PGE2-induced inhibition of potassium currents in rat sensory neurones. J Physiol. 1999;516:163–178. doi: 10.1111/j.1469-7793.1999.163aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbretti E, D'Arco M, Fabbro A, Simonetti M, Nistri A, Giniatullin R. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene-related peptide. J Neurosci. 2006;26:6163–6171. doi: 10.1523/JNEUROSCI.0647-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, McMullan S, Lawson SN, Djouhri L. Electrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivo. J Physiol. 2005;565:927–943. doi: 10.1113/jphysiol.2005.086199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM. Modulation of TTX-R I Na by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro . J Neurosci. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigaliunas A, Bradley RM, MacCallum DK, Mistretta CM. Distinctive neurophysiological properties of embryonic trigeminal and geniculate neurons in culture. J Neurophysiol. 2002;88:2058–2074. doi: 10.1152/jn.2002.88.4.2058. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hess D, Nanou E, El Manira A. Characterization of Na+-activated K+ currents in larval lamprey spinal cord neurons. J Neurophysiol. 2007;97:3484–3493. doi: 10.1152/jn.00742.2006. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, Kaczmarek LK. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat Neurosci. 1998;1:462–469. doi: 10.1038/2176. [DOI] [PubMed] [Google Scholar]
- Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport. 2001;12:3077–3080. doi: 10.1097/00001756-200110080-00019. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Choi JO, Rim HD, Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res. 2002;105:146–152. doi: 10.1016/s0169-328x(02)00388-1. [DOI] [PubMed] [Google Scholar]
- Komagiri Y, Kitamura N. Comparison of effects of PKA catalytic subunit on I(h) and calcium channel currents in rat dorsal root ganglion cells. Biomed Res. 2007;28:177–189. doi: 10.2220/biomedres.28.177. [DOI] [PubMed] [Google Scholar]
- Krafte DS, Bannon AW. Sodium channels and nociception: recent concepts and therapeutic opportunities. Curr Opin Pharmacol. 2008;8:50–56. doi: 10.1016/j.coph.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Q, Su Y, Bao L. Prostaglandin E(2) promotes Na(v)1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway. Traffic. 2010;11:405–417. doi: 10.1111/j.1600-0854.2009.01027.x. [DOI] [PubMed] [Google Scholar]
- Lu S, Das P, Fadool DA, Kaczmarek LK. The Slack sodium-activated potassium channel provides a major outward current in olfactory neurons of Kv1.3−/− super-smeller mice. J Neurophysiol. 2010;103:3311–3319. doi: 10.1152/jn.00607.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Brandon EP, Idzerda RL, Liu H, McKnight GS, Basbaum AI. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci. 1997;17:7462–7470. doi: 10.1523/JNEUROSCI.17-19-07462.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy PW, Lawson SN. Differing action potential shapes in rat dorsal root ganglion neurones related to their substance P and calcitonin gene-related peptide immunoreactivity. J Comp Neurol. 1997;388:541–549. [PubMed] [Google Scholar]
- Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- Momin A, McNaughton PA. Regulation of firing frequency in nociceptive neurons by pro-inflammatory mediators. Exp Brain Res. 2009;196:45–52. doi: 10.1007/s00221-009-1744-2. [DOI] [PubMed] [Google Scholar]
- Nassar MA, Levato A, Stirling LC, Wood JN. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol Pain. 2005;1:24. doi: 10.1186/1744-8069-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Baker MD, Levato A, Ingram R, Mallucci G, McMahon SB, Wood JN. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol Pain. 2006;2:33. doi: 10.1186/1744-8069-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuwer MO, Picchione KE, Bhattacharjee A. cAMP-dependent kinase does not modulate the Slack sodium-activated potassium channel. Neuropharmacology. 2009;57:219–226. doi: 10.1016/j.neuropharm.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Ouseph AK, Khasar SG, Levine JD. Multiple second messenger systems act sequentially to mediate rolipram-induced prolongation of prostaglandin E2-induced mechanical hyperalgesia in the rat. Neuroscience. 1995;64:769–776. doi: 10.1016/0306-4522(94)00397-n. [DOI] [PubMed] [Google Scholar]
- Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci U S A. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM, Liberator P, Iyer LM, Kash SF, Kohler MG, Kaczorowski GJ, MacIntyre DE, Martin WJ. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A. 2005;102:9382–9387. doi: 10.1073/pnas.0501549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roza C, Laird JM, Souslova V, Wood JN, Cervero F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J Physiol. 2003;550:921–926. doi: 10.1113/jphysiol.2003.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi CM, Ferreira G, Yang B, Gazula VR, Butler A, Wei A, Kaczmarek LK, Salkoff L. Opposite regulation of Slick and Slack K+ channels by neuromodulators. J Neurosci. 2006;26:5059–5068. doi: 10.1523/JNEUROSCI.3372-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Dubin AE, Petrus MJ, Earley TJ, Patapoutian A. Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron. 2009;64:498–509. doi: 10.1016/j.neuron.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnizler K, Shutov LP, Van Kanegan MJ, Merrill MA, Nichols B, McKnight GS, Strack S, Hell JW, Usachev YM. Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J Neurosci. 2008;28:4904–4917. doi: 10.1523/JNEUROSCI.0233-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XJ, Wang ZB, Gan Q, Walters ET. cAMP and cGMP contribute to sensory neuron hyperexcitability and hyperalgesia in rats with dorsal root ganglia compression. J Neurophysiol. 2006;95:479–492. doi: 10.1152/jn.00503.2005. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–135. doi: 10.1016/0306-4522(91)90255-m. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32:577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Heller PH, Levine JD. Mediation of serotonin hyperalgesia by the cAMP second messenger system. Neuroscience. 1992;48:479–483. doi: 10.1016/0306-4522(92)90507-x. [DOI] [PubMed] [Google Scholar]
- Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci. 2009;29:5127–5134. doi: 10.1523/JNEUROSCI.0859-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ZY, Donnelly DF, LaMotte RH. Effects of a chronic compression of the dorsal root ganglion on voltage-gated Na+ and K+ currents in cutaneous afferent neurons. J Neurophysiol. 2006;95:1115–1123. doi: 10.1152/jn.00830.2005. [DOI] [PubMed] [Google Scholar]
- Toman RE, Payne SG, Watterson KR, Maceyka M, Lee NH, Milstien S, Bigbee JW, Spiegel S. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J Cell Biol. 2004;166:381–392. doi: 10.1083/jcb.200402016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaragavan K, Boutjdir M, Chahine M. Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J Neurophysiol. 2004;91:1556–1569. doi: 10.1152/jn.00676.2003. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Pan HL. Role of TRPV1 and intracellular Ca2+ in excitation of cardiac sensory neurons by bradykinin. Am J Physiol Regul Integr Comp Physiol. 2007;293:R276–R283. doi: 10.1152/ajpregu.00094.2007. [DOI] [PubMed] [Google Scholar]
- Yang B, Gribkoff VK, Pan J, Damagnez V, Dworetzky SI, Boissard CG, Bhattacharjee A, Yan Y, Sigworth FJ, Kaczmarek LK. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology. 2006;51:896–906. doi: 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Ye S, Li Y, Chen L, Jiang Y. Crystal structures of a ligand-free MthK gating ring: insights into the ligand gating mechanism of K+ channels. Cell. 2006;126:1161–1173. doi: 10.1016/j.cell.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J-H, Walters ET, Song X-J. Dissociation of dorsal root ganglion neurons induces hyperexcitability that is maintained by increased responsiveness to cAMP and cGMP. J Neurophysiol. 2007;97:15–25. doi: 10.1152/jn.00559.2006. [DOI] [PubMed] [Google Scholar]