

Abstract
Alterations in cortical excitability are implicated in the pathophysiology of migraine. However, the relationship between cortical spreading depression (CSD) and headache has not been fully elucidated. We aimed to identify the corticofugal networks that directly influence meningeal nociception in the brainstem trigeminocervical complex (Sp5C) of the rat. Cortical areas projecting to the brainstem were first identified by retrograde tracing from Sp5C areas that receive direct meningeal inputs. Anterograde tracers were then injected into these cortical areas to determine the precise pattern of descending axonal terminal fields in the Sp5C. Descending cortical projections to brainstem areas innervated by the ophthalmic branch of the trigeminal nerve originate contralaterally from insular (Ins) and primary somatosensory (S1) cortices and terminate in laminae I–II and III–V of the Sp5C, respectively. In another set of experiments, electrophysiological recordings were simultaneously performed in Ins, S1 or primary visual cortex (V1), and Sp5C neurons. KCl was microinjected into such cortical areas to test the effects of CSD on meningeal nociception. CSD initiated in Ins and S1 induced facilitation and inhibition of meningeal-evoked responses, respectively. CSD triggered in V1 affects differently Ins and S1 cortices, enhancing or inhibiting meningeal-evoked responses of Sp5C, without affecting cutaneous-evoked nociceptive responses. Our data suggest that “top-down” influences from lateralized areas within Ins and S1 selectively affect interoceptive (meningeal) over exteroceptive (cutaneous) nociceptive inputs onto Sp5C. Such corticofugal influences could contribute to the development of migraine pain in terms of both topographic localization and pain tuning during an attack.
Introduction
Preclinical data suggest that migraine pain is initiated by the activation of meningeal perivascular nociceptors that in turn activate upper cervical and brainstem trigeminovascular neurons (Sp5C) (Strassman et al., 1996; Burstein et al., 1998). Among the variety of internal and external migraine triggers, endogenous, cortical dysexcitability has been proposed as a determinant factor on the susceptibility to migraine headache (Coppola et al., 2007), including the neurophysiological correlate of the preceding sensory aura, known as cortical spreading depression (CSD). CSD is characterized by a slowly propagating wave of neuronal and glial depolarization, followed by a sustained depression of cortical function (Leão, 1944; Smith et al., 2006).
A number of animal studies have suggested that CSD could be a key event in the cascade leading to local neurogenic inflammation and activation of the trigeminovascular system (Bolay et al., 2002; Pietrobon and Striessnig, 2003). In humans, a temporal correlation has been observed between changes in blood oxygenation level-dependent (BOLD) signal of the occipital cortex and the aura perception (Hadjikhani et al., 2001). Magnetoelectroencephalographic recordings have shown changes in the cortical magnetic field related to CSD-like waves in patients with spontaneous and visually triggered aura (Bowyer et al., 2001). Also, interictal electrophysiological abnormalities of the thalamocortical circuitry and thalamocortical dysrhythmia have been proposed at the origin of the cortical hyper-responsiveness observed in migraineurs (Coppola et al., 2007). Taking into account the altered cortical excitability commonly found in migraine sufferers (Pietrobon and Striessnig, 2003; Stankewitz and May, 2009), it is possible that different cortical areas might be involved in the triggering of auras and migraine pain. Also, their contribution to migraine attacks could depend on the degree and localization of altered cortical excitability phenotypes in genetically susceptible individuals. Although both migraine with (MA) and without (MO) aura may result in clinical manifestations of altered cortical excitability, the mechanisms underlying the activation of the trigeminovascular system by cortical dysfunction remain elusive and is still a matter of controversy (Lambert et al., 1999; Ebersberger et al., 2001; Goadsby, 2001; Charles and Brennan, 2009).
The overall aim of this study was to investigate the existence of a direct relationship between cortical excitability disturbances and the activities of brainstem trigeminovascular neurons involved in meningeal sensory processing. For this purpose, we studied the functional architecture of descending cortical projections that could directly modulate brainstem Sp5C neurons. Initially, we examined the topographic organization of corticotrigeminal projections with high-resolution retrograde tracing from Sp5C areas that receive direct meningeal inputs. Anterograde tracers were then injected into these cortical areas to determine the precise pattern of descending axonal terminal fields in the Sp5C. Additional series of experiments were designed to perform simultaneous extracellular recordings in cortical areas at the origin of corticotrigeminal afferents and Sp5C neurons. KCl was microinjected directly within the primary somatosensory (S1) or insular (Ins) cortex to investigate whether top-down influences elicited by CSD modulate meningeal- and/or cutaneous-evoked responses of Sp5C neurons. In a final experiment, descending modulation was evaluated by triggering CSD in the primary visual cortex (V1).
Materials and Methods
General.
Male Sprague Dawley rats weighing 250–350 g were used in this study. All animal experiments were approved by our local animal care committee and were in accordance with the guidelines of the International Association for the Study of Pain (Zimmermann, 1983).
Electrophoretic injections of fluorogold into Sp5C.
The rats were anesthetized with chloral hydrate (400 mg/kg, i.p.) and mounted in a stereotaxic device. The atlanto-occipital membrane and the overlying dura were removed. Glass micropipettes (40-μm-diameter tips) filled with a 2% solution of hydroxystilbamidine [fluorogold (FG); Interchim] were positioned at 2700–4000 μm caudal to the obex and inserted into the ventrolateral aspect of the Sp5C. Multiunitary recordings of cells responding to tactile and noxious pinch stimuli of the ophthalmic dermatome of the trigeminal nerve were performed. Once these cells were encountered, direct positive currents (6–10 μA, on–off cycles of 10 s) were applied for 15 min. After surgery, animals were kept in individual cages for 72 h, reanesthetized with an overdose of chloral hydrate (800 mg/kg, i.p.), and perfused with heparinized saline, followed by 500 ml of a PBS (0.2 m; pH 7.3), containing 4% paraformaldehyde and 0.05% picric acid. The brain and medulla were removed and soaked in the latter solution for 2 h and then cryoprotected in a buffered 30% sucrose solution.
Electrophoretic injections of biotin–dextran or tetramethyl-rhodamine dextran into S1 or Ins cortices.
The rats were anesthetized with chloral hydrate (400 mg/kg, i.p.) and mounted in a stereotaxic device, and a craniotomy was performed. Glass micropipettes (40-μm-diameter tips) filled with a 10% solution of biotin–dextran (BD) (10,000 molecular weight; Invitrogen) or 10% of tetramethyl-rhodamine dextran (TMR) (3000 molecular weight; Invitrogen) were inserted into the S1 or Ins cortical regions. Once the micropipette was in place, direct positive current (2–4 μA) was applied for 20 s every 30 s for 15–20 min. After a postoperative survival period of 1 week, the animals were re anesthetized and perfused as described previously (Noseda et al., 2008).
Histological processing.
Coronal sections (40 μm thick) were rinsed with PBS preincubated for 1 h in a blocking solution of PBS containing 0.3% Triton X-100, 0.3% bovine serum albumin (PBT) and 2% normal goat serum (NGS). They were then incubated overnight in the primary antibody, namely 1:5000 rabbit anti-fluorogold (Millipore Bioscience Research Reagents) in PBT and 2% NGS. After 20 min rinsing with PBS, the sections were incubated for l h in the secondary antiserum, namely l:200 biotinylated goat anti-rabbit IgG (Interchim) in PBT and 2% NGS. After rinsing, the sections were processed for ABC/DAB (Vector Laboratories) to localize the retrograde labeling in different brain structures. For BD injections, 40-μm-thick coronal sections were processed for ABC/DAB histochemistry to localize the anterograde labeling in different brain structures; the brain tissue was counterstained with cresyl violet.
Analysis of the retrograde and anterograde labeling.
In all tract-tracing experiments, the injection sites, labeled cell bodies, fibers, varicosities, and boutons were examined under bright- and dark-field illumination. Each injection site was analyzed using camera lucida drawings of coronal sections processed with DAB. Representative cases were grouped on standard drawings of the caudal medulla and cortical areas (see Figs. 1C, 2A,D). Bright-field photomicrographs of the injection sites and representative labeling were built as digitized grayscale images using computer-assisted reconstruction of labeling.
Figure 1.

Retrograde labeling of trigeminally projecting cortical neurons. A, Extracellular recordings of a dura-sensitive neuron (top) with periocular receptive field (bottom) before an FG injection into the trigeminal nucleus caudalis (B; Sp5C/C1). Camera lucida drawings of coronal sections, illustrating the location and extent of all FG injection sites (C) and retrogradely labeled cells in the cortex (D; dots) (adapted from Paxinos and Watson, 1998). Note that the majority of labeled cells were confined to the contralateral S1, caudal Ins, and, to a lesser extent, the S2, covering from 3.1 to 4.1 mm caudal to bregma. Scale bar, 100 μm. Cu, Cuneate nucleus; Gr, gracile nucleus; CC, corpus callosum; ec, external capsule; pyx, pyramidal decussation; PtA, parietal association cortex; Au, auditory cortex.
Figure 2.
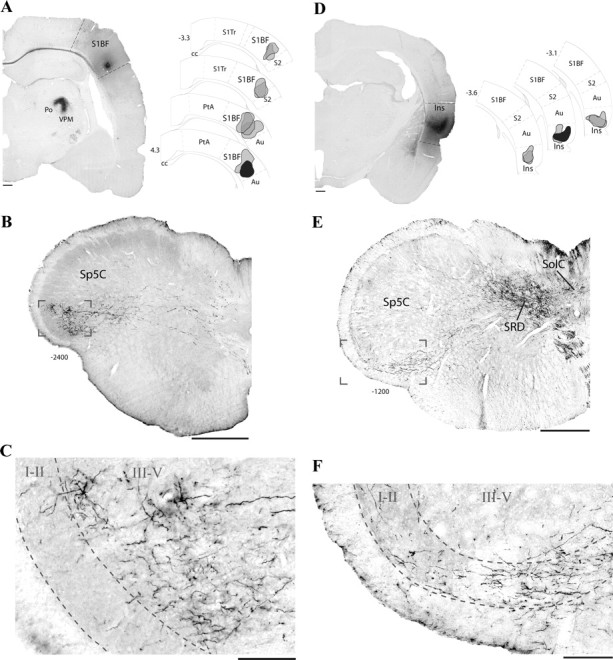
Axonal terminations of trigeminally projecting cortical neurons. A, Coronal view of a BD injection site in the primary somatosensory cortex (S1BF; left) and its schematic representation (selected site in black; right) among other S1 injection sites (gray). B, C, Bright-field images of labeling in a coronal section of the caudal medulla. Note the high density of labeled axons in the pyramidal tract and terminal labeling in the deep layers (III–V) of the contralateral Sp5C. D, Injection site of BD in caudal Ins and selected sites (right). E, F, Bright-field images of labeling in a coronal section of the caudal medulla. Note the high density of labeled axons in the pyramidal tract and terminal labeling exclusively in the superficial layers of the Sp5C (I–II). Scale bars: A, D, 500 μm; B, C, E, F, 100 μm. S1Tr, Primary somatosensory cortex, trunk region; cc, corpus callosum; Au, auditory cortex; PtA, parietal association cortex; SRD, subnucleus reticularis dorsalis; Sol, nucleus of the solitary tract. Numbers indicate distance from bregma.
Unitary and multiunitary extracellular recordings.
In rats anesthetized with urethane (1.5 g/kg), multiunit recordings were performed in layers V–VI of the S1 or Ins cortices, using low-impedance (400 kΩ to 1 MΩ) glass micropipette electrodes. Extracellular single units were simultaneously recorded in the Sp5C using quartz-insulated, platinum–tungsten microelectrodes (4–6 MΩ). In the latter case, a five-channel Eckhorn mini-matrix microdrive system (Thomas Recordings) was used. Microelectrode signals were amplified, filtered, digitized, and discriminated with a personal computer-controlled multichannel neuronal acquisition processor (Plexon) as described previously (Monconduit et al., 2006). Baseline cortical activity and evoked trigeminovascular neuronal responses were obtained and analyzed as cumulative frequency and poststimulus histograms (PSTHs), respectively, to detect the occurrence of statistically significant neuronal responses.
Intracortical administration of KCl or artificial CSF.
We tested the effects of 1 μl, 1 m KCl or 1 μl of artificial CSF (aCSF) microinjections within layers V–VI of the S1, Ins, or V1 on baseline cortical activity and evoked trigeminovascular neuronal responses. KCl or aCSF was injected at a rate of 500 nl/min using a Hamilton syringe (170 μm tip) driven by an electronic-geared syringe pump (Micro4; WPI). The distance between the microinjections and the cortical recordings was ±100 μm. At the conclusion of the experiments, the recording and microinjection sites were marked by deposition of pontamine sky blue.
Experimental design: innocuous and noxious stimulation.
Dura-sensitive Sp5C neurons responding to electrical stimulation of the left transverse sinus (1 Hz; 0.8 ms duration; 1 mA) and innocuous (brushing at 2 Hz frequency for 30 s) or noxious (electrical; 1 Hz; 2 ms duration; twofold C-fiber threshold) stimulation on the periocular dermatome were studied. Three sets of control responses of periocular brushing, followed by meningeal electrical stimuli (1 Hz for 30 s; twofold C-fiber threshold intensity), were obtained before cortical microinjections within 10 min interval. Postinjection tests were performed at different intervals, as shown in Figure 4.
Figure 4.
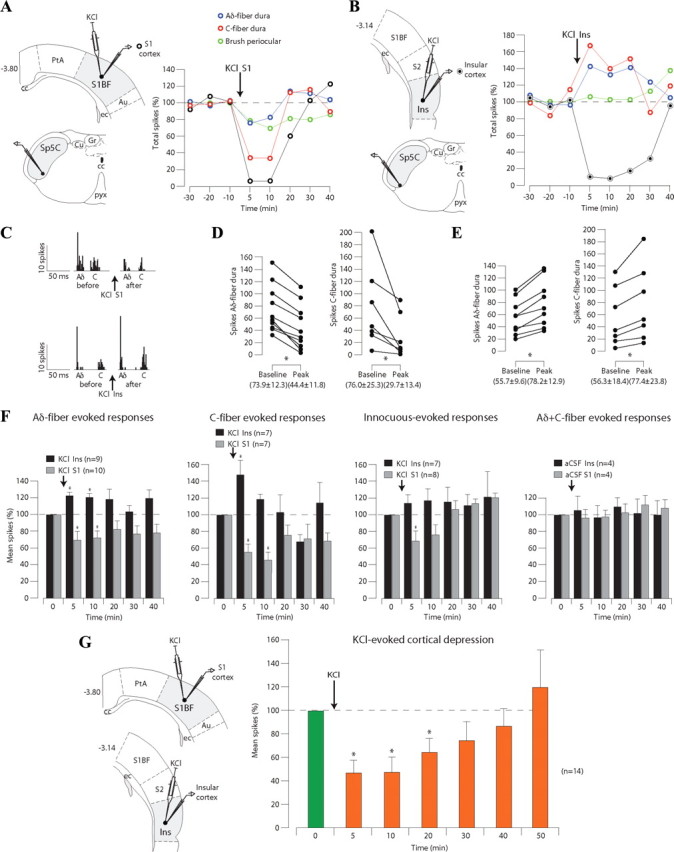
CSD initiated in S1 or Ins cortex differentially modulates the responses of trigeminovascular neurons simultaneously recorded in the Sp5C. A, Example of the effects of a KCl microinjection (1 μl, 1 m) into layer V–VI of S1 cortex on the responses of a neuron in the Sp5C. Note that cortical activity was depressed by the microinjection (black), whereas both cutaneous innocuous-evoked (green) and meningeal nociceptive-evoked (red and blue) responses were depressed. B, Example of the effects of a microinjection of KCl (1 μl, 1 m) into layer V–VI of the Ins cortex on the responses of a neuron in the Sp5C. Cortical activity was depressed by the microinjection (black), whereas meningeal nociceptive-evoked responses (red and blue) were enhanced in the Sp5C. C, Examples of poststimulus histograms showing the effects of KCl into S1 or Ins on Aδ- and C-fiber meningeal responses. D, E, Peak reduction or facilitation of Aδ- and C-fiber meningeal-evoked responses of each Sp5C neuron during depression of S1 or Ins layer V–VI neurons, respectively. F, Cumulative results of the effects of cortical manipulation with KCl or aCSF on the nociceptive- and innocuous-evoked responses of Sp5C neurons. G, Summary of the depressant effects of microinjections of KCl into layers V–VI of both S1 and Ins cortices. Results are expressed as mean ± SEM of the percentage changes with regard to the mean values obtained before the cortical manipulation (*p < 0.05). For abbreviations, see Figure 1.
To obtain a good temporal correlation between cortical spreading depression and trigeminal evoked activities, we designed an additional experimental protocol, as follows: (1) a set of control measurement consisting of 10 min trains of electrical stimulation applied either on the meningeal or cutaneous (stimulation as above) receptive fields; (2) 5 min resting period; (3) a set of 15 min trains of electrical stimulation. During this set, at the end of the third minute of stimulation, KCl was injected into V1 cortex (500 nl/min). Aδ- and C-fiber-evoked activities were measured within 3 min periods after KCl, at different timeframes, as shown in Figure 5.
Figure 5.
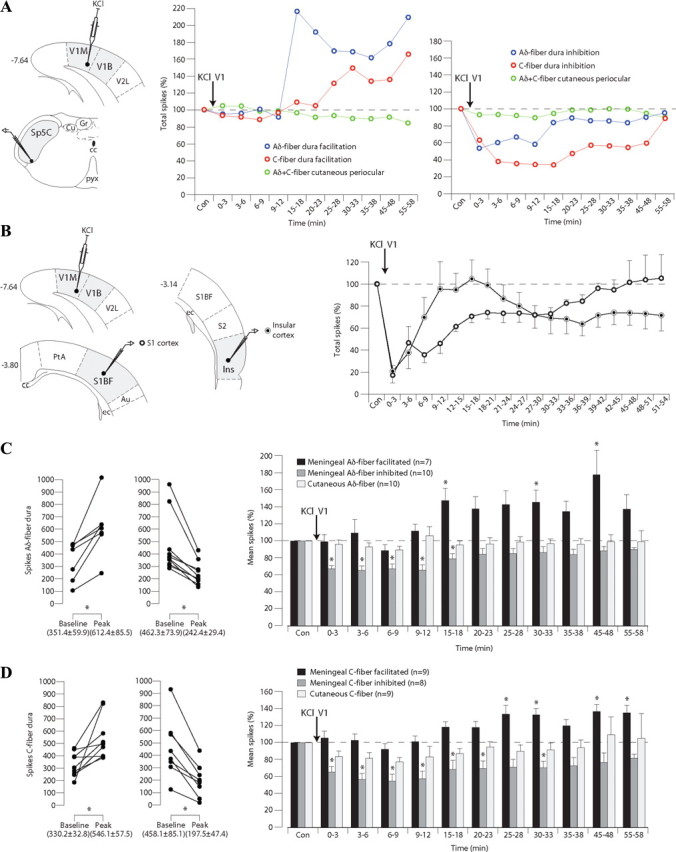
CSD initiated in V1 cortex induces delayed facilitation or immediate inhibition of meningeal-evoked responses of trigeminovascular neurons in the Sp5C. A, Example of the effects of a microinjection of KCl (1 μl, 1 m) into the primary visual cortex (V1M, monocular; V1B, binocular) on the responses of neurons simultaneously recorded in the Sp5C. Note that meningeal-evoked nociceptive responses (red and blue) were either enhanced or reduced in the Sp5C, whereas cutaneous-evoked responses (green) were unaffected. B, Example of the effects of a microinjection of KCl (1 μl, 1 m) into V1 cortex on the activities of neurons simultaneously recorded in layer V–VI of S1 or Ins cortices. C, Peak facilitation or depression of meningeal Aδ-fiber-evoked responses of each Sp5C neuron recorded during KCl injections into V1 (left) and histograms summarizing such effects (right). D, Peak facilitation or depression of meningeal C-fiber-evoked responses of each Sp5C neuron recorded during KCl injections into V1 and summary of such effects. Results are expressed as mean ± SEM of the percentage changes with regard to control responses recorded before the microinjection (*p < 0.05). Cu, Cuneate nucleus; Gr, gracile nucleus; V2L, secondary visual cortex, lateral area; cc, corpus callosum; pyx, pyramidal decussation; PtA, parietal association cortex; S1BF, primary somatosensory cortex, barrel field; ec, external capsule; Au, auditory cortex.
PSTH analysis of noxious, meningeal, or cutaneous electrical stimulation was expressed as the total number of spikes triggered by trains of single electrical pulses (between 0–30 and 30–100 ms timeframe windows for Aδ- and C-fibers, respectively). Innocuous, cutaneous-evoked neuronal discharges were expressed in hertz.
Statistical analysis.
One dura-sensitive neuron was studied per animal. Cumulative results were expressed as mean ± SEM of the percentage changes with regard to the mean values obtained before the cortical manipulation. Analyses were performed using ANOVA for repeated measures with Bonferroni's post hoc correction for multiples comparisons. Wilcoxon's matched-pairs signed-ranks test was also performed to determine significance between control and peak responses. The level of significance was set at p < 0.05.
Results
Topographic organization of corticotrigeminal pathways
With the aim of identifying cortical regions projecting to brainstem areas that contain trigeminovascular neurons, iontophoretic injections of the retrograde tracer FG were performed under electrophysiological control into the functionally characterized area of the Sp5C (Fig. 1A,B). Representative sites were selected and further analyzed on the basis of their small sizes and locations (Fig. 1C). As shown in previous studies (Burstein et al., 1998; Noseda et al., 2008), neurons driven by meningeal and cutaneous periocular inputs are mainly located in the ventral Sp5C and upper cervical segments of the spinal cord (C1/C2), including its lateralmost aspect (laminae I–II) and spreading medially to the deep layers (lamina V). Injections of FG into the area of Sp5C/C1 produced dense retrograde labeling of a band of pyramidal cells located in layer V of the cortex, contralateral to the injection sites (Fig. 1D). These clusters of corticotrigeminal cells occupied the ventral half of the caudal S1 and more rostrally the subjacent secondary somatosensory cortex (S2) and ventral part of the caudal Ins. Labeled cells in S1 and S2/Ins were observed in an area extending between 3.3 and 4.3 mm and between 3.1 and 3.8 mm caudal to bregma, respectively.
To determine precisely the organization and distribution of cortical afferents in Sp5C/C1, anterograde tracers BD or TMR were injected into the cortical areas retrogradely labeled from Sp5C. Based on their restricted sizes and locations, representative cases in S1 and Ins were selected and further analyzed by comparing the pattern of labeling in the caudal medulla.
Injections of BD into S1 (Fig. 2A) resulted in dense labeling of fibers in the pyramidal tract that crossed the midline and terminated in the contralateral caudal medulla (Fig. 2B). Axonal terminal labeling consisting of thin fibers and small varicosities was restricted to the ventrolateral Sp5C throughout its caudalmost aspect, including C1/C2. Terminal labeling was also observed rostrally in the ventral part of the contralateral trigeminal nuclei interpolaris, oralis, and principalis. A detailed view shows that terminal labeling was confined to the deeper laminae (III–V) of the Sp5C (Fig. 2C) in the projection area of the ophthalmic branch of the trigeminal nerve. In addition, dense labeling was observed in the dorsal aspect of the ipsilateral ventral posteromedial (VPM) and posterior (Po) thalamic nuclei (Fig. 2A).
Injections of BD into the caudal insula (Fig. 2D) resulted in a different pattern of labeling in the contralateral Sp5C. Descending fibers from Ins were observed in the pyramidal tract, then crossed the midline, and terminated in the subnucleus reticularis dorsalis, the nucleus of the solitary tract, and Sp5C (Fig. 2E). Sp5C labeling was confined to the projection area of the ophthalmic branch of the trigeminal nerve throughout its caudalmost aspect, including C1/C2. Terminal labeling was also observed more rostrally in the ventral part of the contralateral, caudal trigeminal nuclei interpolaris and oralis. A detailed view shows that terminal labeling within the Sp5C exclusively reach superficial laminae I–II (Fig. 2F). In addition, dense labeling was observed in the caudal part of the ipsilateral VPM, Po, ventral posterior parvicellular, and posterior triangular thalamic nuclei (data not shown).
Corticofugal modulation of Sp5C meningeal-evoked responses is region specific
To study the effects of corticofugal modulation on Sp5C responses to meningeal and cutaneous stimuli, we performed simultaneous single-unit extracellular recordings of Sp5C neurons and multiunit recordings from layers V–VI of S1 or Ins cortex. The effects of KCl-induced cortical depression on Sp5C neuronal activity were tested in 47 rats during innocuous, periocular brushing and noxious, electrical stimulation of the dura, before and after KCl microinjections within S1 or Ins (Fig. 3) (for more details, see Materials and Methods).
Figure 3.

A, Experimental setup for simultaneous electrophysiological recordings, cortical microinjections, and receptive fields stimulations. B, Summary of the histological findings of KCl (black dots) or aCSF (white dots) microinjections in primary visual cortex (V1M, monocular; V1B, binocular), S1, or caudal Ins. S1Tr, Primary somatosensory cortex, trunk region; S1BF, primary somatosensory cortex, barrel field; cc, corpus callosum; ec, external capsule; Au, auditory cortex; PtA, parietal association cortex; V2L, secondary visual cortex, lateral area. Numbers indicate distance from bregma.
In agreement with previous studies (Burstein et al., 1998; Noseda et al., 2008), neurons driven by meningeal electrical stimulation were located in the deep, medial aspect of the ventrolateral medulla, including the caudal aspect of Sp5C and C1/C2. These cells presented a mean ± SEM background activity of 0.3 ± 0.1 Hz and were driven by both electrical stimulation of the dura and periocular brushing (Fig. 1A). Supramaximal electrical stimulation of the dura (mean intensity of 1.1 ± 0.3 mA) produced both an early and late peak of activation. The cumulative results revealed that, for the onset of the earlier and later peaks of activation, the latencies were 5.3 ± 0.3 and 45.4 ± 3.0 ms, respectively. According to the literature (Levy and Strassman, 2002) and taking into account the distance between the stimulated area and the recording electrode, such latencies correspond to peripheral conduction velocities within the Aδ-fiber (5.7 m/s) and C-fiber (0.7 m/s) ranges, respectively. Repetitive, periocular brushing produced a mean tonic activation of 20.3 ± 3.5 Hz.
As shown in the individual example (Fig. 4A), a microinjection of KCl (1 μl; 1 m) within S1 elicited a reduction in cortical activity in the deep layers of S1 that lasted for ∼25 min after the injection (see also multiunit recording in Fig. S3A, available at www.jneurosci.org as supplemental material). Both periocular brushing and dura-evoked responses of the simultaneously recorded Sp5C neuron were strongly depressed for ∼10 min after the cortical microinjection. The reduction of meningeal Aδ- and C-fiber-evoked responses was observed only during the depression of S1 neurons elicited by KCl. An example of PSTH analysis obtained from responses to 30 electrical stimuli of the dura before and after KCl into S1 is shown in Figure 4C (top). Peak responses obtained with the same stimulation paradigm are shown in Figure 4D. Cumulative results showed that meningeal Aδ- and C-fiber-evoked responses were significantly depressed to 69.8 ± 10.6 and 55.7 ± 10.8% of the control values, respectively, 5 min after KCl; the corresponding values 10 min after KCl were 72.3 ± 8.5 and 46.3 ± 10.6% (Fig. 4F). Innocuous-evoked responses were significantly depressed to 68.7 ± 13.2% of the control values, only 5 min after KCl microinjections (Fig. 4F).
In another set of experiments, we tested the effects of microinjections of KCl within the caudal insula. As shown in the individual example (Fig. 4B), there was a KCl-induced reduction of cortical activity in the deep layers of Ins, with a concomitant enhancement of meningeal Aδ- and C-fiber-evoked responses from the Sp5C neuron. The enhancement of meningeal-evoked responses was only observed during the depression of Ins ongoing activity elicited by KCl and affected all the neurons recorded in the Sp5C (Fig. 4E). An example of PSTH analysis of responses to electrical stimulation of the dura before and after KCl into Ins is shown in Figure 4C (bottom). Cumulative results showed that meningeal Aδ- and C-fiber-evoked responses were significantly enhanced to 122.7 ± 4.5 and 147.8 ± 18.1% of the control values, respectively, 5 min after KCl; the corresponding values 10 min after KCl were 120.6 ± 5.1 and 118.6 ± 6.2% (Fig. 4F). Innocuous-evoked responses were not significantly affected by KCl microinjections at any of the measured time points (Fig. 4F).
Cumulative data obtained from aCSF (1 μl) microinjections within S1 and Ins showed no changes in both cortical ongoing activity and Aδ- and C-fiber meningeal-evoked responses (Fig. 4F). Moreover, blood pressure monitoring during and after electrical stimulation of the dura showed no changes in mean arterial blood pressure compared with control values (105.9 ± 3.3 mmHg). Also, no significant changes in blood pressure control values were observed after any of the cortical microinjections (99.6 ± 3.8, 101.4 ± 1.8% and 98.2 ± 1.6, 98.2 ± 3.2% at 5 and 15 min after S1 and Ins KCl microinjections, respectively).
Ongoing multiunitary activity of both S1 and Ins areas at the origin of corticotrigeminal pathways that innervate Sp5C showed a significant depression during the initial 20 min period that followed the microinjections of KCl (Fig. 4G). Cumulative data collected at 5, 10, 20, 30, 40, and 50 min after KCl were 47.5 ± 10.6, 48.9 ± 12.3, 64.6 ± 12.0, 74.8 ± 16.4, 86.9 ± 14.5, and 120.2 ± 32.2%, respectively, compared with control values.
CSD triggered in V1 differentially affects trigeminal-projecting cortical areas and Sp5C-evoked responses
The last experiments were designed to investigate the temporal pattern of the effects induced by CSD in V1 over trigeminal-projecting cortical areas and Sp5C neurons. Electrophysiological recordings from layers V–VI of S1 or Ins and Sp5C neurons were simultaneously performed during meningeal or percutaneous periocular nociceptive electrical stimulation before and after KCl microinjections within V1 (Fig. 3) (see Materials and Methods).
As shown in two individual examples (Fig. 5A), a microinjection of KCl within V1 elicited either a delayed facilitation or an early inhibition of dura-evoked responses of Sp5C neurons, without affecting cutaneous-evoked responses. Cumulative results obtained from cortical recordings showed that KCl within V1 elicited an early reduction of ongoing activities in the deep layers of both S1 and Ins (Fig. 5B). S1 activities returned progressively to control levels in a timeframe of ∼35 min. Ins activities recovered ∼10 min after KCl in V1, and 10 min later such activities were again depressed and did not recover within the 60 min recording period. CSD spread ipsilaterally, as shown by a lack of changes in activities within layers V–VI of S1 and Ins areas recorded in the opposite side of KCl injection. Cumulative results showed that, in approximately half of the Sp5C neurons recorded, meningeal Aδ- and C-fiber-evoked responses undergo a delayed facilitation within a timeframe starting at ∼15 min that persisted during the whole 60 min recording period (Fig. 5C,D). In contrast, reduction of meningeal-evoked responses occurred immediately after the KCl microinjection and recovered progressively within a timeframe of ∼30 min. In both populations, cutaneous Aδ- and C-fiber-evoked responses were unaffected (Fig. 5C,D).
Discussion
The present study provides precise information about the somatotopic organization of corticotrigeminal projections that are anatomically positioned to modulate meningeal nociceptive processing. We propose that cortical dysexcitability and/or CSD-related phenomena selectively affect interoceptive (meningeal) over exteroceptive (cutaneous) nociceptive inputs onto Sp5C neurons via corticofugal “top-down” influences.
Corticotrigeminal projections to brainstem areas of trigeminovascular processing
We have described dense cortical projections to confined zones of the Sp5C innervated by the ophthalmic branch of the trigeminal nerve. These projections originate unilaterally from pyramidal cells of layer V of areas within S1, S2, and caudal insula. In accordance with other studies (Wise and Jones, 1977; Desbois et al., 1999), descending axons from S1 and Ins terminate in deep (III–V) and superficial (I–IIo) laminae of Sp5C, respectively, as confirmed by their relationship with 5-HT1D/calcitonin gene-related peptide (CGRP) primary afferents (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Peripheral and central targets could account for the efficacy of CGRP antagonists in the acute treatment of migraine (Ho et al., 2008). In this respect, trigeminally projecting neurons that closely appose CGRP fibers in the insula (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material) represent a potential pharmacological target for CGRP antagonists.
Corticotrigeminal projections are topographically organized in a caudorostral and dorsoventral manner (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). This would be in agreement with a general, somatotopic and reciprocal organization of corticofugal modulation, in that there is a direct relationship between afferent inputs to and efferent outflow from specific groups of cortical cells (Deschênes et al., 1998). This is also supported by our findings showing that the same S1 and insular areas projecting to the ventral Sp5C also innervate the thalamic recipient zones for ascending ophthalmic branch projections (Noseda et al., 2008). Accordingly, our c-fos studies also confirmed a precise somatotopic organization of Sp5C neurons responding to noxious stimulation of the three branches of the trigeminal nerve (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material).
Corticofugal influences on brainstem trigeminovascular neurons: a link for top-down processing of migraine headache
The gradual spread of migraine aura, as shown by its temporal correlation with BOLD signal and electrophysiological studies in humans, strongly suggest that it is caused by CSD (Bowyer et al., 2001; Hadjikhani et al., 2001). We investigated the effects of KCl microinjections because it constitutes a reliable stimulus inducing the well characterized phenomenon of CSD in animals, which transiently interrupts cortical function in the intact brain (Leão, 1944; Smith et al., 2006) (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). According to the pioneering work of Leão (1944), KCl in V1 initiates a propagating unilateral wave of CSD that differentially interrupted cortical function in S1 and Ins, eliciting bidirectional effects on meningeal-evoked responses in Sp5C neurons. In contrast, cutaneous-evoked responses were unaffected, suggesting that CSD phenomenon selectively affects meningeal over cutaneous nociceptive inputs onto Sp5C neurons. Such effects appeared within a large timeframe, probably depending on the variability in magnitude, diffusion, and duration of cortical inactivation that depend on the density of cellular elements and thus on the site of CSD initiation, with longer depression in superficial, as well as limited diffusion to deep cortical layers (Richter and Lehmenkuhler, 1993). The differential spreading of CSD from V1 could account for the early depression and delayed enhancement of meningeal-evoked responses in Sp5C neuronal subpopulations. Interestingly, the delayed enhancement coincides with the second phase of insular depression. This would be in agreement with the recently described biphasic features of CSD (Chang et al., 2010).
Transient cortical inactivation after KCl microinjections within S1 or Ins was fundamental to determine differential top-down modulatory influences on meningeal inputs conveyed by Sp5C neurons that were observed within a narrower timeframe. These findings raise the possibility that specific tuning mechanisms acting on meningeal nociception, namely S1-mediated inhibition or Ins-mediated facilitation, are probably conveyed via direct corticotrigeminal outflow. The depression of both cutaneous- and meningeal-evoked responses of Sp5C neurons could be sustained by S1 projections to deep laminae, which contain neurons activated by both large- and fine-diameter fiber inputs (Burstein et al., 1998). In contrast, the facilitatory influences on Aδ- and C-fiber meningeal-evoked responses could be mediated by Ins axons innervating the superficial layers of Sp5C, suggesting a selective modulation of nociceptive primary afferents. Nevertheless, insular facilitation was variable and weak, probably because disinhibition of trigeminovascular neurons caused by Ins blockade could be cancelled by a fast spread of CSD to S1, because superficial cortical regions are more sensitive to CSD than Ins (see above) (Richter and Lehmenkuhler, 1993). Such a possibility is also supported by the observation that, after a concomitant depression of Ins and a return to baseline activity of S1 neurons, a more sustained enhancement of meningeal-evoked activities of Sp5C neurons was observed (Fig. 5). In this respect, impaired inhibition as a consequence of aura has been proposed as a major underlying phenomenon of cortical excitability changes in migraine patients (Coppola et al., 2007; Stankewitz and May, 2009). Human studies have implicated the insula as part of the interoceptive system involved in the conscious perception of the physiological state of the body (Craig, 2002; Critchley et al., 2004; Khalsa et al., 2009), which could fit with the implication of this region in headaches that have the features of visceral pain (Moskowitz, 1991). In this respect, electrical stimulation of restricted areas of the human insular cortex can elicit a highly unpleasant headache (Afif et al., 2008).
Previous electrophysiological studies had shown a lack of effect of CSD on Sp5C meningeal activities (Lambert et al., 1999; Ebersberger et al., 2001). It is possible that the experimental setup used in these studies did not allow the detection of effects, because it has been reported recently that a subset of meningeal nociceptors become immediately activated or within a short delay after CSD (Zhang et al., 2010). Moreover, it has been proposed that plasma extravasation and the subsequent activation of meningeal nociceptors by CSD is the trigger event of migraine pain (Bolay et al., 2002). Thus, corticotrigeminal influences on Sp5C neurons could then act as modulators, once the triggering of migraine pain was produced by the direct activation of meningeal nociceptors. In accordance with this view, the perception of visual aura is predominantly contralateral to the pain, as expected if CSD activates only the trigeminovascular system through adjacent pial nociceptors. From the clinical point of view, however, epidemiological studies have shown that 40% of migraine patients also experience bilateral headaches (Rasmussen et al., 1991) and that the headache location appears to have little specificity or diagnostic utility (Bruehl et al., 1999). Other clinical studies have documented ipsilateral visual aura and headache in some migraine patients who experience aura, simultaneous aura at the onset of pain, or even aura without pain (Pietrobon and Striessnig, 2003 and references therein). These observations, in congruence with the fact that most migraine patients do not experience aura, appear to challenge the idea of strict unilateral CSD-like events at the origin of migraine pain (Goadsby, 2001). It is tempting to suggest that corticotrigeminal dysfunction attributable to altered cortical excitability phenotypes in genetically susceptible individuals could contribute to the development of migraine pain in terms of both topographic localization and pain tuning during an attack.
Potentially, transcallosal influences (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material) in addition to our proposed corticotrigeminal mechanism may occur in parallel with indirect (e.g., brainstem) descending modulation (Lambert et al., 2008), but it seems unlikely that periaqueductal gray or rostral ventromedial medulla regions participate in specific migraine processing because descending modulatory pathways from these brainstem areas are not somatotopically organized and influence multiple areas in the spinal and trigeminal dorsal horns (Villanueva and Fields, 2004). In contrast, focal dysfunction originated in direct, strictly topographically organized, descending corticotrigeminal pathways could contribute to provide a specific profile in the pathophysiology of this disorder.
Footnotes
This work was supported by Inserm, Evaluation-Coopération Scientifique France-Chili Programme C08 S03, European Union Programme Alban Scholarship E04D027959CL (R.N.), and Proyecto Fondo Nacional de Desarrollo Científico y Tecnológico Grant 1050099 (L.C.). We are grateful to Drs. S. W. Cadden, R. Burstein, and M. Hamon for advice in the preparation of this manuscript. We thank L. Monconduit and T. Mariqueo for their technical contribution during the early phase of this work and A. H. Ahn and A. I. Basbaum (University of California, San Francisco, San Francisco, CA) for the generous gift of the 5HT1D antibody.
References
- Afif A, Hoffmann D, Minotti L, Benabid AL, Kahane P. Middle short gyrus of the insula implicated in pain processing. Pain. 2008;138:546–555. doi: 10.1016/j.pain.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- Bowyer SM, Aurora KS, Moran JE, Tepley N, Welch KM. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol. 2001;50:582–587. doi: 10.1002/ana.1293. [DOI] [PubMed] [Google Scholar]
- Bruehl S, Lofland KR, Semenchuk EM, Rokicki LA, Penzien DB. Use of cluster analysis to validate IHS diagnostic criteria for migraine and tension-type headache. Headache. 1999;39:181–189. doi: 10.1046/j.1526-4610.1999.3903181.x. [DOI] [PubMed] [Google Scholar]
- Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964–982. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, Brennan KC. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain. 2010;133:996–1012. doi: 10.1093/brain/awp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A, Brennan K. Cortical spreading depression-new insights and persistent questions. Cephalalgia. 2009;29:1115–1124. doi: 10.1111/j.1468-2982.2009.01983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Pierelli F, Schoenen J. Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia. 2007;27:1427–1439. doi: 10.1111/j.1468-2982.2007.01500.x. [DOI] [PubMed] [Google Scholar]
- Craig AD. How do you feel? Interoception: the sense of the physiological condition of the body. Nat Rev Neurosci. 2002;3:655–666. doi: 10.1038/nrn894. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Wiens S, Rotshtein P, Ohman A, Dolan RJ. Neural systems supporting interoceptive awareness. Nat Neurosci. 2004;7:189–195. doi: 10.1038/nn1176. [DOI] [PubMed] [Google Scholar]
- Desbois C, Le Bars D, Villanueva L. Organization of cortical projections to the medullary subnucleus reticularis dorsalis: a retrograde and anterograde tracing study in the rat. J Comp Neurol. 1999;410:178–196. [PubMed] [Google Scholar]
- Deschênes M, Veinante P, Zhang ZW. The organization of corticothalamic projections: reciprocity versus parity. Brain Res Brain Res Rev. 1998;28:286–308. doi: 10.1016/s0165-0173(98)00017-4. [DOI] [PubMed] [Google Scholar]
- Ebersberger A, Schaible HG, Averbeck B, Richter F. Is there a correlation between spreading depression, neurogenic inflammation, and nociception that might cause migraine headache? Ann Neurol. 2001;49:7–13. [PubMed] [Google Scholar]
- Goadsby PJ. Migraine, aura, and cortical spreading depression: why are we still talking about it? Ann Neurol. 2001;49:4–6. doi: 10.1002/1531-8249(200101)49:1<4::aid-ana3>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TW, Ferrari MD, Dodick DW, Galet V, Kost J, Fan X, Leibensperger H, Froman S, Assaid C, Lines C, Koppen H, Winner PK. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet. 2008;372:2115–2123. doi: 10.1016/S0140-6736(08)61626-8. [DOI] [PubMed] [Google Scholar]
- Khalsa SS, Rudrauf D, Feinstein JS, Tranel D. The pathways for interoceptive awareness. Nat Neurosci. 2009;12:1494–1496. doi: 10.1038/nn.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GA, Michalicek J, Storer RJ, Zagami AS. Effect of cortical spreading depression on activity of trigeminovascular sensory neurons. Cephalalgia. 1999;19:631–638. doi: 10.1046/j.1468-2982.1999.019007631.x. [DOI] [PubMed] [Google Scholar]
- Lambert GA, Hoskin KL, Zagami AS. Cortico-NRM influences on trigeminal neuronal sensation. Cephalalgia. 2008;28:640–652. doi: 10.1111/j.1468-2982.2008.01572.x. [DOI] [PubMed] [Google Scholar]
- Leão A. Spreading depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Levy D, Strassman AM. Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol. 2002;88:3021–3031. doi: 10.1152/jn.00029.2002. [DOI] [PubMed] [Google Scholar]
- Monconduit L, Lopez-Avila A, Molat JL, Chalus M, Villanueva L. Corticofugal output from the primary somatosensory cortex selectively modulates innocuous and noxious inputs in the rat spinothalamic system. J Neurosci. 2006;26:8441–8450. doi: 10.1523/JNEUROSCI.1293-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA. The visceral organ brain: implications for the pathophysiology of vascular head pain. Neurology. 1991;41:182–186. doi: 10.1212/wnl.41.2_part_1.182. [DOI] [PubMed] [Google Scholar]
- Noseda R, Monconduit L, Constandil L, Chalus M, Villanueva L. Central nervous system networks involved in the processing of meningeal and cutaneous inputs from the ophthalmic branch of the trigeminal nerve in the rat. Cephalalgia. 2008;28:813–824. doi: 10.1111/j.1468-2982.2008.01588.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386–398. doi: 10.1038/nrn1102. [DOI] [PubMed] [Google Scholar]
- Rasmussen BK, Jensen R, Olesen J. A population-based analysis of the diagnostic criteria of the international headache society. Cephalalgia. 1991;11:129–134. doi: 10.1046/j.1468-2982.1991.1103129.x. [DOI] [PubMed] [Google Scholar]
- Richter F, Lehmenkühler A. Spreading depression can be restricted to distinct depths of the rat cerebral cortex. Neurosci Lett. 1993;152:65–68. doi: 10.1016/0304-3940(93)90484-3. [DOI] [PubMed] [Google Scholar]
- Smith JM, Bradley DP, James MF, Huang CL. Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc. 2006;81:457–481. doi: 10.1017/S1464793106007081. [DOI] [PubMed] [Google Scholar]
- Stankewitz A, May A. The phenomenon of changes in cortical excitability in migraine is not migraine specific: a unifying thesis. Pain. 2009;145:14–17. doi: 10.1016/j.pain.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- Villanueva L, Fields H. Endogenous central mechanisms of pain modulation. In: Villanueva L, Dickenson AH, Ollat H, editors. The pain system in normal and pathological states: a primer for clinicians. Seattle: International Association for the Study of Pain; 2004. pp. 223–243. [Google Scholar]
- Wise SP, Jones EG. Cells of origin and terminal distribution of descending projections of the rat somatic sensory cortex. J Comp Neurol. 1977;175:129–157. doi: 10.1002/cne.901750202. [DOI] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications to migraine with aura. J Neurosci. 2010;30:8807–8814. doi: 10.1523/JNEUROSCI.0511-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]