

Abstract
YAC transgenic mice expressing poly(Q)-expanded full-length huntingtin (mhtt) recapitulate many behavioral and neuropathological features of Huntington disease (HD). We have previously observed a reduction in phosphorylation of mhtt at S421 in the presence of the mutation for HD. In addition, phosphorylation of normal S421-htt is reduced after excitotoxic stimulation of NMDA receptors (NMDARs). To test whether NMDAR stimulation contributes to reduced pS421-htt levels in HD, we determined phosphorylation of htt at Ser421 after NMDA-induced excitotoxicity in neurons from YAC128 mice.
Here, we report that the total level of pS421-htt is reduced in YAC128 primary neurons after excitotoxic NMDAR stimulation. Similarly, the total level of pS421-htt is reduced in YAC128 transgenic mice after quinolinic acid injection into the striatum. In contrast, loss of phosphorylation of pS421-htt is prevented in YAC mice that never develop clinical or neuropathological features of HD [the caspase 6-resistant YAC128 transgene (C6R)].
To gain insight into the mechanisms underlying these findings, we determined that the Ser/Thr protein phosphatases PP1 and PP2A dephosphorylate pS421-htt in situ and after excitotoxic stimulation of NMDARs in neurons. Furthermore, increasing the phosphorylation of htt at S421 by blocking PP1 and PP2A activity protects YAC128 striatal neurons from NMDA-induced cell death. These results, together with the observed modulation of pS421-htt levels by dopamine, the reduced expression of PP1 inhibitor Darpp-32 in the striatum of YAC128 mice, and the reduced phosphorylation of PP1 substrate CreB, point to altered regulation of phosphatase activity in HD and highlight enhancing phosphorylation of htt at S421 as a therapeutic target.
Introduction
Huntington disease (HD) is an adult-onset neurodegenerative disorder characterized by progressive motor dysfunction and cognitive decline (Harper, 1999). Important hallmarks of this disorder are recapitulated in several mouse models of HD demonstrating motor abnormalities, cognitive dysfunction, and striatal neuronal degeneration (Ehrnhoefer et al., 2009). Involvement of htt cleavage by caspases figures prominently among cellular mechanisms mediating neuropathology in HD and mutation of the caspase 6 cleavage site in poly(Q)-expanded full-length htt prevents onset of HD symptoms and signs in a YAC128 mouse model (Graham et al., 2006).
Htt is phosphorylated at Ser421 through stimulation of IGF1Rs and activation of the prosurvival kinase Akt (Humbert et al., 2002; Warby et al., 2005). In the presence of poly(Q) expansion, htt phosphorylation at Ser421 is reduced, resulting in defects in intracellular transport (Gauthier et al., 2004). One mechanism contributing to dysregulation of htt phosphorylation in HD may result from NMDA receptor (NMDAR) overactivation, as excitotoxic stimulation of NMDARs decreases pS421-htt levels in primary neurons (Metzler et al., 2007). However, little is known about the relationship between phosphorylation of htt at Ser421 and NMDA-induced excitotoxicity in HD. Recent evidence suggests that enhanced NMDAR signaling contributes to neuronal dysfunction and cell death in HD (Beal et al., 1986; Fan and Raymond, 2007; Milnerwood et al., 2010). In addition, mimicking phosphorylation at Ser13 and Ser16 of htt prevents features of HD in BAC transgenic mice, which highlights potential benefits of increasing htt phosphorylation at Ser13 and Ser16 of htt (Gu et al., 2009).
NMDARs promote neuronal survival and trigger cell death depending on the age of a neuron, stimulus intensity, subunit composition, and receptor localization leading to the expression of distinct transcriptional responses (Zhang et al., 2007). Stimulation of synaptic NMDARs supports neuronal survival, whereas stimulation of extrasynaptic NMDARs triggers a cell death response by blocking the prosurvival function of cAMP response element-binding protein (CreB) that may involve dephosphorylation by protein phosphatase 1 (PP1) (Bito et al., 1996; Sala et al., 2000; Hardingham et al., 2002).
PP1 belongs to a family of Ser/Thr phosphatases of which PP1, PP2A, and PP2B (calcineurin) are the most abundant in mammalian neurons. PP1, PP2A, and PP2B play important roles in the regulation of ligand-gated ion channels and G-protein-coupled receptors and serve important functions in synaptic plasticity, learning, and memory and, as more recent studies suggest, in cell survival and cell death (Van and Goris, 2003; Gee and Mansuy, 2005; Mansuy and Shenolikar, 2006).
To gain insight into the mechanism leading to neuronal dysfunction and ultimately cell death in HD, we examined the relationship between previously described enhanced NMDA-induced excitotoxicity and altered phosphorylation of htt at S421. These studies demonstrate that loss of htt phosphorylation is directly influenced by enhanced NMDA-induced excitotoxicity in YAC128 neurons and activation of PP1 and PP2A with major contribution from PP1. Furthermore, we show protection from NMDA-induced excitotoxicity by blocking dephosphorylation of htt at S421. These data, together with reduced expression of Darpp-32 and reduced phosphorylation of PP1 substrates in YAC128 striatum, provide an explanation for the reduced phosphorylation of htt at S421 in HD and demonstrate a relationship between excitotoxicity and phosphorylation status of htt at S421. Furthermore, these investigations also highlight restoration of phosphorylation activity at S421 as a means to reduce the enhanced excitotoxicity in HD.
Materials and Methods
Neuronal culture and mice.
Cortical and striatal cultures were established from embryonic day 16.5 embryos as described previously (Metzler et al., 2007). Neurons were seeded onto poly-d-lysine-coated surfaces at a density of ∼1.5 × 105 per cm2. Cultures were maintained at 37°C under 5% CO2 and one-half of the culture media was exchanged once every 4–5 d. At 10–12 d in vitro (DIV), neurons were stimulated with NMDA, 30 μm glycine in ECS (140 mm NaCl, 5.4 mm KCl, 1.3 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, 25 mm glucose at pH 7.35) in the absence of MgCl2 for 10 min at 37°C. Neurons were washed in ECS, cultured for 15 min, and harvested.
All mice used in this study are on a FVB/N background and are of mixed gender. YAC128 (line HD53), YAC18 (line 212), C6R (C6R13), and wild-type (WT) mice were used in all in vivo experiments. Only quinolinic acid (QA) injections were performed with line HD55, which is more susceptible to QA-induced excitotoxicity compared with line HD53 (Graham et al., 2006, 2009). Furthermore, line HD55 was chosen for cell culture experiments because it is susceptible to enhanced NMDA-induced excitotoxicity in vitro and it is homozygous for the htt transgene in contrast to line HD53. This allowed us to set up many neuronal cultures at the same plating density, which facilitates the simultaneous analyses of htt phosphorylation kinetics among genotypes.
All experiments were performed in accordance with protocols (animal protocol A07-0106) approved by the University of British Columbia Committee on Animal Care and the Canadian Council on Animal Care.
Drugs.
MK-801 (Sigma-Aldrich) and memantine (Merz Pharmaceuticals) were dissolved in PBS (10 μm) and ifenprodil (Iwaki Seikayu) in DMSO (10 mm), respectively, and added to the culture medium 1 h before NMDAR stimulation. FK-506 and okadaic acid (OA) (Alexis Biochemicals) were dissolved in DMSO (10 and 50 mm, respectively) and added to the culture media 15 min before NMDAR stimulation. Cyclosporin A (50 mm) and tautomycin (50 μm; Alexis Biochemicals) were dissolved in ethanol and added to the culture media 20 min and 2 h, respectively, before NMDAR stimulation. Dopamine (Sigma-Aldrich) was freshly prepared each time before administration. SKF-81297 was purchased from Tocris Bioscience, and QA was purchased from Sigma-Aldrich. QA was dissolved in PBS (120 mm), and aliquots were kept frozen at −20°C and further diluted in PBS before administration.
Intraperitoneal injections and stereotactic surgery.
Mice (6–10 weeks of age and weighing 20–28 g) were deeply anesthetized with freshly prepared Avertin (Sigma-Aldrich; 4 mg per 60 g of body weight in PBS) and injected unilaterally via stereotactic surgery according to the following coordinates from bregma: 0.8 mm rostral, 1.8 mm lateral, 3.5 mm ventral. Drugs or vehicle were injected into the striatum in a total volume of 1 μl over 2 min. The injection needle remained in position for 1 min before and 2 min after injection. For intraperitoneal injections, drugs or vehicle were injected in a 100 μl volume. Mice were decapitated at the indicated time points, tail samples were taken for genotype determination, and brain regions were isolated on ice and immediately frozen and kept at −80°C.
Cell death assays.
Caspase 3 activation was measured 3 h after NMDA stimulation of cortical neurons at 12 DIV as described previously (Metzler et al., 2007). Neurons were washed in ECS and lysed in 20 mm Tris-HCl, pH 7.5, 140 mm NaCl, 0.5 mm EDTA, 1% Triton X-100, 4 mm PefaBloc. Samples were incubated for 10 min on ice and centrifuged for 10 min at 956 × g. The supernatant was removed, and aliquots were taken to determine protein concentration. Duplicate samples were incubated in a 96-well plate with the fluorogenic caspase 3 substrate N-acetyl-DEVD-7-amino-4-trifluoromethyl coumarin (Ac-DEVD-AFC) (BIOMOL). The amount of cleaved AFC was measured in a multiwell plate reader (Victor; PerkinElmer) equipped with fluorescence capabilities at an excitation at 400 nm and emission at 505 nm, respectively. Measurements were taken every 10 min over a time course of 1 h and compared with an AFC standard curve.
To determine cell death based on nuclear morphology, striatal neurons were fixed in 4% paraformaldehyde for 15 min, permeabilized in 0.3% Triton X-100 for 5 min, and washed in PBS. Neurons were then stained with 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen), washed in PBS, and mounted in Mowiol for detection under the microscope. Approximately, 300 neurons were analyzed per coverslip, treatment condition, and genotype to determine the percentage of dying neurons.
Antibodies.
The following antibodies (Abs) were used for expression analyses: phosphorylation of htt at Ser421 was determined with a rabbit polyclonal Ab (pAb), as described previously (Metzler et al., 2007). Total htt was determined by probing with monoclonal Ab (mAb) 2166 (Millipore Bioscience Research Reagents). Total Akt, pS473-Akt, total Erk, pS44/42-Erk, total CreB, and pS133-CreB were detected with rabbit pAbs from Cell Signaling. Darpp-32 and pT34-Darpp32 were detected with a rabbit pAb (Millipore Bioscience Research Reagents and R&D Systems, respectively). Hsp90 was determined with a rabbit pAb (Santa Cruz). Goat anti-mouse IRDye-800CW (Rockland Immunochemicals) and goat-anti rabbit Alexa Fluor 680 (Invitrogen) were used as secondary Abs.
Quantification of phosphorylation.
To determine changes in phosphorylation of htt, Akt, and Erk, cortical neurons at 12 DIV and striatal neurons at 10 DIV were harvested after NMDA stimulation in ice-cold PBS supplemented with 1 μg/ml aprotinin, 1 μg/ml leupeptin, 4 mm PefaBloc, 2.5 mm NaF, 1 mm NaVO4, and 1 mm β-glycero-PO4 and processed as described previously (Metzler et al., 2007). The pellet was resuspended in lysis buffer [50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% IGEPAL, 1 μg /ml aprotinin, 1 μg/ml leupeptin, 4 mm PefaBloc, 2.5 mm NaF, 1 mm NaVO4, 1 mm β-glycero-PO4, 5 μm Z-VAD (Calbiochem), and Complete protease inhibitor mixture (Roche)] and incubated for 20 min on ice. After centrifugation at 20,817 × g, the protein concentration was measured in the supernatant, and 150–400 μg of protein was used for immunoprecipitation of htt. Protein was incubated with 2–4 μl of mouse mAb 2166 bound to protein A/G-Sepharose 4 Fast Flow (GE Healthcare) in lysis buffer overnight at 4°C. After immunoprecipitation, samples were washed in lysis buffer, and bound protein was eluted in (lithium dodecyl sulfate) LDS-PAGE sample buffer. Proteins were separated by 3–8% Tris-acetate gel electrophoresis (Invitrogen), blotted onto membranes, and probed with a rabbit pAb specifically recognizing pS421-htt; total htt was detected by probing with mAb 2166. After washing and probing with red (goat anti-rabbit Alexa Fluor 680) and green labeled (goat anti-mouse IRDye-800-CW) secondary Abs, immunoreactive bands were quantified with a LI-COR system using Odyssey software. The ratios of pS421-htt and total htt were calculated and expressed relative to control conditions. In all experiments, the relative level of htt phosphorylation at S421 was pooled for mutant and endogenous htt.
To determine the level of pS473-Akt and pS44/42-Erk in neuronal lysates, 30 μg of protein was resuspended in LDS sample buffer and separated by 4–12% Tris-Bis gel electrophoresis (Invitrogen), blotted onto membranes, and probed with Abs recognizing pS473-Akt, total Akt, pS44/42-Erk, and total Erk, respectively, followed by incubation with the appropriate secondary Abs.
To determine protein expression and the relative level of protein phosphorylation in striatum and cerebellum, brain tissue was isolated at the indicated times, homogenized in lysis buffer, and processed for immunoprecipitation of htt using 400 μg of protein from cerebellum and 150 μg of protein from striatum. To determine the level of pS473-Akt, total Akt, pS44/42-Erk, total Erk, pT34-Darpp-32, and total Darpp-32, 30 μg of protein was resuspended in LDS sample buffer and separated by 4–12% Tris-Bis gel electrophoresis. To determine the phosphorylation level of CreB, samples were homogenized in lysis buffer and sonicated in the presence of 2% SDS. Protein expression and the ratios between phosphorylated and total proteins were determined using a LI-COR system and Odyssey software and expressed relative to controls.
In vitro dephosphorylation of pS421-htt.
An N-terminal fragment of htt (Htt-1212-15Q) (Warby et al., 2005) was transiently transfected into COS7 cells, immunoprecipitated with mAb 2166 (Millipore Bioscience Research Reagents), and phosphorylated in the presence of recombinant Akt (Millipore). Equimolar concentrations of in vitro phosphorylated htt-1212-15Q or immunoprecipitated full-length htt from 12 DIV cortical neurons were incubated in phosphatase reaction buffer according to the manufacturer's protocol. The indicated amounts of λ protein phosphatase, PP1 (both from New England Biolabs), or PP2A and PP2B (both from Calbiochem) were added to each reaction mixture and the dephosphorylation of pS421-htt was performed for 30 min at 30°C. Samples were eluted in LDS sample buffer and proteins were separated by Western blotting and probed with Abs detecting pS421-htt and total htt (2166).
Real-time quantitative reverse transcription–PCR.
Total RNA was extracted from striatum of YAC128 (line HD53) mice and littermate controls using the RNeasy Protect Mini kit (QIAGEN). First-strand cDNA was generated by reverse transcription using the Quantitect RT kit (QIAGEN). All reactions were performed in duplicate using 2 μl of generated cDNA. Absolute quantity of the targets in each sample was calculated based on the standard curve method. Primer efficiencies were determined using a dilution series of adult striatum cDNA. Only primer pairs with an efficiency >0.98 were used in subsequent analyses. DARPP-32-specific primers were designed using Primer3 software (forward, 5′-TGGAAGGCAGAGCAACACTA; reverse, 5′-GCCCTAGCAGGTGAAAGACA). Amplification of mouse β-actin (forward, 5′-CCAGCCTTCCTTCTTGGGTAT; reverse, 5′-TGTGTTGGCATAGAGGTCTTTACG) was performed to normalize the amount of sample RNA added to each reaction. Darpp-32 mRNA expression levels were measured using the ABI 7500 real-time PCR system and RQ Study Add On software (ABI).
Phosphatase activity.
Phosphatase activity was determined using a phosphatase assay kit (Calbiochem). Striatal lysates were prepared and 10 μg of protein was assayed for phosphatase activity in the presence of RII phosphopeptide substrate according to the manufacturer's protocol. The activity was determined per 10 μg protein and expressed relative to WT controls.
Data analysis.
For statistical analysis, data were analyzed by Student's t test for comparison between genotypes, and one-way ANOVA was used to determine dose–response effects using GraphPad Prism 5 software. Statistical significance was defined as p < 0.05, and data were expressed as mean ± SEM.
Results
YAC128 primary neurons show loss of phosphorylation of pS421-htt after NMDA-induced excitotoxicity
In a recent study, we observed a reduction in phosphorylation of mhtt (Warby et al., 2005). Moreover, a reduction in phosphorylation of normal htt at S421 occurs after excitotoxic stimulation of NMDARs (Metzler et al., 2007). Together, these data suggest loss of phosphorylation of pS421-htt may occur after NMDAR activation in HD.
To further investigate this, we determined the level of pS421-htt after excitotoxic stimulation of NMDARs in primary cortical neurons from YAC128 and WT mice. For this purpose, we immunoprecipitated and enriched total htt from lysate and determined the level of pS421-htt using a phosphospecific antibody as previously described (Warby et al., 2005; Metzler et al., 2007) and as explained in supplemental Figures 1 and 2 (available at www.jneurosci.org as supplemental material). Comparing neurons from both mutant and WT mice, we observed significant reductions in the level of pS421-htt in neurons after NMDAR stimulation (Fig. 1A, Table 1). The level of pS421-htt after excitotoxic stimulation with 50 μm NMDA was 66.8% in YAC128 neurons and 87.9% in WT neurons (p < 0.0001 for YAC128 and p < 0.003 for WT; n = 10 for both genotypes). Importantly, the level of phosphorylation of pS421-htt was significantly decreased in YAC128 neurons compared with WT by 21.1 and 16.2% after treatment with 50 and 100 μm NMDA, respectively (p < 0.002 for 50 μm NMDA and p < 0.035 for 100 μm NMDA; n = 8).
Figure 1.
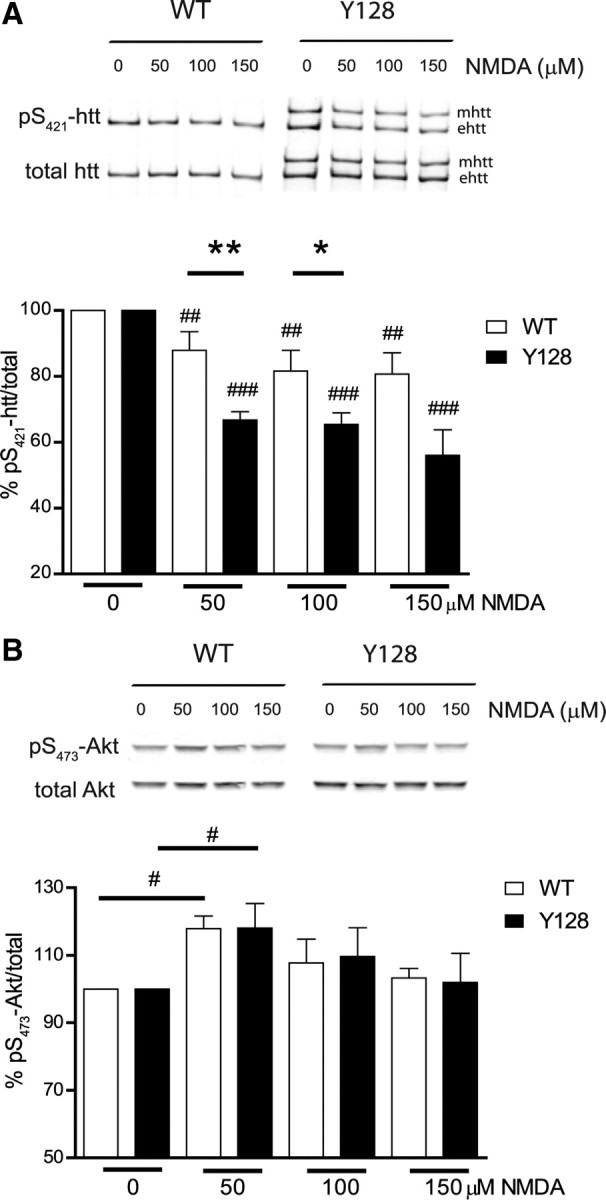
YAC128 primary neurons show loss of phosphorylation of pS421-htt after NMDA-induced excitotoxicity. A, Cortical neurons were treated with varying doses of NMDA, 30 μm glycine for 10 min at 12 DIV. After drug removal, neurons were cultured for 15 min, and protein lysates were either processed for immunoprecipitation of htt or analyzed as described in B. Immunoprecipitated htt was separated by Western blot and membranes were probed with Abs against pS421-htt and total htt (mAb 2166) followed by probing with red- (goat anti-rabbit Alexa Fluor 680) and green-labeled (goat anti-mouse IRDye-800-CW) secondary Abs. Quantification of immunoreactivity was performed with a LI-COR system using Odyssey software. The ratio between pS421-htt and total htt was determined and expressed relative to controls for each condition and genotype in each of eight independent experiments. Relative levels of phosphorylation for mutant htt (mhtt) and endogenous htt (ehtt) were pooled. *p < 0.05, **p < 0.005 compared with WT between genotypes, by t test; ##p < 0.005, ###p < 0.0005 compared with control within genotype, by paired t test. Error bars denote ±SEM. B, Protein lysates, prepared as described in A, were separated by Western blot and probed with Abs against pS473-Akt and total Akt. The ratio between pS473-Akt and total Akt was determined and expressed relative to controls. Ratios for each condition and genotype in each of five independent experiments were determined. #p < 0.05 compared with controls within genotypes, by paired t test. Error bars denote ±SEM.
Table 1.
The level of pS421-htt in WT and YAC128 primary neurons after stimulation of NMDARs with different doses of NMDA
| NMDA (μm) | Percentage of pS421-htt/total after excitotoxic NMDAR stimulation in primary cortical neurons |
|||
|---|---|---|---|---|
| 0 | 50 | 100 | 150 | |
| WT | 100 | 87.88 ± 5.7 | 81.64 ± 6.3 | 80.7 ± 6.4 |
| YAC128 | 100 | 66.76 ± 2.5 | 65.41 ± 3.6 | 56.04 ± 7.7 |
Although we performed most of our experiments in cortical neurons, a limited number of experiments was performed in striatal neurons to verify our findings in cells showing early neuronal loss in HD. Again, we observed a significant decrease in the level of htt phosphorylation at S421 in YAC128 neurons compared with WT after excitotoxic NMDAR stimulation with 100 and 200 μm NMDA (p < 0.05; n = 5) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (data not shown). Together, these studies demonstrate that the level of pS421-htt is significantly reduced in neurons expressing mhtt after excitotoxic stimulation of NMDARs.
Reduced levels of pS421-htt in YAC128 primary neurons after NMDA-induced excitotoxicity are not attributable to differences in kinase activity
Levels of phosphorylation are determined by the relative activity of kinases and phosphatases. A decrease in pS421-htt status could be influenced by reduced kinase activity or increased phosphatase activation. Previous reports have shown phosphorylation of pS421-htt by the prosurvival kinase Akt (Humbert et al., 2002; Warby et al., 2005). To determine whether altered Akt kinase activity influences the loss of phosphorylation of pS421-htt after NMDAR stimulation, we assessed the level of pS473-Akt as a measure of Akt kinase activity (Fig. 1B). At low doses of NMDA, we observed a significant increase in pS473-Akt in both genotypes in agreement with previous reports demonstrating activation of the PI3-kinase pathway as a mediator of NMDAR signaling (Perkinton et al., 2002; Sutton and Chandler, 2002). The level of pS473-Akt at 50 μm doses of NMDA increased by 18.1% in YAC128 neurons (p < 0.027; n = 5) and by 17.9% in WT neurons (p < 0.006; n = 5). The extent of Akt phosphorylation was similar for both genotypes and at all doses of NMDA tested. These results demonstrate that Akt kinase activity does not contribute to the differences in pS421-htt in YAC128 neurons compared with WT after excitotoxic NMDAR stimulation. Moreover, the increase in Akt activity immediately after excitotoxic NMDAR stimulation and the rapid reduction in pS421-htt levels that occur within the same time frame suggested involvement of phosphatases in the dephosphorylation of pS421-htt after NMDAR stimulation (Fig. 1A,B).
In a similar manner, we tested another survival pathway, the mitogen-activated protein (MAP) kinase pathway that is activated by NMDAR stimulation (see supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Again, no significant differences were observed between genotypes after excitotoxic NMDAR stimulation in the level of pS42/44-Erk as a measure of MAP kinase activation. Together, our data demonstrate loss of phosphorylation of pS421-htt after NMDAR stimulation is independent of changes in PI3 kinase and MAP kinase activation.
Enhanced excitotoxic cell death correlates with reduced levels of pS421-htt in YAC128 primary neurons
Previous studies have shown enhanced excitotoxic cell death after NMDAR stimulation in primary striatal neurons from YAC128 mice (Zeron et al., 2002; Graham et al., 2009). To determine whether excitotoxic cell death is also enhanced in primary cortical neurons from YAC128 mice, we analyzed caspase 3 activation 3 h after stimulation with 500 μm NMDA by cleavage of Ac-DEVD-AFC. These studies revealed a significant increase in caspase 3 activation in both YAC128 by 105% and in WT neurons by 68.2% (p < 0.0001 for both genotypes; n = 7). Caspase 3 activity was significantly increased in YAC128 cortical neurons compared with WT littermate controls (36.7%; p < 0.028). This result demonstrates enhanced susceptibility to NMDA-induced excitotoxicity in cortical neurons from YAC128 mice and reveals an association between the extent of NMDA-induced excitotoxicity, caspase-3 activation, and loss of pS421-htt in the presence of mhtt.
The level of pS421-htt is reduced in the striatum of YAC128 mice after QA-induced excitotoxicity but not in YAC18 and C6R mice
We next determined the phosphorylation status of htt at Ser421 in response to NMDAR stimulation after intrastriatal injections of QA in YAC128 mice. QA injection leads to an HD-like pathology resulting from overstimulation of NMDARs and altered NMDAR signaling (Beal et al., 1986; Szatmari et al., 2005). Moreover, QA-induced lesion volume and associated striatal degeneration is enhanced in YAC128 mice at an early stage in HD pathology (Graham et al., 2009).
To test whether the level of pS421-htt correlates with the extent of excitotoxicity in this system in vivo, we first tested the relationship between the level of pS421-htt and the effect of QA in WT mice. We observed a dose-dependent decrease in the level of pS421-htt to 83% after 30 min and to 63% after 2 h after injection of 30 nmol of QA into the striatum of WT mice (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material) (one-way ANOVA treatment, p = 0.001, F(1,6) = 4.9). The most profound change in pS421-htt levels occurred within the first 30 min and is the most suitable time frame to determine whether differences exist between genotypes.
We next compared phosphorylation of htt after QA injection in YAC128 mice at 2 months of age. A significant reduction in the level of pS421-htt occurred in YAC128 striatum (Fig. 2, Table 2; supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). At 15 min, the level of pS421-htt was reduced by 19.4% in YAC128 but by only 4.8% in WT striatum (p < 0.024; n = 7). As observed in primary neurons, no differences in PI3 kinase or MAP kinase activity were evident in YAC128 compared with WT mice (data not shown). These data suggest that the mutation in HD modulates the phosphorylation status of htt after excitotoxic stimulation of NMDARs in vivo.
Figure 2.

The level of pS421-htt is reduced in the striatum of YAC128 mice after QA-induced excitotoxicity but not in YAC18 and C6R mice. A total of 30 nmol of QA or PBS was injected into the striata of YAC128, YAC18, C6R, and WT mice at 6–8 weeks of age. Mice were killed at the indicated time points, and the level of pS421-htt was determined in striatal lysates after immunoprecipitation of htt (see supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Immunoblots were probed with Abs against pS421-htt and total htt (mAb 2166) followed by probing with secondary Abs and quantification of immunoreactivity. The ratio between pS421-htt and total htt was determined and expressed relative to PBS-injected controls for each condition and genotype in each of seven (YAC128 and WT) and four (YAC18 and C6R) independent experiments. Relative levels of phosphorylation for mhtt and ehtt were pooled. *p < 0.05 compared with WT; #p < 0.05, ##p < 0.005, ###p < 0.0005 compared with YAC128, by t test. Error bars denote ±SEM.
Table 2.
The level of pS421-htt at different time points in the striatum after intrastriatal injection of 30 nmol of QA
| NMDA (μm) | Percentage of pS421-htt/total after QA injection into the striatum |
|||
|---|---|---|---|---|
| 0 min | 5 min | 15 min | 30 min | |
| WT | 100 | 98.4 ± 6.9 | 95.2 ± 5.0 | 84.7 ± 3.6 |
| YAC128 | 100 | 92.7 ± 4.4 | 80.6 ± 3.2 | 72.1 ± 2.7 |
| YAC18 | 100 | 111.4 ± 14.1 | 115.4 ± 12.7 | 96.6 ± 17.1 |
| C6R | 100 | 116.8 ± 12.4 | 103.1 ± 6.6 | 94.2 ± 8.0 |
Previous studies have shown protection from NMDA and QA-induced excitotoxicity in mice overexpressing either full-length htt or caspase 6-resistant mhtt (Graham et al., 2006; Leavitt et al., 2006). Therefore, we wanted to assess whether protection from NMDA-induced excitotoxicity correlates with higher levels of pS421-htt in YAC18 and C6R mice after QA injection (Fig. 2; supplemental Fig. 5C, available at www.jneurosci.org as supplemental material). Indeed, the level of pS421-htt was significantly increased in YAC18 and C6R mice compared with YAC128. At 15 min, the level of pS421-htt increased by 22.5% in YAC18 and by 34.84% in C6R striatum compared with YAC128 mice (p < 0.001 for YAC18 and p < 0.018 for C6R; n = 4). These data demonstrate elevated levels of pS421-htt in the striatum of mice that are protected from NMDA-induced excitotoxicity. Interestingly, a significant increase in pS473-Akt levels occurred in YAC18 and C6R mice at 15 min after QA injection (p < 0.041 for YAC18, n = 7, and p < 0.009 for C6R, n = 5, compared with YAC128 by t test) (data not shown). This increase in Akt kinase activity may contribute to protection seen from NMDA-induced excitotoxicity observed in these mice (Leavitt et al., 2006).
Together, these data demonstrate a correlation between the degree of NMDAR-induced excitotoxicity and the level of pS421-htt in mouse models of HD in vivo.
YAC128 mice show a progressive striatal-specific decrease in pS421-htt but normal levels of htt phosphorylation are observed in YAC18 and C6R mice
Our studies have shown loss of pS421-htt levels in YAC128 neurons after acute stimulation of NMDARs in vitro and in vivo. We next wanted to determine the basal level of pS421-htt and the natural history of changes in pS421-htt levels in the striatum by comparing WT, YAC128, YAC18, and C6R mice from 7 weeks to 12 months of age. These studies demonstrate a gradual loss of total pS421-htt levels in the striatum of YAC128 transgenic mice starting at ∼3 months of age and continuing toward 12 months of age (Fig. 3A). At 6 and 12 months of age, the level of total pS421-htt is significantly reduced by 25.7 and 32.95%, respectively, and in agreement with a previous study showing reduced pS421-htt levels of the YAC128 compared with Y18 transgene (p < 0.013 for 6 months and p < 0.012 for 12 months, n = 6, by paired t test) (Warby et al., 2005). No reduction in pS421-htt levels is evident in YAC18 and C6R mice at all time points analyzed. In addition, the reduction in pS421-htt in YAC128 mice compared with WT is region specific as normal levels of pS421-htt were observed in the cerebellum, a tissue not affected in HD (Fig. 3B). The decrease in pS421-htt levels in YAC128 mice is progressive starting as early as 3 months of age and is reminiscent of the progressive phenotype in the YAC128 transgenic mice.
Figure 3.
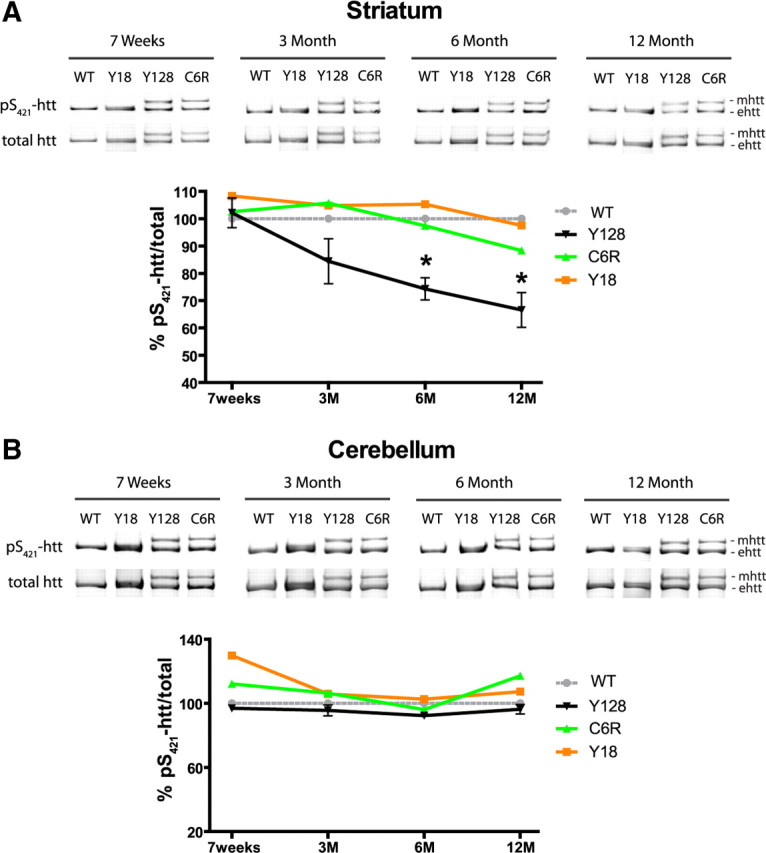
YAC128 mice show a progressive striatal-specific decrease in pS421-htt, but normal levels of htt phosphorylation are observed in YAC18 and C6R mice. A, B, Brain tissue was isolated at the indicated time points and the level of pS421-htt was determined in striatum (A) and cerebellum (B) after immunoprecipitation of htt, Western blot, and probing with Abs against pS421-htt and total htt (mAb 2166) followed by probing with secondary Abs. Quantification of immunoreactivity was performed with a LI-COR system using Odyssey software. The ratio between pS421-htt and total htt was determined and expressed relative to WT for each tissue, time point, and genotype in three independent experiments. Relative levels of phosphorylation for mutant htt (mhtt) and endogenous htt (ehtt) were pooled. *p < 0.05 compared with WT, by t test. Error bars denote ±SEM.
Reduced levels of pS421-htt after excitotoxic NMDAR stimulation are influenced by PP1 and PP2A activity
Stimulation of NMDARs leads to activation of several phosphatases including PP1, PP2A, and PP2B (calcineurin) whose substrate specificity is primarily determined through interaction with regulatory and targeting proteins (Mansuy and Shenolikar, 2006). A small GST (glutathione S-transferase) fragment of pS421-htt encompassing amino acids 384–467 can be dephosphorylated by PP2B in situ (Pardo et al., 2006). However, the role of PP1 and PP2A in the dephosphorylation of pS421-htt has not been explored. In an attempt to investigate the dephosphorylation of htt at S421 by PP1, PP2A, and PP2B, we performed in situ dephosphorylation assays. For this purpose, we expressed an N-terminal fragment of htt (htt-1212-Q15) in COS7 cells that was immunopurified and phosphorylated at S421 in the presence of Akt. Since the endogenous phosphorylation level of pS421-htt-1212-Q15 in COS7 cells is high, the level of phosphorylation in the presence of Akt could only be increased by 13% (p < 0.05; n = 3). Next, we incubated equimolar concentrations of pS421-htt-1212-Q15 with PP1, PP2A, and PP2B to test their activity toward pS421-htt-1212-Q15 (Fig. 4A). The pan-specific λ protein phosphatase was used as control and reduced the level of pS421-htt-1212-Q15 by 97% (p < 0.005; n = 3). PP1 demonstrated the greatest extent of dephosphorylation and reduced the pS421-htt-1212-Q15 level by 84% (p = 0.02; n = 3). PP2A reduced the level of pS421-htt-1212-Q15 by 24% (p = 0.04; n = 3). In contrast, PP2B reduced the level of pS421-htt-1212-Q15 by only 10%, and this reduction was not significant. Next, we analyzed the extent of dephosphorylation in the presence of full-length htt that was immunopurified from cortical neurons. Again, we observed a loss in htt phosphorylation at S421 that was most profound in the presence of PP1 (Fig. 4B).
Figure 4.

Reduced levels of pS421-htt after excitotoxic NMDAR stimulation is attributable to enhanced PP1 and PP2A activity. A, Htt-1212-Q15 was expressed in COS7 cells, immunopurified with mAb 2166, and phosphorylated at S421 in the presence of Akt. Equimolar amounts of htt-1212-Q15 were treated with phosphatases for 30 min at the indicated concentrations and analyzed by Western blot followed by probing with Abs against pS421-htt and total htt (mAb 2166). *p < 0.05 compared without Akt (lane 2); #p < 0.05 compared with Akt (lane 3) by paired t test. B, Full-length htt was immunoprecipitated from cortical lysate harvested at 12 DIV and equimolar amounts of htt were phosphatase-treated as described in A. *p < 0.05, **p < 0.005 compared with control by paired t test. C–E, Cortical neurons at 12 DIV were treated with 250 μm NMDA, 30 μm glycine for 10 min, or left untreated in the presence or absence of phosphatase inhibitors. After receptor stimulation, cultures were incubated for 15 min and processed for immunoprecipitation of htt and Western blot. Membranes were probed with Abs against pS421-htt and total htt (mAb 2166) followed by probing with secondary Abs and quantification. In all experiments, the ratio between pS421-htt and total htt was determined and expressed relative to controls in each of three independent experiments. *p < 0.05, **p < 0.005 compared with control by paired t test; #p < 0.05, ##p < 0.005, and ###p < 0.005 compared with NMDA-treated cultures by t test. Error bars denote ±SEM.
To determine which phosphatases are involved in NMDA-induced dephosphorylation of pS421-htt, we treated WT cortical neurons with inhibitors blocking PP1, PP2A, and PP2B activity independently (Fig. 4C) (Sheppeck et al., 1997). These studies demonstrate a significant increase in the level of pS421-htt in the presence of drugs blocking PP1 and PP2A such as OA and tautomycin (p < 0.0001 compared with NMDA-treated cultures for both drugs; n = 5). Drugs blocking PP2B activity, such as cyclosporin A and FK-506, increased the level of pS421-htt, although these effects did not reach significance (p = 0.3 and p = 0.4 for FK-506 and cyclosporin A, respectively; n = 5). Next, we analyzed whether the same phosphatases are responsible for the NMDA-dependent loss of htt phosphorylation in YAC128 neurons. Again, we observed significant effects in the presence of OA and tautomycin (supplemental Fig. 6A,B, available at www.jneurosci.org as supplemental material). As in WT neurons, FK-506 and cyclosporin A increased the pS421-htt level, although these effects did not reach significance (p = 0.5 for FK-506 and p = 0.3 for cyclosporin A; n = 3).
Together, these data support a major role for PP1 and PP2A in the dephosphorylation of pS421-htt during NMDA-induced excitotoxicity. However, a small effect of PP2B in this process cannot be excluded.
In an attempt to distinguish between PP1 and PP2A, we took advantage of the 50-fold higher selectivity of OA toward PP2A (IC50 for PP2A, 0.02–1 nm; IC50 for PP1, 10–1300 nm) (Gupta et al., 1997) and determined the level of pS421-htt in the presence of varying doses of OA during NMDA stimulation (1–500 nm) (Fig. 4D; supplemental Fig. 6C, available at www.jneurosci.org as supplemental material). These studies demonstrate a significant dose-dependent effect (one-way ANOVA for WT: treatment, p < 0.0002, F(4,21) = 9.3; for YAC128: treatment, p < 0.0001, F(4,16) = 12.6) of OA on pS421-htt level in the presence of NMDA with an IC50 value of 42.7 and 48.3 nm in WT and YAC128 neurons, respectively. Phosphorylation of htt increased significantly in the presence of 100 and 1000 nm OA, concentrations of OA inhibiting both PP1 and PP2A. In contrast, no significant increase occurred in the presence of 10 nm OA, a concentration blocking only PP2A. These data highlight involvement of PP1 in the NMDA-induced dephosphorylation of pS421-htt. To further investigate, we assessed the effect of varying concentrations of tautomycin. This inhibitor is approximately fourfold more selective for PP1 than PP2A (IC50 for PP1, 1–7.5 nm; IC50 for PP2A, 10–23 nm) (Fig. 4E). Using concentrations of tautomycin ranging from 1 to 100 nm, we observed a dose-dependent effect (one-way ANOVA treatment, p < 0.02, F(4,12) = 4.6) with an IC50 value of 5.1 nm in WT neurons, again suggesting PP1 is the more likely phosphatase dephosphorylating pS421-htt after NMDA-induced excitotoxicity.
Increased phosphorylation of htt at S421 protects Y128 striatal neurons from NMDA-induced excitotoxicity
Previous studies have shown that increasing the phosphorylation of htt at S421 protects neurons from poly(Q)-mediated toxicity in a fragment model of HD (Humbert et al., 2002; Pardo et al., 2006). To investigate whether increased phosphorylation of full-length htt protects against NMDA-induced cell death, we treated striatal neurons from YAC128 and WT mice with 100 nm OA during NMDAR stimulation. As observed previously, YAC128 neurons demonstrate a significant increase in NMDA-induced excitotoxicity by 5.2 and 10.3% compared with WT neurons after stimulation with 100 and 500 μm NMDA, respectively (p = 0.03 for 100 μm NMDA and p < 0.0005 for 500 μm NMDA; n = 10) (Fig. 5A,C) (Zeron et al., 2002; Shehadeh et al., 2006; Graham et al., 2009). Furthermore, loss of htt phosphorylation at S421 is blocked in the presence of 100 nm OA in neurons of both genotypes (Fig. 5B). These results are consistent with studies in cortical neurons from WT and YAC128 mice demonstrating increased levels of pS421-htt in the presence of OA after NMDAR stimulation (Fig. 4C,D; supplemental Fig. 6A,C, available at www.jneurosci.org as supplemental material). Importantly, increasing the level of pS421-htt significantly protects neurons from NMDA-induced cell death in YAC128 and WT neurons at 100 and 500 μm NMDA, respectively (at 100 μm NMDA, p = 0.001 for YAC128 and p = 0.03 for WT; at 500 μm NMDA, p < 0.0001 for YAC128 and WT, n = 10) (Fig. 5A,C). Moreover, treatment of neurons with OA alone has no significant effect on cell viability. Together, these data suggest a beneficial effect of increasing htt phosphorylation mitigating the effects of enhanced excitotoxicity in HD.
Figure 5.

Increased levels of pS421-htt through blockade of PP1 and PP2A protects against NMDA-induced cell death. A, Striatal neurons from YAC128 and WT mice at 10 DIV were treated with 100 and 500 μm NMDA, 30 μm glycine for 10 min in the presence or absence of 100 nm OA. After drug removal, neurons were cultured for 5 h. Neurons were processed to visualize nuclear morphology by DAPI stain. The arrows indicate condensed nuclei. Scale bar, 40 μm. B, Striatal neurons were stimulated with 500 μm NMDA, as described in A and cultured for 15 min. Protein lysates were processed for immunoprecipitation of htt and separation of htt alleles by Western blot. Membranes were probed with Abs against pS421-htt and total htt (mAb 2166) followed by probing with secondary Abs. C, The number of condensed nuclei (white arrows) was determined for each genotype and treatment and expressed relative to the total number of cells in three independent experiments. *p < 0.05, **p < 0.005, ***p < 0.0005 compared within genotype, and #p < 0.05 and ###p < 0.0005 compared between genotypes by t test. Error bars denote ±SEM.
D1 receptor stimulation and inhibition of PP1 modulates the level of pS421-htt in the striatum
In the striatum, PP1 activity is regulated by glutamatergic and dopaminergic inputs and Darpp-32 phosphorylation is an important integrator of these activities (Nishi et al., 1997; Snyder et al., 1998). For instance, stimulation of dopamine D1 receptors (D1Rs) leads to a rise in cAMP levels and activation of protein kinase A (PKA) that phosphorylates Darpp-32 at Thr34 and turns Darpp-32 into a potent inhibitor of PP1. This dopaminergic control of PP1 activity may influence the phosphorylation status of S421-htt. To test this hypothesis and to obtain additional evidence for regulation of pS421-htt levels by PP1, we analyzed the effect of dopamine D1R stimulation on phosphorylation of htt in vivo. Interestingly, the level of pS421-htt increased significantly in the striatum after injection of the D1R-specific agonist SKF-81297 (Fig. 6A,B). For example, injection of SKF-81297 into the peritoneum or directly into the striatum increased the level of pS421-htt by 104.1 and 49.4%, respectively (p < 0.003 and p < 0.005; n = 3). This increase in pS421-htt occurred within 15 min of drug injection and correlates well with the observed increase in Darpp-32 phosphorylation at Thr34 after D1R stimulation (Nishi et al., 1997).
Figure 6.

D1R stimulation and inhibition of PP1 modulates the level of pS421-htt in the striatum. A, B, Dopamine or the D1-specific agonist SKF-81297 were either injected into the peritoneum (A) or via stereotactic surgery directly into the striatum (B) of WT mice. Mice were killed 15 min later, and the level of pS421-htt was determined in the striatum after immunoprecipitation of htt and Western blot. In all experiments, membranes were probed with Abs against pS421-htt and total htt followed by probing with secondary Abs and quantification of immunoreactivity. The relative level of htt phosphorylation at S421 was determined and expressed relative to vehicle-injected controls. *p < 0.05, **p < 0.005, by t test. Error bars denote ±SEM.
To obtain additional evidence for regulation of pS421-htt levels by D1Rs and Darpp-32-dependent inhibition of PP1, we injected the PP1 inhibitor OA into the striatum of WT mice. These studies demonstrate an increase in pS421-htt levels by 59% (p = 0.03; n = 3) after inhibition of PP1 and suggest that D1R stimulation and Darpp-32 dependent inhibition of PP1 contributes to the increase in phosphorylation of htt at Ser421.
The level of Darpp-32 decreases in the striatum of YAC128 mice
Previous studies have shown a reduction in total expression of Darpp-32 in the striatum of HD mice that could contribute to loss of pS421-htt levels over time (Bibb et al., 2000; Leavitt et al., 2006). Therefore, we determined changes in Darpp-32 expression over time in YAC128 mice (Fig. 7A). These studies demonstrate significant reductions in Darpp-32 expression (35.1%, p < 0.032 at 6 months, n = 4; and 43%, p < 0.0048 at 12 months, n = 5) but not at earlier time points. No change in expression was apparent in YAC18 and C6R mice at all ages analyzed.
Figure 7.
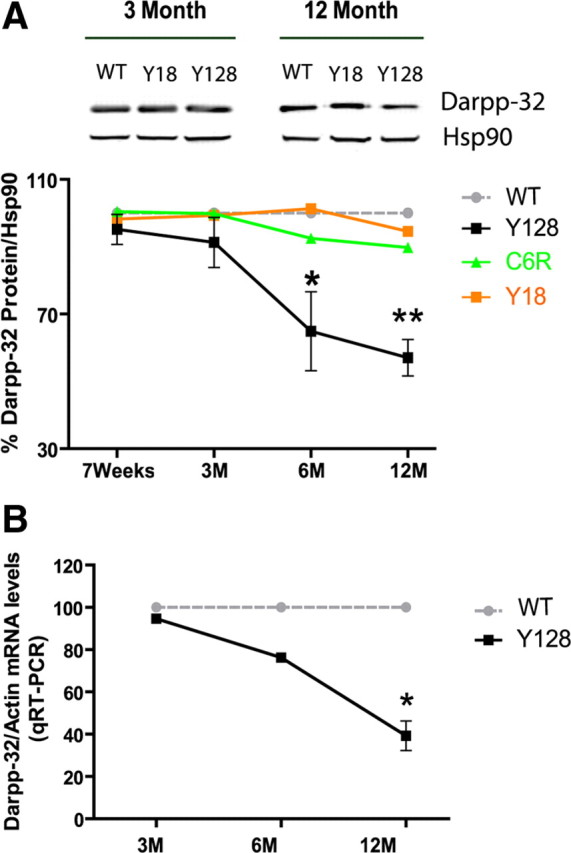
Early decrease in the level of Darpp-32 in the striatum of YAC128 mice. A, Striatal tissue was isolated at the indicated time points and protein lysates were analyzed by Western blot. Membranes were probed with Abs against Darpp-32 and Hsp90. The level of Darpp-32 relative to Hsp90 was determined and expressed relative to WT mice in each of three independent experiments. *p < 0.05, **p < 0.005 compared with WT, by t test. B, mRNA was extracted from the striatum of YAC128 transgenic mice and littermate controls at the indicated time points. Real-time quantitative RT-PCR was performed and the level of Darpp-32 mRNA expressed relative to β-actin. *p < 0.05 compared with WT, by t test. Error bars denote ±SEM.
Transcriptional changes have been observed in HD patients and in HD mice (Luthi-Carter et al., 2000; Hodges et al., 2006). To determine whether reduced expression of Darpp-32 results from altered transcription, we performed quantitative reverse transcription (RT)-PCR analyses (Fig. 7B). These studies demonstrate a trend toward a decrease in relative mRNA levels of striatal Darpp-32 in YAC128 compared with WT between 3 and 6 months of age (p < 0.58 at 3 months, n = 5; p < 0.19 at 6 months, n = 7). This decrease becomes significant at 12 months of age when mRNA levels of Darpp-32 are reduced by 60.8% in YAC128 mice (p < 0.03; n = 7 for YAC128 and n = 11 for WT).
Darpp-32-dependent regulation of PP1 activity is primarily determined by phosphorylation of Darpp-32 at Thr34. An increase in pT34-Darpp32 levels leads to greater inhibition of PP1. Therefore, we determined whether phosphorylation of T34-Darpp-32 is altered in YAC128 and control mice. These studies demonstrate normal pT34-Darpp-32 levels in YAC128 and WT mice at 7 weeks, 3 months, and 12 months of age (data not shown). However, a trend toward increased phosphorylation at T34–Darpp-32 is evident in the striatum of YAC128 mice at 12 months of age (p = 0.13 compared with WT; n = 5).
To analyze phosphatase activity directly, we determined PP1/PP2A and PP2B activity in the striatum of YAC128 and WT mice at 3 and 12 months of age. No significant changes in phosphatase activity were evident with the exception of PP2B activity that was significantly reduced in YAC128 mice at 12 months of age (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). In addition, we observed a small increase in PP1 and PP2A activity of 11% in YAC128 striatum at 12 months of age (p = 0.38; n = 4) (data not shown). However, this increase does not reach statistical significance. In summary, it is difficult to predict to which extent changes in phosphatase activity influence htt phosphorylation because of many confounding factors including early neuronal loss in YAC128 mice (Slow et al., 2003; Lerch et al., 2008).
Together, our findings provide evidence for dopaminergic dysfunction in HD because of loss of Darpp-32 expression at 6 and 12 months of age in YAC128 mice that may contribute to loss of pS421-htt.
Altered phosphorylation of CreB in YAC128 mice
PP1 and PKA regulate the phosphorylation state and activity of several substrates including CreB in response to glutamatergic and dopaminergic inputs into the striatum. Dopamine via stimulation of D1Rs and PKA leads to phosphorylation of CreB at Ser133 in the striatum, an effect that is attenuated in Darpp-32 knock-out mice (Das et al., 1997). At 3 months of age, the level of pS133-CreB was reduced by 17.1% in YAC128 striatum compared with littermate controls (p = 0.1; n = 4) (Fig. 8). This difference between genotypes became significant at 12 months of age when the level of pS133-CreB was reduced by 23% (p = 0.005; n = 3). No significant differences were observed in pS133-CreB levels in the cerebellum between YAC128 and littermate controls (Fig. 8). Again, the loss of phosphorylation of CreB at S133 precedes the reduction in Darpp-32 expression in YAC128 mice. Loss of CreB phosphorylation at Ser133 is developmentally regulated and robustly induced in mature neurons after excitotoxic stimulation of NMDARs (Sala et al., 2000). Therefore, one possible mechanism contributing to early reduction in pS133-CreB could result from enhanced NMDAR activation in neurons expressing mhtt.
Figure 8.
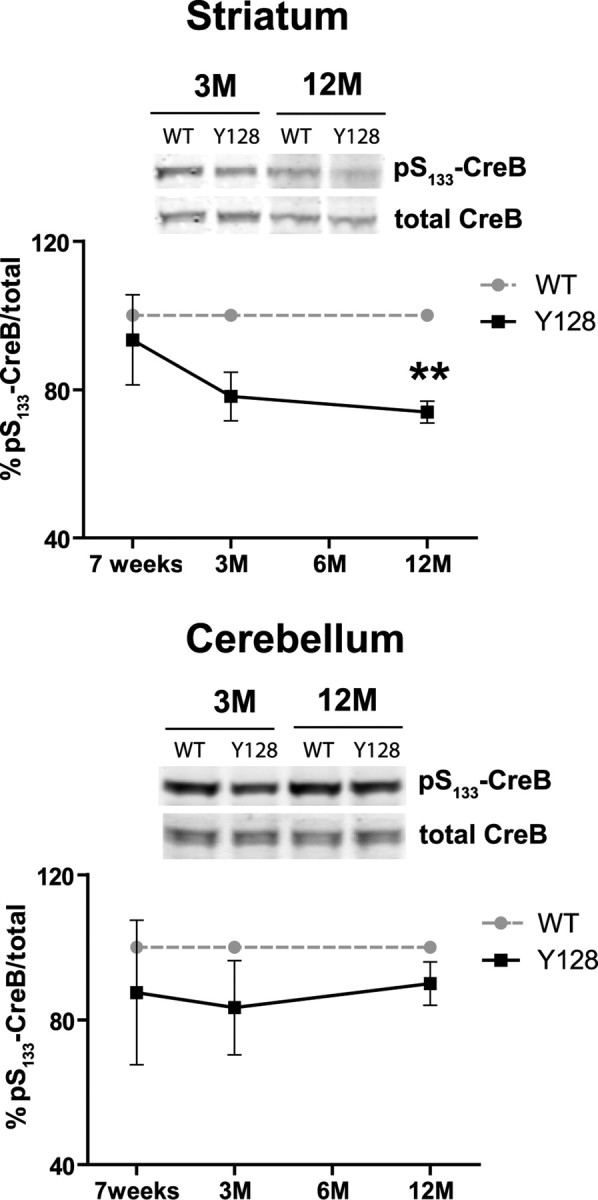
Altered phosphorylation of pS133-CreB in the striatum of YAC128 mice. Brain tissue was isolated at the indicated time points, and the level of pS133-CreB and total CreB was determined in striatal and cerebellar lysate by Western blot. Examples of Western blots for 3 and 12 month time points are shown for both tissues. The ratios of pS133-CreB and total CreB were determined and expressed relative to WT mice in each of three independent experiments. **p < 0.005 compared with WT, by t test. Error bars denote ±SEM.
Discussion
This study demonstrates that the level of htt phosphorylation at Ser421 is regulated through stimulation of NMDA and dopamine receptors and provides evidence that both input pathways are dysfunctional in the striatum of YAC128 mice. Our data demonstrate that the level of pS421-htt is influenced by Ser/Thr protein phosphatase PP1 and PP2A. Furthermore, our studies suggest that enhanced susceptibility to NMDAR-induced excitotoxicity and loss of Darpp-32 in YAC128 mice contributes to the progressive loss of pS421-htt and the decrease in phosphorylation of CreB. As a consequence, the prosurvival function of pS421-htt progressively declines in YAC128 mice and renders neurons increasingly dysfunctional. In contrast, we observe protection from NMDA-induced excitotoxicity by increasing the phosphorylation of htt at S421 through the blockade of PP1 and PP2A, suggesting that increasing the phosphorylation of htt may prove beneficial in protecting vulnerable neurons from enhanced excitotoxicity in vivo.
The underlying mechanism that accounts for the difference in pS421-htt levels between YAC128 and C6R mice is currently unknown. However, it is possible that blockade of cleavage at the caspase-6 site in mhtt restores normal NMDAR function in C6R mice (Graham et al., 2006). As recent studies suggest, it is also possible that htt plays a role as a caspase inhibitor, whose function is diminished in the presence of mhtt (Rigamonti et al., 2001; Hermel et al., 2004; Zhang et al., 2006). Again, through blockade of proteolytic cleavage at the caspase-6 cleavage site in mhtt, the caspase inhibitory function of htt may be restored in C6R mice favoring neuronal survival and the stimulation of survival pathways resulting in increased levels of pS421-htt.
The level of pS421-htt is decreased in YAC128 mice and influenced by enhanced NMDA-induced excitotoxicity
A steady decline in the level of total pS421-htt occurs in the striatum of YAC128 mice that correlates with age and CAG dependence in YAC128 mice. This phenotype is characterized by early cognitive dysfunction at 2 months of age, a deficit in motor performance and hyperactivity at 2–4 months of age, striatal neuronal loss clearly detectable at 8 months of age, and hypoactivity at 12 months of age, which recapitulates many features of the human disease (Slow et al., 2003; Van Raamsdonk et al., 2007; Lerch et al., 2008). At 2–3 months of age, YAC128 mice are more susceptible to excitotoxic stress (Graham et al., 2009) and this is associated with a greater loss of phosphorylation of htt at Ser421 in YAC128 neurons after NMDAR stimulation in vitro. Similarly, the total level of pS421-htt is reduced in YAC128 mice after QA injection into the striatum. Together, these results suggest that early enhanced susceptibility to excitotoxicity in YAC128 mice contributes to the early decline in pS421-htt levels. This is in agreement with the early reduction in CreB phosphorylation at Ser133 in YAC128 striatum and a recent study demonstrating increased extrasynaptic NMDAR signaling in YAC128 mice (Milnerwood et al., 2010).
Increased pS421-htt phosphorylation protects against NMDA-induced excitotoxicity
The loss of pS421-htt in YAC128 neurons during NMDA-induced excitotoxicity is not significantly altered by changes in PI3 kinase and MAP kinase activity but predominantly influenced by changes in PP1 and PP2A activity leading to dephosphorylation of pS133-CreB and reduced neuronal survival. Interestingly, we observe protection from NMDA-induced excitotoxicity in striatal neurons from YAC128 and WT neurons by blocking PP1 and PP2A activity and increasing the level of pS421-htt. This result is consistent with previous studies showing increased pS421-htt levels protect from cell death in a fragment model of HD (Humbert et al., 2002; Pardo et al., 2006). However, we cannot exclude that inhibition of PP1 and PP2A in the presence of OA increases the phosphorylation and activity of other proteins involved in the protection against NMDA-mediated cell death. Both increased phosphorylation of CreB and increased phosphorylation of Gsk3β is neuroprotective and enhanced in the presence of OA during NMDA-induced cell death (Szatmari et al., 2005). It is also possible that other cellular processes are affected in the presence of OA supporting neuronal survival. Importantly, expression of two phosphomimetic mutations of htt at Ser13 and Ser16 in HD BAC transgenic also prevents disease pathogenesis and highlights the potential therapeutic benefit of targeting multiple phosphorylation sites in htt involving distinct mechanisms of protection (Gu et al., 2009).
The NMDA-induced dephosphorylation of pS421-htt is blocked in a dose-dependent manner in the presence of OA and tautomycin, respectively, suggesting involvement of PP1 in this process. This is supported by the efficient dephosphorylation of N-terminal htt-1212-Q15 fragment and full-length htt by PP1 in situ (Fig. 4A,B). One possible mechanism that could explain NMDAR-dependent activation of PP1 is the Ca2+-dependent dephosphorylation of pT34-Darpp-32 by Ca2+-calmodulin-activated PP2B (Halpain et al., 1990). In addition, other PP2B-independent mechanisms of PP1 activation may participate in the NMDA-induced dephosphorylation of htt during excitotoxicity such as a feedback loop involving activation of Gsk3β and PP1 inhibitor 2 (Szatmari et al., 2005). Finally, we cannot rule out participation of PP2A in this process because of the limited specificity of OA and tautomycin and the observation that PP2A significantly dephosphorylates an N-terminal fragment and full-length htt in situ (Fig. 4A,B).
Inhibiting PP2B with either cyclosporin A or FK-506 did not significantly increase phosphorylation of S421-htt in cortical neurons after NMDAR stimulation. These data are in agreement with our finding that PP1 and PP2A dephosphorylate pS421-htt in situ, but no significant effects were observed in the presence of PP2B. Moreover, inhibiting PP1 and PP2A in the presence of OA and tautomycin under basal conditions increased the level of pS421-htt, suggesting that these phosphatases are involved in maintaining pS421-htt levels under normal physiological conditions (Fig. 4D) (data not shown). Although inhibition of PP2B with cyclosporin A increased the basal level of pS421-htt in cortical neurons, this effect was less pronounced compared with blockade of PP1 and PP2A and suggests a predominant role of these phosphatases in the dephosphorylation of pS421-htt.
In this study, we observed a reduction in PP2B activity in the striatum of YAC128 mice at 12 months of age without changes in PP2B expression (supplemental Fig. 7, available at www.jneurosci.org as supplemental material) (data not shown). This observation is in agreement with a previous study showing reduced PP2B activity in the striatum of R6/1 mice at 30 weeks of age but not at an earlier time point (Xifró et al., 2009). However, the significance of these findings is unclear because of many confounding factors including early neuronal loss in YAC128 mice (Slow et al., 2003; Lerch et al., 2008). In contrast, PP2B expression is increased in an immortalized striatal precursor cell line model of HD rendering neurons more susceptible to NMDA-induced toxicity (Xifró et al., 2008). Similarly, a reduction in neuronal cell death occurs after expression of a htt fragment with 68 poly(Q) (htt-480-68Q) in the presence of FK-506 or a catalytically dead dominant-negative form of calcineurin (Pardo et al., 2006). Furthermore, increased PP2B activity is observed in the cortex of HdhQ111/Q111 knock-in mice at 12 months of age (Pineda et al., 2009). However, these mice have a much slower onset and less severe HD phenotype compared with YAC128 mice (Ehrnhoefer et al., 2009).
Based on these observations, it is possible that the pathways leading to pathology in various HD mouse models involve different phosphatases and distinct mechanisms of phosphatase activation. Such mechanisms could involve the activation of a phosphatase through phosphorylation, dissociation of an inhibitor, or recruitment of an activator (Garcia et al., 2003; Mansuy and Shenolikar, 2006; Platholi et al., 2008). It is also possible that an inhibitor turns into an activator after NMDAR-dependent Ca2+ influx, which represents one mechanism of PP2A activation in the striatum (Ahn et al., 2007). Finally, we cannot entirely rule out the possibility that alterations in kinase activity contribute to the observed loss of htt phosphorylation in the striatum of YAC128 mice.
In summary, PP1 and PP2A activation could contribute to the loss of pS421-htt after excitotoxic NMDAR stimulation in HD.
Loss of Darpp-32 may influence the level of pS421-htt in HD
Another possible mechanism contributing to changes in pS421-htt levels in YAC128 mice may involve altered regulation of protein phosphatase activity resulting from loss of Darpp-32 expression in YAC128 mice. Darpp-32 is highly expressed in striatal medium-sized spiny neurons and integrates glutamatergic and dopaminergic inputs (Svenningsson et al., 2004). A substantial portion of PP1 is inhibited after stimulation of D1Rs, activation of PKA, and phosphorylation of Darpp-32 at Thr34 in the striatum (Svenningsson et al., 1998).
Interestingly, we observe that the level of pS421-htt increases in the striatum after stimulation with the D1R-specific agonist SKF-81297. This suggests regulation of pS421-htt levels through the dopaminergic system that results in all likelihood from Darpp-32-mediated inhibition of PP1. This is supported by the increase in pS421-htt level after injection of OA into the striatum and inhibition of PP1. These data suggest that the reduction in Darpp-32 expression at 6 months of age in YAC128 mice may contribute to an additional decrease in the level of pS421-htt as a consequence of loss of Darpp-32-mediated inhibition of PP1. However, PP1 and PP2A activity increased by only 11% in YAC128 striatum at 12 months of age. This increase did not reach significance and its effect on htt phosphorylation remains uncertain.
Interestingly, altered regulation of PP1 may not be limited to the CNS since PP1 activation in response to insulin promotes liver glycogen synthesis and dysfunction in glucose metabolism has been observed in a fragment model of HD (Hurlbert et al., 1999; Josefsen et al., 2008).
In the present study, we have shown regulation of pS421-htt levels by the glutamatergic and dopaminergic system, both of which are dysfunctional in HD. The resulting reduction in pS421-htt levels may increase cleavage of htt by caspase 6 and the generation of a toxic N-terminal htt fragment. This hypothesis is supported by the recent observation that htt cleavage is increased in the presence of phosphoresistant S421-htt (Warby et al., 2009). It is also possible that loss of htt phosphorylation influences the direction of intracellular vesicle transport and favors retrograde movement toward the nucleus reducing the secretion of important growth factors such as BDNF. This is in agreement with a recent study suggesting that dephosphorylation of htt at Ser421 functions as a molecular switch by releasing the anterograde motor kinesin-1 from microtubules and promoting vesicle transport along microtubules in retrograde direction (Colin et al., 2008).
Here, we have shown increased phosphorylation of pS421-htt protects YAC128 striatal neurons from NMDA-induced cell death, an early feature of HD. Similarly, phosphomimetic mutations of htt at Ser13 and Ser16 in HD BAC transgenic mice prevent disease pathogenesis (Gu et al., 2009). Together, these data provide evidence for phosphorylation as an important target for neuroprotection in HD.
Footnotes
This work was supported by Canadian Institutes of Health Research Grant MT-9133 (M.R.H.), the Huntington Disease Society of America, and a TREAT grant from the Cure Huntington Disease Initiative. R.K.G. is a recipient of a Michael Smith Foundation for Health Research fellowship. M.R.H., a Killam University Professor, holds a Canada Research Chair in Human Genetics. We are grateful to Lynn E. Raymond and Isabelle M. Mansuy for their comments on this manuscript. We thank Simon Warby for the pS421-htt antibody.
References
- Ahn JH, Sung JY, McAvoy T, Nishi A, Janssens V, Goris J, Greengard P, Nairn AC. The B″/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc Natl Acad Sci U S A. 2007;104:9876–9881. doi: 10.1073/pnas.0703589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P. Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci U S A. 2000;97:6809–6814. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Colin E, Zala D, Liot G, Rangone H, Borrell-Pagès M, Li XJ, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Grunert M, Williams L, Vincent SR. NMDA and D1 receptors regulate the phosphorylation of CREB and the induction of c-fos in striatal neurons in primary culture. Synapse. 1997;25:227–233. doi: 10.1002/(SICI)1098-2396(199703)25:3<227::AID-SYN1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Butland SL, Pouladi MA, Hayden MR. Mouse models of Huntington disease: variations on a theme. Dis Model Mech. 2009;2:123–129. doi: 10.1242/dmm.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-Methyl-d-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Garcia A, Cayla X, Guergnon J, Dessauge F, Hospital V, Rebollo MP, Fleischer A, Rebollo A. Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie. 2003;85:721–726. doi: 10.1016/j.biochi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gee CE, Mansuy IM. Protein phosphatases and their potential implications in neuroprotective processes. Cell Mol Life Sci. 2005;62:1120–1130. doi: 10.1007/s00018-005-5008-4. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Graham RK, Pouladi MA, Joshi P, Lu G, Deng Y, Wu NP, Figueroa BE, Metzler M, André VM, Slow EJ, Raymond L, Friedlander R, Levine MS, Leavitt BR, Hayden MR. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J Neurosci. 2009;29:2193–2204. doi: 10.1523/JNEUROSCI.5473-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Ogawa AK, Du X, Houk KN, Armstrong RW. A model for binding of structurally diverse natural product inhibitors of protein phosphatases PP1 and PP2A. J Med Chem. 1997;40:3199–3206. doi: 10.1021/jm960873x. [DOI] [PubMed] [Google Scholar]
- Halpain S, Girault JA, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harper PS. Huntington's disease: a clinical, genetic and molecular model for polyglutamine repeat disorders. Philos Trans R Soc Lond B Biol Sci. 1999;354:957–961. doi: 10.1098/rstb.1999.0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 2004;11:424–438. doi: 10.1038/sj.cdd.4401358. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Hurlbert MS, Zhou W, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington's disease gene develop diabetes. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- Josefsen K, Nielsen MD, Jørgensen KH, Bock T, Nørremølle A, Sørensen SA, Naver B, Hasholt L. Impaired glucose tolerance in the R6/1 transgenic mouse model of Huntington's disease. J Neuroendocrinol. 2008;20:165–172. doi: 10.1111/j.1365-2826.2007.01629.x. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–1129. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- Lerch JP, Carroll JB, Spring S, Bertram LN, Schwab C, Hayden MR, Henkelman RM. Automated deformation analysis in the YAC128 Huntington disease mouse model. Neuroimage. 2008;39:32–39. doi: 10.1016/j.neuroimage.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Shenolikar S. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci. 2006;29:679–686. doi: 10.1016/j.tins.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Metzler M, Gan L, Wong TP, Liu L, Helm J, Liu L, Georgiou J, Wang Y, Bissada N, Cheng K, Roder JC, Wang YT, Hayden MR. NMDA receptor function and NMDA receptor-dependent phosphorylation of huntingtin is altered by the endocytic protein HIP1. J Neurosci. 2007;27:2298–2308. doi: 10.1523/JNEUROSCI.5175-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo R, Colin E, Régulier E, Aebischer P, Déglon N, Humbert S, Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain. 2009;2:33. doi: 10.1186/1756-6606-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platholi J, Heerdt PM, Lim Tung HY, Hemmings HC., Jr Activation of brain protein phosphatase-1(I) following cardiac arrest and resuscitation involving an interaction with 14-3-3 gamma. J Neurochem. 2008;105:2029–2038. doi: 10.1111/j.1471-4159.2008.05300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Sipione S, Goffredo D, Zuccato C, Fossale E, Cattaneo E. Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J Biol Chem. 2001;276:14545–14548. doi: 10.1074/jbc.C100044200. [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M. Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci. 2000;20:3529–3536. doi: 10.1523/JNEUROSCI.20-10-03529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Sheppeck JE, 2nd, Gauss CM, Chamberlin AR. Inhibition of the Ser-Thr phosphatases PP1 and PP2A by naturally occurring toxins. Bioorg Med Chem. 1997;5:1739–1750. doi: 10.1016/s0968-0896(97)00146-6. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Lindskog M, Rognoni F, Fredholm BB, Greengard P, Fisone G. Activation of adenosine A2A and dopamine D1 receptors stimulates cyclic AMP-dependent phosphorylation of DARPP-32 in distinct populations of striatal projection neurons. Neuroscience. 1998;84:223–228. doi: 10.1016/s0306-4522(97)00510-1. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Szatmari E, Habas A, Yang P, Zheng JJ, Hagg T, Hetman M. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 2005;280:37526–37535. doi: 10.1074/jbc.M502699200. [DOI] [PubMed] [Google Scholar]
- Van Hoof C, Goris J. Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta. 2003;1640:97–104. doi: 10.1016/s0167-4889(03)00029-6. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J, Graham RK, Leavitt BR, Hayden MR. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- Warby SC, Doty CN, Graham RK, Shively J, Singaraja RR, Hayden MR. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol Cell Neurosci. 2009;40:121–127. doi: 10.1016/j.mcn.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Xifró X, García-Martínez JM, Del Toro D, Alberch J, Pérez-Navarro E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J Neurochem. 2008;105:1596–1612. doi: 10.1111/j.1471-4159.2008.05252.x. [DOI] [PubMed] [Google Scholar]
- Xifró X, Giralt A, Saavedra A, García-Martínez JM, Díaz-Hernández M, Lucas JJ, Alberch J, Pérez-Navarro E. Reduced calcineurin protein levels and activity in exon-1 mouse models of Huntington's disease: role in excitotoxicity. Neurobiol Dis. 2009;36:461–469. doi: 10.1016/j.nbd.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schütz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Leavitt BR, van Raamsdonk JM, Dragatsis I, Goldowitz D, MacDonald ME, Hayden MR, Friedlander RM. Huntingtin inhibits caspase-3 activation. EMBO J. 2006;25:5896–5906. doi: 10.1038/sj.emboj.7601445. [DOI] [PMC free article] [PubMed] [Google Scholar]