

Abstract
Developing neural circuits face the dual challenge of growing in an activity-induced fashion and maintaining stability through homeostatic mechanisms. Compared to our understanding of homeostatic regulation of excitatory synapses, relatively little is known about the mechanism mediating homeostatic plasticity of inhibitory synapses, especially that following activity elevation. Here, we found that elevating neuronal activity in cultured hippocampal neurons for 4 h significantly increased the frequency and amplitude of mIPSCs, before detectable change at excitatory synapses. Consistently, we observed increases in presynaptic and postsynaptic proteins of GABAergic synapses, including GAD65, vGAT, and GABAARα1. By suppressing activity-induced increase of neuronal firing with expression of the inward rectifier potassium channel Kir2.1 in individual neurons, we showed that elevation in postsynaptic spiking activity is required for activity-dependent increase in the frequency and amplitude of mIPSCs. Importantly, directly elevating spiking in individual postsynaptic neurons, by capsaicin activation of overexpressed TRPV1 channels, was sufficient to induce increased mIPSC amplitude and frequency, mimicking the effect of elevated neuronal activity. Downregulating BDNF expression in the postsynaptic neuron or its extracellular scavenging prevented activity-induced increase in mIPSC frequency, consistent with a role of BDNF-dependent retrograde signaling in this process. Finally, elevating activity in vivo by kainate injection increased both mIPSC amplitude and frequency in CA1 pyramidal neurons. Thus, spiking-induced, cell-autonomous upregulation of GABAergic synaptic inputs, through retrograde BDNF signaling, represents an early adaptive response of neural circuits to elevated network activity.
Introduction
During development, the formation of functional connections between neurons is accompanied by increased neuronal activity, from a combination of spontaneous and synaptic events, as well as secreted trophic factors (Katz and Shatz, 1996). This elevated activity, through correlated presynaptic and postsynaptic spiking, leads to strengthening of synaptic connections via Hebbian-type plasticity (Bi and Poo, 2001). However, the same mechanism, if unchecked, could lead to overexcitation of neurons (Abbott and Nelson, 2000). Homeostatic synaptic plasticity serves as a compensatory negative feedback mechanism for keeping neuronal spiking within the operating range following change in overall neural activity (Burrone and Murthy, 2003; Turrigiano and Nelson, 2004; Davis, 2006; Rich and Wenner, 2007).
According to homeostatic rules, elevation of neuronal activity results in compensatory reduction of excitatory neurotransmission and enhancement of inhibition. Compared to our knowledge of mechanisms underlying homeostatic regulation of excitatory synaptic strength (Burrone and Murthy, 2003; Davis, 2006; Rich and Wenner, 2007; Turrigiano, 2008; Yu and Goda, 2009), relatively little is known about those regulating inhibitory synapses (Mody, 2005; Erickson et al., 2006), especially following activity elevation. Previous work showed that during chronic activity blockade, homeostatic regulation of inhibitory synapses required change in the activity level of multiple neurons within the network (Hartman et al., 2006), suggesting that homeostatic regulation of inhibitory synapses following activity blockade is non-cell-autonomous. This is in contrast to recent reports of local homeostatic regulation of excitatory synapses following activity blockade (Burrone et al., 2002; Ibata et al., 2008; Yu and Goda, 2009). Whether homeostatic plasticity of inhibitory synapses could occur more locally under certain circumstances, such as following activity elevation, remains unknown.
Using dissociated hippocampal neuronal cultures, we found that elevating global neuronal activity for 4 h significantly increased the amplitude and frequency of mIPSCs, before detectable change at excitatory synapses. Importantly, activity-induced increase in the spiking activity of individual postsynaptic neurons was necessary and sufficient for inducing homeostatic upregulation of GABAergic synaptic inputs. We further demonstrated that brain-derived neurotrophic factor (BDNF) likely serves as a retrograde messenger for spiking-induced signaling to presynaptic GABAergic terminals. Finally, our main findings in cultured neurons could be replicated following in vivo injection of kainic acid (KA) in juvenile rats. Together, these results demonstrated that during elevated neuronal activity, an individual neuron, by sensing its own increased firing, can rapidly upregulate its inhibitory synaptic inputs through BDNF-dependent retrograde signaling, thereby self-compensating for the increased excitation within the circuit.
Materials and Methods
Hippocampal neuronal culture preparation and transfection.
High-density mixed neuronal–glial cultures were prepared from postnatal day 0 (P0) Sprague Dawley rat pups as previously described (Yu and Malenka, 2003) and according to procedures approved by the Institutional Animal Care and Use Committee of the Institute of Neuroscience, Chinese Academy of Sciences (Shanghai, China). Briefly, hippocampal preparations were plated on Matrigel (BD Biosciences)-coated glass coverslips (Assistent) at 35,000–50,000 cells/cm2 in medium consisting of Neurobasal medium (Invitrogen), B-27 (Invitrogen), and Glutamax-I (Invitrogen). On the third day in vitro (DIV 3), when astrocytes had formed a monolayer over the entire coverslip, cells were treated with the mitotic inhibitor 5-fluoro-2′-deoxyuridine (FUDR, Sigma). Calcium phosphate transfections were performed at DIV 7–9 using 2–4 μg of DNA per well in a 24-well plate. A plasmid encoding GFP (0.2–0.7 μg) was included in all mixtures for identification of transfected neurons. The low efficiency of this method ensures that the transfected neuron is surrounded by untransfected presynaptic contacts. For experiments in Figure 5O–R, to overexpress TrkB-T1 in a large percentage of cells, neurons were electroporated with GFP or GFP plus TrkB-T1 using Amaxa Nucleofector Device II (Lonza) at the time of plating. All electrophysiological and immunocytochemical experiments were performed at DIV 12–14.
Figure 5.
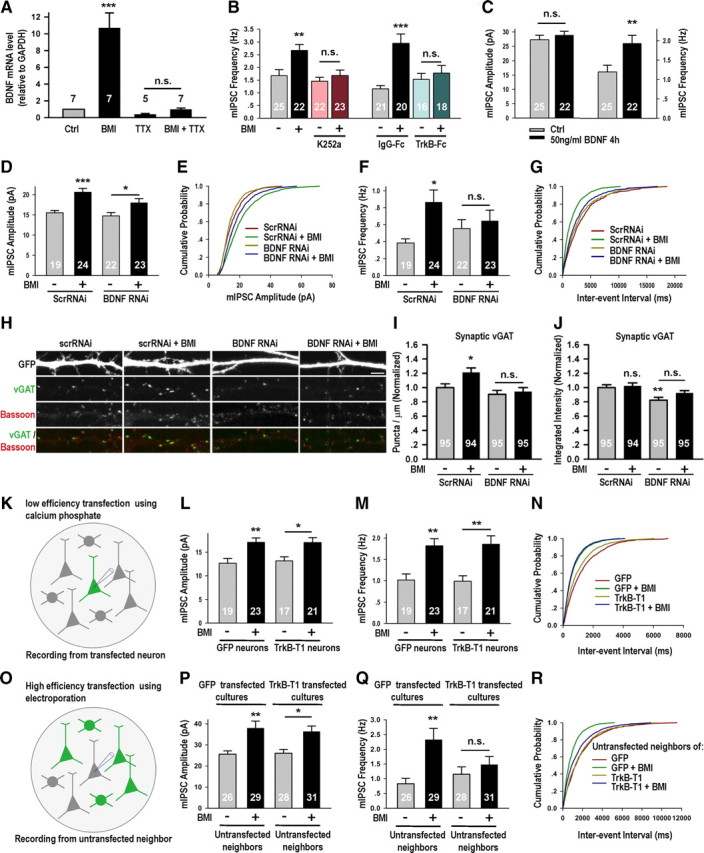
BDNF signaling was critical for activity-induced presynaptic changes at GABAergic synapses. A, BMI-induced upregulation of BDNF mRNA (10.70 ± 1.78-fold, p < 0.001) was blocked by coapplication of TTX (TTX: 0.40 ± 0.09-fold, p > 0.05 vs Ctrl; TTX + BMI: 0.97 ± 0.17, p > 0.05 vs TTX). B, K252a blocked BMI-induced increase in mIPSC frequency (Ctrl: 1.68 ± 0.23 Hz; BMI: 2.67 ± 0.24 Hz, p < 0.01 vs Ctrl; K252a: 1.45 ± 0.16 Hz, p > 0.05 vs Ctrl; BMI + K252a: 1.68 ± 0.22 Hz, p > 0.05 vs K252a); TrkB-Fc also blocked BMI-induced increase in mIPSC frequency (IgG-Fc: 1.16 ± 0.12 Hz; IgG-Fc + BMI: 2.95 ± 0.37 Hz, p < 0.001 vs IgG-Fc; TrkB-Fc: 1.53 ± 0.23 Hz, p > 0.05 vs IgG-Fc; TrkB-Fc + BMI, 1.78 ± 0.30 Hz, p > 0.05 vs TrkB-Fc). C, Four hour BDNF treatment did not affect mIPSC amplitude, but significantly increased its frequency (Ctrl: 1.19 ± 0.17 Hz, BDNF: 1.93 ± 0.21 Hz, p < 0.01). D, BMI increased the mIPSC amplitude of both neurons transfected with scrambled RNAi (ScrRNAi: 15.52 ± 0.58 pA; ScrRNAi + BMI: 20.67 ± 0.91 pA, p < 0.001) and those transfected with BDNF RNAi (BDNF RNAi: 14.75 ± 0.83 pA, p > 0.05 vs ScrRNAi; BDNF RNAi + BMI, 18.02 ± 1.01 pA, p < 0.05 vs BDNF RNAi). E, Corresponding cumulative distributions: ScrRNAi versus ScrRNAi + BMI, p < 0.001; BDNF RNAi versus BDNF RNAi + BMI, p < 0.01. F, BMI increased the mIPSC frequency of neurons transfected with ScrRNAi (ScrRNAi: 0.38 ± 0.05 Hz; ScrRNAi + BMI: 0.87 ± 0.14 Hz, p < 0.05), an effect blocked by BDNF RNAi (BDNF RNAi: 0.55 ± 0.10 Hz; BDNF RNAi + BMI: 0.64 ± 0.13 Hz, p > 0.05 vs BDNF RNAi). G, Corresponding cumulative distributions: ScrRNAi versus ScrRNAi + BMI, p < 0.001; BDNF RNAi versus BDNF RNAi + BMI, p = 0.41. H–J, Representative images and quantifications of the average density and integrated intensity of synaptic vGAT in neurons transfected with ScrRNAi or BDNF RNAi, in Ctrl or BMI conditions. Scale bar is 5 μm. I, BMI treatment increased the density of synaptically localized vGAT puncta (1.21 ± 0.07, p < 0.05), an effect blocked by BDNF RNAi (BDNF RNAi: 0.91 ± 0.05, p > 0.05 vs ScrRNAi; BDNF RNAi + BMI: 0.94 ± 0.06, p > 0.05 vs BDNF RNAi). J, BMI treatment did not affect the integrated intensity of synaptic vGAT in neurons transfected with ScrRNAi (ScrRNAi + BMI 1.02 ± 0.04, p > 0.05) or BDNF RNAi (BDNF RNAi: 0.82 ± 0.04, p < 0.01 vs ScrRNAi; BDNF RNAi + BMI: 0.92 ± 0.04, p > 0.05 vs ScrRNAi, p > 0.05 vs BDNF RNAi). K–N, Postsynaptic TrkB-T1 did not block BMI-induced increase in mIPSC amplitude or frequency. K, An illustration showing that recording was made from a low-density transfected neuron surrounded by many untransfected neurons. L, BMI increased the mIPSC amplitude of both neurons transfected with GFP (Ctrl: 12.66 ± 1.00 pA; Ctrl + BMI: 17.11 ± 0.92 pA, p < 0.01) and those transfected with TrkB-T1 (TrkB-T1: 13.16 ± 0.86 pA, p > 0.05 vs Ctrl; TrkB-T1 + BMI, 17.06 ± 1.04 pA, p < 0.05 vs TrkB-T1). M, BMI increased the mIPSC frequency of both neurons transfected with GFP (Ctrl: 1.02 ± 0.14 Hz; Ctrl + BMI: 1.82 ± 0.17 Hz, p < 0.01), and those transfected with TrkB-T1 (TrkB-T1: 0.99 ± 0.13 Hz, p > 0.05 vs Ctrl; TrkB-T1 + BMI: 1.86 ± 0.20 Hz, p < 0.01 vs TrkB-T1). N, Cumulative distributions of mIPSC frequency: Ctrl versus BMI, p < 0.001; TrkB-T1 versus TrkB-T1 + BMI, p < 0.001. O–R, Interfering with presynaptic BDNF signaling with high-efficiency TrkB-T1 transfection significantly blocked the BMI-induced increase of mIPSC frequency. O, An illustration showing that recording was made from an untransfected neuron surrounded by several transfected neurons. P, BMI increased the mIPSC amplitude of both untransfected neurons neighboring GFP-expressing neurons (GFP neighbor: 25.62 ± 1.57 pA; GFP neighbor + BMI: 37.98 ± 3.34 pA, p < 0.01), and untransfected neurons neighboring TrkB-T1-expressing neurons (TrkB-T1 neighbor: 25.95 ± 1.75 pA, p > 0.05 vs GFP neighbor; TrkB-T1 neighbor + BMI: 36.36 ± 2.59 pA, p < 0.05 vs TrkB-T1 neighbor). Q, BMI-induced increase in mIPSC frequency (GFP neighbor: 0.83 ± 0.18 Hz; GFP neighbor + BMI: 2.33 ± 0.39 Hz, p < 0.01) was significantly blocked by expression of TrkB-T1 in neighboring neurons (TrkB-T1 neighbor: 1.16 ± 0.25 Hz, p > 0.05 vs GFP neighbor; TrkB-T1 neighbor + BMI: 1.48 ± 0.28 Hz, p > 0.05 vs TrkB-T1 neighbor). R, Corresponding cumulative distributions: GFP neighbor versus GFP neighbor + BMI, p < 0.001; TrkB-T1 neighbor versus TrkB-T1 neighbor + BMI, p = 0.10. *p < 0.05, **p < 0.01, ***p < 0.001.
DNA constructs and pharmacological treatments.
The coding sequences of Kir2.1, mKir2.1 (gifts of Dr. Chun-lei Wang, Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China) (Wang et al., 2007), TRPV1 (Caterina et al., 1997) (gift from Dr. David Julius, University of California, San Francisco, San Francisco, CA), and TrkB-T1 (Gonzalez et al., 1999) (gift from Dr. Rita Balice-Gordon, University of Pennsylvania, Philadelphia, PA) were subcloned into pCS2. BDNF RNAi, directed against sequence GGCACTGGAACTCGCAATG, and its scrambled control GGATATGGGCTCACTGAGA were generated using published sequences (Zhou et al., 2007) and cloned into pSuper (Zhang et al., 2007). FLAG-TrkB-GFP (Zhao et al., 2009) was obtained from Dr. Z.-G. Luo (Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China) with permission from Dr. Z.-Y. Chen (Shandong University, Jinan, China). Pharmacological treatments included the following: 20 μm bicuculline methobromide (BMI, Tocris Bioscience), 0.5 μm tetrodotoxin (TTX, Fisheries Science and Technology Development), 5 μm 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX, Tocris Bioscience), 10 μm kainic acid (Tocris Bioscience), 50 nm capsaicin (Sigma), 50 ng/ml BDNF (Alomone Labs), 100 nm K252a (Sigma), 2 μg/ml TrkB-Fc (R&D systems), and 2 μg/ml IgG-Fc (Jackson ImmunoResearch). For TrkB-Fc treatment, neurons were preincubated with TrkB-Fc or IgG-Fc for 30 min before BMI treatment. To significantly increase the firing rates of TRPV1-expressing neurons, compared to untransfected neighboring neurons, 5 μm NBQX was added together with 50 nm capsaicin or vehicle to reduce the basal firing rates in all experimental conditions.
Electrophysiological recordings in cultured neurons.
Whole-cell patch-clamp recordings in hippocampal neuronal cultures were performed at room temperature on DIV 12–14 neurons with Multiclamp 700B amplifier (Molecular Devices) using low-resistance pipettes (2–5 MΩ). Neurons were visualized with a phase-contrast inverted microscope (Nikon TE2000-S). Pyramidal neurons were identified morphologically, based on the classically pyramidal shape and an apical-like (larger diameter) dendrite from the soma. The extracellular solution contained (in mm) NaCl 129, KCl 5, glucose 30, HEPES 25, CaCl2 2, and MgCl2 1 (pH 7.3, 310 mOsm). For mIPSC recordings, the intracellular solution contained (in mm) CsCl 110, HEPES 40, NaCl 10, MgCl2 5, EGTA 0.6, MgATP 2, and Na3GTP 0.2 (pH 7.25, 291 mOsm); 0.5 μm TTX and 5 μm NBQX were added to the extracellular solution. For mEPSC and spontaneous firing recordings, the intercellular solution contained (in mm) K-gluconate 110, HEPES 20, KCl 20, MgCl2 5, EGTA 0.6, MgATP 2, and Na3GTP 0.2 (pH 7.3, 290 mOsm); 0.5 μm TTX and 50 μm picrotoxin were added to the extracellular solution for mEPSC recordings. Firing rates were recorded following direct application of 20 μm BMI or 50 nm capsaicin to the extracellular solution. For paired-pulse ratio measurements, pairs of pulses (at 25–200 ms intervals) were generated with Master 8 (A.M.P.I.), and IPSCs were locally evoked with a concentric bipolar electrode (FHC). For recording GABA-induced currents, 100 μm GABA was focally applied to the soma through a micropipette.
Neurons were voltage clamped at −60 mV or −70 mV for mIPSC and mEPSC recordings, respectively. Data were filtered at 2 kHz and sampled at 10 kHz using Digidata 1322A (Molecular Devices). Analysis of miniature PSCs was performed blindly using Mini Analysis Program (Synaptosoft) with an amplitude threshold of 3 pA for mEPSCs and 5 pA for mIPSCs. Cumulative distributions were generated using 150 consecutive mIPSCs (100 for Fig. 5E,G) averaged across all cells, and compared using the Kolmogorov–Smirnov two-sample test. Cellular input and series resistances were monitored by giving a hyperpolarizing pulse (−5 mV, 20 ms) through the patch-clamp electrode every 10 s. Recordings where either parameter altered by >20% during the course of the recording were excluded from the analyses.
All recordings were performed with interleaved controls (vehicle, transfection control, or untreated) from sister culture preparations. At least two independent culture preparations were used for each experimental condition. For experiments presented in Figure 1A–K, to keep the duration of BMI treatment precise, recordings for each time point were interleaved with their corresponding controls from sister cultures. In all experiments, neurons were perfused with extracellular solution for at least 3 min to wash out remaining pharmacological reagents, and no more than 3 neurons were recorded from per coverslip.
Figure 1.

Global elevation of neuronal activity for 4 h enhanced mIPSC amplitude and frequency. A–C, L, Representative mIPSC recordings and average waveforms for conditions as indicated. The scale bars are 20 pA and 1 s for the sweeps, and 5 pA and 20 ms for the averaged traces. D, Graph of average mIPSC amplitudes (normalized to respective controls) from neurons treated with BMI for 2 h (1.11 ± 0.05, p = 0.14), 4 h (1.21 ± 0.04, p < 0.001), or 12 h (1.28 ± 0.06, p < 0.001). For each time point, values were normalized to those of control neurons from sister cultures, n ≥ 20 neurons for each control condition. E–G, Cumulative distributions of mIPSC amplitude plotted versus respective controls, p = 0.08 (E), p < 0.001 (F), p < 0.001 (G). The insets (F, G) are the scaled BMI distributions as compared with Ctrl (p = 1.0 for both). H–J, Cumulative distributions of mIPSC frequency plotted versus respective controls, p = 0.42 (H), p < 0.001(I), p < 0.001 (J). K, Graph of normalized mIPSC frequency from neurons treated with BMI for 2 h (1.35 ± 0.19, p = 0.21), 4 h (2.52 ± 0.37, p < 0.001), or 12 h (2.50 ± 0.55, p < 0.05). M, K+ treatment significantly increased mIPSC amplitude (Ctrl: 23.26 ± 1.45 pA; 10 mm K+: 30.51 ± 1.87 pA, p < 0.01); KA treatment significantly increased mIPSC amplitude (Ctrl: 15.46 ± 0.57 pA, 10 μm KA: 19.43 ± 0.85 pA, p < 0.001). N, K+ treatments significantly increased mIPSC frequency (Ctrl: 1.75 ± 0.27 Hz; 10 mm K+: 4.01 ± 0.38 Hz, p < 0.001), KA treatment significantly increased mIPSC frequency (Ctrl: 1.63 ± 0.21 Hz, 10 μm KA: 3.03 ± 0.24 Hz, p < 0.001). In this and subsequent graphs, error bars represent SEM; number of neurons as indicated in bar graphs. *p < 0.05, **p < 0.01, ***p < 0.001.
Immunocytochemistry.
Cultured neurons were thoroughly washed in PBS before 20 min fixation in 4% paraformaldehyde (Sigma) in PBS at room temperature. Following 5 min of membrane permeabilization with 0.1% Triton X-100 in PBS, neurons were blocked with 3% bovine serum albumin (BSA, Calbiochem) in PBS for 1 h at 37°C. Primary and secondary antibody incubations (both in 3% BSA) were, respectively, overnight at 4°C and 2 h at 37°C. Primary antibodies included the following: Bassoon (mouse, Stressgen, 1:1000), Bassoon (guinea pig, Synaptic Systems, 1:500), GABAARα1 (rabbit, Alomone Labs, 1:200), GAD-6 (mouse monoclonal specific for GAD65, Developmental Studies Hybridoma Bank, 1:500), vGAT (rabbit, Synaptic Systems, 1:1000), MAP2 (chick, Millipore, 1:1000), and synapsin I (rabbit, Millipore, 1:1000). Alexa Fluor 488, 568, or 633 secondary antibodies (Invitrogen) were used at 1:1000. Coverslips were mounted in Fluoromount-G (Electron Microscopy Sciences).
Image acquisition and analysis.
Images (Z-stacks) were acquired on a Zeiss LSM PASCAL laser scanning confocal microscope with a 63× oil-immersion Plan-Apochromat objective (N.A. = 1.4) at 2× optical zoom. Pyramidal neurons were identified morphologically, based on the classically pyramidal shape and an apical-like (larger diameter) dendrite from the soma. For each neuron, a dendritic segment close to the soma and of comparable diameter, typically 50–200 μm in length, was selected for analysis with the help of GFP fluorescence or MAP2 immunostaining: puncta were included if they had at least one pixel of overlap with GFP or MAP2. Maximal projections were analyzed with Image-Pro Plus (Media Cybernetics) software and customized macros. Briefly, images for each channel were thresholded, and only puncta having at least one pixel of overlap with the active zone protein Bassoon were considered synaptically localized. Synapse density corresponded to the number of puncta per micrometer of dendrite, while integrated intensity measured the summed intensity of all pixels above threshold in each punctum, averaged across all puncta on the selected dendritic branch.
All image analyses were performed blind to the experimental condition. Each experiment was performed using at least two independent culture preparations, with three coverslips per condition, and an average of 6–9 cells per coverslip. An untreated, vehicle, or transfection control was included in each experiment to normalize the data across culture preparations. Images were not adjusted for brightness or contrast for data analyses; for example images, when such adjustments were made, images of all parallel experimental conditions were equally adjusted.
Kainic acid treatment of juvenile rats and electrophysiological recordings in acute hippocampal slices.
Littermate P12–P14 Sprague Dawley rats (30–40 g) were intraperitoneally injected with 4 mg/kg KA or as control, an equal volume of saline (0.9% NaCl) according to procedures approved by the Institutional Animal Care and Use Committee of the Institute of Neuroscience, Chinese Academy of Sciences (Shanghai, China). This intermediate KA dose (Lothman and Collins, 1981) induced acute behavior changes, including mouth and facial movements, wet-dog shakes, and forelimb jerks starting 15 min after KA injection. By 4 h, rodents in the KA group had mostly recovered, but were hyperactive compared to saline-injected controls, which remained dormant. Previous work has demonstrated that KA treatment of juvenile rats induces seizures without apparent neuronal damage (Ben-Ari, 1985, 2008). One or four hours after injections, rats were deeply anesthetized with 0.7% sodium pentobarbital, and slices were prepared as previously described (van Praag et al., 2002). Briefly, brains were rapidly removed and immersed in ice-cold dissection buffer containing the following (in mm): choline-Cl 110, KCl 2.5, NaH2PO4 1.3, MgCl2 7, CaCl2 0.5, NaHCO3 25, and glucose 15, bubbled with 95% O2/5% CO2. Hippocampal slices were cut at 350 μm using a Vibratome 3000 (Leica) microslicer and allowed to recover in a submersion holding chamber with artificial CSF (ACSF, in mm: NaCl 125, KCl 2.5, NaH2PO4 1.3, MgCl2 1.3, CaCl2 2, NaHCO3 25, and glucose 15) bubbled with 95% O2/5% CO2 mixture for at least 1 h before recordings. Slices were visualized with an upright microscope (Nikon FN1) equipped with infrared differential interference contrast. IPSCs were recorded from CA1 pyramidal neurons at −60 mV and 25°C; 5 μm NBQX and 0.5 μm TTX were added for mIPSC recordings. IPSCs were evoked using a bipolar tungsten stimulating electrode (WPI Instruments), with the stimulating electrode placed in the stratum radiatum, ∼50 μm away from the recorded cell. Responses were evoked with a Master-8 pulse generator (A.M.P.I.) coupled through an Iso-flex isolator. Spontaneous spiking activity was recorded at 35°C in current-clamp mode and with modified ACSF containing the following (in mm): NaCl 125, KCl 3.5, CaCl2 1, MgCl2 0.5, NaH2PO4 1.3, NaHCO3 25, and glucose 15. A small amount of bias current was injected to bring to −60 mV the resting membrane potential, which was then allowed to fluctuate freely in response to spontaneous synaptic events (Maffei and Turrigiano, 2008). Internal solutions were the same as those used for cultured neurons (see above).
Western blot analysis.
Four hours after injections with KA or saline (see above), rats were deeply anesthetized with 0.7% sodium pentobarbital. Hippocampi were dissected out and homogenized in ice-cold HEPES-buffered sucrose (0.32 m sucrose and 4 mm HEPES, pH 7.4), containing protease inhibitor cocktail tablets (Roche). Western blot was performed according to standard protocols, and results were quantified using ImageJ software (NIH Image). Primary antibodies included the following: GAPDH (1:10,000, Kangchen Bio-tech) and BDNF (1:1000, Santa Cruz Biotechnology).
Quantitative real-time PCR.
Total RNA from DIV 12 neurons was isolated using the TRIzol reagent (Invitrogen) and reverse transcribed by oligodT priming using SuperScriptIII reverse transcriptase (Invitrogen) following the manufacturer's protocols. Specific primers for BDNF (forward AGCGTGTGTGACAGTATTAGCGAGT, reverse CTATCTTCCCCTTTTAATGGTCAGT) and the housekeeping gene GAPDH (forward CTGCCCAGAACATCATCCCT, reverse TGAAGTCGCAGGAGACAACC) were used. Real-time PCRs were performed using SYBR Green Master Mix (TaKaRa) on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems). All reactions were performed in triplicates, and the relative amount of mRNA was calculated using the comparative CT method (Applied Biosystems, 2001).
Statistical analysis.
For all electrophysiology and immunocytochemistry experiments, statistical analyses were performed using two-tailed unpaired Student's t test (for sample pairs), or one-way ANOVA (for three or more conditions), followed by Tukey's multiple-comparison test. Cumulative distributions were compared using the Kolmogorov–Smirnov two-sample test. Results are shown as mean ± SEM, and “n” represents number of cells as indicated. For Western blots, “n” represents the number of sample pairs (2–4 rats per condition). All results statistically different from the control condition are marked. *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Elevation of neuronal activity increased GABAergic synaptic function within 4 h
Global elevation of neuronal activity through prolonged incubation (48 h) with the GABAA receptor (GABAAR) antagonist bicuculline homeostatically downregulated excitatory synaptic strength (Turrigiano et al., 1998) and upregulated inhibitory synaptic strength (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Given the potential detrimental effect of prolonged neuronal hyperexcitation, we surmised that some other homeostatic regulatory mechanism may come into action more rapidly. To investigate the time course of synaptic changes following increased neuronal activity, we treated dissociated hippocampal neuronal cultures (DIV 12–14) for different durations with BMI (20 μm), a water soluble form of bicuculline, and examined its effect on the mIPSCs of pyramidal neurons by whole-cell recording. To keep the duration of BMI treatment precise, for each time point, we recorded from sister cultures of BMI-treated neurons and interleaved controls, and normalized across culture preparations using the control values of each experiment. We found that incubation with BMI for 4 or 12 h, but not 2 h, was sufficient to significantly elevate both the amplitude and frequency of mIPSCs, as shown by the average values and corresponding cumulative probability distributions (Fig. 1A–K). No significant changes in the kinetics of mIPSCs were detected (supplemental Table 1, available at www.jneurosci.org as supplemental material). Importantly, mIPSC amplitude scaled multiplicatively following both 4 and 12 h of BMI treatment (Fig. 1F,G, inset), consistent with uniform change in synaptic strength across all inhibitory synapses. Since 4 h BMI treatment robustly and consistently induced increases in mIPSC amplitude and frequency, we focused on this time point for the rest of our study. In contrast to change in inhibitory synaptic transmission, 4 h BMI treatment did not significantly affect the amplitude and frequency of mEPSCs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), suggesting that change in inhibitory synaptic transmission might act as a first homeostatic responder to increase in global activity level.
To determine whether BMI-induced increase in inhibitory synaptic transmission represented a general effect of global activity elevation, we used two alternative means of increasing neuronal activity. Similar to BMI treatment, depolarizing neurons with elevated extracellular K+ (10 mm) increased neuronal firing (supplemental Table 2, available at www.jneurosci.org as supplemental material) and significantly elevated the amplitude and frequency of mIPSCs after 4 h treatment (Fig. 1L–N). Elevated mIPSC amplitude and frequency were also observed when neurons were treated with the excitatory amino acid analog kainic acid (10 μm) (Fig. 1L–N) for 4 h. Thus, elevation of neuronal activity by three independent manipulations, including depolarization, reducing inhibitory synaptic transmission and increasing excitatory synaptic transmission, all resulted in homeostatic upregulation of inhibitory synaptic transmission within 4 h.
Elevated neuronal activity increased GABAergic synapse density
To investigate whether presynaptic and/or postsynaptic changes underlie activity-induced enhancement of inhibitory synaptic function, we examined the effect of 4 h BMI treatment on the level of GABAergic presynaptic and postsynaptic components by quantitative immunocytochemistry. Defining synaptic puncta by colocalization with the active zone marker Bassoon, we found significant increases in both the density (number per unit length of dendrite) and integrated puncta intensity of the GABA-synthesizing enzyme GAD65 following activity elevation (Fig. 2A,D,E; supplemental Fig. 3, available at www.jneurosci.org as supplemental material), suggesting enhanced presynaptic function at GABAergic synapses. The synaptic puncta density of the vesicular GABA transporter vGAT also increased, although its integrated puncta intensity did not alter significantly (Fig. 2B,D,E). This is consistent with a previous report of increased vGAT expression following more prolonged activity elevation (De Gois et al., 2005). On the postsynaptic side, we found significant increases in both the density and integrated intensity of synaptic GABAARα1 puncta (Fig. 2C–E). Enhancement in the integrated intensity of individual GABAARα1 puncta, together with increased whole-cell responses to extracellularly applied GABA (supplemental Fig. 4A,B, available at www.jneurosci.org as supplemental material), suggested that an increase in GABAAR number at individual synapses could account for the observed increase in mIPSC amplitude. Increase in the density of GABAergic presynaptic and postsynaptic components, together with only a trend toward reduced paired-pulse ratio of IPSCs (supplemental Fig. 4C,D, available at www.jneurosci.org as supplemental material), suggested that the increase in mIPSC frequency mostly resulted from an increase in synapse number, without excluding additional change in the probability of transmitter release.
Figure 2.

Elevated neuronal activity increased the level of presynaptic and postsynaptic components of GABAergic synapses. A–C, Representative images showing dendrites (labeled with MAP2) of control and BMI-treated neurons, coimmunolabeled for Bassoon and presynaptic and postsynaptic components of inhibitory synapses as indicated; scale bar is 5 μm. D, BMI treatment increased the density of GAD65 (1.39 ± 0.09, p < 0.001), vGAT (1.29 ± 0.07, p < 0.01), and GABAARα1 (1.28 ± 0.09, p < 0.05) puncta colocalizing with Bassoon, as normalized to untreated neurons. E, BMI treatment increased the integrated intensity of synaptically localized GAD65 (1.24 ± 0.06, p < 0.01) and GABAARα1 (1.19 ± 0.07, p < 0.05) puncta, but did not affect that of synaptically localized vGAT puncta (1.03 ± 0.05, p = 0.58). F, BMI treatment increased the percentage of inhibitory synapses as measured by the percentage (normalized) of total Bassoon puncta number colocalizing with GAD65 (1.19 ± 0.06, p < 0.01), vGAT (1.14 ± 0.04, p < 0.05), or GABAARα1 (1.17 ± 0.05, p < 0.05). *p < 0.05, **p < 0.01, ***p < 0.001.
For all components of GABAergic synapses examined, significant increase in their contribution to total synapse number (as marked by Bassoon) was observed following BMI treatment (Fig. 2F). This result was consistent with increased mIPSC frequency (Fig. 1K) and GABAergic synapse density (Fig. 2D) following elevated neuronal activity.
Postsynaptic spiking was required for change at GABAergic synapses
Since spiking activity was significantly and rapidly elevated upon bicuculline application (Turrigiano et al., 1998) (supplemental Table 2, available at www.jneurosci.org as supplemental material), we asked whether this change was required for activity-induced enhancement of inhibitory synaptic transmission. We found that 4 h coincubation of BMI with TTX (0.5 μm), which completely abolished firing, prevented BMI-induced increases in both mIPSC amplitude and frequency (Fig. 3A,B). Although TTX application for 2 d reduced mIPSC amplitude (Kilman et al., 2002), we did not observe significant effect of TTX treatment per se on mIPSC amplitude or frequency at the 4 h time point. In a similar experiment, coapplication of the AMPA receptor antagonist NBQX (5 μm), which significantly reduced neuronal firing (supplemental Table 2, available at www.jneurosci.org as supplemental material), also effectively blocked the increase in mIPSC amplitude and frequency induced by BMI treatment (Fig. 3A,B). Together, the results of these global manipulations suggested that neuronal firing is essential for activity-induced increase in GABAergic synaptic transmission.
Figure 3.

Postsynaptic spiking was required for changes at GABAergic synapses. A, TTX treatment blocked BMI-induced increase of mIPSC amplitude (Ctrl: 16.64 ± 0.78 pA; BMI: 20.10 ± 1.09 pA, p < 0.05; TTX: 14.93 ± 0.68 pA; BMI + TTX: 15.76 ± 0.82 pA); NBQX also blocked BMI-induced increase of mIPSC amplitude (Ctrl: 17.79 ± 0.82 pA; BMI: 22.74 ± 1.47 pA, p < 0.05; NBQX: 16.86 ± 0.71 pA; BMI + NBQX: 18.05 ± 1.14 pA). B, TTX treatment blocked BMI-induced increase of mIPSC frequency (Ctrl: 0.88 ± 0.12 Hz; BMI: 1.62 ± 0.19 Hz, p < 0.01; TTX: 0.77 ± 0.18 Hz; BMI + TTX: 0.60 ± 0.09 Hz). NBQX also blocked mIPSC frequency change (Ctrl: 1.22 ± 0.26 Hz; BMI: 3.42 ± 0.62 Hz, p < 0.001; NBQX: 1.23 ± 0.21 Hz; BMI + NBQX: 1.39 ± 0.17 Hz). C, Representative recordings of spontaneous firing for conditions as indicated. The scale bars are 20 mV and 2.5 s. D, The average firing rate of mKir-expressing neurons recorded in extracellular solution containing BMI (mKir + BMI: 2.57 ± 0.39 Hz, p < 0.01) is significantly higher than that without (mKir, 1.06 ± 0.19 Hz); no such differences were observed in Kir-expressing neurons (Kir: 1.15 ± 0.26 Hz, p > 0.05 vs mKir; Kir + BMI: 0.72 ± 0.25 Hz, p > 0.05 vs Kir). E, Representative mIPSC recordings and average waveforms for conditions as indicated. The scale bars are 20 pA and 1 s for the sweeps, 5 pA and 20 ms for the averaged traces. F, BMI treatment significantly increased the mIPSC amplitude of mKir-expressing neurons (mKir: 17.33 ± 1.00 pA; mKir + BMI: 21.75 ± 1.35 pA, p < 0.05), without affecting those of Kir-expressing neurons (Kir: 15.41 ± 1.02 pA, p > 0.05 vs mKir; Kir + BMI 15.10 ± 0.77 pA, p > 0.05 vs Kir). G, Corresponding cumulative distributions: mKir versus mKir + BMI, p < 0.001; Kir versus Kir + BMI, p = 0.58. H, BMI treatment significantly increased average mIPSC frequency of mKir-expressing neurons (mKir: 1.43 ± 0.15 Hz; mKir + BMI: 2.34 ± 0.23 Hz, p < 0.05), without affecting that of Kir-expressing neurons (Kir: 1.36 ± 0.30 Hz, p > 0.05 vs mKir; Kir + BMI: 1.27 ± 0.18 Hz, p > 0.05 vs Kir). I, Corresponding cumulative distributions: mKir versus mKir + BMI, p < 0.001; Kir versus Kir + BMI, p = 0.96. J–L, Representative images and quantification of the average density and integrated intensity of vGAT puncta colocalizing with Bassoon, conditions as indicated. Scale bar is 5 μm. BMI treatment increased the density of synaptic vGAT puncta in mKir-expressing neurons (mKir: 1 ± 0.06; mKir + BMI: 1.27 ± 0.08, p < 0.05), an effect blocked by Kir expression (Kir: 0.94 ± 0.08, p > 0.05 vs mKir; Kir + BMI: 0.95 ± 0.07, p > 0.05 vs mKir, p > 0.05 vs Kir) (K). None of the treatments affected vGAT puncta intensity (L). *p < 0.05, **p < 0.01, ***p < 0.001.
The requirement for neuronal firing could be in an individual neuron or within the entire network. We first tested the requirement for spiking activity in individual postsynaptic neurons by overexpressing the inward rectifier potassium channel Kir2.1 (Kubo et al., 1993), or as a control mKir2.1, its nonconducting mutant form (Burrone et al., 2002), in cultured hippocampal neurons from DIV 8 to 12. Due to the low transfection efficiency of the calcium phosphate transfection method, Kir 2.1 expression did not disturb the overall network activity. Consistent with previous reports (Holt et al., 1999; Burrone et al., 2002), neurons expressing Kir2.1 had more hyperpolarized resting membrane potentials, lower input resistances, and significantly larger inward-rectifying currents (supplemental Table 3, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). After 4 d of overexpression, Kir2.1-expressing neurons generally recovered their basal spiking rates to the control level (Burrone et al., 2002) (Fig. 3C,D), but failed to respond to BMI application with increased neuronal spiking (Fig. 3C,D). Concurrent with this lack of change in neuronal firing activity, Kir2.1 expression also completely blocked the increase in mIPSC amplitude and frequency induced by 4 h BMI treatment (Fig. 3E–I). This result demonstrated that activity-induced increase in postsynaptic spiking is necessary for the enhancement of inhibitory synaptic transmission. Consistently, overexpression of Kir2.1, compared to mKir2.1, effectively prevented activity-induced increase in the density of presynaptic vGAT protein (Fig. 3J–L), suggesting that activity-induced presynaptic change requires elevated postsynaptic spiking. Together, these results demonstrated that activity-induced change at GABAergic synapses requires elevated spiking of individual postsynaptic neurons.
Elevated postsynaptic spiking was sufficient for increasing inhibitory synaptic transmission
Having demonstrated a requirement for postsynaptic neuronal firing in activity-dependent upregulation of inhibitory synaptic transmission, we next asked whether an increase in the spiking activity of the postsynaptic neuron was sufficient to induce change at inhibitory synapses. To this end, we overexpressed the ligand-activated cation channel TRPV1 (Caterina et al., 1997) in a small percentage of cultured neurons during DIV 8–12. Consistent with a previous report (Zemelman et al., 2003), application of the TRPV1 ligand capsaicin (50 nm) specifically increased neuronal firing in TRPV1-transfected cells (TRPV1 + Cap.), compared to TRPV1-expressing neurons treated with vehicle (Fig. 4A,B). Untransfected neurons neighboring TRPV1-expressing ones (Untrans + Cap.) did not show increased firing in response to capsaicin application (Fig. 4A,B), demonstrating the specificity of our molecular manipulation in affecting only spiking of the transfected neuron, but not that of the local circuit.
Figure 4.

Enhancement of inhibitory synaptic function by capsaicin-activation of overexpressed TRPV1. A, Representative spontaneous firing recordings for conditions as indicated. The scale bars are 20 mV and 5 s. B, Capsaicin (Cap.) application specifically increased the firing rates of TRPV1-transfected neurons [TRPV1 + Cap. 0.95 ± 0.22 Hz, p < 0.01 vs TRPV1 0.27 ± 0.08 Hz, p < 0.01 vs untransfected neurons treated with Cap. (Untrans + Cap.) 0.31 ± 0.09 Hz]. C, Four hour capsaicin treatment of untransfected neurons did not significantly affect mIPSC amplitude or frequency. D, Representative mIPSC recordings and average waveforms for condition as indicated. The scale bars are 20 pA and 1 s for the sweeps, and 5 pA and 20 ms for the averaged traces. E, The average mIPSC amplitude of TRPV1 neurons treated with Cap. was significantly higher than that treated with vehicle (TRPV1: 15.98 ± 0.61 pA; TRPV1 + Cap. 20.34 ± 1.04 pA, p < 0.001) or neighboring untransfected neurons treated with Cap. (16.09 ± 0.61 pA, p < 0.01 vs TRPV1 + Cap.). F, Corresponding cumulative distributions: TRPV1 + Cap. versus TRPV1, p < 0.001; TRPV1 + Cap. versus Untrans + Cap., p < 0.001; inset scaled TRPV1 + Cap. versus TRPV1, p = 1.0. G, TRPV1 neurons treated with Cap. (1.49 ± 0.25 Hz) had higher mIPSC frequency than TRPV1 (0.65 ± 0.10 Hz, p < 0.01) or untransfected neurons treated with Cap. (0.65 ± 0.07 Hz, p > 0.05 vs TRPV1, p < 0.01 vs TRPV1 + Cap.). H, Corresponding cumulative distributions: TRPV1 + Cap. versus TRPV1, p < 0.001; TRPV1 + Cap. versus Untrans + Cap., p < 0.001. I, J, Representative images and quantifications of the average density and integrated intensity of immunostained vGAT puncta colocalizing with Bassoon in TRPV1-transfected neurons treated with vehicle (Ctrl) or Cap. Scale bar is 5 μm. Significant increase in synaptic vGAT density was detected in TRPV1 neurons treated with Cap. (1.25 ± 0.08, p < 0.05). No significant changes in the integrated intensity of vGAT puncta were detected (p = 0.77). *p < 0.05, **p < 0.01, ***p < 0.001.
Since endogenous TRPV1 is expressed in the hippocampus and has been shown to affect Hebbian plasticity through presynaptic mechanisms (Gibson et al., 2008; Kauer and Gibson, 2009), we excluded the possibility that endogenous TRPV1 participated in homeostatic regulation of inhibitory synapses by showing that capsaicin treatment (50 nm) of untransfected neuronal cultures did not affect neuronal firing activity (supplemental Table 2, available at www.jneurosci.org as supplemental material) or inhibitory synaptic transmission (Fig. 4C). In TRPV1-transfected neurons, however, 4 h capsaicin treatment (50 nm) significantly increased both the amplitude and frequency of mIPSCs (Fig. 4D–H), compared to untreated controls. Importantly, the mIPSC amplitude and frequency of untransfected neurons adjacent to those expressing TRPV1 were unaffected (Fig. 4D–H), demonstrating neuron specificity during firing-induced enhancement of GABAergic synaptic transmission. Similar to the effect of globally elevating neuronal activity, the amplitude of mIPSCs from capsaicin-treated TRPV1-transfected neurons also scaled multiplicatively (Fig. 4F, inset).
Consistent with our electrophysiological measurements, we detected increased puncta density of the presynaptic protein vGAT protein in TRPV1-transfected neurons treated with capsaicin (Fig. 4I,J). Thus, manipulating the firing activity of an individual neuron within a circuit, without altering that of its neighbors, is sufficient to mimic global activity-induced enhancement of mIPSC amplitude and frequency, as well as increase the density of presynaptic vGAT puncta. These results demonstrated the sufficiency of postsynaptic spiking in inducing changes in the number and strength of GABAergic synaptic inputs.
Retrograde BDNF signaling was required for presynaptic modification
Our observation that elevation of postsynaptic spiking can mimic activity-induced presynaptic and postsynaptic changes (Fig. 4D–J) suggested retrograde signaling from the postsynaptic cell to GABAergic presynaptic terminals. BDNF is a well known activity-regulated retrograde signal capable of potentiating GABAergic synaptic transmission (Poo, 2001; Vicario-Abejón et al., 2002; Kohara et al., 2007; Kuczewski et al., 2009). We first assayed BDNF mRNA level following various treatments by quantitative real-time PCR (qPCR) and found it to be significantly elevated following BMI treatment, an effect completely blocked by TTX coapplication (Fig. 5A). Thus, activity-induced increase in BDNF expression, like the enhancement of inhibitory synaptic transmission (Fig. 3A,B), required neuronal spiking.
To investigate the requirement of BDNF signaling for activity-induced increase in inhibitory synaptic transmission, we coapplied BMI with the membrane-permeable tyrosine kinase inhibitor K252a (100 nm) (Koizumi et al., 1988), a high-affinity inhibitor of the BDNF receptor TrkB (Tapley et al., 1992). We found that it completely blocked activity-induced increase in mIPSC frequency (Fig. 5B). Application of TrkB-Fc (2 μg/ml), a scavenger of extracellular BDNF, had a similar effect (Fig. 5B). In a complementary experiment, incubation with BDNF protein (50 ng/ml) for 4 h elevated mIPSC frequency without affecting its amplitude (Fig. 5C). Together, these results suggested that BDNF signaling is necessary and sufficient for activity-induced enhancement of mIPSC frequency.
To directly examine the requirement for postsynaptically expressed BDNF, we reduced endogenous BDNF level in individual neurons by RNA interference (RNAi) from DIV 8 to 12. We found that expression of the BDNF RNAi construct (Zhou et al., 2007) in the postsynaptic neuron completely blocked activity-induced increase in mIPSC frequency, without affecting mIPSC amplitude, compared to expression of the control, scrambled RNAi construct (Fig. 5D–G). Thus, postsynaptic expression of BDNF is cell-autonomously required for activity-induced elevation in mIPSC frequency. On examining the effect of BDNF RNAi on vGAT level, we found reduced integrated intensity of vGAT puncta in BDNF RNAi-transfected neurons (Fig. 5H–J), consistent with a developmental role of BDNF in promoting GABAergic synaptic formation (Vicario-Abejón et al., 2002). Importantly, BDNF RNAi effectively blocked the activity-induced increase in vGAT puncta density, compared to expression of the scrambled RNAi (Fig. 5H–J). Thus, BDNF expression in postsynaptic neurons is required for activity-induced increase in mIPSC frequency and in the density of presynaptic vGAT puncta, consistent with its potential role as the retrograde messenger in this process. We noted that the effect of BDNF is very local, as the synaptic defects of BDNF RNAi neurons could not be compensated by BDNF secreted from neighboring neurons.
To examine in detail whether postsynaptically secreted BDNF signaled presynaptically or postsynaptically to regulate inhibitory synaptic transmission, we used TrkB-T1 (Gonzalez et al., 1999), a truncated form of TrkB that functions as a dominant negative to endogenous receptors (supplemental Fig. 6A,B, available at www.jneurosci.org as supplemental material). TrkB-T1 overexpression at low efficiency using calcium phosphate transfection, such that each recorded neuron was surrounded only by untransfected neurons, did not affect activity-induced increase in mIPSC amplitude or frequency (Fig. 5K–N). This result suggested that even though BDNF was secreted postsynaptically, its downstream signaling in the postsynaptic neuron was not required for activity-induced change at presynaptic inhibitory terminals at 4 h. We next examined, in a complementary experiment, whether BDNF signaling was presynaptically required. By electroporating neurons with TrkB-T1 at the time of plating, we were able to overexpress TrkB-T1 in a large percentage of neurons in the culture dish (see Materials and Methods), up to 70–80% in some cases. Recording from untransfected neurons neighboring those transfected with TrkB-T1, as illustrated in Figure 5O and shown in supplemental Figure 6C, we found that BMI-induced increase of mIPSC frequency was specifically blocked in untransfected neurons neighboring those expressing TrkB-T1 (Fig. 5Q,R). Consistent with our other results, mIPSC amplitude was not affected by this manipulation (Fig. 5P). Collectively, these results demonstrated that activity-induced upregulation of mIPSC frequency depends on both BDNF secretion from postsynaptic neurons and its signaling through presynaptic TrkB receptors, thereby suggesting a retrograde signaling role of BDNF in this process.
Spiking-induced increase of mIPSC frequency by TRPV1/capsaicin required BDNF secretion
Putting together our findings of a postsynaptic requirement for neuronal firing (Fig. 4D–J) and for BDNF-dependent retrograde signaling (Fig. 5B–J) in the activity-induced strengthening of inhibitory synapses, we surmised that firing-induced BDNF secretion might be required for the activity-dependent increase in mIPSC frequency. By coapplying the BDNF scavenger TrkB-Fc together with capsaicin in TRPV1-transfected neurons, we found that, indeed, capsaicin-induced increase in mIPSC frequency (Fig. 6D,E), but not its amplitude (Fig. 6B,C), was completely blocked by TrkB-Fc. These results were consistent with those obtained following global activity elevation (Fig. 5B) and postsynaptic BDNF knockdown (Fig. 5D–G). Thus, spiking-induced BDNF secretion is necessary and sufficient for mediating activity-induced increase of inhibitory inputs in a cell-autonomous and neuron-specific manner.
Figure 6.

TrkB-Fc specifically blocked capsaicin-induced increase of mIPSC frequency in TRPV1-expressing neurons. A, Representative mIPSC recordings and average waveforms for conditions as indicated. The scale bars are 20 pA and 1 s for the sweeps, and 5 pA and 20 ms for the averaged traces. B, Quantitation of mIPSC amplitude (IgG-Fc: 16.32 ± 0.86 pA; IgG-Fc + Cap.: 20.18 ± 0.87 pA, p < 0.05; TrkB-Fc: 17.36 ± 0.99 pA; TrkB-Fc + Cap.: 21.64 ± 1.09 pA, p < 0.05 vs TrkB-Fc). C, Cumulative distributions of mIPSC amplitudes: IgG versus IgG + Cap., p < 0.01; TrkB-Fc versus TrkB-Fc + Cap., p < 0.001. D, Quantitation of mIPSC frequency (IgG-Fc 1.12 ± 0.13 Hz; IgG-Fc + Cap.: 2.05 ± 0.31 Hz, p < 0.05; TrkB-Fc: 1.53 ± 0.24 Hz; TrkB-Fc + Cap.: 1.46 ± 0.17 Hz, p > 0.05 vs TrkB-Fc). E, Cumulative distributions of mIPSC frequency: IgG-Fc versus IgG-Fc + Cap., p < 0.001; TrkB-Fc versus TrkB-Fc + Cap., p = 0.41. *p < 0.05, **p < 0.01, ***p < 0.001.
The effect of in vivo kainate injection on neuronal spiking, BDNF, and inhibitory synaptic transmission
Having demonstrated that elevating neuronal activity for 4 h through BMI, high K+, or KA treatment increased the amplitude and frequency of mIPSCs in cultured neurons, and shown that it mechanistically required elevated postsynaptic neuronal spiking and BDNF signaling, we asked if similar phenomena could be observed following in vivo manipulations. As a paradigm for elevating neuronal excitability in vivo, we systemically administered an intermediate dose of KA (4 mg/kg) (Lothman and Collins, 1981) to P12–P14 rats. Recording from CA1 pyramidal neurons of acute brain slices prepared from rats after KA injection for 1 or 4 h, we found gradual and continuing increase in spontaneous firing rates (Maffei et al., 2004) (see Materials and Methods) reaching statistical significance at 4 h (Fig. 7A,B). No epileptic forms of spikes were observed. Consistent with a previous report (Wetmore et al., 1994) and with our qPCR results (Fig. 5A) in culture, we detected a significant increase in BDNF protein level in the hippocampus of KA-treated animals 4 h following injection, compared to saline controls (Fig. 7C) (1.55 ± 0.09-fold compared to control, p < 0.05).
Figure 7.

In vivo kainic acid injection increased neuronal spiking, BDNF level and inhibitory synaptic transmission in juvenile rats. A, Representative recordings of spontaneous firing activity from CA1 pyramidal neurons in hippocampal slices for conditions as indicated, the scale bars are 20 mV and 5 s. B, Average firing rates from Ctrl (1.42 ± 0.40 Hz) rats, and those injected with KA for 1 h (2.58 ± 0.54 Hz, p > 0.05) or 4 h (4.05 ± 0.69 Hz, p < 0.01). C, Example of BDNF protein level from Ctrl and KA-injected rats. D, Representative recordings of mIPSCs for conditions as indicated. The scale bars are 20 pA and 1 s for the sweeps, and 5 pA and 20 ms for the averaged traces. E, Average mIPSC amplitude from Ctrl (26.72 ± 0.73 pA) and KA-injected rats (29.05 ± 0.71 pA, p < 0.05); average mIPSC frequency from Ctrl (0.94 ± 0.10 Hz) and KA-injected rats (1.69 ± 0.20 Hz, p < 0.01). F, Cumulative distributions of mIPSC amplitude: p < 0.05, inset is the scaled distribution, p = 1.0. G, Cumulative distributions of mIPSC frequency: p < 0.001. H, Representative eIPSC recordings from control and KA rats. The scale bars are 0.2 nA and 50 ms. I, Evoked IPSC amplitudes plotted against stimulus intensity, n ≥ 11. *p < 0.05, **p < 0.01.
Most importantly, we found significant increase in both mIPSC amplitude and frequency (Fig. 7D–G) of CA1 pyramidal neurons from animals injected with KA for 4 h, an effect which scaled multiplicatively (Fig. 7F, inset). No differences in mIPSC kinetics, membrane capacitance, or input resistance were detected in neurons from KA-treated animals, compared to saline controls (supplemental Table 4, available at www.jneurosci.org as supplemental material). By stimulating nearby interneurons, we showed that evoked GABAergic synaptic responses were significantly elevated in pyramidal neurons from KA-treated rats (Fig. 7H,I). In summary, elevating neuronal activity in vivo by KA injection significantly increased neuronal firing, BDNF protein level, mIPSC amplitude and frequency, and evoked GABAergic responses.
Discussion
Using a variety of manipulations to increase neuronal activity in cultured hippocampal neurons, we found that 4 h elevation of neuronal activity homeostatically upregulated mIPSC amplitude and frequency, before detectable change in mEPSCs. These functional changes correlated with increased density and/or intensity of presynaptic and postsynaptic components of GABAergic synapses. By increasing the firing of individual neurons through ligand activation of overexpressed TRPV1, we showed that postsynaptic spiking is sufficient for inducing enhancement of inhibitory synaptic transmission. Changes in mIPSC frequency and synapse density required BDNF-mediated retrograde signaling. Importantly, salient features of this form of activity-induced plasticity were observed following in vivo KA administration, suggesting that such changes could occur in vivo and may play important roles in the maintenance of neural circuit stability.
Cell-autonomous mIPSC elevation was induced by postsynaptic spiking elevation
Here we found that elevating the spike activity of a pyramidal neuron is sufficient to increase the strength of its inhibitory synaptic inputs (Fig. 4). The cell autonomy of this homeostatic regulation is similar to previous reports of firing-dependent upregulation of mEPSCs during activity blockade (Burrone et al., 2002; Ibata et al., 2008). However it is very different from the mechanism required for downregulating mIPSCs following activity blockade, which involves change in the firing of a group of neurons (Hartman et al., 2006). In other words, alteration in neuronal firing activity can cell-autonomously induce upregulation of mIPSCs following activity elevation and of mEPSCs following activity blockade, but is not sufficient for the downregulation of mIPSCs following activity blockade, which requires change in the firing activity of multiple neurons in the network. These observations likely also extend to other mechanistic aspects of homeostatic plasticity. For instance, there is a growing body of evidence for homeostatic upregulation of excitatory synapses through local changes in neuronal subcompartments (Yu and Goda, 2009). Our results, showing that the effect of TRPV1 activation did not extend to neighboring neurons (Fig. 4A–H) and that the effect of BDNF RNAi could not be rescued by BDNF from adjacent cells (Fig. 5D–J), also suggested neuron specificity and very local change during activity-induced homeostatic plasticity of inhibitory synapses. While it may be too early to draw conclusions from these limited observations, it is interesting to note that the mechanism of homeostatic plasticity seems not to depend on the type of synapse modified (excitatory or inhibitory), but to depend on the direction of regulation activated (upregulation or downregulation). Whether these observations hold true more generally remains to be explored.
BDNF as a candidate retrograde signal in inhibitory homeostatic plasticity
Retrograde signaling during homeostatic regulation was first described at the Drosophila neuromuscular junction (Petersen et al., 1997; Davis et al., 1998). BDNF is a well known retrograde signaling molecule (Regehr et al., 2009) that can be secreted from dendrites in an activity-dependent fashion (Hartmann et al., 2001; Kojima et al., 2001). Previous reports have shown that BDNF could upregulate inhibitory synaptic function (Murphy et al., 1998; Huang et al., 1999; Bolton et al., 2000; Marty et al., 2000; Yamada et al., 2002; Ohba et al., 2005), while reducing its level in single postsynaptic neurons (Kohara et al., 2007) had converse effects.
In terms of homeostatic plasticity, previous work has shown that BDNF application can prevent the effect of activity blockade (TTX application) on reducing inhibitory synaptic transmission (Swanwick et al., 2006). However, whether BDNF functions as a retrograde messenger during synaptic scaling has not been previously examined. Putting our results together with published work, an all-inclusive interpretation is that BDNF-dependent signaling mediates retrograde, activity-induced homeostatic upregulation of inhibitory synapses, an effect parallel to and counteracting the effect of TTX. Evidence supporting our hypothesis includes the following: (1) BDNF level increased significantly following elevated activity (Regehr et al., 2009) (Fig. 5A), but remained essentially unaltered following TTX treatment (Fig. 5A), making regulation of BDNF level by activity elevation more likely; (2) both activity-induced increase in inhibitory synaptic transmission and BDNF level required neuronal firing (Figs. 3, 5A); (3) postsynaptically reducing BDNF level effectively blocked activity- or firing-induced presynaptic changes, including mIPSC frequency and vGAT density (Figs. 5D–J, 6A–E); and (4) presynaptic interference with BDNF signaling through TrkB-T1 expression effectively blocked activity-induced increase in mIPSC frequency (Fig. 5O–R), while its postsynaptic interference had no effect (Fig. 5K–N). Based on our results, we propose the following model: elevated neuronal activity, through increasing postsynaptic firing activity, induces increased secretion of BDNF, which retrogradely activates GABAergic synaptic inputs, resulting in an increase in their number (Fig. 8).
Figure 8.

Model for homeostatic upregulation of inhibitory inputs by elevated neuronal activity. Elevated activity rapidly increased neuronal firing, which in turn increased the secretion of BDNF from the dendrites of postsynaptic neurons. BDNF then retrogradely modulated presynaptic GABAergic inputs, resulting specifically in an increase in the number of GABAergic synapses by 4 h.
The effect of in vivo activity elevation on inhibitory synaptic transmission
Using kainate injection to induce activity elevation in vivo, we replicated the most salient features of our findings, including increased neuronal firing, elevated BDNF expression and increased inhibitory synaptic transmission (Fig. 7). These results demonstrated the relevance of our findings to the in vivo context. That is, CA1 pyramidal neurons have the ability to adaptively respond to activity elevation in vivo by increasing their inhibitory synaptic strength before detectable change at excitatory synapses. This cell-autonomous and local upregulation of inhibitory synapses by elevated activity would allow developing neural circuits to only put the safety brakes on those neurons at risk of overexcitation without affecting activity-dependent development of other neurons within the same circuit.
Footnotes
This work was supported by grants from the Ministry of Science and Technology (2006CB806600, 2006CB943903), the National Natural Science Foundation of China (30721004), and the Chinese Academy of Sciences (Hundred Talent Program and KSCX2-YW-R-103). We thank Drs. Z.-F. Chen, R. Balice-Gordon, D. Julius, Z.-G. Luo, and C.-L. Wang for gifts of constructs. The GAD-6 antibody developed by Dr. D. Gottlieb was obtained from the Developmental Studies Hybridoma Bank, maintained at the University of Iowa. We thank W. Zhang and Z.-F. Wan for excellent technical assistance. We are grateful to Prof. M.-M. Poo and G. Chen for comments and critical reading of the manuscript, and colleagues at the Institute of Neuroscience and members of the Yu laboratory for suggestions and discussions.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Applied Biosystems. Rev B. Foster City, CA: Applied Biosystems; 2001. User bulletin no. 2. [Google Scholar]
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Epilepsies and neuronal plasticity: for better or for worse? Dialogues Clin Neurosci. 2008;10:17–27. doi: 10.31887/DCNS.2008.10.1/ybenari. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G, Poo M. Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Bolton MM, Pittman AJ, Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 1998;20:305–315. doi: 10.1016/s0896-6273(00)80458-4. [DOI] [PubMed] [Google Scholar]
- De Gois S, Schäfer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci. 2005;25:7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JD, De Gois S, Varoqui H, Schafer MK, Weihe E. Activity-dependent regulation of vesicular glutamate and GABA transporters: a means to scale quantal size. Neurochem Int. 2006;48:643–649. doi: 10.1016/j.neuint.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci. 2006;9:642–649. doi: 10.1038/nn1677. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JR, Johns DC, Wang S, Chen ZY, Dunn RJ, Marban E, Corey DP. Functional expression of exogenous proteins in mammalian sensory hair cells infected with adenoviral vectors. J Neurophysiol. 1999;81:1881–1888. doi: 10.1152/jn.1999.81.4.1881. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Gibson HE. Hot flash: TRPV channels in the brain. Trends Neurosci. 2009;32:215–224. doi: 10.1016/j.tins.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J Neurosci. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi S, Contreras ML, Matsuda Y, Hama T, Lazarovici P, Guroff G. K-252a: a specific inhibitor of the action of nerve growth factor on PC 12 cells. J Neurosci. 1988;8:715–721. doi: 10.1523/JNEUROSCI.08-02-00715.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H. Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity-dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res. 2001;64:1–10. doi: 10.1002/jnr.1080. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa JL. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol. 2009;39:37–49. doi: 10.1007/s12035-009-8050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman EW, Collins RC. Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981;218:299–318. doi: 10.1016/0006-8993(81)91308-1. [DOI] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci. 2008;28:4377–4384. doi: 10.1523/JNEUROSCI.5298-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci. 2004;7:1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrlé R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I. Aspects of the homeostaic plasticity of GABAA receptor-mediated inhibition. J Physiol. 2005;562:37–46. doi: 10.1113/jphysiol.2004.077362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Cole NB, Segal M. Brain-derived neurotrophic factor mediates estradiol-induced dendritic spine formation in hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:11412–11417. doi: 10.1073/pnas.95.19.11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba S, Ikeda T, Ikegaya Y, Nishiyama N, Matsuki N, Yamada MK. BDNF locally potentiates GABAergic presynaptic machineries: target-selective circuit inhibition. Cereb Cortex. 2005;15:291–298. doi: 10.1093/cercor/bhh130. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 2009;63:154–170. doi: 10.1016/j.neuron.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Wenner P. Sensing and expressing homeostatic synaptic plasticity. Trends Neurosci. 2007;30:119–125. doi: 10.1016/j.tins.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Swanwick CC, Murthy NR, Kapur J. Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol Cell Neurosci. 2006;31:481–492. doi: 10.1016/j.mcn.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejón C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nat Rev Neurosci. 2002;3:965–974. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- Wang CL, Zhang L, Zhou Y, Zhou J, Yang XJ, Duan SM, Xiong ZQ, Ding YQ. Activity-dependent development of callosal projections in the somatosensory cortex. J Neurosci. 2007;27:11334–11342. doi: 10.1523/JNEUROSCI.3380-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore C, Olson L, Bean AJ. Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci. 1994;14:1688–1700. doi: 10.1523/JNEUROSCI.14-03-01688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci. 2002;22:7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu LM, Goda Y. Dendritic signalling and homeostatic adaptation. Curr Opin Neurobiol. 2009;19:327–335. doi: 10.1016/j.conb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- Zemelman BV, Nesnas N, Lee GA, Miesenbock G. Photochemical gating of heterologous ion channels: remote control over genetically designated populations of neurons. Proc Natl Acad Sci U S A. 2003;100:1352–1357. doi: 10.1073/pnas.242738899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhu J, Yang GY, Wang QJ, Qian L, Chen YM, Chen F, Tao Y, Hu HS, Wang T, Luo ZG. Dishevelled promotes axon differentiation by regulating atypical protein kinase C. Nat Cell Biol. 2007;9:743–754. doi: 10.1038/ncb1603. [DOI] [PubMed] [Google Scholar]
- Zhao L, Sheng AL, Huang SH, Yin YX, Chen B, Li XZ, Zhang Y, Chen ZY. Mechanism underlying activity-dependent insertion of TrkB into the neuronal surface. J Cell Sci. 2009;122:3123–3136. doi: 10.1242/jcs.047712. [DOI] [PubMed] [Google Scholar]
- Zhou P, Porcionatto M, Pilapil M, Chen Y, Choi Y, Tolias KF, Bikoff JB, Hong EJ, Greenberg ME, Segal RA. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55:53–68. doi: 10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]