

Abstract
Repetitive firing of neurons at a low frequency often leads to a decrease in synaptic strength. The mechanism of this low-frequency depression (LFD) is poorly understood. Here, LFD was studied at Aplysia cholinergic synapses. The absence of a significant change in the paired-pulse ratio during LFD, together with the facts that neither the time course nor the extent of LFD were affected by the initial release probability, suggests that LFD is not related to a depletion of the ready-to-fuse synaptic vesicles (SVs) or to a decrease in the release probability, but results from the silencing of a subpopulation of release sites. A subset of SVs or release sites, which acquired a high release probability status during LFD, permits synapses to rapidly and temporarily recover the initial synaptic strength when the stimulation is stopped. However, the recovery of the full capacity of the synapse to sustain repetitive stimulations is slow and involves spontaneous reactivation of the silent release sites. Application of tetanic stimulations accelerates this recovery by immediately switching on the silent sites. This high-frequency-dependent phenomenon underlies a new form of synaptic plasticity that allows resetting of presynaptic efficiency independently of the recent history of the synapse. Microinjection of a mutated Aplysia synapsin that cannot be phosphorylated by cAMP-dependent protein kinase (PKA), or a PKA inhibitor both prevented high-frequency-dependent awakening of release sites. Changes in the firing pattern of neurons appear to be able to regulate the on–off status of release sites via a molecular cascade involving PKA-dependent phosphorylation of synapsin.
Introduction
The frequency modulation during the firing of action potentials by neurons is a key element for information processing within neural networks. At the presynaptic level, one of the most common consequences of increasing the spiking frequency is a decrease in the quantity of neurotransmitter released at each stimulus. This ubiquitous form of homosynaptic presynaptic plasticity is referred to as synaptic depression. However, this term covers multiple situations distinguishable by the frequency of stimulation that induces them and/or the mechanisms involved. Synaptic depression has been shown to result from depletion of the readily releasable pool (RRP) of synaptic vesicles (SVs) (Foster and Regehr, 2004), inhibition of presynaptic calcium channel (Xu et al., 2007), or failure of action potential propagation (Brody and Yue, 2000). Moreover, increases in the frequency of neuronal activity can also convert presynaptic release sites from an active to a silent status in a reversible manner (Royer et al., 2000; Gover et al., 2002; Moulder et al., 2006, 2008).
How the functional status of a release site can be blocked on a change in the firing rate remains an open question. At Aplysia synapses, disabling the function of synaptic vesicle proteins (such as the GTPase Rac or synaptobrevin) and plasma membrane-associated proteins (such as the phospholipase D1) inhibits exocytosis by decreasing in the number of active release sites and not by changing the probability of release (Doussau et al., 2000; Humeau et al., 2001b, 2002, 2007). This suggests that the fusiogenic status of docked SVs as well as that of the plasma membrane at the fusion site determines the “on–off” functional status of the release site. Along the same line, although the number of docked SVs is not affected in boutons that had been silenced by chronic depolarization at hippocampal synapses, the number of ready-to-fuse SVs is reduced, suggesting that blockade of a step during SV priming underlies synapse silencing (Moulder et al., 2006). Moreover, awakening of the release sites previously silenced by impairing Rac function could be produced by the application of a short burst of high-frequency stimuli (Humeau et al., 2007), indicating that silencing/awakening relies on the frequency and timing of the neuronal firing pattern.
Here, we investigated the mechanisms underlying low-frequency-induced depression (LFD) at identified cholinergic synapses of Aplysia. We found that LFD is not related to a depletion of ready-to-fuse SVs or to a decrease in the probability of release, but is attributable to switching off of the functional status of release sites. Spontaneous awakening of release sites silenced during LFD occurs within ∼40 min, but the application of short bursts of stimulation can almost immediately switch on the sites. This burst-dependent reactivation of silent synapses is a new form of synaptic plasticity that resets the presynaptic strength to the level observed before LFD induction. The presynaptic phosphoproteins synapsins have been implicated in the control of synaptic strength when the firing patterns of neurons changes (Fdez and Hilfiker, 2006). Here, we found that the burst-dependent awakening of the sites silenced during LFD requires phosphorylation of synapsin by protein kinase A (PKA).
Materials and Methods
Acetylcholine release and electrophysiological recordings at Aplysia synapses
Aplysia californica were purchased from the University of Florida (Miami, FL). Electrophysiological experiments were performed at identified, chloride-dependent, inhibitory cholinergic synapses in dissected buccal ganglia of Aplysia as described previously (Doussau et al., 2000; Humeau et al., 2001a,b, 2002). Presynaptic neurons (B4 and B5) and one postsynaptic neuron (either B3 or B6) were impaled with two glass microelectrodes (3 m KCl, Ag/AgCl2, 2–10 MΩ). Action potentials were alternately evoked every 20 s in B4 or B5 using an AxoClamp 2B amplifier (Molecular Devices) (i.e., each presynaptic neuron was stimulated every 40 s; 0.025 Hz). The ensuing evoked, chloride-dependent IPSCs were recorded using the conventional two-electrode voltage-clamp technique (Geneclamp-500 amplifier and Digidata 1322A from Molecular Devices). Their reversal potential Vrev, was determined every 5 min. IPSCs were recorded at a holding potential Vh hyperpolarized 30 mV from Vrev. To express the IPSC amplitude as a value proportional to the amount of released ACh, but independent of the driving force for Cl− ions, IPSC amplitude (I) was converted to apparent membrane conductance changes (G), according to the equation G = I/(Vh − Vrev). Although this has the dimension of a membrane conductance (nanosiemens), we refer to it in this paper as the IPSC amplitude.
Extracellular media and intraneuronal injection
Dissected buccal ganglia were maintained at 22°C using a Peltier-plate system and superfused continuously (10 ml · h−1) with a physiological medium containing the following (in mm): 460 NaCl, 10 KCl, 33 CaCl2, 78 MgCl2, 10 HEPES, pH 7.5, as previously described (Humeau et al., 2001b, 2002, 2007). The very high concentration in divalent cations that we used prevented spontaneous neuron firing activity, which otherwise may induce undesired forms of short-term plasticity. This medium corresponds to a [Ca2+]/[Mg2+]e ratio of 0.42. When necessary, [Ca2+]/[Mg2+]e was modified as described previously (Humeau et al., 2001b). Intraneuronal injection of recombinant proteins and peptides was performed as detailed previously (Humeau et al., 2001b, 2002, 2007). Since the injected volume was in the range of 1–2% of the cell body volume, we assumed that the final intraneuronal concentration was ∼1% of that in the injection micropipette.
Quantal analysis parameters
Consider a synapse or a group of synapses as an array of n independent release sites (i.e., morphological sites), each potentially capable of releasing only one quantum at a time, and with its own individual p, and consider that p is compound resulting from the product of two terms: pA (the chance that a readily releasable quantum is available at the site), which defines the eligibility status of the site for exocytosis (i.e., its functional status), and po, the output probability (the chance that the site releases a quantum if a quantum is available) (for review, see Quastel, 1997). According to this view, the product N = n · pA corresponds to the number of functional release sites. Thus, any event modifying the number of morphologically defines sites or eligibility status is susceptible to induce changes in N. We discussed previously the possibility that the fusogenic status of docked SVs may determine the eligibility or functional status of the release site, and pA is likely not a stochastic parameter (Humeau et al., 2007).
If the release of one SV content produces a postsynaptic response of fixed amplitude q, according to the binomial statistics, at a release site, the average postsynaptic response amplitude is as follows: μ = p · q, with variance, Var = p · (1 − p) · q2. Now, consider the N functional release sites, P, the average of individual output probability po, and Q, the average amplitude of the quantum of response, and assume that the release process at each site is independent of that at the other sites and quanta sum up linearly. The mean amplitudes and variances at the N sites add linearly, and the mean amplitude of the compound response is as follows: Imean = Σi = 1→Nμi. This can be rearranged as follows: Imean = N · P · Q. The fluctuations of the responses around the mean have a variance of the form: Var = N · P · (1 − P) · Q2. Analyzing the relationship between Imean and Var allows getting insights into N, P, and Q (Silver et al., 1998; Reid and Clements, 1999; Humeau et al., 2001b, 2002, 2007; Scheuss et al., 2002; Clements, 2003; Silver, 2003; Foster and Regehr, 2004).
Although the above simplified expressions do not take into account the intrasite and intersite variability in quantum amplitude and the heterogeneity in p at the N sites, which contribute to the fluctuation in amplitude of the postsynaptic response (Clements, 2003; Silver, 2003), the general form of the relationship between Var and Imean is preserved (Humeau et al., 2002, 2007). Therefore, from the two above equations, it can be deduced that when only P is modified, Var = f(Imean) is a parabola: Var = Q · Imean − Imean2/N. This can be used to estimate P (see below). Our previous work at the studied cholinergic Aplysia synapse has shown that the Var = f(Imean), obtained by manipulating [Ca2+]/[Mg2+]e, is almost a simple parabola, indicating that the other sources of variability have no or modest effect at this preparation. Note that parameter N deduced by variance–mean analysis is a functional parameter and refers to the number of release sites able to experience SV fusion, as defined above (Humeau et al., 2001b, 2002, 2007). Thus, N does not take into account long-lasting silent sites.
Determination of the average release probability at various [Ca2+]/[Mg2+]e
The Var = f(Imean) was built at the studied Aplysia cholinergic synapses by manipulating [Ca2+]/[Mg2+]e (Humeau et al., 2002) (see Results). To pool the Var and Imean data obtained from distinct experiments, individual Var = f(Imean) plots were normalized to the maximum Var (Varmax) and the corresponding Imean (Imean-to-Varmax) was determined by fitting the Var = f(Imean) plot from each experiment by a quadratic function of the following form: y = y0 + a · x + b · x2, with all parameters left free, using the nonlinear regression procedure running with SigmaPlot 10 (Systat Software) (Humeau et al., 2007). Although the above described normalization of Var and Imean data did not permit a determination of Q and N from initial slope and parabola extent, the corresponding P could be evaluated. Indeed, when the normalized Var = f(Imean) is a parabola, similar as in non-normalized plots, Varmax is reached at p = 0.5, and thus P can be determined at each point of the parabola (e.g., when Imean = Imax/4, p = 0.25, and when Imean = Imax/2, p = 0.5).
Application of short trains at high frequency
Typically, posttetanic potentiation (PTP) was initiated by two trains (50 Hz for 2 s) (Humeau et al., 2001a). Twenty seconds after the second 50 Hz train terminated, the stimulation rate was returned to the initial rate (i.e., 0.025 Hz). The extent of PTP was expressed by normalizing the amplitude of the maximal IPSC during PTP to the “basal IPSC amplitude” as determined by averaging the amplitudes of the 10 IPSCs preceding the application of the conditioning 50 Hz trains.
Approximation of the time course of LFD, LFD after rest, and posttetanic potentiation during depression
Time constants of LFD, LFD after rest (LFDAR), and posttetanic potentiation during depression (PTPLFD) were obtained by fitting the I = f(t) plots using SigmaPlot (Systat Software) using the following equations: I(t) = y0 + ωI · exp(−t/τI) + ωII · exp(−t/τII) (for LFD) or I(t) = y0 + ωfast · exp(−t/τfast) + ωI · exp(−t/τI) + ωII · exp(−t/τII) (for LFDAR and PTPLFD), where y0 is either the control plateau at 0.025 Hz or the plateau of LFD, τI and τII, the time constants of depression during LFD and LFDAR, and τfast, the time constant of the disappearance of the superimposed facilitation observed at depressed synapses during LFDAR. We imposed the following constraints to the weighting factors: ωI + ωII = 100 (for LFD); ωfast + ωI + ωII = 100 (for LFDAR and PTPLFD).
Use of the paired-pulse ratio as a measure of release probability
Rationale.
The analysis of the paired-pulse ratio (PPR) between the amplitudes of two postsynaptic responses induced by twin stimuli separated by a short (<1 s) time interval (PPR = I2/I1) allows one to gain insight into changes in release probability. Albeit in certain preparations, other mechanisms can produce PPR changes, such as fast inactivation of postsynaptic receptors or of presynaptic P/Q-type Ca2+ channels (Bellingham and Walmsley, 1999; Heine et al., 2008), at the studied Aplysia synapse, the extent of PPR is mostly, if not exclusively, determined by P (Doussau et al., 1998; Humeau et al., 2007). The rationale for using PPR to probe changes in P is the following: the extent of the I2/I1 ratio is determined by the balance between a facilitatory component and a depression component both activated by the conditioning stimulus. The facilitatory component, which affects P, is probably the consequence of a Ca2+-dependent conditioning of the release machinery (Zucker and Regehr, 2002) and the saturation of cytosolic Ca2+ buffers (Felmy et al., 2003). The depression component is likely to result from incomplete refilling of the SV pool consumed by the conditioning stimulus (Zucker and Regehr, 2002; Singer and Diamond, 2006).
PPR determination.
Paired postsynaptic responses were evoked by presynaptically applying two brief depolarizing pulses of 5 ms separated by an interval of 40 ms. Since the mean decay time of the evoked postsynaptic responses recorded at 22°C at the cholinergic synapses used in this study varies with the postsynaptic cell and the preparation, the postsynaptic responses partially overlapped. Thus, the amplitude measured at the peak of IPSC2 was corrected by the exponential residual of I1, as described previously; this corrected amplitude is referred as to I2 (Doussau et al., 1998).
PPR modeling.
Let us say that the number of quanta released at the first stimulus of a pair is N1 · P1, where N1 is the number of active (i.e., eligible) sites, and P1 is the output probability of release at stimulus 1. Thus, on average, I1 = N1 · P1 · Q1. At the time of stimulus 2, N2 is the number of release-ready SVs not consumed at stimulus 1 [i.e., N1 · (1 − P1)] plus the number of SVs that have been replaced (refilling) and/or acquired fusion competence during the interpulse interval. This fraction depends on the number of SVs (N1 · P1) that fused at stimulus 1. Therefore, N2 = N1 · ((1 − P1) + (k(t) · N1 · P1) = N1 · (1 + ((k(t) − 1) · P1)), where k(t) is the fraction of refilling with the interpulse. Because of the accumulation of Ca2+ ions at stimulus 1, P2 ≥ P1. On average, I2 = N2 · P2 · Q2, and when expressed as a function of N1 and P1, I2 = N1 · (1 + ((k(t) − 1) · P1)) · P2 · Q2. If we assume that quantal size Q does not change between the two paired stimuli, (Q1 = Q2), I2/I1 = (1 + ((k(t) − 1) · P1)) · P2/P1. This means that PPR is independent of N. Accordingly, any change in PPR should indicates a modification of the release probability P. Conversely, no change in PPR when synaptic strength is modified should indicate that modification of N is responsible for changing the amplitude of the postsynaptic responses.
If (but see below) P2/P1 and k(t) are independent of [Ca2+]e, the relationship between I2/I1 and P1 (made by changing [Ca2+]e) is simple and has the following form: y = y0+ a · x with y0 = P2/P1, and a negative slope, a = −[(P2/P1) · (1 − k(t))]. Also, the relationship between I2/I1 and I1 becomes linear with same y0, but with a negative slope, a′ = −[(P2/P1) · (1 − k(t))]/I1max, where I1max is the amplitude of I1 when P1 = 1 (i.e., I1max = N1 · Q).
However, it is rather unlikely that P2/P1 and k(t) are independent of [Ca2+]e over a wide range of [Ca2+]e and the linear function provided above corresponds to the tangent of PPR = f(I1) at a given [Ca2+]e. This relationship can be also used to provide a linear approximation of a limited portion of the PPR = f(I1) plot. At the studied Aplysia cholinergic synapse, PPR = f(I1) plots made by a 10-fold change in the [Ca2+]/[Mg2+]e (from 2.1 to 0.21) display a large almost linear portion, albeit they significantly deviate from linearity at the lowest and highest [Ca2+]/[Mg2+]e [Humeau et al. (2007), their Fig. 1] (see also Fig. 2). This suggests that, at this Aplysia synapse and under the physiological conditions used ([Ca2+]/[Mg2+]e between 0.21 and 2.1), P2/P1 and k(t) can be considered to be nearly independent of [Ca2+]e.
Figure 1.
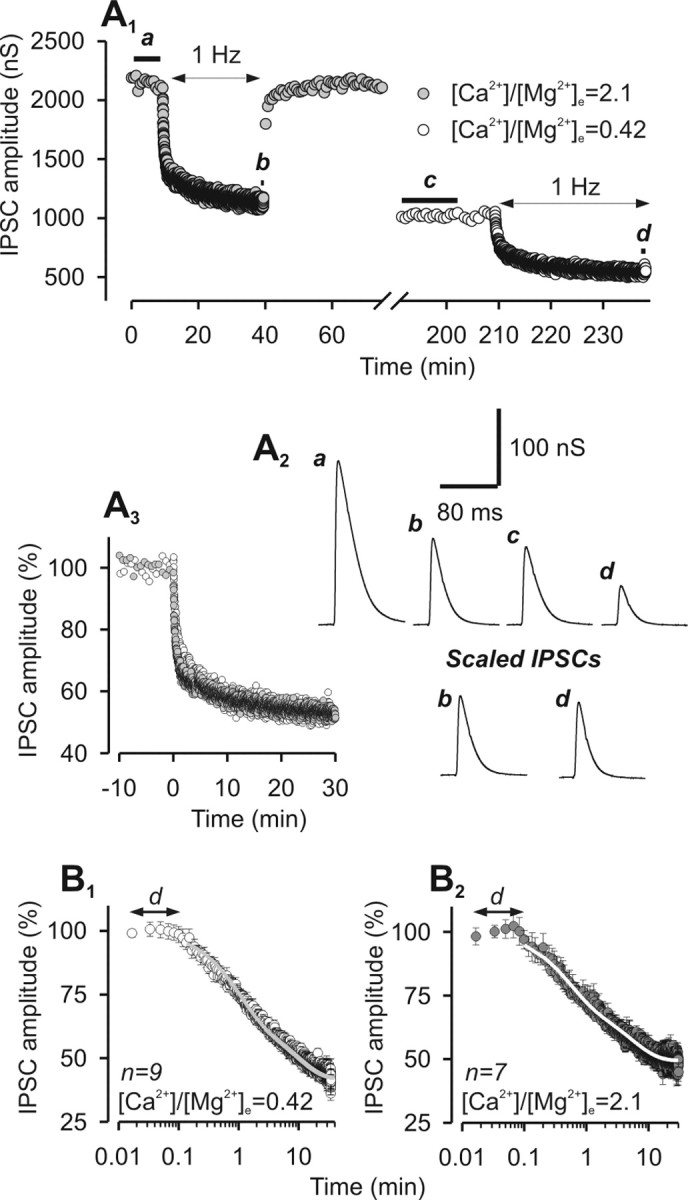
Time course and plateau of LFD are independent of the initial release probability. A1, Representative experiment of IPSC amplitude plotted against time before and after the induction of LFD at high and low [Ca2+]/[Mg2+]e. A2, Top traces (a–d), Average traces of at least 10 successive IPSCs recorded during the corresponding period indicated by thick horizontal lines on A1. Bottom traces, Traces b and d corresponding to the plateau of LFD at high and low [Ca2+]/[Mg2+]e, respectively, were normalized with respect to the mean value of IPSCs recorded during the plateau at 0.025 Hz. A3, Superimposition of normalized IPSCs amplitude during LFD monitored at low and high Ca2+-containing medium (same symbols as in A1). IPSC amplitudes were normalized with respect to the mean IPSC value recorded during the plateau at 0.025 Hz. Time 0 denotes the start of 1 Hz stimulation. B, Time course of LFD at low (B1) and high (B2) Ca2+-containing medium was determined by averaging normalized IPSCs from n experiments as indicated on the graphs. The solid gray and white lines represent the biexponential decay curves fitted to the data. The duration of the delay (d) before the actual induction of depression is represented by the black arrows. Error bars indicate SEM.
Figure 2.

LFD is not attributable to a decrease in P. A, B, PPR is inversely proportional to the probability of release at Aplysia cholinergic synapses. A, Stationary analysis of the fluctuations in IPSC amplitude when release probability is modified by changes in [Ca2+]/[Mg2+]e. The Var = f(Imean) plot was obtained by pooling the results from five different experiments during which ACh release was evoked by paired-pulse stimulations and under different [Ca2+]/[Mg2+]e (0.14, 0.21, 0.42, 0.84, and 2.1). After each change in [Ca2+]/[Mg2+]e and stabilization of IPSC amplitude, Imean PPR and Var were determined of at least 20 subsequent IPSCs. For more detail, see Materials and Methods. The probability of release was deduced from each point in this plot, and associated with the corresponding value of PPR. B, Plots of PPR against the probability after changes in [Ca2+]/[Mg2+]e. The probability was calculated from the Var = f(Imean) plot (A). The solid black line represents the linear function fitted to the data. C, D, Comparison of the effects of a change in [Ca2+]/[Mg2+]e with LFD on PPR and IPSC amplitude. C, Typical experiment during which PPR (gray-filled symbols) and IPSC amplitude (open squares and filled circles, respectively) were probed successively during a change in [Ca2+]/[Mg2+]e (left) or during LFD (right). The values of the [Ca2+]/[Mg2+]e and of the stimulation frequency are represented by the horizontal gray bars and the horizontal black bars, respectively. Note that the extent of the blockage of ACh release was approximately identical after the two treatments. The thin solid black lines correspond to the mean value of PPR calculated from three successive paired stimuli. D, Plots of PPR against I1 amplitude after changes in [Ca2+]/[Mg2+]e or during LFD. The solid black line represents the linear function fitted to the data after changes in [Ca2+]/[Mg2+]e. E, Averaged values of Var/Imean obtained at the plateau of 0.025 Hz stimulation (open bar), at the plateau of LFD (black-filled bar) and after a change in [Ca2+]/[Mg2+]e (from 2.1 to 0.42; gray-filled bar). The dashed lines represent the expected values of Var/Imean that should be observed if the ∼43% decrease in IPSC amplitude during LFD or after a change in [Ca2+]/[Mg2+]e were simply attributable to a change in Q, N, or P. ***p < 0.001; n.s. denotes no significant difference (i.e., p > 0.05). F, Minor PPR changes during LFD; each point corresponds to the mean value of I1 (black-filled circles) or PPR (gray-filled circles) from 11 experiments. The black-filled square and the gray-filled square correspond to the mean values of IPSC amplitude and PPR respectively calculated during the plateau at 0.025 Hz. Stimulus 1 corresponds to the first response at 1 Hz. Error bars indicate SEM.
Data presentation
In some cases, values obtained from various neurons were averaged and, unless otherwise stated, presented as mean ± SEM. In several experiments, data were normalized using the average value observed under control conditions (i.e., before any treatment). When appropriate (normally distributed data with equal variance), the significance between mean differences was tested by a one-way ANOVA or the Student's t test. Asterisks denote the following: *p < 0.05, **p < 0.01, and ***p < 0.001. n.s. denotes no significant difference (i.e., p > 0.05). If not stated otherwise, the number n, mentioned when analyzing the statistical significance of the comparison, refers to the number of independent sets of experiments.
Results
Low-frequency depression does not result from a depletion of ready-to-fuse synaptic vesicles
In the buccal ganglion of Aplysia, the twin B4 and B5 cholinergic neurons make well established inhibitory synapses on the same set of postsynaptic neurons. Evoked release level remains stable for very long time when the stimulation rate is maintained at a given frequency, but any increase, even moderate, in the stimulation frequency reproducibly decreases synaptic efficacy (Doussau et al., 1998; Humeau et al., 2001a,b, 2002). As shown in Figure 1A, increasing the stimulation rate from 0.025 to 1 Hz quickly induced an onset of depression reaching a plateau (at 43.16 ± 1.79% from control amplitude) in ∼30 min. Because of its induction at a low frequency (i.e., 1 Hz), we termed it LFD and investigated the underlying mechanisms.
At many synapses, synaptic depression occurring on repetitive activity arises from depletion of the RRP of SVs, possibly because of incomplete refilling at each stimulus (for review, see Kavalali, 2006). Accordingly, provided the interpulse interval is shorter than the time needed to refill all the empty release sites (we expected this is the case using a 1 Hz frequency), an increase in the output release probability P should produce faster depletion of the RRP on repetitive stimulation. Experimentally, P can be easily manipulated by changing the concentration of extracellular Ca2+ (Silver et al., 1998; Humeau et al., 2002). Under such conditions, we determined (see Materials and Methods, and below) an average p = 0.26 when [Ca2+]/[Mg2+]e = 0.42 and p = 0.62 when [Ca2+]/[Mg2+]e = 2.1. LFD was elicited under two conditions, namely in low and high [Ca2+]/[Mg2+]e-containing medium (Fig. 1A1). Although the average IPSC amplitude was greatly modified by ∼2.4-fold (at 0.025 Hz) because of the increase in P between the low and high [Ca2+]/[Mg2+]e conditions, the time courses of LFD were very similar (Fig. A3): in both conditions, the delay (d) before the actual induction of depression (8.4 s at low P and 7.4 s at high P) and the relative extents of depression (43.16 ± 1.79% at low P and 44.22 ± 1.39% at high P) were similar. In addition, the fast (τI) and slow (τII) time constants, and the relative weight of the decay components were all in the same range of magnitude (Table 1, Fig. 1B). Overall, these findings indicate that the kinetics and extent of LFD are independent of initial P, suggesting that changes in refilling of empty release sites or replacement of consumed SVs is unlikely to be the main cause of LFD.
Table 1.
Time constants of LFD, LFDAR, and PTPLFD were obtained by fitting mean amplitude values of IPSC (I) at any stimulus from n experiments (see Materials and Methods)
| Stim. | n | ωfast (%) | τfast (s) | ωI (%) | τI (s) | ωII (%) | τII (min) | |
|---|---|---|---|---|---|---|---|---|
| LFD low P | S | 9 | 54.6 ± 4.9 | 54.5 ± 6.9 | 45.4 ± 2.3 | 8.9 ± 1.0 | ||
| LFD high P | S | 7 | 51.9 ± 3.3 | 39.1 ± 9.7 | 48.1 ± 5.5 | 8.1 ± 0.9 | ||
| Stat. with LFD low P | * | * | NS | NS | ||||
| LFD low P + ApSynAla | S | 4 | 64.3 ± 2.1 | 39.1 ± 7.1 | 35.7 ± 3.9 | 10.2 ± 2.8 | ||
| Stat. versus LFD low P | * | NS | NS | NS | ||||
| LFD low P + PKAi6-22 | S | 4 | 57.2 ± 3.8 | 18.7 ± 2.9 | 42.8 ± 3.9 | 6.8 ± 1.7 | ||
| Stat. with LFD low P | NS | ** | NS | NS | ||||
| LFD high P | PP | 10 | 43.3 ± 4.1 | 26.8 ± 4.5 | 56.7 ± 5.8 | 10.4 ± 1.1 | ||
| LFDAR rest 10 s | PP | 4 | 100 | 1.2 ± 0.2 | ||||
| LFDAR rest 15 s | PP | 4 | 100 | 1.6 ± 0.4 | ||||
| LFDAR rest 20 s | PP | 6 | 85.8 ± 5.8 | 1.0 ± 0.1 | 14.2 ± 2.0 | 19.8 ± 6.1 | ||
| Stat. with LFD high P | — | — | — | NS | ||||
| LFDAR rest 40 s | PP | 5 | 86.3 ± 13.5 | 1.5 ± 0.2 | 13.7 ± 1.7 | 70.8 ± 12.0 | ||
| Stat. with LFD high P | — | — | — | ** | ||||
| LFDAR rest 60 s | PP | 4 | 76.7 ± 9.7 | 1.4 ± 0.4 | 23.3 ± 10.3 | 34.3 ± 10.2 | ||
| Stat. with LFD high P | — | — | — | NS | ||||
| LFDAR rest 90 s | PP | 6 | 69.0 ± 12.0 | 1.0 ± 0.2 | 18.4 ± 7.8 | 13.2 ± 3.9 | 12.6 ± 0.6 | 2.3 ± 0.6 |
| Stat. with LFD high P | — | — | — | NS | — | *** | ||
| LFDAR rest 120 s | PP | 5 | 65.0 ± 12.2 | 1.4 ± 0.2 | 22.1 ± 6.0 | 24.3 ± 8.3 | 12.1 ± 0.5 | 2.2 ± 0.1 |
| Stat. with LFD high P | — | — | — | NS | — | ** | ||
| LFDAR rest 240 s | PP | 4 | 63.1 ± 3.6 | 1.6 ± 0.4 | 23.3 ± 6.1 | 22.8 ± 5.5 | 13.6 ± 1.4 | 3.4 ± 0.3 |
| Stat. with LFD high P | — | — | — | NS | — | ** | ||
| LFDAR rest 900 s | PP | 7 | 49.0 ± 8.9 | 1.3 ± 0.2 | 29.2 ± 6.1 | 17.8 ± 2.7 | 21.8 ± 3.0 | 4.3 ± 0.3 |
| Stat. with LFD high P | — | — | — | NS | — | *** | ||
| PTPLFD rest 20 s | PP | 5 | 60.3 ± 12.7 | 1.0 ± 0.1 | 27.7 ± 5.3 | 6.6 ± 0.1 | 12.0 ± 2.7 | 5.5 ± 0.8 |
| Stat. with LFD high P | — | — | — | ** | — | ** | ||
| PTPLFD rest 120 s | PP | 3 | 59.1 ± 6.9 | 1.6 ± 0.4 | 26.1 ± 4.1 | 9.4 ± 3.8 | 14.8 ± 2.7 | 3.4 ± 1.2 |
| Stat. with LFD high P | — | — | — | ** | — | ** | ||
| Stat. with PTPLFD rest 20 s | NS | NS | — | NS | — | NS |
Low P and high P correspond to [Ca2+]/[Mg2+]e = 0.42 and [Ca2+]/[Mg2+]e = 2.1, respectively. LFDAR and PTPLFD were probed at [Ca2+]/[Mg2+]e = 2.1. LFD, LFDAR, and PTPLFD were probed using either a single stimulus (denoted as S) or a paired-pulse stimulation (interval, 40 ms; denoted as “PP”). The statistical differences (mentioned as “Stat.”) in the weights (ωI and ωII) and in the time constants (τI and τII) of the exponential decays were tested for two different conditions using either the t test or the Mann–Whitney rank sum test. The statistical difference in τfast calculated during LFDAR and PTPLFD at any rest period was probed using a one-way ANOVA. This test did not detect any statistical difference in τfast (p = 0.64).
*p < 0.05,
**p < 0.01, and
***p < 0.001. NS denotes no significant difference (i.e., p > 0.05).
Changes in the release probability or in quantal size do not mediate LFD
The use of the PPR as an index to detect change in P, although widely used in many works, has been challenged by several studies showing that modification of postsynaptic receptors properties may also induce change in PPR (Akopian and Walsh, 2002; Heine et al., 2008). At Aplysia cholinergic synapses, PPR has been shown to be inversely proportional to [Ca2+]/[Mg2+]e (Doussau et al., 1998; Humeau et al., 2001a, 2007) and a change in [Ca2+]/[Mg2+]e induces a change in P (Humeau et al., 2002). However, a direct correlation between PPR and P has never been established. In a series of five experiments, ganglia were superfused using different [Ca2+]/[Mg2+]e ratios (0.14, 0.21, 0.42, 0.84, and 2.1) and ACh release was evoked using twin stimuli (interval, 40 ms). When a stable plateau was reached, PPR, Imean, and Var were determined for at least 20 subsequent IPSCs [for a rationale, see Materials and Methods and Humeau et al. (2002, 2007)]. To pool the data, Var = f(Imean) data were fitted using a simple parabola and then normalized to the fitted maximum Var and the corresponding Imean (Fig. 2A). From the pooled parabola, average P were deduced from each point in this plot and associated with the corresponding PPR value. As shown in Figure 2B, a linear relationship between PPR and P can be established as follows: PPR = 1.6 − 1.2 · P (r2 = 0.86).
These results confirm that, at these synapses, PPR is actually proportional to P. Therefore, PPR was used to determine whether LFD results from a decrease in P. The mean values of IPSC amplitude and PPR were determined before LFD by applying twin stimuli (40 ms interval) at 0.025 Hz, and after the induction of LFD by increasing the paired-pulse stimulation frequency to 1 Hz. To allow a comparison with the effect of manipulating P, we also monitored PPR when [Ca2+]/[Mg2+]e was progressively reduced. Although IPSC amplitude was markedly reduced at the plateau of LFD (I1 of the pair reduced by ∼40%), PPR was only slightly increased (Fig. 2C, right) compared with the PPR enhancement observed on decreasing ACh release to a similar extent (by ∼40%) by lowering [Ca2+]/[Mg2+]e (Fig. 2C, left). The PPR = f(I1) relationship obtained by manipulating [Ca2+]/[Mg2+]e was satisfactorily fitted using a linear function with a negative slope of the following form: PPR = 1.4 − 4.9*I1 (r2 = 0.91) (Fig. 2D). In contrast, changes in I1 were not correlated with changes in PPR (a linear relationship between PPR and I1 cannot be established, r2 = 0.15) when PPR was determined during LFD (Fig. 2D). This lack of a significant change in PPR during the course of LFD indicates that a reduction in P cannot directly contribute to LFD.
Next, we investigated whether changes in the number of the functional release sites, N, or in the quantal size, Q, may underlie LFD. Assuming that Imean = N · P · Q and Var = N · P · (1 − P) · Q2 (see Materials and Methods), the ratio Var/Imean = Q · (1 − P) between the initial conditions (i.e., at 0.025 Hz, before LFD) and plateau of LFD (i.e., at 1 Hz) can be used to identify which of N, Q, and P is modified and responsible for the reduction in Imean during LFD. Figure 2E presents the calculated values of Var/Imean that we should have observed if the ∼44% decrease in IPSC amplitude during LFD were attributable to a pure change in Q, N, or P (dashed lines). Note that modification of N leaves Var/Imean unmodified, whereas this ratio changes in opposite ways when Q or P is modified. We found no significant difference between the Var/Imean determined before LFD or at its plateau (Fig. 2E, compare open and black-filled bars). However, when [Ca2+]/[Mg2+]e was decreased from 2.1 to 0.42 (i.e., a P decrease from 0.62 to 0.26), Var/Imean was significantly enhanced (Fig. 2E, compare open and gray-filled bars). These findings pinpoint that LFD mainly results from a decrease in N, indicating a silencing of release sites. However, these data cannot distinguish whether this results from a permanent or long-lasting (1) block of the refilling at certain release sites, (2) blockage of the priming process for a subpopulation of SVs, (3) or from an inactivation of the fusion competence of plasma membrane at a subpopulation of release sites.
Minor PPR changes during LFD indicate concomitant expression of opposite forms of synaptic plasticity
Although, overall, PPR does not change at the plateau of LFD compared with the control before LFD, minor changes in PPR (ranging between 0.97 and 1.12) reproducibly occurred at the same stimulus number during the course of LFD (Fig. 2F). When the stimulation rate was raised from 0.025 to 1 Hz, PPR was slightly, but significantly (p = 0.01), increased at the first stimulus, whereas the depression in I1 amplitude was barely apparent. Surprisingly, over the subsequent ∼200–250 stimuli, PPR decreases concomitantly with I1 (Fig. 2F) pinpointing a puzzling situation during which the decline in I1 is associated with an increase in P. Then, during the last phase of LFD (after stimulus 250), there was again a slight increase in PPR, whereas the amplitude of I1 continued to decrease. Overall, this suggests that several opposite forms of synaptic plasticity are operating during the early phase of LFD; after an initial decrease in P observed at the first stimulus, the reduction in N, which underlies LFD, is temporarily compensated by a concomitant short-term synaptic enhancement because of an increase in P (decrease of both I1 and PPR for the first 200–250 stimuli). This is likely to account for the observed delay that occurs after the 1 Hz stimulation is initiated. This may also explain why, in some preparations, a transient potentiation of IPSC amplitude could be observed during the earliest phase of LFD (data not shown).
Recovery from depression after variable periods of inactivity implicates the awakening of silenced release sites
To investigate the recovery from LFD, we first examined the kinetics by which depressed synapses at rest (i.e., without stimulation) recovered their initial release capacity. LFD was induced, and after the plateau was reached (after ∼30 min at 1 Hz), the stimulation was stopped for varying periods of rest (of duration Δt), before resuming the stimulation (paradigm displayed on Fig. 3A). After a rest period of ∼60 s, the amplitude of the first IPSC reached the value of the plateau determined before induction of LFD (Fig. 3B). However, compared with control conditions (i.e., when LFD was elicited after a plateau at 0.025 Hz), the capacity to sustain repetitive stimulation after a rest period was very low. Indeed, for the shortest rest periods, resuming the 1 Hz stimulation led to a depression with a very fast decay (τfast ranging from 1 to 1.6 s) (Table 1), which reached the plateau of LFD observed before rest with just few stimuli (Fig. 3B) (see below). We will refer to this as LFDAR. LFDAR displayed a fast monoexponential decay for Δt <20 s, and clearly a triexponential decay for Δt >40 s (Fig. 3C,D). The two slowest decay components of LFDAR were in the same order of magnitude (tens of seconds for τI and several minutes for τII) as those determined for control LFD. Overall, regardless of the rest periods, the time constant τI of LFDAR was not significantly changed as compared with τI of LFD (Table 1). In contrast, the time constant τII of LFDAR was markedly faster at all rest periods tested (Fig. 3E, Table 1). Even if τII of LFDAR tended to increase proportionally with the length of the rest period (Fig. 3E), at the longest rest period tested (15 min) this kinetic component was more than twofold faster than the τII of LFD. This indicates that a residual form of depression induced during LFD was not still present after 15 min of rest.
Figure 3.
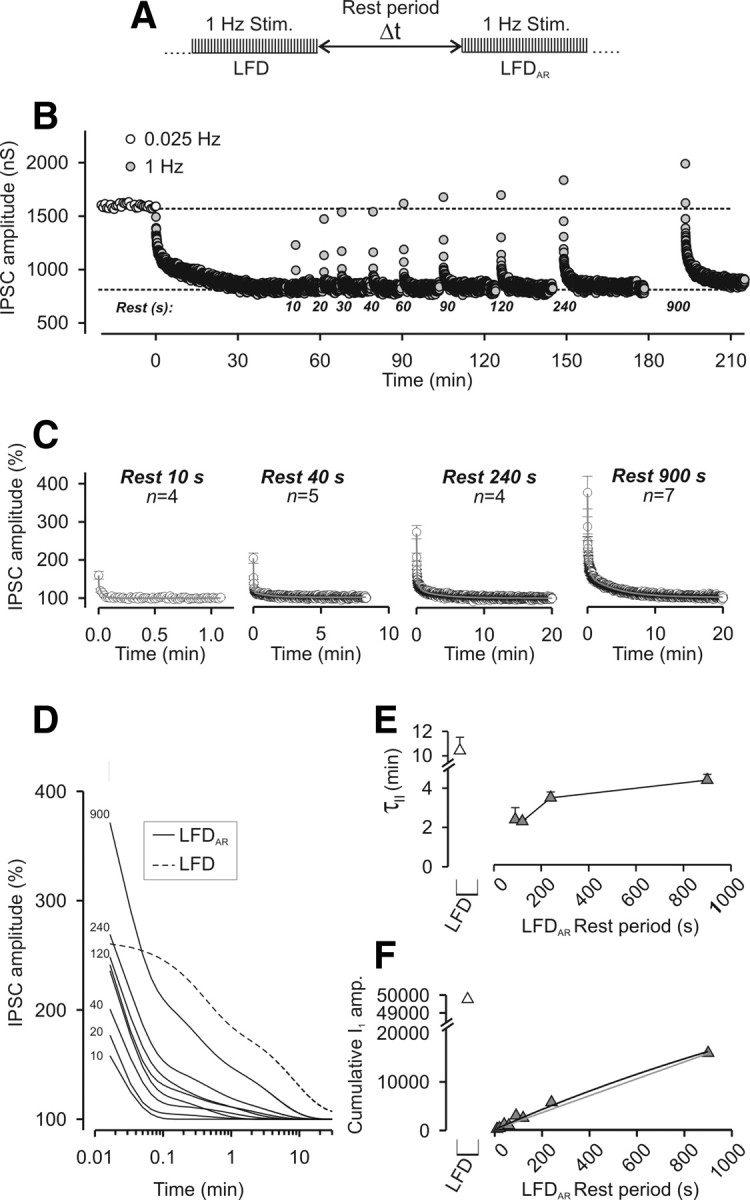
Recovery from LFD after variable rest periods. A, Paradigm of stimulation used to elicit LFDAR. B, Typical experiment performed at the same synapse during which LFDAR was probed after different duration of the rest periods. Time 0 denotes the start of 1 Hz stimulation. Note that the plateaus of LFD and LFDAR are identical. C, The time course of LFDAR was determined by averaging IPSCs at any stimulus from n experiments as indicated on the graphs. The solid gray lines represent the exponential decay curves fitted to the data. Only four representative rest periods are shown. D, Superimposition of the fitted decay curves to the data obtained for all the tested rest periods. The number indicated on the left of curves corresponds to the respective duration of the rest period. For C and D, IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau of LFD. E, The slowest time constants (τII) of LFDAR (gray-filled triangles) that were obtained by fitting mean amplitude values of IPSC (Table 1) were plotted against the length of the rest periods and compared with the slowest time constant of LFD (open triangle). F, Cumulative plots of IPSC amplitude were calculated from the fits obtained in D over a period of 30 min (same symbols as in E). Error bars indicate SEM.
The cumulative IPSC amplitude can be used as an index of the total number of quanta released during a sustained stimulation (Schneggenburger et al., 1999). Since the above data suggest that, after short rest periods, the capacity to sustain ACh release at depressed synapses is far from being fully recovered, we calculated the cumulative IPSC amplitude over the first 30 min of LFD or LFDAR (either from LFDAR data or extrapolating their fit to 30 min). Figure 3F shows that the initial release capacity calculated during LFD (Fig. 3F, gray-filled triangle) was not yet recovered after rest periods as long as Δt = 900 s. Extrapolation of the “cumulative IPSC amplitude = f(Δt)” relationship using either a monoexponential function rising to a maximum (Fig. 3F, solid black line) or a linear function (Fig. 3F, solid gray line) indicated that spontaneous full recovery of release capacity is expected between 40 min and 3 h. Similarly as the deduction made when examining τII of LFDAR, these data indicate that recovery from LFD is a slow process, which is not complete after 15 min of rest (Fig. 3, compare E, F). Together, this suggests that LFD is a form of presynaptic long-term depression.
To investigate the mechanisms underlying the spontaneous recovery from LFD, we analyzed the PPR changes that occurred during LFDAR. Although increasing the duration of the rest period at the plateau of LFD greatly increased the extent of recovery in the IPSC amplitude (see above) (Fig. 4A1), the average PPR remained unchanged (Fig. 4A2). Except for the PPR determined for the first two IPSCs corresponding to the very fast phase of LFDAR, the time course of minor PPR changes occurring during LFDAR and LFD were indistinguishable (Fig. 4B2). Together, these findings suggest that, except for the early phase, LFDAR and LFD and their underlying mechanisms are similar. The fact that the changes in IPSC amplitude during LFD or LFDAR were not associated with PPR changes (except for stimuli 1 and 2 corresponding to the fast early phase) indicates that synaptic depression is in both cases attributable to a decrease in N (i.e., release site silencing). By inference, this suggests that the recovery of synaptic efficacy during the rest periods is attributable to the reactivation of a fraction of release sites that were turned off during LFD, and that the number of sites that switch on during rest is somehow proportional to the duration of the rest period. However, both the persistence of a faster kinetic component and an incomplete restoration of the release capacity (Fig. 3E,F) indicate that the functioning of these sites was not reinitiated to initial conditions at the end of the rest periods tested.
Figure 4.

Effects of the duration of the rest period on IPSC amplitude and PPR. A, PPR change during LFDAR is independent of the duration of the rest period. A1, Mean values of I1 from n experiments plotted against the stimulus number during LFDAR after three distinct rest periods. A2, Corresponding values of PPR. B, Minor PPR changes during LFDAR are similar to those observed during LFD (except for the first response) and appear fixed to stimulus number. B1, Mean values of I1 from n experiments plotted against the stimulus number during LFD (same data as in Fig. 2D) and LFDAR (rest, 900 s; same data as in Fig. 5A1). B2, Corresponding values of PPR. For A1 and B1, IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau of LFD. Error bars indicate SEM.
Next, we investigated the early fast depression phase that characterizes LFDAR and contributes mostly to the first two IPSCs recorded after resuming the 1 Hz stimulation. The fastest exponential decay component of LFDAR (τfast) was isolated by a fitting procedure (Fig. 5A) and the number of quanta released during this phase was estimated in function of the duration of the rest period (Fig. 5B). The function “cumulative IPSC amplitude = f(Δt)” could be fitted by a biexponential rising a maximum with time constants of 7.9 s and 4 min. This fit indicates that the capacity to sustain release during the fast phase promptly reached saturation. In contrast to the slower decay components of LFDAR and LFD (τI and τII), the early fast decay phase of LFDAR was associated with a very low value of PPR (Fig. 5C), which indicates that the probability of release (P) is high during this early short-lived phase. Importantly, the particular value of PPR determined at stimulus 1 (denoted as a, b, or c in Fig. 5C2) was constant and independent of the duration of the rest period (Fig. 5D). After several minutes of rest, cytosolic [Ca2+] should have returned to basal levels and thus the acquisition of a high P status cannot be accounted for by a high amount of Ca2+ in the presynaptic terminal at the time of resuming the 1 Hz stimulation. However, this does not rule out a role for Ca2+ in this process. Indeed, a rise in cytosolic [Ca2+], which is likely to occur at naive synapses during the first stimulation at 1 Hz, may induce a switch of active releases sites to a high P status that persists for minutes (at least 15 min) after stopping stimulation. In any cases, the fast saturation of the early fast phase (Fig. 5B) clearly indicates that the high P status is acquired for rest periods too short to allow the awakening of silenced release sites. Therefore, the acquired high P is likely to apply to release sites that had not been turned off during LFD (i.e., release sites still active at the plateau of depression, or a subset of them). Overall, this high P phase is the expression of a peculiar form of presynaptic plasticity that superimposes on the awakening of release sites. This superimposed facilitation required a rest period to be expressed, is preserved whatever the length of the rest period is, and fades only after reactivation of the release machinery. The fact that the kinetics of LFDAR was the result of the superimposition of two different forms of synaptic depression (the fast decaying component τfast and the components τI and τII of LFD) probably explains the absence of a delay in the onset of depression observed at naive synapse (Fig. 1B).
Figure 5.

Characterization of the fast decay phase of LFDAR. A, Exponential decay curves of LFDAR for all rest period tested could be decomposed into a monoexponential decay function (solid black line) with a fast component τfast (0.9 s < τfast < 1.7 s) and a biexponential decay function (solid gray line) with two components τI and τII (15 s < τI < 70 s and τII > 2 min). Only three representative rest periods are displayed. IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau of LFD. B, Cumulative plots of IPSCs during the fast phases of LFDAR were calculated from the fits of these fast phases (represented by the solid black lines in A) were plotted against the duration of the rest period. Values of the cumulative plots were normalized with respect to the value of the cumulative plot of IPSC amplitude calculated during LFD. C, Representative experiments showing the time course of I1 (C1) and of the corresponding values of PPR (C2) during LFDAR elicited after three distinct rest periods. C3, The left traces represent the first pairs of IPSCs recorded after the indicated rest period and correspond to the black arrow in C1. The right traces represent the same pairs of IPSCs normalized with respect to the amplitude of I1. For C1, IPSC amplitudes were normalized with respect to the mean IPSC value recorded during the plateau at 0.025 Hz. D, Mean values of the amplitude of the first response and the corresponding PPR as a function of the duration of the rest period. The dashed line represents the mean value of PPR of the first response calculated independently of the duration of the rest period. IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau of LFD. Error bars indicate SEM.
An application of high-frequency trains rapidly switches on the release sites silenced during LFD and allows fast resetting of neurotransmitter release
We previously reported that silencing of release sites because of inactivation of Rac-GTPase can be relieved by a high-frequency stimulation (or “tetanus”), which activates a bypass pathway (Doussau et al., 2000; Humeau et al., 2002, 2007). We examined whether a similar stimulation paradigm accelerates the reactivation of the sites switched off by the 1 Hz stimulation. However, as a tetanus induces PTP (Doussau et al., 1998, 2000; Humeau et al., 2001a, 2007), the concomitant acceleration of recovery from LFD must be separated from the overlapping PTP. First, PTP characteristics were determined under control conditions before induction of LFD (Fig. 6A, paradigm P1). This classical form of PTP is termed PTPcont. Consistent with previous reports (Doussau et al., 1998; Humeau et al., 2001a, 2002, 2007), PTPcont peaked at 73% above the basal amplitude and recovered to basal levels with a monoexponential decay (with a time constant of 11 min) (Fig. 6B). After LFD was induced and had reached plateau, the PTP paradigm was applied (Fig. 6A, paradigm P2), inducing a PTP termed PTPLFD (Fig. 6B). The 1 Hz stimulation was then resumed to induce LFD. With a delay of 20 s between the 50 Hz trains and this stimulation, the IPSC amplitudes at peaks of both PTPLFD and PTPcont were found to be identical (peak of PTPcont = 822 nS and peak of PTPLFD = 827 nS) (Fig. 6B). Since the stimulation rates used to monitor PTPLFD and PTPcont were different (0.025 vs 1 Hz), their time courses were also different. This apparent recovery from LFD after the 50 Hz trains (Fig. 6B) is probably the consequence of a combination of different forms of synaptic plasticity that act either additively or synergistically. For release sites silenced during LFD, 50 Hz trains may act by immediately switching them on by either bypassing the mechanism underlying their silencing or accelerating their spontaneous recovery. Fifty hertz trains may also induce a classical form of PTP (i.e., PTPcont) that acts on release sites still active at the plateau of LFD, on release sites that were reactivated by these trains, and on release sites with a high intrinsic P. Interestingly after delays longer than 10 s, the peak and time courses of PTPLFD were found to be independent of the duration of the rest period (Fig. 6C1,C2). Note the perfect superimposition of IPSC amplitudes for PTPLFD elicited after rest periods of 1, 20, and 120 s on Figure 6C2. When PTPLFD was probed 1 s after the 50 Hz train, a marked reduction of IPSC amplitude at stimuli 1 and 2 was observed (Fig. 6C2,C3), but the amplitudes of IPSCs at any other stimulus number were identical with the amplitudes of corresponding IPSCs measured during PTPLFD with longer rest periods (Fig. 6C3). This short-term depression, which overlaps PTPLFD only during the first 5 s that follow the 50 Hz trains, may be attributable to a 50-Hz-induced depletion of the RRP as previously reported for PTPcont at these synapses (Humeau et al., 2007). Alternatively, this short-term depression may be the consequence of a transient activation of a Ca2+-activated K+ current induced by Ca2+ accumulation during the tetanus as previously shown for PTP at another Aplysia synapse (Kretz et al., 1982). In any case, regardless of the mechanisms underlying PTPLFD, the process is initiated during or immediately after the 50 Hz train.
Figure 6.

Recovery from LFD after a tetanus. A, Paradigms of stimulation used to elicit PTPcont, PTPLFD, and PTP1→0.025. Tetanus (T) corresponds to two trains of APs at 50 Hz for 1 s each produced at 1 s interval. B, Representative experiment showing that the peaks of PTPcont and PTPLFD are identical. PTPcont and PTPLFD were elicited at the times indicated by the black arrows. P1 and P2 denote the stimulation paradigm illustrated in A. For B and C1, the times mentioned above the peaks of PTPLFD indicate the duration of the rest period after the conditioning trains (i.e., Δt in the paradigm P2). C1, Typical experiment performed at the same synapse during which PTPLFD was probed after different duration of the rest period after the conditioning trains. Conditioning trains were applied at the times indicated by the black arrows. C2, The superimposition of IPSC amplitude from C1 obtained during PTPLFD elicited after rest periods after the conditioning train of 1 s (open circles), 20 s (black-filled squares), and 120 s (gray-filled triangles) shows that the time course of PTPLFD was independent of the duration of the rest period after the conditioning trains. C3, The time courses of PTPLFD probed after rest periods after the conditioning trains of 1 s (open circles) and 20 s (black-filled squares) were obtained by averaging IPSCs amplitude recorded at the same stimulus number from four and five experiments, respectively. D, E, Resetting of ACh release after a tetanus. D, Representative experiment from a series of four, showing that the peak and time course of PTPcont and PTP1→0.025 are identical. Traces a, c, d, and f represent average traces of at least 10 successive IPSCs recorded during the period indicated by the thick horizontal lines. Traces b and e correspond to IPSCs recorded at the peaks of PTPcont and PTP1→0.025, respectively, as indicated by the thin black arrow. E1, Superimposition of the time courses of PTPcont (open circles) and PTP1→0.025 (gray-filled circles) elicited at low and high [Ca2+]/[Mg2+]e (left and right panels, respectively). Conditioning trains were applied at the times denoted by the black arrows. Mean values of IPSCs (from n experiments as indicated on the graphs) were expressed as a percentage of basal values (i.e., mean values of at least 10 successive IPSCs monitored during 0.025 Hz stimulation). E2, Corresponding values of PPR. Note that the values of PPR were slightly higher at the plateau of LFD (gray-filled circles, before tetanus) as already shown in Figure 2C. Error bars indicate SEM.
Since PTPLFD and PTPcont were not monitored at the same stimulation frequency, an evaluation of the release capacity of the synapses (as defined above) could not be performed. Thus, we compared the time course of PTPcont with the time course of a PTP elicited during a plateau of LFD, but this time probed with a 0.025 Hz stimulation frequency (Fig. 6A, paradigm P3). This PTP was termed PTP1→0.025 (Fig. 6D). Since we previously established that PTPcont at these Aplysia synapses is attributable to a transient increase in P, we also monitored PPR. As shown in Figure 6E, the amplitudes of IPSCs at any stimulus number, as well as the extent of PPR (monitored in the same neuron) were identical during PTPcont and PTP1→0.025. This was also independent of the initial release probability P (i.e., at various [Ca2+]/[Mg2+]e) (Fig. 6E). These experiments indicate that the 50 Hz stimulus can relieve LFD, and suggest that, after a train of high-frequency stimulation, the release parameters are fixed to values that are independent of the history of the synapse.
Next, we determined the release component contributing to PTPLFD. Given the paradigm used to induce PTPLFD, this latter is likely to result from a combination of PTPcont and LFDAR. Decay of PTPLFD recorded after a delay ≥20 s after the 50 Hz train was well fitted by a triexponential function with time constants close to those reported for LFDAR (Table 1). To separate the contribution of PTPLFD from spontaneous recovery from depression attributable to the rest period between the 50 Hz train and resuming stimulation (Fig. 6A), the amplitudes of IPSCs recorded during LFDAR were subtracted from those corresponding to the same stimulus number during PTPLFD (Fig. 7A1 for Δt = 20 s; B1 for Δt = 120 s). These differences were still well fitted by a triexponential function, confirming that the 50 Hz trains act on all release sites participating in LFDAR.
Figure 7.
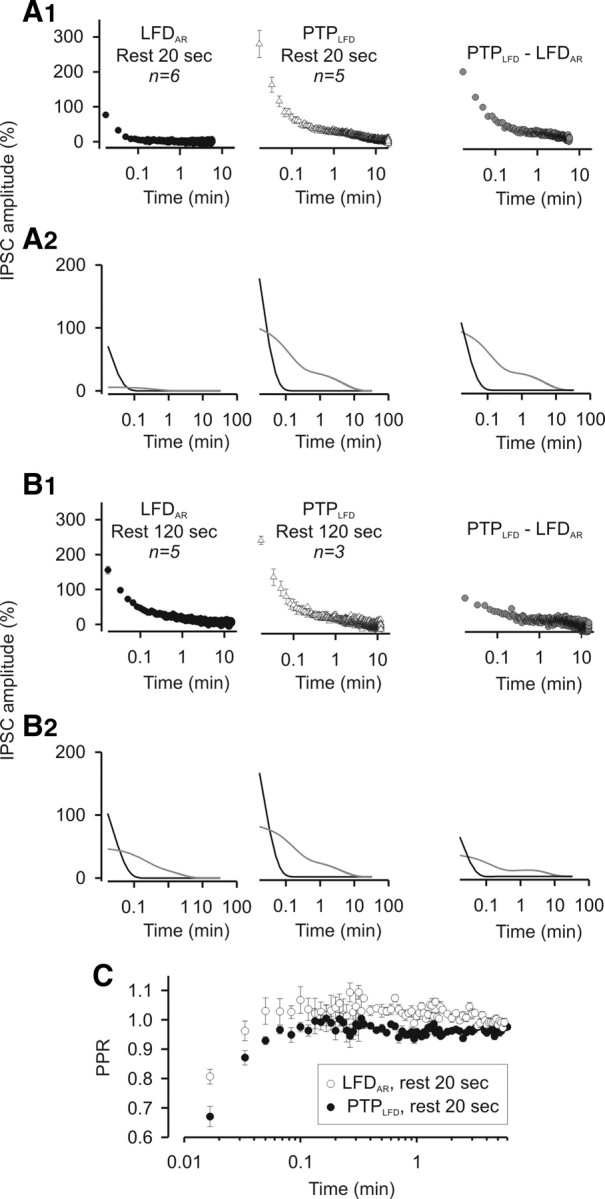
High-frequency stimulations accelerate the reactivation of the same population of release sites that were reactivated during period of inactivity. A1, Left and middle, Mean values of IPSCs recorded during LFDAR and PTPLFD after a rest period of 20 s. Each point corresponds to the mean value of n experiments as indicated on the graphs. Right, Subtraction of the mean values of IPSCs recorded during LFDAR after a rest period of 20 s from the corresponding IPSCs (i.e., at the same stimulus number) recorded during PTPLFD after a rest period of 20 s. A2, The time courses of LFRAR, PTPLFD, and the difference between PTPLFD and LFRAR could be fitted by a triexponential decay curve, which can be decomposed into a monoexponential decay with a fast component (0.9 s < τfast < 1.7 s; black lines) and a biexponential decay with time constants τI and τII close to the ones found in LFD (gray lines). B, Same measurements as in A, but for a rest period of 120 s. C, Superimposition of the mean values of PPR calculated for each stimulus during PTPLFD and during LFDAR after a rest period of 20 s. For A1 and B1, IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau of LFD. Error bars indicate SEM.
Determination of the relative weights of the three decay components revealed that, after the 50 Hz tetanus, the extents of the very fast phase (Fig. 7A2,B2, black line) and phases I and II (Fig. 7A2,B2, gray line) were increased in comparison with spontaneous recovery after a period of rest. This effect was less marked after a Δt = 120 s (Fig. 7B2), suggesting that the subsets of release sites reactivated by 50 Hz trains and release sites that spontaneously switched on during a period of inactivity are the same.
If a transient increase in P also contributes to PTPLFD (as in PTPcont), PPR should transiently decrease as observed during PTP1→0.025 or PTPcont. To visualize an eventual increase in P during PTPLFD, we superimposed the mean values of PPR obtained during PTPLFD from the corresponding PPR values obtained during LFDAR. Indeed, an increase in P was found to contribute to a much higher extent to PTPLFD than during LFDAR because PPR was significantly lower during PTPLFD than during LFDAR (Fig. 7C).
Together, these data indicate that trains of high-frequency stimulation are able to induce a rapid, nearly complete recovery from LFD, notably by accelerating the kinetics of spontaneous recovery from LFD. This raises the question of the synaptic mechanisms that allow immediate “resetting” of release characteristics to the level observed before LFD was induced.
Phosphorylation of synapsin at its P-site 1 is implicated in LFD and resetting
Phosphorylation of synapsin at its phosphorylation site 1 (P-site 1) by PKA has been shown to be essential in several forms of short-term plasticity including synaptic depression and PTP (Angers et al., 2002; Chi et al., 2003; Fiumara et al., 2004, 2007; Bonanomi et al., 2005; Hilfiker et al., 2005; Menegon et al., 2006).
We tried to determine whether phosphorylation of synapsin by PKA plays a role in LFD, PTP, and the resetting of synaptic strength at Aplysia cholinergic synapses. Presynaptic neurons were injected with either a recombinant Aplysia synapsin bearing a Ser9→Ala mutation (ApSynAla) that suppresses its ability to be phosphorylated by PKA (Fiumara et al., 2004, 2007) or a highly specific inhibitor of PKA (peptide 6–22 amide; PKAi6–22) (Hilfiker et al., 2001).
As shown in Figure 8A, neither ApSynAla nor PKAi6-22 had any effect on the basal release of ACh monitored at 0.025 Hz, but they both strongly affected the amplitude and the time course of LFD elicited at 1 Hz (Fig. 8B). The delay (d) that precedes the onset of depression was virtually abolished, and a significant depression was detected (by ∼20%) already at the first stimulus at 1 Hz. Analysis of the decay of LFD revealed that, after intracellular application of ApSynAla or PKAi6-22, LFD was best fitted by a biexponential decay with phases I and II similar to phase I and II measured under control conditions (Table 1). Moreover, the plateau of LFD was diminished (26.32 and 30.26% with ApSynAla and PKAi6-22, respectively), compared with the control plateau (43.16 ± 1.79%).
Figure 8.

Phosphorylation of synapsin at P-site 1 by PKA is involved in LFD, PTPcont, and PTPLFD. A1, A2, Representative experiment from a series of three during which PTPcont (paradigm P1) (Fig. 6A) and PTPLFD (paradigm P2) (Fig. 6A) (rest period of 20 s) were elicited in a mock-injected presynaptic neuron (A1) or in a neuron injected with ApSynAla (A2). Conditioning trains were applied at times indicated by the thick black arrows. A3, PTPcont and PTPLFD probed before and after microinjection of PKAi6-22, an inhibitor of PKA. ApSynAla (A2) and PKAi6-22 (A3) were injected at the time indicated by the white arrows. Traces a, c, e, g, i, k, m, o, and q correspond to the mean traces of at least 10 successive IPSCs recorded before the conditioning train as indicated by the thick horizontal black lines, whereas traces b, d, f, h, j, l, n, p, and r were recorded at the peak of PTPcont or PTPLFD as indicated by the thin black arrows. B, Phosphorylation of synapsin at P-site 1 by PKA is involved in LFD. Comparison of the time course of LFD (mean values of IPSCs from n experiments as indicated on the graph) elicited in control condition (open circles, same values as in Fig. 1B1) and neurons injected with either ApSynAla (black-filled triangles) or PKAi6-22 (gray-filled squares), respectively. C, D, Changes in IPSC amplitude during PTPcont (C) and PTPLFD probed after a rest period of 20 s (D). PTP was determined in control neurons (open circles) and in neurons injected with either ApSynAla (black-filled triangles) or PKAi6-22 (gray-filled squares). Each point corresponds to mean values of IPSCs from n experiments as indicated on the graphs. Note that intracellular injections of ApSynAla and PKAi6-22 greatly altered the PTPcont and PTPLFD. For A–C, IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau at 0.025 Hz. For D, IPSC amplitudes were normalized to the average IPSC amplitude determined during the plateau of LFD. Error bars indicate SEM.
Intracellular injections of ApSynAla and PKAi6-22 greatly altered both PTPcont (Fig. 8C) and PTPLFD (Fig. 8D). In this series of experiments ([Ca2+]/[Mg2+]e = 0.42), the PTPcont peaked at 92% (±7.6%; n = 7) above basal IPSC amplitude, and decayed monoexponentially (time constant of 11 min). After ApSynAla microinjections, PTP was abolished, creating instead a marked posttetanic depression (by 22.1 ± 8.9%; n = 3) (Fig. 8A2,C). PKAi6-22 microinjections reduced and delayed the peak of PTPcont; indeed, the peak was reached only after five to six stimulations and PTPcont peaked only at 38% (±6%; n = 4) above basal release (Fig. 8A3,C). Consistent with their effects on PTPcont, ApSynAla also abolished PTPLFD and PKAi6-22 strongly reduced it (Fig. 8D). This difference in the efficacy of PKAi6-22 and ApSynAla to affect PTPLFD may be attributable to a progressive degradation of PKAi6-22 in the cell. Indeed, we observed that, several hours after its injection, PKAi6-22 lost its effects on both PTPcont and LFD. Since PTPLFD was elicited at least 1 h after PKAi6-22 injection, we cannot exclude that the lack of efficacy of this peptide is attributable to its partial degradation in the cell. In any case, these data indicate that, at this Aplysia synapse, the fast recovery from LFD induced by tetanus requires phosphorylation of synapsin at its P-site 1.
Discussion
Comparison of LFD with other forms of homosynaptic depression
The form of LFD described here is clearly reminiscent of the synaptic fatigue or LFD reported at other invertebrate and mammalian synapses (Silver et al., 1998; Saviane et al., 2002; Abrahamsson et al., 2005; Silverman-Gavrila et al., 2005; Silverman-Gavrila and Charlton, 2009). In all cases, LFD is induced at the same rate of stimulation (1 Hz) and stabilizes at a plateau-fixed level. However, the underlying mechanisms of LFD probably depend on the type of synapse. At Aplysia cholinergic synapses, several important findings indicate that LFD mainly results from an activity-dependent silencing of a fraction of release sites. First, LFD is independent of the initial synaptic strength. In addition, there are changes neither in Var/Imean nor in PPR during the course of LFD. Finally, at rest, the restoration of the full capacity of release is achieved with a time constant much larger than the time constant of SV recycling. Interestingly, during the first minutes at 1 Hz, the depression attributable to the silencing of release sites is temporally counterbalanced by a quasi-immediate increase in P induced by the acceleration in stimulation frequency. Our results clearly implicate the PKA–synapsin pathway in this form of short-term synaptic enhancement as both ApSynAla and PKA inhibitor eliminated the delay in the onset of LFD. This is consistent with previous studies showing that a synapsin-dependent mobilization of SVs supports exocytosis during repetitive activity (Humeau et al., 2001a; Chi et al., 2003; Fassio et al., 2006; Menegon et al., 2006).
LFD differs from depression induced by high-frequency stimulation, which is the consequence of the rate-limiting kinetics of the refilling mechanisms and therefore related to RRP depletion (Wang and Kaczmarek, 1998; Wu and Borst, 1999; Schneggenburger et al., 2002; Zucker and Regehr, 2002; Foster and Regehr, 2004). LFD shares obvious similarities with homosynaptic depression described at Aplysia sensorimotor synapses (Royer et al., 2000; Gover et al., 2002). Both are induced at a low rate of stimulation, are independent of the release probability, and are the consequence of synapse silencing (Jiang and Abrams, 1998; Gover et al., 2002). In addition, these forms of depression are not related to RRP depletion. Full recovery of synaptic strength after LFD or after homosynaptic depression requires a very long rest period (Gover et al., 2002; this study). However, the finding of a release component characterized by a high intrinsic release probability and a limited capacity that allows fast, yet labile (lost in two to three stimuli at 1 Hz), recovery from LFD is a unique feature of the synapse studied here. At sensorimotor synapses, homosynaptic depression is relieved by serotonin application (Gover et al., 2002), whereas in our model serotonin by itself induces depression (data not shown), and very fast recovery can be induced by trains of high-frequency stimulation.
Mechanisms underlying the switching off of release sites
The mechanisms involved in the silencing of release sites during LFD were not identified. In theory, any event controlling the availability of readily releasable SVs on a release site in an all-or-none manner may determine the functional status of the site (see also Introduction and Materials and Methods). Since coactivation of several Ca2+ channels is likely to be required for inducing SV fusion in our model (Fossier et al., 1999; Humeau et al., 2007), we can exclude an activity-dependent inactivation of presynaptic Ca2+ channels during LFD, because this inactivation would have been detected as a decrease in P, rather than site silencing. Our data do not distinguish between selective long-lasting blockage of refilling at certain release sites, inactivation of fusion competence of plasma membrane at subpopulation of release sites, or complete blockage of priming events at a subset of sites or docked SVs. Interestingly, at mammalian synapses, the all-or-none feature of presynaptic adaptation has been proposed to result from changes in SV priming (Moulder et al., 2006). Moreover, at phasic synapses of a crayfish motoneuron, LFD is induced by an activity-dependent dephosphorylation of proteins involved in neurotransmitter release (Silverman-Gavrila et al., 2005). Therefore, a progressive and activity-dependent dephosphorylation of proteins involved in priming may lead to a silencing of presynaptic release sites.
Mechanisms underlying the switching on of release sites and resetting
Release site silencing is reversible. Indeed, sites can be reactivated either spontaneously with a slow time constant (∼40 min) or in an accelerated manner after a burst of high-frequency activity. PKA inhibition or microinjection of a nonphosphorylable synapsin mutated at its P-site 1 does not affect spontaneous recovery but compromises the accelerated recovery from LFD after application of 50 Hz trains. Hence, tetanus acts by activating the PKA–synapsin pathway probably via an elevation of cAMP concentration in the vicinity of release sites. Additional investigations should be done to establish the link between an application of 50 Hz trains and the activation of a synaptic adenylate cyclase. At its P-site 1, Aplysia synapsin can also be phosphorylated by Ca2+/calmodulin-dependent protein kinases I and IV (Angers et al., 2002; Fiumara et al., 2004); thus, our results do not permit to exclude the recruitment of these kinases in the pathway that controls the functional status of release sites even if all calmodulin inhibitors tested in our model were unable to affect LFD (data not shown).
Once again, this study is consistent with the finding that serotonin recruits the cAMP–PKA–synapsin pathway to switch on release sites silenced by repetitive activity at Aplysia sensorimotor synapses (Angers et al., 2002; Gover et al., 2002; Fioravante et al., 2007). Since synapsin also operates during the post-docking/priming events of exocytosis at invertebrate and vertebrate synapses (Hilfiker et al., 1998, 2005; Humeau et al., 2001a; Fassio et al., 2006), it may activate a pathway that bypasses the molecular events underlying silencing of release sites. The recruitment of this cascade for the reactivation of silent release sites at Aplysia synapses may be the functional equivalent of the cAMP–PKA–RIM1α pathway involved in presynaptic forms of long-term potentiation at both the hippocampal mossy fiber–CA3 synapses (Lonart et al., 2003) and the corticolateral amygdala synapses (Fourcaudot et al., 2008). Interestingly, the cAMP–PKA–RIM1α pathway is also implicated in the recovery from long-term depression at the mossy fiber-to-stratum lucidum synapses in the hippocampus (Pelkey et al., 2008).
Strikingly, bursts of high-frequency activity occurring at depressed synapses are able to reset all parameters of neurotransmitter release to the values observed before induction of LFD. At depressed synapses, resetting is illustrated by the induction of a form of PTP (PTP1→0.025), which is indistinguishable from PTP induced before LFD (PTPcont). The fact that the PKA–synapsin pathway is also involved in PTPcont does not necessarily mean that the mechanisms underlying PTP1→0.025 (or PTPLFD) and PTPcont are identical. PTPcont is related to an increase in P (which is detected as a decrease in PPR) (Fig. 6E) (Humeau et al., 2007), whereas PTPLFD and PTP1→0.025 are related to synapse awakening combined with an increase in P that applies to all classes of release sites (i.e., release sites with a high intrinsic P, release sites reactivated by the tetanus, and release sites that remained active before the tetanus). The mechanisms underlying this reactivation and the increase in P are probably different even if they are initiated by the same molecular cascades.
In addition to the activation of the PKA–synapsin pathway, the key event for resetting is the involvement of a subset of release sites that have acquired a high release probability status (as detected by a very small PPR) during LFD. This subset of release sites, which cannot by itself reconstitute full release capacity, can be maintained without any decline during long rest periods, and shapes the peak and the first phase of PTPLFD and PTP1→0.025. Along the same line, cAMP also selectively increases P at the calyx of Held synapses (Sakaba and Neher, 2001). In addition, at hippocampal synapses, the phosphorylation of synapsin by PKA counteracts synaptic depression and accelerates recovery from depression when neurons are challenged with sustained stimulation (Menegon et al., 2006).
In conclusion, we have explored the interplay between various forms of short-term synaptic plasticity involving either an activity-dependent regulation of the on/off functional status of release sites or a change in the probability of release. Although the molecular mechanisms involved are not completely elucidated, the PKA–synapsin pathway appears to be involved. The timing and the interplay of multiple forms of short-term plasticity allow fine-tuning of synaptic efficacy in response to changes in the stimulation rate and also confer surprising properties to the synapse. The most striking is the possibility of the synapse to “forget” its recent history (i.e., history of synaptic activity) and reset synaptic strength to a value that is fixed and independent of the status of release sites.
Footnotes
This work was supported by the European Neuroscience Institutes Network. We thank Dr. Nancy Grant for critically reading this manuscript and Dr. Jean-Luc Rodeau for his help in statistical analysis.
References
- Abrahamsson T, Gustafsson B, Hanse E. Synaptic fatigue at the naive perforant path-dentate granule cell synapse in the rat. J Physiol. 2005;569:737–750. doi: 10.1113/jphysiol.2005.097725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian G, Walsh JP. Corticostriatal paired-pulse potentiation produced by voltage-dependent activation of NMDA receptors and L-type Ca2+ channels. J Neurophysiol. 2002;87:157–165. doi: 10.1152/jn.00115.2001. [DOI] [PubMed] [Google Scholar]
- Angers A, Fioravante D, Chin J, Cleary LJ, Bean AJ, Byrne JH. Serotonin stimulates phosphorylation of Aplysia synapsin and alters its subcellular distribution in sensory neurons. J Neurosci. 2002;22:5412–5422. doi: 10.1523/JNEUROSCI.22-13-05412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, Menegon A, Miccio A, Ferrari G, Corradi A, Kao HT, Benfenati F, Valtorta F. Phosphorylation of synapsin I by cAMP-dependent protein kinase controls synaptic vesicle dynamics in developing neurons. J Neurosci. 2005;25:7299–7308. doi: 10.1523/JNEUROSCI.1573-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Clements JD. Variance-mean analysis: a simple and reliable approach for investigating synaptic transmission and modulation. J Neurosci Methods. 2003;130:115–125. doi: 10.1016/j.jneumeth.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Doussau F, Clabecq A, Henry JP, Darchen F, Poulain B. Calcium-dependent regulation of Rab3 in short-term plasticity. J Neurosci. 1998;18:3147–3157. doi: 10.1523/JNEUROSCI.18-09-03147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doussau F, Gasman S, Humeau Y, Vitiello F, Popoff M, Boquet P, Bader MF, Poulain B. A Rho-related GTPase is involved in Ca2+-dependent neurotransmitter exocytosis. J Biol Chem. 2000;275:7764–7770. doi: 10.1074/jbc.275.11.7764. [DOI] [PubMed] [Google Scholar]
- Fassio A, Merlo D, Mapelli J, Menegon A, Corradi A, Mete M, Zappettini S, Bonanno G, Valtorta F, D'Angelo E, Benfenati F. The synapsin domain E accelerates the exoendocytotic cycle of synaptic vesicles in cerebellar Purkinje cells. J Cell Sci. 2006;119:4257–4268. doi: 10.1242/jcs.03194. [DOI] [PubMed] [Google Scholar]
- Fdez E, Hilfiker S. Vesicle pools and synapsins: new insights into old enigmas. Brain Cell Biol. 2006;35:107–115. doi: 10.1007/s11068-007-9013-4. [DOI] [PubMed] [Google Scholar]
- Felmy F, Neher E, Schneggenburger R. Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron. 2003;37:801–811. doi: 10.1016/s0896-6273(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Liu RY, Netek AK, Cleary LJ, Byrne JH. Synapsin regulates basal synaptic strength, synaptic depression, and serotonin-induced facilitation of sensorimotor synapses in Aplysia. J Neurophysiol. 2007;98:3568–3580. doi: 10.1152/jn.00604.2007. [DOI] [PubMed] [Google Scholar]
- Fiumara F, Giovedì S, Menegon A, Milanese C, Merlo D, Montarolo PG, Valtorta F, Benfenati F, Ghirardi M. Phosphorylation by cAMP-dependent protein kinase is essential for synapsin-induced enhancement of neurotransmitter release in invertebrate neurons. J Cell Sci. 2004;117:5145–5154. doi: 10.1242/jcs.01388. [DOI] [PubMed] [Google Scholar]
- Fiumara F, Milanese C, Corradi A, Giovedì S, Leitinger G, Menegon A, Montarolo PG, Benfenati F, Ghirardi M. Phosphorylation of synapsin domain A is required for post-tetanic potentiation. J Cell Sci. 2007;120:3228–3237. doi: 10.1242/jcs.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossier P, Tauc L, Baux G. Calcium transients and neurotransmitter release at an identified synapse. Trends Neurosci. 1999;22:161–166. doi: 10.1016/s0166-2236(98)01307-1. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, Lüthi A. cAMP/PKA signaling and RIM1alpha mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gover TD, Jiang XY, Abrams TW. Persistent, exocytosis-independent silencing of release sites underlies homosynaptic depression at sensory synapses in Aplysia. J Neurosci. 2002;22:1942–1955. doi: 10.1523/JNEUROSCI.22-05-01942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Czernik AJ, Greengard P, Augustine GJ. Tonically active protein kinase A regulates neurotransmitter release at the squid giant synapse. J Physiol. 2001;531:141–146. doi: 10.1111/j.1469-7793.2001.0141j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Benfenati F, Doussau F, Nairn AC, Czernik AJ, Augustine GJ, Greengard P. Structural domains involved in the regulation of transmitter release by synapsins. J Neurosci. 2005;25:2658–2669. doi: 10.1523/JNEUROSCI.4278-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short term plasticity in Aplysia. J Neurosci. 2001a;21:4195–4206. doi: 10.1523/JNEUROSCI.21-12-04195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Vitale N, Chasserot-Golaz S, Dupont JL, Du G, Frohman MA, Bader MF, Poulain B. A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci U S A. 2001b;98:15300–15305. doi: 10.1073/pnas.261358698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Popoff MR, Kojima H, Doussau F, Poulain B. Rac GTPase plays an essential role in exocytosis by controlling the fusion competence of release sites. J Neurosci. 2002;22:7968–7981. doi: 10.1523/JNEUROSCI.22-18-07968.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Popoff MR, Benfenati F, Poulain B. Fast changes in the functional status of release sites during short-term plasticity: involvement of a frequency-dependent bypass of Rac at Aplysia synapses. J Physiol. 2007;583:983–1004. doi: 10.1113/jphysiol.2007.139899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XY, Abrams TW. Use-dependent decline of paired-pulse facilitation at Aplysia sensory neuron synapses suggests a distinct vesicle pool or release mechanism. J Neurosci. 1998;18:10310–10319. doi: 10.1523/JNEUROSCI.18-24-10310.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. Synaptic vesicle reuse and its implications. Neuroscientist. 2006;12:57–66. doi: 10.1177/1073858405281852. [DOI] [PubMed] [Google Scholar]
- Kretz R, Shapiro E, Kandel ER. Post-tetanic potentiation at an identified synapse in Aplysia is correlated with a Ca2+-activated K+ current in the presynaptic neuron: evidence for Ca2+ accumulation. Proc Natl Acad Sci U S A. 1982;79:5430–5434. doi: 10.1073/pnas.79.17.5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao HT, Benfenati F, Baldelli P, Valtorta F. Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci. 2006;26:11670–11681. doi: 10.1523/JNEUROSCI.3321-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Olney JW, Mennerick S. Physiological activity depresses synaptic function through an effect on vesicle priming. J Neurosci. 2006;26:6618–6626. doi: 10.1523/JNEUROSCI.5498-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Chang C, Taylor AA, Benz AM, Conti AC, Muglia LJ, Mennerick S. A specific role for Ca2+-dependent adenylyl cyclases in recovery from adaptive presynaptic silencing. J Neurosci. 2008;28:5159–5168. doi: 10.1523/JNEUROSCI.5317-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Yuan XQ, Lacaille JC, McBain CJ. State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron. 2008;60:980–987. doi: 10.1016/j.neuron.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quastel DM. The binomial model in fluctuation analysis of quantal neurotransmitter release. Biophys J. 1997;72:728–753. doi: 10.1016/s0006-3495(97)78709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Clements JD. Postsynaptic expression of long-term potentiation in the rat dentate gyrus demonstrated by variance-mean analysis. J Physiol. 1999;518:121–130. doi: 10.1111/j.1469-7793.1999.0121r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Coulson RL, Klein M. Switching off and on of synaptic sites at Aplysia sensorimotor synapses. J Neurosci. 2000;20:626–638. doi: 10.1523/JNEUROSCI.20-02-00626.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Preferential potentiation of fast-releasing synaptic vesicles by cAMP at the calyx of Held. Proc Natl Acad Sci U S A. 2001;98:331–336. doi: 10.1073/pnas.021541098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, Savtchenko LP, Raffaelli G, Voronin LL, Cherubini E. Frequency-dependent shift from paired-pulse facilitation to paired-pulse depression at unitary CA3-CA3 synapses in the rat hippocampus. J Physiol. 2002;544:469–476. doi: 10.1113/jphysiol.2002.026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of Held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol. 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman-Gavrila LB, Charlton MP. Calcineurin and cytoskeleton in low-frequency depression. J Neurochem. 2009;109:716–732. doi: 10.1111/j.1471-4159.2009.05973.x. [DOI] [PubMed] [Google Scholar]
- Silverman-Gavrila LB, Orth PM, Charlton MP. Phosphorylation-dependent low-frequency depression at phasic synapses of a crayfish motoneuron. J Neurosci. 2005;25:3168–3180. doi: 10.1523/JNEUROSCI.4908-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Vesicle depletion and synaptic depression at a mammalian ribbon synapse. J Neurophysiol. 2006;95:3191–3198. doi: 10.1152/jn.01309.2005. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Xu J, He L, Wu LG. Role of Ca2+ channels in short-term synaptic plasticity. Curr Opin Neurobiol. 2007;17:352–359. doi: 10.1016/j.conb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]