

Abstract
An increasing body of evidence points to a key role of endoplasmic reticulum (ER) stress in acute and chronic neurodegenerative conditions. Extensive ER stress can trigger neuronal apoptosis, but the signaling pathways that regulate this cell death remain unclear. In the present study, we demonstrate that PUMA, a Bcl-2 homology 3 (BH3)-only member of the Bcl-2 family, is transcriptionally activated in cortical neurons by ER stress and is essential for ER-stress-induced cell death. PUMA is known to be a key transcriptional target of p53, but we have found that ER stress triggers PUMA induction and cell death through a p53-independent mechanism mediated by the ER-stress-inducible transcription factor ATF4 (activating transcription factor 4). Specifically, we demonstrate that ectopic expression of ATF4 sensitizes mouse cortical neurons to ER-stress-induced apoptosis and that ATF4-deficient neurons exhibit markedly reduced levels of PUMA expression and cell death. However, chromatin immunoprecipitation experiments suggest that ATF4 does not directly regulate the PUMA promoter. Rather, we found that ATF4 induces expression of the transcription factor CHOP (C/EBP homologous protein) and that CHOP in turn activates PUMA induction. Specifically, we demonstrate that CHOP binds to the PUMA promoter during ER stress and that CHOP knockdown attenuates PUMA induction and neuronal apoptosis. In summary, we have identified a key signaling pathway in ER-stress-induced neuronal death involving ATF4–CHOP-mediated transactivation of the proapoptotic Bcl-2 family member PUMA. We propose that this pathway may be an important therapeutic target relevant to a number of neurodegenerative conditions.
Introduction
DNA damage and endoplasmic reticulum (ER) stress are believed to be central initiators of neuronal death in acute and chronic neurodegenerative conditions. Indeed, DNA strand breaks and markers of ER stress are common features of neurons destined to die in models of cerebral ischemia and neurodegenerative disease (Chen et al., 1997; DeGracia and Montie, 2004; Paschen and Mengesdorf, 2005; Martin, 2008). Although the signaling pathways mediating DNA damage and ER-stress-induced neuronal death are starting to be appreciated, the complexities of these signaling networks have yet to be fully defined.
One of the central players critical for the DNA damage response in neurons is the tumor suppressor protein p53 because it regulates several pathways involved in DNA repair and cell death (Vousden and Prives, 2009). Importantly, we and others have shown that p53 is essential for neuronal death induced by DNA damage (Xiang et al., 1996; D'Sa-Eipper et al., 2001; Morris et al., 2001). PUMA, a Bcl-2 homology 3 (BH3)-only family member, has been implicated as a proapoptotic p53-target gene (Han et al., 2001; Nakano and Vousden, 2001; Yu et al., 2001). We have demonstrated previously that PUMA is a critical downstream effecter of p53 in DNA-damage-induced apoptosis in neurons (Cregan et al., 2004). Although the p53–PUMA pathway is clearly critical for DNA-damage-induced neuronal death, its relevance to ER-stress-induced apoptosis is unclear.
ER stress is known to activate a series of signals that comprise the unfolded protein response (UPR). The UPR includes at least three signaling pathways initiated by the kinases IRE1 and PERK and the transcription factor ATF6 (activating transcription factor 6) (Ron and Walter, 2007). These signals coordinate the cellular response to unfolded proteins, which includes (1) downregulation of protein translation, (2) enhanced expression of ER chaperone proteins that promote protein refolding, and (3) activation of proteases involved in the degradation of misfolded proteins. Conversely, prolonged or severe ER stress can lead to the activation of apoptotic cell death. However, the mechanisms regulating ER-stress-induced cell death have not been clearly defined.
Of relevance to the present study, the transcription factors ATF4 and CHOP (C/EBP homologous protein) have been implicated in the ER stress response. ATF4 is a member of the basic leucine zipper family of transcription factors (Hai and Hartman, 2001). During ER stress, ATF4 is believed to be induced in a translationally dependent manner by the PERK–eIF2α pathway (Harding et al., 2000a; Scheuner et al., 2001). ATF4, in turn, can regulate the expression of a number of factors, including the transcription factor CHOP (Fawcett et al., 1999; Harding et al., 2003). Although it has been established that ATF4 and CHOP are induced by ER stress, the functional relevance of this induction and the mechanism by which this pathway regulate cell death is less clear.
In the present study, we define a critical signaling pathway involved in the regulation of ER-stress-induced apoptosis. Specifically, we demonstrate that Puma induction is critical for ER-stress-mediated neuronal death and establish that Puma expression is regulated by an ATF4–CHOP signaling axis.
Materials and Methods
Animals.
p53 mice and Bax mice were obtained from The Jackson Laboratory, mice harboring null mutations of Puma and Bim genes were generated in the laboratory of Dr. Andreas Strasser (Walter and Eliza Hall Institute of Medical Research, Bundoora, Victoria, Australia), and mice carrying an ATF4 null mutation were obtained from Drs. Tim Townes and Joe Sun (University of Alabama at Birmingham, Birmingham, AL). All transgenic strains were maintained on a C57BL/6 background and genotyped as described previously (Bouillet et al., 1999; Cregan et al., 1999; Fortin et al., 2001; Masuoka and Townes, 2002; Villunger et al., 2003). Wild-type and knock-out littermates (experimental mice) were generated by breeding heterozygous mice.
Primary cortical neuron cultures.
Cortical neurons were dissociated from embryonic days 14.5–15.5 male and female mouse embryos and cultured in Neurobasal media containing N2 and B27 supplements (Invitrogen) as described previously (Cregan et al., 2002). Drug treatments were initiated 2–3 d after plating. Stock solutions of tunicamycin, thapsigargin, camptothecin, and etoposide were prepared in DMSO (all from Sigma-Aldrich) and diluted in culture media immediately before adding to cultures.
Calcium phosphate transfection.
The pCMV–ATF4 plasmid was a kind gift from Dr. Amy S. Lee (University of Southern California, Los Angeles, CA). A CHOP cDNA cloned into pCMV–Sport6 was purchased from Invitrogen and subcloned into pcDNA3. Cortical neurons were transiently transfected using calcium phosphate method as described previously (Aleyasin et al., 2004). Cortical neurons were plated in 24-well plates (4 × 105 cells per well) and after 2–3 d were cotransfected with 0.25 μg of pGFP (as a reporter) and 0.75 μg of either pCMV–ATF4, pCMV–CHOP, or pcDNA3 as an empty vector control.
Recombinant adenoviral infection.
Human ATF4 cDNA was a kind gift from Dr. Amy S. Lee. Recombinant adenovirus expressing ATF4 was generated by subcloning ATF4 cDNA (HindIII–EcoRV fragment) into the pAdTrack vector under a cytomegalovirus (CMV) promoter. The pAdTrack vector also contains a second CMV promoter, which controls expression of green fluorescent protein (GFP). This construct was used to generate recombinant adenovirus, as described previously (He et al., 1998). Adenovirus expressing only GFP was used as control. Cortical neurons were plated at 4 × 105 cells per well and transduced with recombinant adenoviruses at a multiplicity of infection (MOI) of 50 at the time of plating.
Nucleofection.
Cortical neurons were transiently transfected with small interfering RNA (siRNAs) using the Nucleofection system according to the instructions of the manufacturers (Amaxa Biosystems). Briefly, 50 pmol of siRNA was added to freshly dissociated cortical neurons suspended in Amaxa Mouse Neuron Nucleofector solution (5 × 107 cells/ml). In rescue experiments, 2.5 μg of the expression plasmids pCMV–GFP or pCMV–GFP–CHOP were also added to the cell suspension. The cell suspension was then electroporated with the Nucleofector device using program setting O-05, and then cells were immediately transferred to tubes containing DMEM supplemented with 10% FBS. Neurons were plated at a cell density of 1 × 106 cells/ml in DMEM/FBS, and the following day the media was changed to standard Neurobasal media containing N2/B27 supplements. Drug treatments were initiated after an additional 24 h incubation. HPLC purified siRNAs were obtained from Ambion (Applied Biosystems) and sequences are available on request.
Cell death and survival assays.
Neuronal apoptosis was assessed by examining nuclear morphology in Hoechst 33258 stained cells as described previously (Cregan et al., 2002). Briefly, neurons were fixed in 4% paraformaldehyde (containing 0.2% picric acid in 0.1 m phosphate buffer, pH 7.1) for 30 min, washed in PBS, and stained with Hoechst 33258 (0.25 μg/ml) dye. Neurons were visualized by fluorescence microscopy, and images were captured using a CCD camera (Q-imaging) and Northern Eclipse software (Empix Imaging). The fraction of cells exhibiting an apoptotic nuclear morphology characterized by pyknotic and/or fragmented nuclei containing condensed chromatin was scored by an individual blinded to the treatments. In certain experiments, the fraction of apoptotic cells was also determined by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay as described previously (Cregan et al., 1999).
Neuronal survival was assessed as described previously (Farinelli et al., 1998). Briefly, cells were lysed in 200 μl of lysis solution (10× PBS, 10% Triton X-100, 1 m MgCl2, and 5% cetyldimethylethylammonium bromide) and loaded on a hemocytometer, and the number of intact nuclei was evaluated using phase-contrast microscopy. Percentage survival was calculated as the ratio of live (intact) cells in drug-treated versus nontreated wells from the same embryo and multiplied by 100%. When indicated, neuronal survival was also assessed by Live-Dead Cytotoxicity assay according to the instructions of the manufacturer (Invitrogen) and described previously (Fortin et al., 2001). Survival is reported as the percentage of cells exhibiting calcein-AM fluorescence. In all survival and apoptotic assays, a minimum of 500 cells were scored for each well.
Caspase activity assay.
Neurons were harvested in caspase lysis buffer (10 mm HEPES, pH 7.4, 1 mm KCl, 1.5 mm MgCl2, 1 mm DTT, 1 mm PMSF, 10% glycerol, 5 μg/ml leupeptin, and 2 μg/ml aprotinin), and 10 μg of protein was used in caspase-3-like activity assay as described previously (Cregan et al., 1999). Briefly, protein samples were added to caspase reaction buffer (25 mm HEPES, pH 7.4, 10 mm DTT, 10% sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylamm onio]-1-propanesulfonate, and 10 μm caspase-3 substrate Ac-DEVD-AFC), and fluorescence produced by cleavage of Ac-DEVD-AFC (acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl-coumaryl) substrate was measured on a SpectraMax M5 spectrofluorimeter (400 nm excitation, 505 nm emission) over a 1 h interval. Caspase activity is reported as ratio of fluorescence output in treated samples relative to corresponding nontreated controls.
Quantitative reverse transcription-PCR.
RNA was isolated using Trizol reagent as per the instructions of the manufacturers (Invitrogen), and RNA concentration was measured on a spectrophotometer. Quantitative reverse transcription (RT)-PCR was performed using the QuantiFast SYBR Green RT-PCR kit (Qiagen) and 20 ng of RNA as described previously (Steckley et al., 2007). RT-PCR was performed on a Chromo4 detection system (MJ Research/Bio-Rad), and changes in gene expression were determined using the Δ(ΔCt) method using the ribosomal S12 transcript for normalization. Data are reported as fold increase in mRNA levels in treated samples relative to corresponding untreated control samples for each transcript. All PCRs exhibited high amplification efficiencies (>90%), and the specificity of the PCR products were confirmed by sequencing. Primer sequences used for gene specific amplification are available on request.
Western blotting.
Whole-cell extracts (50 μg of protein) were loaded and separated on 10–12% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Blots were probed with primary antibodies to ATF4/cAMP response element-binding protein-2 (CREB-2) (1:500; Santa Cruz Biotechnology), Gadd153/CHOP (1:500; Santa Cruz Biotechnology), PUMA (1:500; Prosci), or BIM (1:500; Stressgen) in blocking buffer overnight. Membranes were washed in TBST buffer (10 mm Tris, 150 mm NaCl, 0.1% Tween 20) and probed with HRP secondary (1:10,000), and immunoreactive bands proteins were visualized using SuperSignal ECL (Pierce). Blots were stripped and reprobed for β-actin (1:10,000; Santa Cruz Biotechnology) as a loading control.
Luciferase reporter assay.
A DNA fragment encompassing the PUMA promoter region −828 to +89 bp relative to the transcription start site was amplified from mouse genomic DNA using the primers 5′-GATCGGTACCCAAGCCATTTAGCAGGAACC and 5′-CTAGGCTAGCTGCTGCTTCTGGAGTCTTCG. The Puma(0.9 kb)–LUC reporter construct was generated by inserting the 0.9 kb Puma promoter fragment into the KpnI/NheI restriction sites of the pGL3basic vector (Promega). The 0.3 and 0.18 kb Puma promoter fragments were generated by PCR amplification of the Puma(0.9 kb)–LUC using the primers 5′-GATCGGTACCTGCTCCTCCTGCCTGGACCAG and 5′-GATCGGTACCCGTGGGAGCCAGCGAGAG, respectively, and the common reverse primer 5′-CTAGGCTAGCTGCTGCTTCTGGAGTCTTCG. The corresponding PCR products were digested and inserted into the KpnI/NheI restriction sites of pGL3b vector to generate the Puma(0.3 kb)–LUC and Puma(0.18 kb)–LUC reporter constructs. Neurons were cotransfected with 0.5 μg of the Puma–LUC constructs or pGL3b and 0.25 μg of either pcDNA3 or pCMV–ATF4, and, after 4 h, neurons were treated with 200 nm thapsigargin. Luciferase activity was measured after 24 h using the Dual-Luciferase Reporter Assay System (Promega) according to the instructions of the manufacturers. The Renilla Luciferase vector phRL–simian virus 40 was included in all transfections and used to normalize the reporter luciferase activity. Reporter-based luciferase activity is reported as the ratio of luciferase activity produced in cells transfected with pCMV–ATF4 or pCMV–CHOP relative to pcDNA3 for each reporter construct.
Chromatin immunoprecipitation assay.
Neuronal cultures (20 × 106 cells) were crosslinked with 0.5% formaldehyde and then harvested in SDS lysis buffer and sonicated to fragment DNA. Extracts were incubated with antibodies to p53 (CM5; Novocastra), ATF4/CREB-2 (C-20X; Santa Cruz Biotechnology), Gadd153/CHOP (sc-575; Santa Cruz Biotechnology), or rabbit IgG control and immunoprecipitated with protein-G Sepharose beads. Immunocomplexes were eluted and decrosslinked at 65°C, and DNA was recovered by phenol-chloroform extraction. Quantitative PCR was performed using the Quantitect SYBER Green PCR kit according to the instructions of the manufacturer (Qiagen). Relative DNA binding of p53, ATF4, and CHOP was determined as the ΔCt values between treated and untreated samples immunoprecipitated with the same antibody. ΔCt values were corrected for any changes in nonspecific interactions as determined by corresponding IgG pull-down samples, and data are reported as fold enrichment (2ΔCt) in DNA binding. Primer sequences used for amplification of the PUMA promoter were 5′-CTGTCCCCACGCTGTC (forward) and 5′-GCTTGCTTGCTGGTGTCG (reverse) and for the CHOP promoter were 5′-GACAAGTTCAGGAAGGACAGC (forward) and 5′-CGGAGGAGGTGAGTGAGTCA (reverse).
Data analysis.
Data are reported as mean and SEM. The value n represents the number of independent neuron cultures or number of embryos of indicated genotype from which independent neuron cultures were prepared involving at least three independent experiments. Differences between groups were determined by ANOVA and post hoc Tukey's test and were considered statistically significant when p < 0.05.
Results
ER stress triggers neuronal apoptosis through a PUMA-mediated pathway that is activated independently of p53
Several studies have indicated that ER stress induces cell death through an intrinsic/mitochondrial pathway regulated by Bax/Bak (Wei et al., 2001; Smith and Deshmukh, 2007). BH3-only Bcl-2 family proteins are known to be critical regulators of Bax/Bak activation during apoptosis (Bouillet and Strasser, 2002). Previous studies have suggested that the BH3-only family members Bim, Noxa, and Puma can be induced by agents that induce ER stress (Reimertz et al., 2003; Li et al., 2006; Puthalakath et al., 2007). Therefore, we examined the expression of these family members in cortical neurons treated with the ER stressors tunicamycin (N-glycosylation inhibitor) and thapsigargin (sarcoendoplasmic reticulum Ca2+-ATPase, Ca2+-ATPase inhibitor). We found that both of the ER stressors triggered a marked induction in Puma expression and a modest increase in Bim mRNA levels (Fig. 1A). However, Noxa mRNA levels were not induced in response to either of these agents. PUMA and to a lesser extent BIM protein levels were also found to be induced by ER stress, although this was only evident in Bax-deficient neurons, presumably as a result of the rapid cell death that occurs in wild-type neurons (Fig. 1B). We next examined whether these BH3-only members were required for ER-stress-induced apoptosis in neurons. To address this, we treated Puma+/+ and Puma−/− cortical neurons with the ER stressors tunicamycin and thapsigargin and assessed apoptosis by examining nuclear morphology. For comparison, sister cultures were treated with the DNA-damaging agents camptothecin and etoposide. As shown in Figure 2, treatment of wild-type neurons with ER stressors (Fig. 2A) or DNA-damaging agents (Fig. 2B) induced a substantial increase in the fraction of cells (>80%) exhibiting an apoptotic morphology characterized by chromatin condensation and/or nuclear fragmentation. In contrast, in Puma-deficient neurons, the ER stressors and DNA-damaging agents induced only a modest increase in apoptosis (<20%). Consistent with the observed protection afforded by Puma deficiency, caspase-3 activity induced by both ER stressors and DNA-damaging agents in wild-type neurons was dramatically attenuated in Puma-deficient neurons (Fig. 2C). These results indicate that, in addition to its key role in DNA-damage-induced cell death, PUMA is also a critical regulator of neuronal apoptosis induced by ER stress. Because BIM expression was also modestly upregulated in response to ER stress, we examined tunicamycin- and thapsigargin-induced apoptosis in wild-type and Bim-deficient neurons. However, unlike Puma-deficient neurons, Bim-deficient neurons were not protected against thapsigargin- or tunicamycin-induced apoptosis, suggesting that BIM is not required for ER-stress-induced apoptosis in neurons (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Figure 1.

Puma expression is induced in cortical neurons during ER stress. A, Cortical neurons were treated with tunicamycin (3 μg/ml) or thapsigargin (1 μm), and Bim, Noxa, and Puma mRNA levels were assessed by real-time RT-PCR. Expression was normalized to ribosomal S12 levels and is reported as fold increase over untreated controls (n = 4). B, Bax-deficient cortical neurons were treated with tunicamycin (TM), and Puma and Bim protein levels were assessed by Western blot at 24 h. NT, Nontreated.
Figure 2.

PUMA is required for DNA- damage-induced and ER-stress-induced apoptosis in cortical neurons. A, B, Cortical neurons derived from Puma wild-type and knock-out littermates were treated with 1 μm thapsigargin (TG, Tg), 3 μg/ml tunicamycin (TM, Tm), 10 μm camptothecin (CPT), or 10 μm etoposide (ETP), and the fraction of apoptotic cells was determined at 24 and 48 h by assessing nuclear morphology after Hoechst staining. The fraction of apoptotic neurons was significantly decreased in Puma+/+ versus Puma−/− cultures (n ≥ 7; *p < 0.001). C, Puma wild-type and knock-out cortical neurons were treated with DNA-damaging agents or ER stressors as above. Cell lysates obtained 20 h after treatment were assayed for caspase-3 activity (n = 5; *p < 0.001).
We and others have previously demonstrated that the transcriptional activator p53 is essential for PUMA induction and neuronal apoptosis induced by DNA damage (Cregan et al., 2004; Wyttenbach and Tolkovsky, 2006; Uo et al., 2007). Accordingly, we next investigated whether a p53–PUMA pathway is also involved in ER-stress-mediated neuronal death. To address this, we examined Puma expression in p53+/+ and p53−/− cortical neurons after treatment with the ER stressors tunicamycin and thapsigargin. As shown in Figure 3A, Puma mRNA levels were increased by the ER stressors to a similar extent in p53+/+ and p53−/− neurons, whereas, as expected, DNA-damage-induced Puma expression was abolished in the absence of p53. Furthermore, we found that p53 deficiency did not affect the level of ER-stress-induced neuronal apoptosis but, consistent with previous studies, dramatically reduced DNA-damage-induced cell death (Fig. 3B). Together, our data indicate that ER stress induces neuronal apoptosis through a PUMA-mediated pathway that is activated independently of p53.
Figure 3.

ER stress triggers Puma induction and neuronal apoptosis through a p53-independent mechanism. Cortical neurons derived from p53 wild-type and p53 knock-out littermates were treated with thapsigargin (1 μm; TG), tunicamycin (3 μg/ml; TM), camptothecin (10 μm; CPT), or etoposide (10 μm; ETP). A, RNA was extracted 12 h after treatment, and Puma mRNA levels were quantified by real-time RT-PCR. Puma expression was normalized to S12 levels and is reported as fold increase over untreated controls (n ≥ 6; *p < 0.01). B, Neurons were stained with Hoechst dye 30 h after treatment, and the fraction of apoptotic cells was determined by assessing nuclear morphology (n = 5; *p < 0.01). Veh, Vehicle.
ATF4 promotes ER-stress-induced neuronal apoptosis
The above results clearly indicate that a transcriptional activator other than p53 is required for Puma induction during ER stress. The transcription factor ATF4 has been implicated in ER stress, but its functional role in the ER stress response and relevance to cell death remain unclear. Accordingly, we examined the potential role of ATF4 in ER-stress- and DNA-damage-mediated death pathways. Indeed, we observed a robust induction in ATF4 protein levels after treatment with the ER stressor tunicamycin but not in response to the DNA-damaging agent camptothecin (Fig. 4A). ATF4 was also rapidly induced in response to thapsigargin treatment, and increased protein levels were evident within 2 h of treatment (Fig. 4B).
Figure 4.

ATF4 expression is induced by ER stress but not DNA damage. A, Cortical neurons derived from ATF4−/− mice and wild-type littermates were treated with tunicamycin (3 μg/ml) or camptothecin (10 μm), and ATF4 protein levels were assessed by Western blot. A nonspecific (ns) band was detected in extracts from both wild-type and ATF4-deficient neurons. NT, Nontreated. B, Cortical neurons were untreated or treated with thapsigargin (1 μm) for the indicated times, and ATF4 protein levels were assessed by Western blot.
We next examined whether gain or loss of function of ATF4 affects neuronal death induced by DNA damage or ER stress. In the first set of experiments, cortical neurons were cotransfected with plasmids expressing GFP and either ATF4 or an empty vector as control. After 24 h, transfected neurons were challenged with either tunicamycin or camptothecin, and the percentage of GFP-positive neurons exhibiting an apoptotic nuclear morphology was determined. As shown in Figure 5A, enforced expression of ATF4 sensitized neurons to tunicamycin-induced death. In contrast, ectopic expression of ATF4 protected neurons against camptothecin-induced apoptosis evident by an increase in neuronal survival from ∼40 to 60% (Fig. 5A). To ensure that these results were not an artifact of the transfection protocol, we repeated these experiments using recombinant-adenovirus-mediated ATF4 expression (Fig. 5B). Consistent with the transfection results, we found that ectopic expression of ATF4 markedly enhanced cell death induction by the ER stressors tunicamycin (84.7 vs 40% survival) and thapsigargin (71.7 vs 21.8% survival) at 24 h after treatment (Fig. 5B).
Figure 5.

Ectopic expression of ATF4 sensitizes neurons to ER-stress-induced but not DNA-damage-induced apoptosis. A, Cortical neurons were cotransfected with pGFP and either pcDNA3 [empty vector (EV)] or pcDNA3–ATF4, and, after 24 h, neurons were treated with camptothecin (10 μm), tunicamycin (2 μg/ml), or DMSO (0.1%) as a vehicle control. At the indicated times, neurons were fixed and stained with Hoechst 33258, and the fraction of apoptotic and nonapoptotic GFP-positive cells was assessed by fluorescence microscopy (n = 3; *p < 0.05). B, Cortical neurons were infected with recombinant adenovirus expressing GFP (Ad–GFP) or coexpressing ATF4 and GFP (Ad–ATF4/GFP) at 50 MOI and, after 48 h, treated with tunicamycin (2 μg/ml) or thapsigargin (1 μm). After 24 h, cells were fixed and stained with Hoechst 33258, and the fraction of apoptotic and nonapoptotic GFP-positive neurons was determined (n = 3; **p < 0.01).
To address the role of endogenous ATF4, we assessed neuronal survival in wild-type and ATF4-deficient neuronal cultures. As demonstrated in Figure 6A, ATF4-deficient neurons were found to be significantly more resistant than wild-type neurons to ER-stress-induced cell death. For example, whereas only ∼20% of ATF4+/+ and ATF4+/− neurons remain alive 36 h after treatment with tunicamycin or thapsigargin, ∼60–70% of ATF4-deficient neurons survive under the same conditions (Fig. 6A). Similar results were found when neuronal survival and apoptosis was measured by live–dead and TUNEL assays, respectively (Fig. 6C,D). In contrast, ATF4-deficient neurons were not resistant to DNA-damage-induced cell death (Fig. 6B). In fact, ATF4-deficient neurons exhibited a modest increase in cell death compared with wild-type neurons. Together, these results suggest that ATF4 promotes neuronal apoptosis during ER stress but can antagonize DNA-damage-induced cell death.
Figure 6.

ATF4-deficient neurons are protected against ER-stress-induced but not DNA-damage-induced apoptosis. A, B, Cortical neurons derived from ATF4−/− embryos and wild-type littermates were treated with the ER stressors tunicamycin (3 μg/ml) and thapsigargin (1 μm) or the DNA-damaging agents camptothecin (10 μm) and etoposide (10 μm). At the indicated times, cells were lysed, and the number of intact nuclei (viable cells) was counted. Survival was calculated as the ratio of viable drug-treated neurons to viable untreated neurons for each embryo (n ≥ 5; *p < 0.05, **p < 0.01). C, D, Wild-type and ATF4-deficient neurons were treated with thapsigargin (1 μm; TG) or tunicamycin (3 μg/ml; TM), and the percentage of calcein-AM-positive (live) cells and TUNEL-positive cells was measured after 36 h (n = 3; **p < 0.01).
ATF4 regulates Puma induction during ER stress
Given our observation that ATF4 promotes ER-stress-induced neuronal apoptosis, we investigated whether ATF4 is required for Puma induction by assessing Puma mRNA levels in wild-type and ATF4-deficient neurons. As shown in Figure 7, Puma induction by both tunicamycin and thapsigargin was significantly reduced in ATF4-deficient neurons compared with wild-type neurons at both 8 and 12 h after treatment. In contrast, Puma expression induced by DNA damage was not affected by ATF4 deletion. Thus, ATF4 promotes Puma induction during ER stress but not in response to DNA damage.
Figure 7.

ATF4 regulates Puma induction during ER stress but not DNA damage. Cortical neurons derived from ATF4−/− and ATF4+/+ littermates were treated with the ER stressors tunicamycin (3 μg/ml; TM) and thapsigargin (1 μm; TG) or the DNA-damaging agents camptothecin (10 μm; CPT) and etoposide (10 μm; ETP), and RNA was extracted after 8 and 12 h. Puma mRNA levels were quantified by real-time RT-PCR and are reported as fold increase over corresponding untreated controls (n ≥ 5; *p < 0.01).
We next examined whether ATF4 can activate the Puma promoter. To address this, we generated a reporter construct, Puma(0.9 kb)–LUC, in which we inserted a ∼0.9 kb mouse DNA fragment homologous to the previously reported human Puma promoter region upstream of luciferase in the pGL3b vector (Han et al., 2001; Yu et al., 2001). To identify the ATF4-responsive region, we also generated the truncated Puma reporter constructs Puma(0.3 kb)–LUC and Puma(0.18 kb)–LUC that contained only the proximal ∼300 and ∼180 bp regions of the 0.9 kb Puma promoter fragment. As shown in Figure 8A, cotransfection of the Puma(0.9 kb)–LUC reporter construct with an expression plasmid for ATF4 (pCMV–ATF4) resulted in an approximately threefold increase in luciferase activity relative to empty vector control (pcDNA3). Ectopic expression of ATF4 caused a similar level of luciferase induction when cotransfected with the Puma(0.3 kb)–LUC but did not significantly activate luciferase expression from the Puma(0.18 kb)–LUC or pGL3b vectors. These results suggest that an ATF4-responsive element is present in the Puma promoter and is located in the nonoverlapping region of the 0.3 and 0.18 kb Puma–LUC constructs (corresponding to a region ∼90–210 bp upstream of the transcription start site of Puma). Therefore, we examined this region for potential ATF4 binding sites using the Genomatix promoter analysis software and identified a conserved element exhibiting significant homology to the consensus ATF/CRE binding sequence and located ∼25 bp downstream of the previously identified p53 binding site (Fig. 8B). To determine whether ATF4 binds to the Puma promoter during ER stress in situ, we performed quantitative chromatin immunoprecipitation (ChIP) assays using PCR primers flanking the ATF4-responsive region and encompassing the putative ATF4 and p53 binding sites (Fig. 8B). As expected, p53 binding to this region was enriched ∼4.5 fold after camptothecin treatment (Fig. 8C). In contrast, p53 binding was not increased in neurons treated with ER stressors, consistent with the lack of p53 involvement in Puma expression in this death paradigm (Fig. 8C). Curiously, we did not detect an increase in ATF4 binding in this region after either DNA damage or ER stress treatments (Fig. 8D). However, ATF4 binding was significantly enriched at a previously described ATF4 response element located in the CHOP promoter (Ma et al., 2002), confirming that the ATF4 ChIP assay worked (Fig. 8D). These results suggest that ATF4 likely regulates Puma expression during ER stress via an indirect mechanism.
Figure 8.

ATF4 does not directly activate the Puma promoter during ER stress. A, Neurons were cotransfected with the pGL3b or Puma–LUC reporter constructs and either pcDNA3 or pcDNA3–ATF4 and, after 4 h, treated with 200 nm thapsigargin. Luciferase activity was measured after 24 h and is reported as the ratio of luciferase activity produced in cells transfected with pcDNA3–ATF4 relative to empty pcDNA3 vector for each reporter construct (n = 4; *p < 0.05). B, Schematic of the Puma promoter showing the location of the putative ATF/CRE and p53 binding sites. Arrows indicate the approximate positions of the PCR primers used in the ChIP assays. C, Cortical neurons were treated with camptothecin (10 μm; CPT), tunicamycin (3 μg/ml; TM), or thapsigargin (1 μm; TG), and p53 binding to the Puma promoter was assessed after 8 h by ChIP assay. The level of p53 binding was quantified by real-time PCR and is reported as fold enrichment over untreated controls (Ctrl) (n = 5; *p < 0.01). D, Cortical neurons were treated as above, and binding of ATF4 was assessed by ChIP assay and real-time PCR using primers specifically targeting the putative ATF4 response elements (RE) in the Puma and Chop promoters. Data are reported as fold increase over untreated control samples for each promoter region (n = 5; *p < 0.01).
ATF4 regulates Puma expression indirectly through CHOP induction during ER stress
The above results suggest that an intermediary factor is involved in ATF4-mediated Puma induction. Previous studies have suggested a relationship between ATF4 and the transcription factor CHOP, and indeed we observed enhanced binding of ATF4 to the CHOP promoter in neurons during ER stress (Fig. 8D). Therefore, we hypothesized that ATF4 may regulate Puma induction through CHOP. Indeed, we found CHOP mRNA and protein levels to be markedly upregulated in neurons after treatment with tunicamycin or thapsigargin but not in response to DNA damage (Fig. 9A,B). This increase in CHOP expression by ER stress was markedly reduced in ATF4-deficient neurons, consistent with its activation primarily by ATF4 in this paradigm (Fig. 9C,D).
Figure 9.
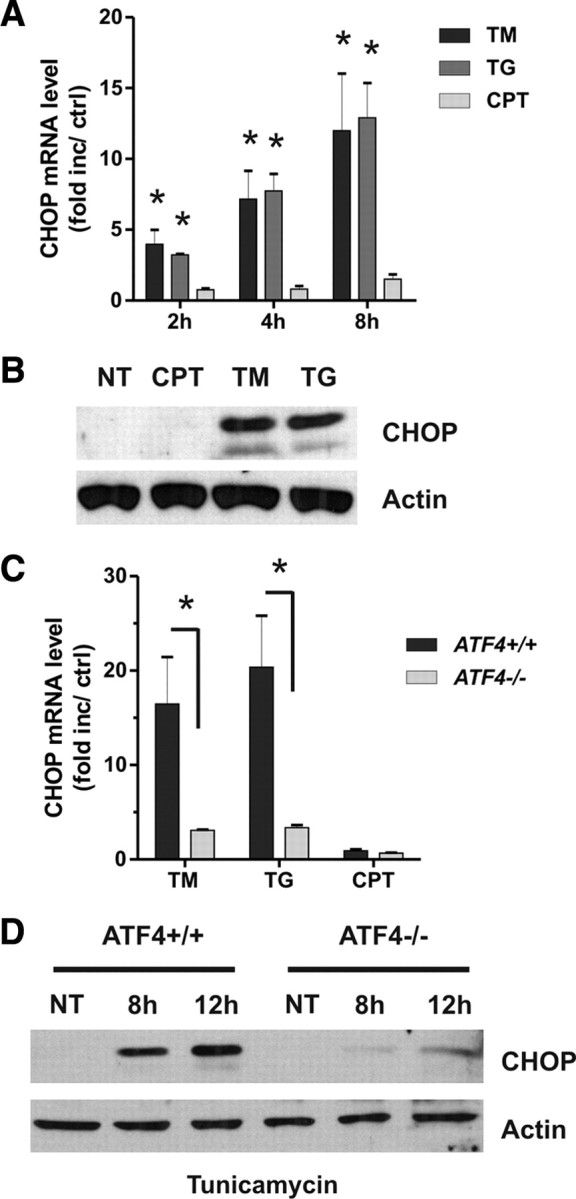
ATF4 regulates CHOP induction in response to ER stress but not DNA damage. A, Cortical neurons were treated with camptothecin (10 μm; CPT), tunicamycin (3 μg/ml; TM), or thapsigargin (1 μm; TG), and CHOP mRNA levels were quantified by real-time RT-PCR after 2, 4, and 8 h (n = 4; *p < 0.01). Ctrl, Control. B, CHOP protein levels were assessed by Western blot 10 h after treatment with camptothecin, tunicamycin, or thapsigargin. NT, Nontreated. C, Cortical neurons derived from ATF4+/+ and ATF4−/− littermates were treated with camptothecin, tunicamycin, or thapsigargin, and, after 8 h, CHOP mRNA levels were quantified by real-time RT-PCR (n ≥ 4; *p < 0.01). D, ATF4+/+ and ATF4−/− cortical neurons were treated with tunicamycin and after 8 and 12 h CHOP protein levels were assessed by Western blot.
To determine whether CHOP contributes to Puma induction by ER stress, we transfected cortical neurons with siRNAs directed against CHOP or nontargeting control siRNA using nucleofection technology. Neurons transfected with the CHOP targeting siRNAs exhibited a marked reduction in ER-stress-induced CHOP expression (Fig. 10A). Importantly, we found that CHOP knockdown reduced Puma mRNA induction by tunicamycin and thapsigargin by ∼40% (Fig. 10B). In contrast, Puma induction by the DNA-damaging agent camptothecin was not affected by transfection with the CHOP siRNAs (Fig. 10B). Furthermore, neurons transfected with siRNAs targeting CHOP exhibited a significant decrease in ER-stress-induced neuronal apoptosis but not DNA-damage-induced neuronal cell death (Fig. 10C). Similarly, we found that CHOP knockdown significantly increased neuronal survival after tunicamycin or thapsigargin treatment as measured by the live–dead cytotoxicity assay (Fig. 10D). To confirm the specificity of the CHOP siRNA knockdown effect, we nucleofected neurons with the Chop–siRNA-1 that targets the 5′ untranslated region of the CHOP mRNA and a GFP fused CHOP expression plasmid lacking the 5′ untranslated region (pCMV–GFP–CHOP) or the control vector pCMV–GFP. As shown in Figure 10E, in contrast to pCMV-GFP, ectopic expression of the siRNA-resistant GFP–CHOP protein partially restored tunicamycin-induced Puma expression and neuronal cell death in Chop–siRNA-transfected neurons (Fig. 10E,F). It was noted that the rescue of Puma induction was not complete, which is likely attributable to the lower transfection efficiency (∼60% GFP-positive cells) of the expression plasmids compared with siRNA (>85%).
Figure 10.

CHOP knockdown diminishes Puma expression and neuronal apoptosis induced by ER stress but not DNA damage. Cortical neurons were nucleofected with two different siRNAs directed against CHOP or a nontargeting control siRNA (Ctrl si), and, 24 h later, the neurons were treated with camptothecin (10 μm; CPT), tunicamycin (3 μg/ml; Tm), or thapsigargin (1 μm; Tg). A, Protein was extracted 12 h after treatment, and CHOP protein was assessed by Western blot. Ctrl, Control. B, RNA was extracted 12 h after treatment, and Puma mRNA levels were quantified by real-time RT-PCR (n = 4; *p < 0.05). C, Neurons were stained with Hoechst after 36 h, and the fraction of apoptotic cells was determined by assessing nuclear morphology (n = 4; *p < 0.05). D, Cortical neurons nucleofected with Chop–siRNA-1 or negative control siRNA were treated with thapsigargin (1 μm) or tunicamycin (3 μg/ml), and the percentage of calcein-AM-positive (live) cells was determined at 36 h (n = 3; *p < 0.05). E, F, Cortical neurons were nucleofected with Chop–siRNA-1 or negative control siRNA and either pCMV–GFP or pCMV–GFP–CHOP, and, after 18 h, neurons were challenged with tunicamycin (3 μg/ml). E, The fraction of apoptotic GFP-positive neurons was assessed by Hoechst staining 30 h after treatment with tunicamycin (n = 3). F, Puma mRNA levels were quantified by real-time RT-PCR 10 h after tunicamycin treatment and are expressed as fold increase over corresponding untreated controls (n = 3).
We next performed reporter assays to determine whether CHOP can activate the Puma promoter from the ATF4-responsive region. As shown in Figure 11A, cotransfection of the Puma(0.3 kb)–LUC reporter construct with an expression plasmid for CHOP (pCMV–CHOP) resulted in a >2.5-fold increase in luciferase activity relative to empty vector control (pcDNA3). In contrast, pCMV–CHOP did not induce luciferase activity when cotransfected with the Puma(0.18 kb)–LUC or pGL3b reporter constructs (Fig. 11A). Interestingly, we found that enforced expression of CHOP was sufficient to induce apoptosis in wild-type neurons but not Puma−/− neurons, suggesting that PUMA is required for CHOP-induced cell death (Fig. 11B). We then performed ChIP assays to determine whether CHOP binds to the Puma promoter in situ during ER stress. As shown in Figure 11C, CHOP binding to the Puma promoter increased approximately threefold after treatment with tunicamycin or thapsigargin but was not increased by camptothecin treatment. These results suggest that CHOP can directly regulate the Puma promoter during ER stress but not in response to DNA damage.
Figure 11.

CHOP activates the Puma promoter during ER-stress-induced apoptosis. A, Neurons were cotransfected with the pGL3b or Puma–LUC reporter constructs and either pcDNA3 or pcDNA3–CHOP, and luciferase activity was measured after 24 h. Luciferase activity is reported as the ratio of luciferase activity produced in cells transfected with pcDNA3–CHOP relative to empty pcDNA3 vector for each reporter construct (n = 4; *p < 0.05). B, Cortical neurons derived from Puma+/+ and Puma−/− littermates were nucleofected with pGFP and either pcDNA3 [empty vector (EV)] or pcDNA3–CHOP. Neurons were Hoechst stained 48 h after transfection, and the fraction of GFP-positive neurons exhibiting an apoptotic nuclear morphology was determined (n = 3; *p < 0.05). C, Cortical neurons were treated with camptothecin (10 μm; CPT), tunicamycin (3 μg/ml; TM), or thapsigargin (1 μm; TG), and CHOP binding to the Puma promoter was assessed after 12 h by ChIP assay. The level of CHOP binding was quantified by real-time PCR and is reported as fold enrichment over untreated controls (n = 4; *p < 0.05).
Together, our results indicate that ER stress induces neuronal apoptosis through a p53-independent pathway involving ATF4- and CHOP-mediated transcriptional induction of the proapoptotic Bcl-2 family member PUMA.
Discussion
An increasing body of evidence points to a key role of ER stress in a wide variety of acute and chronic neurodegenerative conditions. Indeed, protein aggregates and markers of the ER stress response have been observed in dying neurons in animal models of cerebral ischemia (Hu et al., 2001; Kumar et al., 2001) and Alzheimer's, Parkinson's, and Huntingtin's diseases (Ryu et al., 2002; Katayama et al., 2004; Reijonen et al., 2008). Therefore, it has become essential to identify the signaling pathways that regulate ER-stress-induced neuronal death.
ER stress induces neuronal apoptosis via a PUMA-dependent but p53-independent pathway
Recent studies suggest that ER stress induces apoptosis through an intrinsic (mitochondrial) pathway (Masud et al., 2007). The BCL-2 family proteins are key regulators of mitochondrial permeabilization, and the proapoptotic family members Bax and Bak have been implicated in ER-stress-induced cell death (Wei et al., 2001; Smith and Deshmukh, 2007). The BH3-only subfamily of Bcl-2 proteins are activated in a cell-type- and stimulus-dependent manner and function by directly or indirectly promoting Bax/Bak activation (Bouillet and Strasser, 2002). In this study, we demonstrate that transcriptional activation of the BH3-only family member PUMA is required for both ER-stress- and DNA-damage-induced apoptosis in neurons but that the upstream regulators of Puma induction are distinct in the two paradigms. We and others have shown previously that DNA-damage-induced Puma expression in neurons is mediated by the tumor suppressor p53 (Cregan et al., 2004; Wyttenbach and Tolkovsky, 2006). However, this is clearly not the case with ER stress because p53 deletion had no significant effect on Puma induction or cell death. Consistent with the lack of involvement of p53, we did not detect an increase in expression of the p53-responsive BH3-only member Noxa (Oda et al., 2000). Nonetheless, the involvement of p53 in ER stress may be impacted by the cellular context. For example, it has been reported previously that tunicamycin can induce Puma expression in Saos-2 cells that lack p53 function (Reimertz et al., 2003). Conversely, PUMA induction in response to ER stress in mouse embryonic fibroblasts has been reported to be dependent on p53 (Li et al., 2006).
ATF4 regulates ER-stress-induced Puma expression and neuronalapoptosis
We have determined that ATF4 is rapidly induced in response to ER stress but not DNA damage and that ATF4 is a key regulator of ER-stress-induced neuronal apoptosis. Interestingly, ATF4 has also been shown to regulate the expression of genes involved in amino acid metabolism and redox functions, suggestive of a cellular protective function (Harding et al., 2003). The reason for this apparent dichotomy is not clear, but ATF4 is known to dimerize with a number of transcription factors, including FIAT, Nrf2, NF-IL6, as well as AP-1 and Maf family members, and it is likely that this can alter its biological function (Tanaka et al., 1998; Hai and Hartman, 2001; He et al., 2001). The dual nature of ATF4 is reflected in several other proteins that can play both pro-survival and death roles, such as p53 a protein involved in both DNA repair and cell death (Offer et al., 2002) and nuclear factor-κB (NF-κB) (Aleyasin et al., 2004). Importantly, we have determined that ATF4 promotes ER-stress-induced neuronal apoptosis by regulating the transcriptional induction of Puma. Thus, ER stress and DNA damage induce Puma expression via distinct pathways involving ATF4 and p53, respectively. It has been reported recently that ATF4 also plays a proapoptotic role in oxidative-stress-induced apoptosis in neurons (Lange et al., 2008). Interestingly, we have demonstrated recently that PUMA plays a critical role in regulating oxidative-stress-induced neuronal apoptosis and that Puma induction in this context involves both p53 and p53-independent mechanisms (Steckley et al., 2007). Thus, it would be interesting to determine whether ATF4 also contributes to Puma induction during oxidative-stress-induced neuronal death.
CHOP regulates PUMA induction in neurons during ER stress
Despite our finding that ATF4 is an important regulator of PUMA induction, we were unable to detect a direct interaction of ATF4 with the PUMA promoter, suggesting that an ATF4-responsive intermediate may be involved. The stress-inducible transcription factor CHOP is known to be activated by a number of pathways, including ATF4, ATF2, ATF6, and Ire1/XBP-1 (Yoshida et al., 2000; Ma et al., 2002; Averous et al., 2004). We demonstrate that CHOP is induced in response to ER stress but not DNA damage and that at least in this context its induction occurs predominately through ATF- mediated transactivation. Previous studies have implicated CHOP as a proapoptotic factor. For example, enforced expression of CHOP has been shown to induce cell death (Maytin et al., 2001), and CHOP-deficient cells have been reported to be resistant to ER-stress-induced cell death (Zinszner et al., 1998; Oyadomari et al., 2002). However, the mechanism by which CHOP engages the apoptotic pathway has remained unclear. The involvement of the Bcl-2 family has been suggested by the findings that CHOP-induced cell death is associated with translocation of Bax to the mitochondria and that enforced expression of Bcl-2 can block CHOP-induced cell death (Matsumoto et al., 1996; Gotoh et al., 2004). Importantly, we have determined that CHOP knockdown decreases ER-stress-induced Puma expression and neuronal apoptosis. Consistent with this, we found that CHOP can activate the Puma promoter and that it binds to the Puma promoter in situ during ER stress. Furthermore, we show that enforced expression of CHOP can induce apoptosis in wild-type but not Puma-deficient neurons, indicating that PUMA is required for CHOP-induced neuronal death. Paradoxically, we found that ectopic expression of ATF4 was not sufficient to induce neuronal cell death but rather sensitized neurons to ER-stress-induced apoptosis. This likely reflects a requirement for ER stress signals in the activation of ATF4. Indeed, it has been shown previously that ectopic expression of ATF4 is not sufficient to induce CHOP expression in the absence of an ER stress signal (Harding et al., 2000b). The half-life of ATF4 under nonstressed conditions is very short because it is rapidly targeted for proteasomal degradation (Lassot et al., 2001). Thus, it is assumed that ER stress signals directly or indirectly affect the stability of the ATF4 protein. It is also possible that ER stress leads to the induction of an additional factor required for efficient ATF4-mediated transactivation. It should be noted, however, that even ectopic expression of CHOP only resulted in a relatively modest increase in neuronal apoptosis (∼30% cell death after 48 h). Thus, it is likely that ER stress signaling can potentiate the proapoptotic functions of CHOP as well.
Although our study clearly demonstrates the important involvement of ATF4–CHOP signaling in ER-stress-induced neuronal apoptosis, it is evident that inhibition of this pathway does not completely abrogate Puma induction or cell death. This suggests that additional factors contribute to Puma induction during ER-stress-induced apoptosis. In addition to ATF4, ER stress activates the Ire1α and ATF6 arms of the unfolded protein response, and it is possible that these pathways may also contribute to Puma induction. Indeed, both proapoptotic and anti-apoptotic functions have been attributed to these pathways (Lin et al., 2007; Yokouchi et al., 2007). Interestingly, during activation by ER stress, Ire1α exhibits dual functions in inducing the expression of the prosurvival transcription factor XBP1 and activating the proapoptotic JNK signaling pathway (Urano et al., 2000; Yoshida et al., 2001; Calfon et al., 2002). Thus, it is possible that Ire1–JNK signaling could contribute to Puma induction through activation of the transcription factor c-jun. Furthermore, several transcription factors, including E2F1, Foxo3a, p73, and NF-κB, have been implicated in the transcriptional regulation of Puma, but their relevance in ER stress has not yet been examined (Hershko and Ginsberg, 2004; Melino et al., 2004; You et al., 2006; Wang et al., 2009).
ATF4–CHOP–PUMA pathway in neurodegenerative conditions
In the present study, we have identified a key signaling pathway in ER-stress-induced neuronal apoptosis involving the transcription factors ATF4 and CHOP and their downstream target PUMA. Several lines of evidence suggest that this pathway may be relevant and important in brain injury and neurodegenerative conditions. For example, induction of ATF4, CHOP, and Puma levels have been reported in affected brain regions in models of cerebral ischemia (Paschen et al., 1998; Jin et al., 2001; Morimoto et al., 2007; Niizuma et al., 2009). Furthermore, both ATF4-deficient and CHOP-deficient mice have been reported to exhibit smaller infarcts after ischemia–reperfusion injury (Tajiri et al., 2004; Lange et al., 2008). Similarly, both CHOP and PUMA have been implicated in dopaminergic neuronal death in Parkinson's disease models (Holtz and O'Malley, 2003; Biswas et al., 2005; Silva et al., 2005). Finally, it has been reported recently that Puma expression is induced in degenerating motor neurons in a mouse model of amyotrophic lateral sclerosis and that Puma deletion reduces motor neuron apoptosis in this model (Kieran et al., 2007).
In summary, our results demonstrate that an ATF4–CHOP–PUMA signaling pathway plays a key role in the regulation of ER-stress-induced neuronal apoptosis. We suggest that this pathway may be an important therapeutic target relevant to a variety of neurodegenerative conditions.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and Heart and Stroke Foundation of Ontario (HSFO) (S.P.C.) and the CIHR, HSFO, Canadian Stroke Network and Parkinson's Research Consortium, Neuroscience Canada Brain Repair Program, Parkinson's Disease Foundation, Parkinson's Society Canada, and The World Class University program through the National Research Foundation of Korea (Grant R31-2008-000-20004-0) (D.S.P.). S.P.C. is a Canada Research Chair. We thank Drs. Tim Townes, Joe Sun, and Amy Lee for providing critical reagents as noted in text.
References
- Aleyasin H, Cregan SP, Iyirhiaro G, O'Hare MJ, Callaghan SM, Slack RS, Park DS. Nuclear factor-κB modulates the p53 response in neurons exposed to DNA damage. J Neurosci. 2004;24:2963–2973. doi: 10.1523/JNEUROSCI.0155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem. 2004;279:5288–5297. doi: 10.1074/jbc.M311862200. [DOI] [PubMed] [Google Scholar]
- Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Strasser A. BH3-only proteins: evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci. 2002;115:1567–1574. doi: 10.1242/jcs.115.8.1567. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci. 2004;24:10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- D'Sa-Eipper C, Leonard JR, Putcha G, Zheng TS, Flavell RA, Rakic P, Kuida K, Roth KA. DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development. 2001;128:137–146. doi: 10.1242/dev.128.1.137. [DOI] [PubMed] [Google Scholar]
- Farinelli SE, Greene LA, Friedman WJ. Neuroprotective actions of dipyridamole on cultured CNS neurons. J Neurosci. 1998;18:5112–5123. doi: 10.1523/JNEUROSCI.18-14-05112.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol. 2001;155:207–216. doi: 10.1083/jcb.200105137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L, Chittenden T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci U S A. 2001;98:11318–11323. doi: 10.1073/pnas.201208798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000a;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000b;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276:20858–20865. doi: 10.1074/jbc.M101198200. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem. 2004;279:8627–8634. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjö BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–875. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- Jin K, Mao XO, Eshoo MW, Nagayama T, Minami M, Simon RP, Greenberg DA. Microarray analysis of hippocampal gene expression in global cerebral ischemia. Ann Neurol. 2001;50:93–103. doi: 10.1002/ana.1073. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer's disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci U S A. 2007;104:20606–20611. doi: 10.1073/pnas.0707906105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem. 2001;77:1418–1421. doi: 10.1046/j.1471-4159.2001.00387.x. [DOI] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassot I, Ségéral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, Benarous R, Margottin-Goguet F. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol Cell Biol. 2001;21:2192–2202. doi: 10.1128/MCB.21.6.2192-2202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol. 2008;67:377–387. doi: 10.1097/NEN.0b013e31816ff780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R, Flavell RA. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J Biol Chem. 2007;282:14132–14139. doi: 10.1074/jbc.M700077200. [DOI] [PubMed] [Google Scholar]
- Masuoka HC, Townes TM. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood. 2002;99:736–745. doi: 10.1182/blood.v99.3.736. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. 1996;395:143–147. doi: 10.1016/0014-5793(96)01016-2. [DOI] [PubMed] [Google Scholar]
- Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res. 2001;267:193–204. doi: 10.1006/excr.2001.5248. [DOI] [PubMed] [Google Scholar]
- Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–8083. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- Morimoto N, Oida Y, Shimazawa M, Miura M, Kudo T, Imaizumi K, Hara H. Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience. 2007;147:957–967. doi: 10.1016/j.neuroscience.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Keramaris E, Rideout HJ, Slack RS, Dyson NJ, Stefanis L, Park DS. Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J Neurosci. 2001;21:5017–5026. doi: 10.1523/JNEUROSCI.21-14-05017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40:618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Offer H, Erez N, Zurer I, Tang X, Milyavsky M, Goldfinger N, Rotter V. The onset of p53-dependent DNA repair or apoptosis is determined by the level of accumulated damaged DNA. Carcinogenesis. 2002;23:1025–1032. doi: 10.1093/carcin/23.6.1025. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W, Mengesdorf T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium. 2005;38:409–415. doi: 10.1016/j.ceca.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Paschen W, Gissel C, Linden T, Althausen S, Doutheil J. Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res Mol Brain Res. 1998;60:115–122. doi: 10.1016/s0169-328x(98)00180-6. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Reijonen S, Putkonen N, Nørremølle A, Lindholm D, Korhonen L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res. 2008;314:950–960. doi: 10.1016/j.yexcr.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kögel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MI, Deshmukh M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 2007;14:1011–1019. doi: 10.1038/sj.cdd.4402089. [DOI] [PubMed] [Google Scholar]
- Steckley D, Karajgikar M, Dale LB, Fuerth B, Swan P, Drummond-Main C, Poulter MO, Ferguson SS, Strasser A, Cregan SP. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J Neurosci. 2007;27:12989–12999. doi: 10.1523/JNEUROSCI.3400-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells. 1998;3:801–810. doi: 10.1046/j.1365-2443.1998.00230.x. [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS. Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J Neurosci. 2007;27:12198–12210. doi: 10.1523/JNEUROSCI.3222-05.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang P, Qiu W, Dudgeon C, Liu H, Huang C, Zambetti GP, Yu J, Zhang L. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death Differ. 2009;16:1192–1202. doi: 10.1038/cdd.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Tolkovsky AM. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J Neurochem. 2006;96:1213–1226. doi: 10.1111/j.1471-4159.2005.03676.x. [DOI] [PubMed] [Google Scholar]
- Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokouchi M, Hiramatsu N, Hayakawa K, Kasai A, Takano Y, Yao J, Kitamura M. Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ. 2007;14:1467–1474. doi: 10.1038/sj.cdd.4402154. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]