

Abstract
Copper-catalyzed atom transfer radical polymerization (Cu-ATRP) is one of the most widely used controlled radical polymerization techniques. Notwithstanding the extensive mechanistic studies in the literature, the transition states of the activation/deactivation of the growing polymer chain, a key equilibrium in Cu-ATRP, have not been investigated computationally. Therefore, the understanding of the origin of ligand and initiator effects on the rates of activation/deactivation is still limited. Here, we present the first computational analysis of Cu-ATRP activation transition states to reveal factors that affect the rates of activation and deactivation. The Br atom transfer between the polymer chain and the Cu catalyst occurs through an unusual bent geometry that involves pronounced interactions between the polymer chain end and the ancillary ligand on the Cu catalyst. Therefore, the rates of activation/deactivation are determined by both the electronic properties of the Cu catalyst and the ligand-initiator steric repulsions. In addition, our calculations revealed the important role of ligand backbone flexibility on the activation. These theoretical analyses led to the identification of three chemically meaningful descriptors, namely HOMO energy of the catalyst (EHOMO), percent buried volume (Vbur%), and distortion energy of the catalyst (ΔEdist), to describe the electronic, steric, and flexibility effects on reactivity, respectively. A robust and simple predictive model for ligand effect on reactivity is thereby established by correlating these three descriptors with experimental activation rate constants using multivariate linear regression. Validation using a structurally diverse set of ligands revealed the average error is less than ±2kcal/mol compared to the experimentally derived activation energies. The same approach was also applied to develop a predictive model for reactivity of different alkyl halide initiators using R–X bond dissociation energy (BDE) and Cu–X halogenophilicity as descriptors.
Graphical Abstract
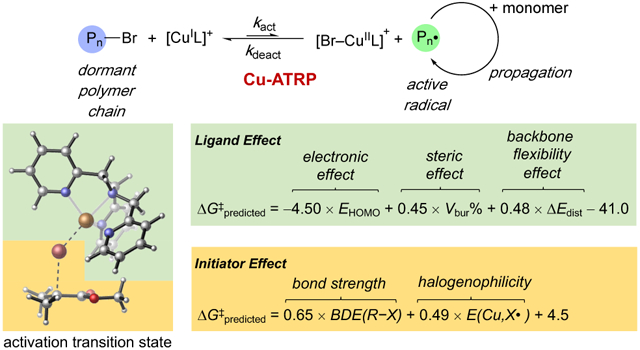
1. Introduction
Atom transfer radical polymerization (ATRP) is among the most powerful and robust controlled radical polymerization techniques that facilitate macromolecular engineering by synthesis of polymers with precise molecular weights, low dispersities, and well-controlled architectures.1 Although ATRP has been achieved with diverse transition metal2 and photoredox catalysts,3,4 copper-catalyzed ATRP (Cu-ATRP) is still the most extensively used and investigated ATRP1a,5 with a wide range of compatible monomers, initiators, and solvents. Control of polymer chain growth via Cu-ATRP is largely attributed to the dynamic activation/deactivation equilibrium between a [CuIL]+/[Br-CuIIL]+ couple where L represents a multidentate nitrogen-donor ligand (Figure 1).6,7 The [CuIL]+ catalyst activates the dormant alkyl bromide chain end (Pn–Br) to form the [Br-CuIIL]+ complex and a propagating alkyl radical (Pn•). The alkyl radical continues to grow by adding a few monomers before it abstracts the Br from [Br-CuIIL]+ to reform an alkyl bromide. A successful ATRP catalyst system should have a large activation rate constant (kact) and an even larger deactivation rate constant (kdeact) to provide good control over the polymerization while maintaining a reasonable polymerization rate.
Figure 1.

Ligand effects on the activation/deactivation of Cu-ATRP.
Previous mechanistic studies from Coote, Gennaro, and Matyjaszewski indicated that Cu-ATRP occurs via a concerted inner-sphere electron transfer (ISET) process in which the Br atom is transferred from the alkyl bromide to the [CuIL]+ catalyst (Figure 1a), because the predicted outer-sphere electron transfer (OSET) barriers are much higher than experimental data.8 However, the geometry and energy of this Br atom transfer transition state have not been explored by computations so far. Although a few computational studies demonstrated that the reactivity of alkyl halide initiators is affected by their bond dissociation energies8,9 and LUMO energies,10 it remains challenging to understand and computationally predict the reactivities of Cu-ATRP catalysts with the structurally-diverse ligands (Figure 1b). The lack of theoretical insights into the origins of ligand effects hinders the rational catalyst design for Cu-ATRP. Herein, we describe a workflow to establish mechanistically guided predictive models for the reactivity of Cu-ATRP catalysts. Inspired by the elegant work from Sigman,11 Doyle,12 and Paton13‘s groups that utilizes a multivariate regression approach to predict ligand effects in transition metal catalyzed reactions using steric and electronic parameters, we surmised such parameterization approach may be facilitated by mechanistic insights from DFT calculations. In particular, in-depth analysis of factors that stabilize the rate-determining transition state may offer the theoretical basis for the rational selection of ligand parameters.14 Our workflow to establish the predictive mathematical equation is summarized in Figure 2. First, we applied DFT calculations to obtain the geometries and energies of Cu-ATRP activation transition sates. Detailed investigations of the transition states revealed key factors that control catalyst and initiator reactivities. These theoretical insights were then utilized to rationally select a set of chemically meaningful descriptors to define effects of different factors on the reactivity. Finally, a simple mathematical equation for predicting ligand effect was established by correlating these descriptors with experimental reactivities using a multivariate regression approach. Similarly, a predictive equation was developed for the reactivity of different alkyl halide initiators. These predictive models can be used to guide rational catalyst design15 and computational ligand discovery for Cu-ATRP reactions.
Figure 2.

Workflow of establishing predictive models for reactivity of Cu-ATRP catalysts.
2. Computational Methods
All geometry optimizations and single point energy calculations were performed with the Gaussian 09 software package.16 Geometries were optimized in the gas phase using the ω-B97XD17 functional and a mixed basis set of SDD18 for Cu and 6-31G(d)19 for other atoms. Single point energies were calculated using ω-B97XD and the def2-TZVP20 basis set in acetonitrile using the CPCM solvation model.21 Reported Gibbs free energies and enthalpies in solution include thermal corrections computed at 298 K and are computed at the standard concentration (1 mol/L). The activation free energies of the outer-sphere single electron transfer reactions were calculated using modified Marcus theory (see SI for details).22,23 Parameters used in the predictive models for initiator and ligand effects (eqs. 3 and 4), including bond dissociation energies (BDE), halogenophilicity (E(Cu, X•)), HOMO energies (EHOMO), and catalyst distortion energies (ΔEdist(TS) and ΔEdist(BrCuIIL)), were also calculated at the ω-B97XD/def2-TZVP level of theory in acetonitrile using the CPCM solvation model. Distortion/interaction model and energy decomposition analysis (EDA) calculations in Figure 5 were performed to dissect the computed gas-phase activation energy (ΔE‡). The activation energy was first decomposed into the distortion energy of the two reactive fragments, i.e. the [CuIL]+ catalyst and the alkyl halide, to reach their transition state geometries (ΔEdist = ΔEdist(CuL) + ΔEdist(RX)) and the interaction energy (ΔEint) between these two fragments (eq. 1).24,25
| (eq.1) |
Figure 5.

The origin of the bent geometry in TS1.
Here, the distortion energies were calculated from the energy difference between the distorted fragment in the transition state geometry and the same fragment in the fully optimized ground state geometry. Then, ΔEint is calculated from ΔEint = ΔE‡ – ΔEdist. Next, ΔEint was further dissected into chemically meaningful terms using the second-generation EDA based on absolutely-localized molecular orbitals (ALMO-EDA2)26,27 in Q-Chem 5.0 (eq. 2).28
| (eq.2) |
Here, ΔEpauli is the Pauli repulsion, ΔEelstat is the electrostatic interactions, ΔEdisp is the dispersion interaction, and ΔEorb is the orbital interaction energy that consists of interfragment charge transfer energy and intrafragment polarization energy.
3. Results and Discussion
3.1. Activation mechanisms in Cu-ATRP
In previous studies, several possible mechanisms have been proposed for the activation/deactivation process in Cu-ATRP (Figure 3a), including inner-sphere single electron transfer (ISET), stepwise outer-sphere single electron transfer (OSET-SW), and dissociative electron transfer (DET).8 Here, we used DFT and Marcus theory to investigate these previously proposed pathways, as well as the oxidative addition (OA) of alkyl bromide to the CuI catalyst to form a CuIII intermediate (Figure 3b). The barrier for the concerted DET to form the MMA radical 7 and a bromide was estimated using the “sticky model”, a modification of the Marcus theory.8,23 The calculations were performed using a model system that consists of [CuI(TPMA)]+ (TPMA: tris(2-pyridylmethyl)amine), one of the most popular ATRP catalysts, and a widely used initiator α-bromoisobutyrate (i.e. a mimic of a dormant chain end in ATRP of methyl methacrylate, MMA-Br, 2). In agreement with previous computational studies,8 the OSET-SW and DET processes both involve very high barriers that are inconsistent with experimental data. The oxidative addition of the sterically crowded tertiary alkyl bromide (TS2) also requires a very high barrier.29 The most favorable activation mechanism is the concerted ISET (TS1), which requires a substantially lower barrier (ΔG‡ = 16.6 kcal/mol in MeCN) than other processes considered.30
Figure 3.

Computed energy profiles of Cu-ATRP activation pathways in MeCN at 25 °C with tris(2-pyridylmethyl)amine (TPMA) ligand.
The optimized geometries, Mulliken atomic charges, and spin densities of the CuI and CuII complexes and transition state involved in the ISET pathway are shown in Figure 4.31 The analysis of charge and spin density at different C(MMA)–Br bond distances (Figure 4b) indicates a dramatic change of charge and spin density before TS1, while both quantities remain almost constant after the transition state. The computed Mulliken charge of the CuL fragment in TS1 is +1.33, indicating a considerable amount (0.33 e) of charge transfer from the [CuIL]+ catalyst to the alkyl bromide in the ISET transition state. Nonetheless, since the charge transfer in TS1 is much less than unity and the Cu is less positively charged than that in the [Br-CuIIL]+ complex 8, the Cu...Br interaction in TS1 has a significant covalent character. The much-shortened Cu–Br bond distance (2.39 Å) and the elongated C–Br bond distance (2.59 Å) in TS1, along with the relatively large spin densities on the Cu catalyst and the MMA, indicate the ISET process involves a late transition state that structurally resembles the [Br-CuIIL]+ product. Furthermore, the computed Wiberg bond indices32 of the Cu–Br bond in TS1 and 8 are 0.386 and 0.574, respectively, which is consistent with a late transition state with a substantial bonding interaction between the Cu and Br in TS1.
Figure 4.

Optimized geometries, energies, Mulliken charges, and spin densities of computed structures in the ISET pathway.
The optimized geometry of TS1 has a substantially bent Cu–Br–C bond angle (141.9°).33 This is rather surprising considering most known halogen atom transfer processes involve a linear transition state structure.3,34,35 The bent geometry means a closer distance between the ancillary ligand and the substrate (i.e. the alkyl halide initiator or the dormant chain end). As such, the non-covalent interactions between the ligand and the substrate, a previously underappreciated effect, may play a significant role on the activation/deactivation reactivity. Because of its potential impacts on the catalyst reactivity, we have undertaken a detailed computational analysis to investigate the origin of the bent geometry. We performed a constrained optimization of the ISET transition state by forcing a linear geometry, fixing the Cu–Br–C bond angle at 179° (Figure 5a). It was found that the freely optimized bent transition state was 2.3 kcal/mol lower in energy than the linear transition state. The distortion/interaction model analysis indicates the bent TS geometry has 3.0 kcal/mol more favorable interaction energy (ΔEint) between the CuL catalyst and the MMA-Br. We then applied the energy decomposition analysis methods to dissect the interaction energy (Figure 5b). Although the bent transition state is disfavored by Pauli repulsion (ΔΔEPauli = +9.4 kcal/mol), it has stronger attractive electrostatic interaction (ΔΔEelstat = −8.2 kcal/mol) and dispersion interaction (ΔΔEdisp = −4.3 kcal/mol) between the catalyst and the substrate than linear transition state. While the increased dispersion is expected to partially compensate the Pauli repulsion effect, the stronger electrostatic interaction is attributed to the attraction between the positively charged Cu catalyst and the partial negatively charged ester group of the substrate. DFT calculations using other alkyl halides also provided bent ISET transition state structures (see Figure S6 in the SI). The bent TS structures are stabilized by either electrostatic attraction with the Cu catalyst when an electron-withdrawing group is present in the chain end or attractive London dispersion forces in reactions with benzylic halides (see SI for details).
Collectively, the computational data indicate the ISET involves a late, open-shell singlet transition state that is consistent with a concerted bromine atom transfer3,34,35 process. The substantial C–Br bond stretch and Cu–Br interactions in the late TS implies that the BDE of the carbon-halogen bond and the halogen binding energy to Cu both play a significant role on the reaction rate.36 In addition, the charge transfer from the Cu catalyst to the alkyl bromide in the ISET TS suggests a more electron-rich ligand would promote the reaction. The bent transition state geometry suggests the ligand-substrate non-bonding interactions are expected to affect the stability of the transition state. These mechanistic insights were used to develop the predictive models for reactivity between different alkyl halide initiators and the Cu ATRP catalysts (vide infra).
3.2. Predictive Model for Initiator Effects on Reactivity
Over the past decades, Cu-ATRP was successfully employed with a variety of initiators, which activation rate constants (kact) have been determined by experimental kinetic studies with different catalysts (Figure 6a).37,38 Herein, we report the first computational prediction of activation barriers for a representative set of alkyl bromide and alkyl chloride initiators (R–X, Figure 6a) using transition state calculations. The DFT-computed barriers of the ISET transition states (ΔG‡DFT) with the [CuI(TPMA)]+ catalyst in acetonitrile provided a good agreement with the experimentally determined reactivity trend (ΔG‡exp)39 for both alkyl bromide and alkyl chloride initiators, which validates the robustness and reliability of the computational methods and computed transition state models. To further explore the origin of reactivity of the different initiators, we performed distortion/interaction model analysis for the computed ISET transition states. The results indicate the activation energies correlates well with the distortion energies of the R–X initiators (see Figure S1 in SI). Furthermore, ISET transition states with alkyl bromides are stabilized by stronger interaction energies between the Cu catalyst and the halide compared to those with alkyl chlorides (see Figure S2 in SI). Therefore, the distortion/interaction model analysis reaffirmed the significant roles of both the R–X bond strength and Cu–X interactions on the stability of the transition state.
Figure 6.

Initiator effect in Cu-ATRP with the [CuI(TPMA)]+ catalyst.
Because the transition state analysis discussed above indicated the R–X bond strength and the Cu–X covalent interactions are both important factors for the transition state energy, we surmised the R–X BDE and the halogenophilicity8 (i.e. the binding ability of X• to [CuIL]+) can be used as two appropriate descriptors for the initiator reactivity model. Although it has been widely recognized that the R–X BDE plays a significant role on the activation rate,8,9 the computed BDEs do not have a good overall correlation with experimental barriers for the entire set of alkyl bromide and chloride initiators (Figure 6c, dashed line). However, within the same type of halides (X =Br or C1), R–X BDEs are good indicators of their reactivities (Figure 6c, solid red and blue lines). This indicates the halogenophilicity is another important factor impacting the reactivity. Indeed, when combining R–X BDE with Cu–X halogenophilicity (E(Cu, X•)), the two-parameter equation (eq. 3) provides an excellent correlation with the experimentally observed activation rate
| (eq.3) |
constants. (Figure 7). Previous studies have revealed a good correlation between the activation rate constant and the activation/deactivation equilibrium constant (KATRP).38,40c Because KATRP is determined by both R–X BDE and Cu–X halogenophilicity, our two-parameter equation is consistent with the experimentally observed effects of KATRP on the activation rate.
Figure 7.

Predictive model for initiator reactivity in Cu-ATRP.
3.3. Predictive Model for Ligand Effects on Reactivity
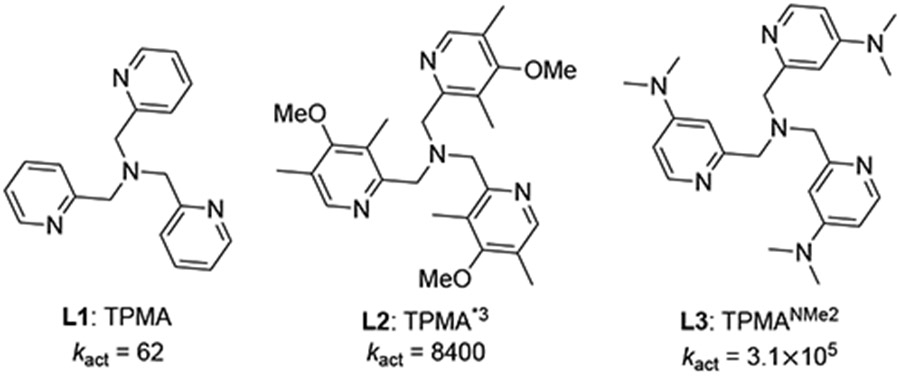
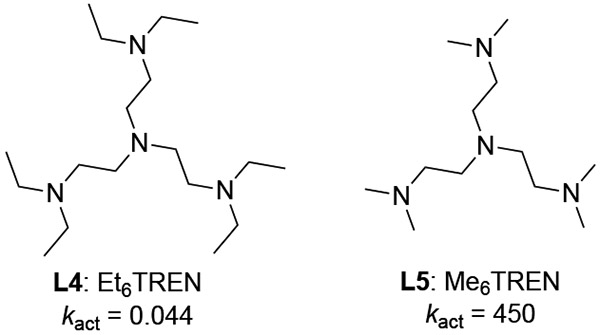
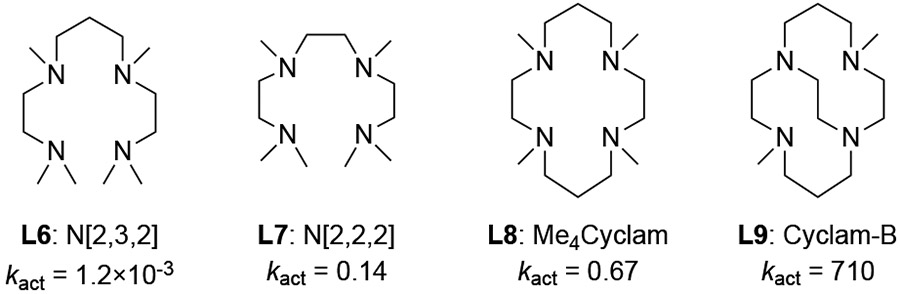
The reactivities of Cu-ATRP catalysts can be significantly impacted by altering the structures of their N-donor ligands. In the past decades, a structurally diverse set of multidentate N-donor ligands have been used experimentally, including bi-, tri-, and tetradentate ligands with either sp2 or sp3 N atoms (Figure 8).40 The reactivity of the Cu-ATRP catalyst appears to be affected by a combination of a few different types of ligand effects, including electronic, steric, denticity, hybridization, and other factors. As such, rational ligand design for efficient Cu-ATRP catalysts remains challenging. Here, we use the three-step approach described in the Introduction (Figure 2) to establish a simple mathematical equation for the prediction of the activation rate of Cu-ATRP catalysts. We selected 9 representative ligands with distinct electronic and steric properties (blue squares in Figure 8) as a training set to understand the factors controlling the ATRP activation rate constants, and to identify suitable descriptors for the predictive model. The rest of the ligands (red squares, circles, and triangles) are used as test sets to validate the reliability of the predictive model. All of ISET transition states were calculated with the same initiator MMA-Br.
Figure 8.

Representative ligands for Cu-ATRP catalyst. Experimentally determined activation rate constants are provided in parentheses.40
3.3.1. Ligand Electronic Effect
Three TPMA derivatives (L1-L3) with similar steric properties were chosen to investigate the electronic effects of the ancillary ligands. Recently, Matyjaszewski group discovered that incorporation of electron-donating groups in the pyridine ring of TPMA profoundly increased the ATRP catalyst activity (Table 1).40c Notably, the dimethyl amino (−NMe2) substituted TPMA (L3, TPMANMe2) ligand forms the most reactive Cu-ATRP catalyst reported to date. We performed DFT calculations to locate the ISET transition states for these three Cu-ATRP catalysts in Figure 9. The DFT-predicted activation barriers (ΔG‡DFT) were consistent with the experimental reactivity trend. The computed percent buried volume (Vbur%)41 and the catalyst distortion energy in the transition state (ΔEdist(TS)) are similar for all three catalysts, which confirmed the ligand steric properties and the distortion of the Cu catalysts are comparable among this group of ligands. As such, their reactivity difference is expected to be mainly controlled by inductive electronic effects of the ligand. A good correlation between the activation energies and the computed HOMO energy of the CuI catalyst (EHOMO) was observed (see Figure S3 in SI). Thus, EHOMO was chosen as an appropriate descriptor to describe the electronic effects of the ancillary ligand.13a,b
Table1.
Electronic effects of the TPMA family ligands.a
 | ||||||
|---|---|---|---|---|---|---|
| ligand | ΔG‡exp | ΔG‡DFT | EHOMOb | Vbur%c | ΔEdist(TS)d | ΔEdist(BrCuIIL)e |
| TPMA | 15.0 | 16.6 | −7.66 | 39.2 | 2.1 | 4.0 |
| TPMA*3 | 12.0 | 14.6 | −7.41 | 40.3 | 2.0 | 3.6 |
| TPMANMe2 | 10.0 | 13.0 | −7.36 | 39.0 | 1.8 | 3.7 |
kact values are in M−1 s−1. All Gibbs free energies and distortion energies are in kcal/mol; EHOMO is in eV. All energies and EHOMO are computed in acetonitrile using the CPCM solvation model.
HOMO energy of [CuIL]+.
Percent buried volume of the ligand computed from the DFT-optimized geometry of [CuIL]+.41
Distortion energy of the CuL catalyst in the ISET transition state with respect to the ground state [CuIL]+.
Distortion energy of the CuL catalyst in the [BrCuIIL]+ complex with respect to the ground state [CuIL]+.
Figure 9.

Optimized geometries of ISET transition states with TPMA family ligands.
3.3.2. Ligand Steric Effect
Me6TREN (L5) is another highly effective and commonly employed ligand in Cu-ATRP.42 When replacing all N-methyl substituents with ethyl groups (L4, Et6TREN), the ATRP reactivity dramatically decreases (Table 2). Considering their similar electronic properties, this series of ligands was chosen to study the ligand steric effect on reactivity. The optimized ISET transition states with both ligands have a greater Cu—Br—C bond angle than that of TS1, indicating greater steric repulsions than with the TPMA ligand (Figure 10). The computationally predicted barrier with Et6TREN is more than 4 kcal/mol higher than that with Me6TREN (Table 2), in agreement with the experimental reactivity trend (ΔΔGexp‡ = 5.5 kcal/mol).40a The decreased reactivity with the Et6TREN ligand is mostly due to the greater steric hindrance of the ligand as evidenced by the less bent geometry in the ISET transition state (Figure 10). As shown in Table 2, the widely used percent buried volume (Vbur%)41 is an appropriate descriptor to distinguish the ligand steric properties. Et6TREN has a much larger Vbur% than Me6TREN (56.5% versus 45.7%), while the less hindered TPMA has a smaller Vbur% (39.2%) than both ligands. As expected, the computed HOMO energies for L4 and L5 are similar, which confirmed the similar electronic properties of these ligands. Although the transition state with Et6TREN has greater distortion energy (ΔEdist(TS)) than that with Me6TREN, the relatively small distortion energy difference between the two transition states suggests the ligand distortion may not strongly correlate with the steric effects of ligands. Effects of ligand distortion on reactivity are discussed in more detail below.
Table2.
Steric effects of the TREN family ligands.a
 |
||||||
|---|---|---|---|---|---|---|
| ligand | ΔG‡exp | ΔG‡DFT | EHOMOb | Vbur%c | ΔEdist(TS)d | ΔEdist(BrCuIIL)e |
| Et6TREN | 19.3 | 18.5 | −7.33 | 56.5 | 3.5 | 5.1 |
| Me6TREN | 13.8 | 14.3 | −7.34 | 45.7 | 2.2 | 4.0 |
kact values are in M−1 s−1. All Gibbs free energies and distortion energies are in kcal/mol; EHOMO is in eV. All energies and EHOMO are computed in acetonitrile using the CPCM solvation model.
HOMO energy of [CuIL]+.
Percent buried volume of the ligand computed from the DFT-optimized geometries of [CuIL]+.41
Distortion energy of the CuL catalyst in the ISET transition state with respect to the ground state [CuIL]+.
Distortion energy of the CuL catalyst in the [BrCuIIL]+ complex with respect to the ground state [CuIL]+.
Figure 10.

Optimized geometries of ISET transition states with TREN family ligands.
3.3.3. Ligand Backbone Flexibility Effect
Finally, we considered a series of tetradentate Cyclam family ligands (L6-L9) to investigate the effect of ligand backbone flexibility (Table 3). Albeit their similar donor abilities, these ligands have significantly different reactivities in Cu-ATRP. Experimental kinetic data showed that the reactivity decreases with augmented backbone flexibility.40a For example, the Cu complex with the most rigid ligand, Cyclam-B, is among the most active catalysts for Cu-ATRP, while the complex with structurally-similar acyclic ligand N[2,3,2] is six orders of magnitude less reactive. This reactivity trend was successfully reproduced by the computed transition state energies (ΔG‡DFT). The computed distortion energies (ΔEdist(TS)) indicated the low reactivities with the more flexible ligands (L6 and L7) are mainly due to the high distortion energy of the CuL catalyst in the ISET transition state. In reactions with L6, L7, and L8, the CuL catalyst is distorted from a tetrahedral geometry in the CuI ground state to a square pyramidal geometry in the ISET transition state (Figure 11 and Figure S4 in the SI). In contrast, the ligand conformation remains similar in reaction with the more rigid ligand L9, leading to a much smaller distortion energy.
Table3.
Flexibility effects of the Cyclam family ligands.
 | ||||||
|---|---|---|---|---|---|---|
| ligand | ΔG‡exp | ΔG‡DFT | EHOMOb | Vbur%c | ΔEdist(TS)d | ΔEdist(BrCuIIL)e |
| N[2,3,2] | 21.5 | 27.5 | −7.12 | 48.4 | 11.9 | 14.4 |
| N[2,2,2] | 18.6 | 26.4 | −7.26 | 45.6 | 14.2 | 17.0 |
| Me4Cyclam | 17.7 | 20.3 | −6.92 | 49.6 | 7.0 | 9.0 |
| Cyclam-B | 13.6 | 16.8 | −6.81 | 47.6 | 4.0 | 5.2 |
kact values are in M−1 s−1. All free energies and distortion energies are in kcal/mol; EHOMO is in eV. All energies and EHOMO are computed in acetonitrile using the CPCM solvation model.
HOMO energy of [CuIL]+.
Percent buried volume of the ligand computed from the DFT-optimized geometries of [CuIL]+.41
Distortion energy of the CuL catalyst in the ISET transition state with respect to the ground state [CuIL]+.
Distortion energy of the CuL catalyst in the [BrCuIIL]+ complex with respect to the ground state [CuIL]+.
Figure 11.

Optimized geometries of CuI catalysts and ISET transition states with Cyclam family ligands.
Because of its significant role on reactivity, a specific parameter should be included in the predictive model to describe the effect of ligand backbone flexibility.5a Here, we found the computed distortion energy of CuL in the ISET transition state (ΔEdist(TS)) and in the [Br-CuIIL]+ product (ΔEdist(BrCuIIL)) can both adequately describe the ligand flexibility effect. Because the ISET transition states are late (vide supra) and product-like, an excellent linear correlation between ΔEdist(TS) and ΔEdist(BrCuIIL) for all 9 aforementioned ligands is observed (See Figure S5 in SI). Because the computation of ΔEdist(BrCuIIL) does not require optimization of the ISET transition state geometry and thus is much more feasible, ΔEdist(BrCuIIL) was chosen as a parameter to describe the ligand flexibility effect.
3.3.4. Predictive Model for Ligand Effect on Reactivity
Through the detailed analysis of computed transition state structures and energies, we not only uncovered the factors that affect reactivity, but also identified appropriate parameters to describe each type of ligand effects. Therefore, these theoretical insights allowed us to establish a simple mathematical equation using only three parameters to predict the reactivity of the structurally diverse Cu-ATRP catalysts, namely, the HOMO energy of the [CuIL]+ catalyst (EHOMO), the percent buried volume (Vbur%), and the distortion energy of CuL in the [BrCuIIL]+ complex (ΔEdist). Using the nine ligands discussed above (L1-L9) as the training set, eq. 4 was developed using multivariate linear regression by correlating the experimental activation free energies with these three calculated parameters (Figure 12). The rest of the ligands in Figure 8 were then used as test sets to validate the predictive model.
Figure 12.

Predictive model for ligand effect on Cu-ATRP reactivity.
Overall, a good correlation between the predicted activation free energies (ΔG‡predicted) and the experimentally derived activation free energies (ΔG‡exp). For all 18 ligands studied, the mean unsigned error (MUE) of the predicted activation free energy is 1.1 kcal/mol. Although the training set only contains tetradentate ligands, eq. 4
| (eq.4) |
performed well for tetradentate as well as tridentate ligands in the test set. The barriers for all tetradentate and tridentate ligands from either the training set or the test set were correctly predicted within ±2 kcal/mol error ranges (between the two dash lines). This is very promising to Cu-ARTP ligand discovery since tetra- and tridentate ligands form the most reactive Cu catalysts for ATRP. The less accurate prediction for the bidentate bpy ligand43 might be due to the fact that a tetradentate coordinated Cu catalyst with two bpy ligands is considered in the modeling while other Cu complexes with one bpy ligand may also exist under the reaction conditions.
To further understand the impact of each factor on the reactivity, the computed parameters of all 18 ligands are shown using the green-yellow-red color scale in Table 4. Here, green indicates a positive effect to enhancing the reactivity, while red indicates a negative effect as compared with other investigated ligands. In terms of electronic effect, ligands with a greater number of chelating sp3 N atoms are generally more effective due to their stronger donicity. For example, the all-sp3 N cyclam-family ligands (Cyclam-B, Me4Cylam, N[2,2,2], N[2,3,2], and N[3,2,3])) have HOMO values between −6.81 eV to −7.26 eV, while the sp3/sp2 hybrid ligands such as TPMA ligands have lower HOMO values ranging from −7.36 eV to −7.66 eV. Similarly, two TREN-type ligands (Me6TREN and Et6TREN) have higher HOMO than TPMA ligands. Although not among the most electronically activated ligands, TPMA ligands are advantageous due to their less steric repulsions and high rigidity with low Vbur% and ΔEdist values. Therefore, five of the seven most reactive ligands (ΔG‡exp≤15.0 kcal/mol) are TPMA derivatives. In fact, we found steric effect (Vbur%) and backbone flexibility effect (ΔEdist) have greater contribution to the reactivity than the electronic effect (EHOMO) from the standardized multivariate linear equation (see eq. S6 in SI). Furthermore, the electronic properties of TPMA ligands can be easily tuned by substitution on the pyridine rings. As such, TPMANMe2 is the most reactive ligand discovered so far because it has low steric repulsion and distortion penalty, and favorable electronic effect via incorporation of strong electron-donating groups.
Table 4.
Calculated electronic (EHOMO), steric (Vbur%), and backbone flexibility (ΔEdist) parameters for all 18 ligands.a
| ligand | ΔG‡exp | ΔG‡predicted | EHOMO | Vbur% | ΔEdist |
|---|---|---|---|---|---|
| TPMANMe2 | 10.0 | 11.5 | −7.36 | 39.0 | 3.7 |
| TPMA*3 | 12.1 | 12.2 | −7.41 | 40.3 | 3.6 |
| TPMA*2 | 13.4 | 12.6 | −7.49 | 40.0 | 3.8 |
| Cyclam-B | 13.6 | 13.6 | −6.81 | 47.6 | 5.2 |
| TPMA*1 | 13.7 | 12.9 | −7.57 | 39.6 | 4.0 |
| Me6TREN | 13.8 | 14.5 | −7.34 | 45.7 | 4.0 |
| TPMA | 15.0 | 13.0 | −7.66 | 39.2 | 4.0 |
| BPED | 16.6 | 18.5 | −7.48 | 41.8 | 14.7 |
| PMDTA | 16.9 | 18.2 | −7.46 | 40.9 | 14.9 |
| Me4Cyclam | 17.7 | 16.8 | −6.92 | 49.6 | 9.0 |
| N[2,2,2] | 18.6 | 20.3 | −7.26 | 45.6 | 17.0 |
| BPMPA | 18.8 | 17.7 | −7.82 | 37.5 | 13.9 |
| bpy | 19.1 | 16.3 | −7.78 | 38.6 | 10.2 |
| Et6TREN | 19.3 | 19.9 | −7.33 | 56.5 | 5.1 |
| TPMAPh | 19.4 | 18.1 | −7.62 | 47.7 | 7.0 |
| TMEDA | 20.0 | 20.5 | −7.39 | 47.4 | 14.4 |
| N[3,2,3] | 20.6 | 21.0 | −7.21 | 51.1 | 13.6 |
| N[2,3,2] | 21.5 | 19.7 | −7.12 | 48.4 | 14.4 |
All calculated parameters are color-coded. Green indicates quantities that are favorable for reactivity, and red indicates quantities that lead to lower reactivity. All energies are in kcal/mol. EHOMO are in eV.
4. Conclusion
In this study, we demonstrate a computational approach to develop predictive mathematical equations for the reactivity of catalysts and initiators in Cu-ATRP, a widely used controlled radical polymerization reaction. We report the first DFT calculations of the inner-sphere electron transfer transition state, which involves a halogen atom transfer between a dormant alkyl halide chain end and the CuI catalyst. An in-depth computational analysis revealed a few key factors controlling the stability of the ISET transition state, and thus the activation rate constant. The ISET transition state involves a bent geometry of the Cu∙∙∙Br∙∙∙R bond, which leads to an enhanced non-covalent interaction between the ancillary N-donor ligand and the alkyl halide chain end. As such, the stability of the ISET transition state is sensitive to the steric properties of the ligand. A substantial amount of charge transfer from the CuI catalyst to the alkyl halide is observed in the transition state, which indicates the ISET process can be facilitated by stronger donor ligands. In addition, ISET transition states with flexible multidentate ligands, such as the acyclic ligand N[2,3,2], suffer from a high catalyst distortion energy due to the different ligand conformations in the [CuIL]+ resting state and the ISET transition state. With these mechanistic insights into the factors controlling reactivity, we identified three parameters, namely the percent buried volume, HOMO energy of the [CuIL]+ catalyst, and the catalyst distortion energy of the [Br-CuIIL]+ complex, to describe the steric, electronic, and flexibility effects of ligand, respectively. We developed a three-variable linear equation using these three DFT-computed parameters to predict the reactivity of a structurally diverse set of Cu-ATRP catalysts. The predicted activation barriers are within ±2 kcal/mol of the experimental values for 17 out of 18 ligands used in the training and test sets.
The same computational approach was used to develop a predictive equation for initiator effects on reactivity. Since the DFT calculations indicated a late ISET transition state with a substantial R–X bond stretch and a strong X∙∙∙Cu interaction, the stability of the transition state is expected to be affected by both the R–X bond dissociation energy (BDE) and the halogenophilicity of the [CuIL]+ complex. These conclusions were reaffirmed by a distortion/interaction model analysis, which indicated the activation energy is affected by both the R–X distortion energies and the interaction energies between R–X and Cu in the ISET transition state. Using these mechanistic insights, a two-variable linear equation was developed using R–X BDE and Cu–X halogenophilicity as parameters to describe the effects of R–X bond stretch and Cu–X interactions, respectively.
Taken together, we demonstrated the use of mechanistic insights derived from transition state calculations to guide the development of predictive mathematical equations for reactivities of Cu-ATRP. We expect our models to shed light on the rational ligand design and discovery for Cu-ATRP. Similar approaches may be employed to benefit the ligand design in other transition metal-catalyzed reactions.
Supplementary Material
ACKNOWLEDGMENT
We thank the NIH (GM128779) and NSF(CHE1707490) for financial support for this work. MLC gratefully acknowledges a Georgina Sweet ARC Laureate Fellowship (FL170100041) and generous allocations of supercomputing time on the National Facility of the Australian National Computational Infrastructure. PL acknowledges supercomputer resources provided by the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF. We also thank contributions of Dr. Ching Yeh Lin at the early stages of the project.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional computational results, Cartesian coordinates (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1 (a).Matyjaszewski K Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039; [Google Scholar]; (b) Braunecker WA; Matyjaszewski K Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci 2007, 32, 93–146; [Google Scholar]; (c) Matyjaszewski K; Tsarevsky NV Macromolecular Engineering by Atom Transfer Radical Polymerization. J. Am. Chem. Soc 2014, 136, 6513–6533; [DOI] [PubMed] [Google Scholar]; (d) Matyjaszewski K, Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater 2018, 30, 1706441. [DOI] [PubMed] [Google Scholar]
- 2 (a).O’Reilly RK; Shaver MP; Gibson VC Nickel(II) α-diimine catalysts for the atom transfer radical polymerization of styrene. Inorg. Chim. Acta 2006, 359, 4417–4420; [Google Scholar]; (b) Zhengbiao Z; Wei Z; Xiulin Z; Zhenping C; Jian Z “Living”/controlled free radical polymerization of MMA in the presence of cobalt(II) 2-ethylhexanoate: A switch from RAFT to ATRP mechanism. J. Polym. Sci., Part A: Polym. Chem 2007, 45, 5722–5730; [Google Scholar]; (c) Nishiura C; Williams V; Matyjaszewski K Iron and copper-based catalysts containing anionic phenolate ligands for atom transfer radical polymerization. Macromol. Res 2017, 25, 504–512; [Google Scholar]; (d) Cao J; Jin C; Keda Z Novel catalyst system of MCl2/FeCl3.6H2O/PPh3 (M = Ni, Co, or Mn) for the atom transfer radical polymerization of methyl methacrylate. J. Polym. Sci., Part A: Polym. Chem 2005, 43, 2625–2631; [Google Scholar]; (e) Sebastien M; Tadeusz B; Rinaldo P; Przemysaw K Atom transfer radical polymerization of methyl acrylate with molybdenum halides as catalysts in an ionic liquid. J. Appl. Polym. Sci 2007, 105, 278–281; [Google Scholar]; (f) Kato M; Kamigaito M; Sawamoto M; Higashimura T Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(triphenylphosphine) ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules 1995, 28, 1721–1723; [Google Scholar]; (g) Xue Z; He D; Xie X Iron-catalyzed atom transfer radical polymerization. Polym. Chem 2015, 6, 1660–1687; [Google Scholar]; (h) di Lena F; Matyjaszewski K Transition metal catalysts for controlled radical polymerization. Prog. Polym. Sci 2010, 35, 959–1021. [Google Scholar]
- 3.Pan X; Fang C; Fantin M; Malhotra N; So WY; Peteanu LA; Isse AA; Gennaro A; Liu P; Matyjaszewski K Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J. Am. Chem. Soc 2016, 138, 2411–2425. [DOI] [PubMed] [Google Scholar]
- 4 (a).Pan X; Lamson M; Yan J; Matyjaszewski K Photoinduced Metal-Free Atom Transfer Radical Polymerization of Acrylonitrile. ACS Macro Letters 2015, 4, 192–196; [DOI] [PubMed] [Google Scholar]; (b) Treat NJ; Sprafke H; Kramer JW; Clark PG; Barton BE; Read de Alaniz J; Fors BP; Hawker CJ Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc 2014, 136, 16096–16101; [DOI] [PubMed] [Google Scholar]; (c) Fors BP; Hawker CJ Control of a Living Radical Polymerization of Methacrylates by Light. Angew. Chem. Int. Ed 2012, 51, 8850–8853; [DOI] [PubMed] [Google Scholar]; (d) Pan X; Tasdelen MA; Laun J; Junkers T; Yagci Y; Matyjaszewski K Photomediated controlled radical polymerization. Prog. Polym. Sci 2016, 62, 73–125 [Google Scholar]
- 5 (a).Zerk TJ; Bernhardt PV Redox-coupled structural changes in copper chemistry: Implications for atom transfer catalysis. Coord. Chem. Rev 2017, 375, 173–190; [Google Scholar]; (b) Pintauer T; Matyjaszewski K Structural aspects of copper catalyzed atom transfer radical polymerization. Coord. Chem. Rev 2005, 249, 1155–1184. [Google Scholar]
- 6.Matyjaszewski K; Patten TE; Xia J Controlled/“Living” Radical Polymerization. Kinetics of the Homogeneous Atom Transfer Radical Polymerization of Styrene. J. Am. Chem. Soc 1997, 119, 674–680. [Google Scholar]
- 7.De Paoli P; Isse AA; Bortolamei N; Gennaro A New insights into the mechanism of activation of atom transfer radical polymerization by Cu(I) complexes. Chem. Commun 2011, 47, 3580–3582. [DOI] [PubMed] [Google Scholar]
- 8.Lin CY; Coote ML; Gennaro A; Matyjaszewski K Ab Initio Evaluation of the Thermodynamic and Electrochemical Properties of Alkyl Halides and Radicals and Their Mechanistic Implications for Atom Transfer Radical Polymerization. J. Am. Chem. Soc 2008, 130, 12762–12774. [DOI] [PubMed] [Google Scholar]
- 9 (a).Gillies MB; Matyjaszewski K; Norrbyx P-O; Pintauer T; Poli R; Richard P A DFT Study of R–X Bond Dissociation Enthalpies of Relevance to the Initiation Process of Atom Transfer Radical Polymerization. Macromolecules 2003, 36, 8551–8559; [Google Scholar]; (b) Matyjaszewski K; Poli R Comparison of Bond Dissociation Energies of Dormant Species Relevant to Degenerative Transfer and Atom Transfer Radical Polymerization. Macromolecules 2005, 38, 8093–8100; [Google Scholar]; (c) Guliashvili T; Percec V A comparative computational study of the homolytic and heterolytic bond dissociation energies involved in the activation step of ATRP and SET-LRP of vinyl monomers. J. Polym. Sci., Part A: Polym. Chem 2007, 45, 1607–1618. [Google Scholar]
- 10.Isse AA; Gennaro A; Lin CY; Hodgson JL; Coote ML; Guliashvili T Mechanism of Carbon–Halogen Bond Reductive Cleavage in Activated Alkyl Halide Initiators Relevant to Living Radical Polymerization: Theoretical and Experimental Study. J. Am. Chem. Soc 2011, 133, 6254–6264. [DOI] [PubMed] [Google Scholar]
- 11 (a).Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301; [DOI] [PubMed] [Google Scholar]; (b) Zhao S; Gensch T; Murray B; Niemeyer ZL; Sigman MS; Biscoe MR Enantiodivergent Pd-catalyzed C–C bond formation enabled through ligand parameterization. Science 2018, 362, 670–674; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Niemeyer ZL; Pindi S; Khrakovsky DA; Kuzniewski CN; Hong CM; Joyce LA; Sigman MS; Toste FD Parameterization of Acyclic Diaminocarbene Ligands Applied to a Gold(I)-Catalyzed Enantioselective Tandem Rearrangement/Cyclization. J. Am. Chem. Soc 2017, 139, 12943–12946; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Niemeyer ZL; Milo A; Hickey DP; Sigman MS Parameterization of phosphine ligands reveals mechanistic pathways and predicts reaction outcomes. Nat. Chem 2016, 8, 610–617. [DOI] [PubMed] [Google Scholar]
- 12.Wu K; Doyle AG Parameterization of phosphine ligands demonstrates enhancement of nickel catalysis via remote steric effects. Nat. Chem 2017, 9, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13 (a).Ardkhean R; Mortimore M; Paton RS; Fletcher SP Formation of quaternary centres by copper catalysed asymmetric conjugate addition to β-substituted cyclopentenones with the aid of a quantitative structure–selectivity relationship. Chem. Sci 2018, 9, 2628–2632; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ardkhean R; Roth PMC; Maksymowicz RM; Curran A; Peng Q; Paton RS; Fletcher SP Enantioselective Conjugate Addition Catalyzed by a Copper Phosphoramidite Complex: Computational and Experimental Exploration of Asymmetric Induction. ACS Catalysis 2017, 7, 6729–6737; [Google Scholar]; (c) Piou T; Romanov-Michailidis F; Romanova-Michaelides M; Jackson KE; Semakul N; Taggart TD; Newell BS; Rithner CD; Paton RS; Rovis T Correlating Reactivity and Selectivity to Cyclopentadienyl Ligand Properties in Rh(III)-Catalyzed C–H Activation Reactions: An Experimental and Computational Study. J. Am. Chem. Soc 2017, 139, 1296–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14 (a).Orlandi M; Coelho JAS; Hilton MJF; Toste FD; Sigman MS Parametrization of Non-covalent Interactions for Transition State Interrogation Applied to Asymmetric Catalysis J. Am. Chem. Soc 2017, 139, 6803–6806; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Orlandi M; Hilton MJ; Yamamoto E; Toste FD; Sigman MS Mechanistic Investigations of the Pd(0)-Catalyzed Enantioselective 1,1-Diarylation of Benzyl Acrylates. J. Am. Chem. Soc 2017, 139, 12688–12695; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morales-Rivera CA; Floreancig PE; Liu P Predictive Model for Oxidative C–H Bond Functionalization Reactivity with 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone. J. Am. Chem. Soc 2017, 139, 49, 17935–17944. [DOI] [PubMed] [Google Scholar]
- 15 (a).Straker RN; Mekareeya A; Paton RS; Anderson EA Computational ligand design in enantio- and diastereoselective ynamide [5+2] cycloisomerization. Nat. Commun. 2016, 7, 10109–10118; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kwon D; Fuller JT; Kilgore UJ; Sydora OL; Bischof SM; Ess DH Computational Transition-State Design Provides Experimentally Verified Cr(P,N) Catalysts for Control of Ethylene Trimerization and Tetramerization. ACS Catal 2018, 8, 1138–1142; [Google Scholar]; (c) Nielsen MC; Bonney KJ; Schoenebeck F Computational Ligand Design for the Reductive Elimination of ArCF3 from a Small Bite Angle PdII Complex: Remarkable Effect of a Perfluoroalkyl Phosphine. Angew. Chem., Int. Ed 2014, 53, 5903–5906; [DOI] [PubMed] [Google Scholar]; (d) Bernales V; League AB; Li Z; Schweitzer NM; Peters AW; Carlson RK; Hupp JT; Cramer CJ; Farha OK; Gagliardi L Computationally Guided Discovery of a Catalytic Cobalt-Decorated Metal–Organic Framework for Ethylene Dimerization. J. Phys. Chem. C 2016, 120, 23576–23583; [Google Scholar]; (e) Sinha I; Lee Y; Bae C; Tussupbayev S; Lee Y; Seo M; Kim J; Baik M; Lee Y; Kim H Computer-aided rational design of Fe(III)-catalysts for the selective formation of cyclic carbonates from CO2 and internal epoxides. Catal. Sci. Technol 2017, 7, 4375–4387. [Google Scholar]; (f) Rosales AR; Wahlers J; Limé E; Meadows RE; Leslie KW; Savin R; Bell F; Hansen E; Helquist P; Munday RH; Wiest O; Norrby P-O Rapid virtual screening of enantioselective catalysts using CatVS. Nat. Catal, 2019, 2, 41–45. [Google Scholar]
- 16.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich A; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; J. A. Montgomery J; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- 17.Chai J-D; Head-Gordon M Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- 18.Fuentealba P; Preuss H; Stoll H; Von Szentpály L A proper account of core-polarization with pseudopotentials: single valence-electron alkali compounds. Chem. Phys. Lett 1982, 89, 418–422. [Google Scholar]
- 19.Rassolov VA; Ratner MA; Pople JA; Redfern PC; Curtiss LA 6-31G* basis set for third-row atoms. J. Comput. Chem 2001, 22, 976–984. [Google Scholar]
- 20.Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 21.Barone V; Cossi M Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- 22.Marcus RA On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys 1956, 24, 966–978. [Google Scholar]
- 23.Saveant JM A simple model for the kinetics of dissociative electron transfer in polar solvents. Application to the homogeneous and heterogeneous reduction of alkyl halides. J. Am. Chem. Soc 1987, 109, 6788–6795. [Google Scholar]
- 24.Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ess DH; Houk KN Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc 2007, 129, 10646–10647. [DOI] [PubMed] [Google Scholar]
- 26.Khaliullin RZ; Cobar EA; Lochan RC; Bell AT; Head-Gordon M Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals. J. Phys. Chem. A 2007, 111, 8753–65 [DOI] [PubMed] [Google Scholar]
- 27.Horn PR; Mao Y; Head-Gordon M Probing non-covalent interactions with a second generation energy decomposition analysis using absolutely localized molecular orbitals. Phys. Chem. Chem. Phys 2016, 18, 23067–79. [DOI] [PubMed] [Google Scholar]
- 28.Shao Y; Gan Z; Epifanovsky E; Gilbert ATB; Wormit M; Kussmann J; Lange AW; Behn A; Deng J; Feng XT; Ghosh D; Goldey M; Horn PR; Jacobson LD; Kaliman I; Khaliullin RZ; Kus T; Landau A; Liu J; Proynov EI; Rhee YM; Rich-ard RM; Rohrdanz MA; Steele RP; Sundstrom EJ; Wood-cock HL; Zimmerman PM; Zuev D; Albrecht B; Alguire E; Austin B; Beran GJO; Bernard YA; Berquist E; Brandhorst K; Bravaya KB; Brown ST; Casanova D; Chang CM; Chen YQ; Chien SH; Closser KD; Crittenden DL; Diedenhofen M; DiStasio RA; Do H; Dutoi AD; Edgar RG; Fatehi S; Fus-ti-Molnar L; Ghysels A; Golubeva-Zadorozhnaya A; Gomes J; Hanson-Heine MWD; Harbach PHP; Hauser AW; Hohen-stein EG; Holden ZC; Jagau TC; Ji HJ; Kaduk B; Khistyaev K; Kim J; Kim J; King RA; Klunzinger P; Kosenkov D; Kow-alczyk T; Krauter CM; Lao KU; Laurent AD; Lawler KV; Levchenko SV; Lin CY; Liu F; Livshits E; Lochan RC; Lu-enser A; Manohar P; Manzer SF; Mao SP; Mardirossian N; Marenich AV; Maurer SA; Mayhall NJ; Neuscamman E; Oana CM; Olivares-Amaya R; O’Neill DP; Parkhill JA; Perrine TM; Peverati R; Prociuk A; Rehn DR; Rosta E; Russ NJ; Sharada SM; Sharma S; Small DW; Sodt A; Stein T; Stuck D; Su YC; Thom AJW; Tsuchimochi T; Vanovschi V; Vogt L; Vydrov O; Wang T; Watson MA; Wenzel J; White A; Wil-liams CF; Yang J; Yeganeh S; Yost SR; You ZQ; Zhang IY; Zhang X; Zhao Y; Brooks BR; Chan GKL; Chipman DM; Cramer CJ; Goddard WA; Gordon MS; Hehre WJ; Klamt A; Schaefer HF; Schmidt MW; Sherrill CD; Truhlar DG; Warshel A; Xu X; Aspuru-Guzik A; Baer R; Bell AT; Besley NA; Chai JD; Dreuw A; Dunietz BD; Furlani TR; Gwaltney SR; Hsu CP; Jung YS; Kong J; Lambrecht DS; Liang WZ; Ochsenfeld C; Rassolov VA; Slipchenko LV; Subotnik JE; Van Voorhis T; Herbert JM; Krylov AI; Gill PMW; Head-Gordon M Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys 2014, 113, 184–215. [Google Scholar]
- 29 (a).Giri R; Brusoe A; Troshin K; Wang JY; Font M; Hartwig JF Mechanism of the Ullmann Biaryl Ether Synthesis Catalyzed by Complexes of Anionic Ligands: Evidence for the Reaction of Iodoarenes with Ligated Anionic CuI Intermediates. J. Am. Chem. Soc 2018, 140, 793–806; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu H-Z; Jiang Y-Y; Fu Y; Liu L Alternative Mechanistic Explanation for Ligand-Dependent Selectivities in Copper-Catalyzed N- and O-Arylation Reactions. J. Am. Chem. Soc 2010, 132, 18078–18091; [DOI] [PubMed] [Google Scholar]; (c) Jones GO; Liu P; Houk KN; Buchwald SL Computational Explorations of Mechanisms and Ligand-Directed Selectivities of Copper-Catalyzed Ullmann-Type Reactions. J. Am. Chem. Soc 2010, 132, 6205–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The computed reaction Gibbs free energy to form 7 and 8 is underestimated when compared with the experimentally-derived ΔG of this reaction. Nonetheless, this level of theory provided the best agreement with the experimental activation rate constants, kact (see SI for detailed benchmark studies), and thus is chosen to investigate the initiator and catalyst effects on kact.
- 31.The TPMA ligand in [CuIL]+, the ISET transition state, and [Br-CuIIL]+ remains tetradentate-coordinated. Our calculations indicate the partial dissociation of TPMA is disfavored (see the Supporting Information for details).
- 32.Wiberg KB Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron, 1968, 24, 1083–1096. [Google Scholar]
- 33.Our calculations indicated the ISET TS structures optimized in the gas phase and in solution are both bent with similar Cu-Br-C angles (see Figure S7 in the Supporting Information).
- 34 (a).Matsubara H; Ryu I; Schiesser CH An ab initio and DFT study of some halogen atom transfer reactions from alkyl groups to acyl radical. Org. Biomol Chem 2007, 5, 3320–3324; [DOI] [PubMed] [Google Scholar]; (b) Yorimitsu H; Shinokubo H; Matsubara S; Oshima K Triethylborane-Induced Bromine Atom-Transfer Radical Addition in Aqueous Media: Study of the Solvent Effect on Radical Addition Reactions. J. Org. Chem 2001, 66, 7776–7785; [DOI] [PubMed] [Google Scholar]; (c) Yamamoto K; Li J; Garber JAO; Rolfes JD; Boursalian GB; Borghs JC; Genicot C; Jacq J; van Gastel M; Neese F; Ritter T Palladium-catalysed electrophilic aromatic C–H fluorination. Nature 2018, 554, 511–514. [DOI] [PubMed] [Google Scholar]
- 35.In a recent computational study on the mechanism of Rh-catalyzed atom transfer radical addition reaction, a bent Rh∙∙∙Br∙∙∙C geometry in a bromide atom transfer transition state was observed: Chen B; Fang C; Liu P; Ready JM Rhodium-Catalyzed Enantioselective Radical Addition of CX4 Reagents to Olefins. Angew. Chem., Int. Ed 2017, 56, 8780–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F; Yang Z; Yu Y; Mei Y; Houk KN Bimodal Evans—Polanyi Relationships in Dioxirane Oxidations of sp3 C—H: Non-perfect Synchronization in Generation of Delocalized Radical Intermediates. J. Am. Chem. Soc 2017, 139, 16650–16656. [DOI] [PubMed] [Google Scholar]
- 37.Tang W; Matyjaszewski K Effects of Initiator Structure on Activation Rate Constants in ATRP. Macromolecules 2007, 40, 1858–1863. [Google Scholar]
- 38.Fantin M; Isse AA; Bortolamei N; Matyjaszewski K; Gennaro A Electrochemical approaches to the determination of rate constants for the activation step in atom transfer radical polymerization. Electrochimica Acta 2016, 222, 393–401. [Google Scholar]
- 39.The experimental activation free energies (ΔG‡exp) were calculated using the Erying equation: , in which kact values were determined by the most recent electrochemical measurements in ref 35. See: Eyring H, The Activated Complex in Chemical Reactions. J. Chem. Phys 1935, 3, 107–115. [Google Scholar]
- 40.The experimental rate constants (kact) for different Cu-ATRP catalysts were taken or derived from the following references to ensure the experimental values were obtained under similar conditions. See the Supporting Information for a detailed discussion about the experimental conditions for the kact measurements,Tang W; Matyjaszewski K Effect of Ligand Structure on Activation Rate Constants in ATRP. Macromolecules, 2006, 39, 4953–4959;Schröder K; Mathers RT; Buback J; Konkolewicz D; Magenau AJD; Matyjaszewski K Substituted Tris(2-pyridylmethyl)amine Ligands for Highly Active ATRP Catalysts. ACS Macro Lett. 2012, 1, 1037–1040;Tang W; Kwak Y; Braunecker W; Tsarevsky NV; Coote ML; Matyjaszewski K Understanding Atom Transfer Radical Polymerization: Effect of Ligand and Initiator Structures on the Equilibrium Constants. J. Am. Chem. Soc 2008, 130, 10702–10713;Ribelli TG; Fantin M; Daran J-C; Augustine KF; Poli R; Matyjaszewski K Synthesis and Characterization of the Most Active Copper ATRP Catalyst Based on Tris[(4-dimethylaminopyridyl)methyl]amine. J. Am. Chem. Soc 2018, 140, 1525–1534.;dos Santos NAC; Lorandi F; Badetti E; Wurst K; Isse AA; Gennaro A; Licini G; Zonta C Tuning the reactivity and efficiency of copper catalysts for atom transfer radical polymerization by synthetic modification of tris(2-methylpyridyl)amine. Polymer, 2017, 128, 169–176.
- 41.The percent buried volume (Vbur%) values were calculated using SambVca 2. See: Falivene L; Credendino R; Poater A; Petta A; Serra L; Oliva R; Scarano V; Cavallo L SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293.A radius of 5.0 Å was used to define the coordination sphere around the metal center. The larger radius is necessary to properly describe the steric properties of the N-donor ligands, because the substituents involved in the steric interactions (e.g. the ethyl substituents on Et6TREN) are not directly coordinated to the metal center. See the Supporting Information of detailed discussions about the radius used for the Vbur% calculations.
- 42.Queffelec J; Gaynor SG; Matyjaszewski K Optimization of Atom Transfer Radical Polymerization Using Cu(I)/Tris(2-(dimethylamino)ethyl)amine as a Catalyst. Macromolecules, 2000, 33, 8629–8639. [Google Scholar]
- 43.Knuehl B; Pintauer T; Kajiwara A; Fischer H; Matyjaszewski K, Characterization of Cu(II) Bipyridine Complexes in Halogen Atom Transfer Reactions by Electron Spin Resonance, Macromolecules 2003, 36, 8291–8296. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.