

Supplemental Digital Content is available in the text
Keywords: coreceptor usage, circulating recombinant form 01_AE, disease progression, HIV-1, N-linked glycosylation site, variable region
Abstract
Objective:
The current study aimed to understand epidemiological feature and critical factors associated with pathogenesis of circulating recombinant form (CRF) 01_AE strains in Northeast China.
Design:
Compared analysis was made between CRF01_AE and non-CRF01_AE samples to understand the pathogenicity features of CRF01_AE. Further analyses between CRF01_AE samples with high or low CD4+ cell counts and between samples with different coreceptor usages were done to explore the possible factors correlating to the pathogenesis of CRF01_AE viruses.
Methods:
The genotypes of newly identified strains were determined by phylogenetic analyses using Mega 6.06. Coreceptor usage was predicted by Geno2Pheno algorithm. Potential N-linked glycosylation site (PNGS) number was calculated using the online N-glycosite software. The properties of amino acid sequences were analyzed by the online ProtParam tool.
Results:
CRF01_AE become the main HIV-1 genotype since 2010. Compared with non-CRF01_AE group, the CRF01_AE group showed a higher proportion of samples with CD4+ cell count less than 200 cells/μl. Shorter amino acid length, fewer PNGSs and the presence of a basic motif R/KNXT or NR/KT in V4 correlated to a lower CD4+ cell count, and existence or coexistence of Thr12, Arg13, Val21 and Lys33, presence of more than 4 of net charges and lack of the PNGS within V3 favored to the X4/R5X4 coreceptor usage of CRF01_AE viruses.
Conclusion:
CRF01_AE has dominated HIV-1 genotype in Northeast China. Infection with CRF01_AE exhibited a fast disease progression, which may be associated with specific amino acid residues and PNGSs in V3 and V4 regions as well as amino acid length of V4 region.
Introduction
The wide and fast transmission of circulating recombinant form (CRF) 01_AE strain played a critical role in diversity and complexity of HIV-1 infection in China. CRF01_AE strain was introduced into China in late 1980s [1]. The national HIV-1 epidemiology surveillance showed the prevalence of CRF01_AE increased from 9.6% of HIV-1 infections [2] during 1996–1998 to 42.5% during 2007–2015 [3]. The rapid transmission of CRF01_AE further complicated the HIV-1 epidemic situation in China [4–8]. Nowadays, CRF01_AE strains have formed several stable transmission clusters in different regions of China, and one cluster was epidemic in Northeast China (Heilongjiang, Jilin and Liaoning provinces) [1,9]. This CRF01_AE cluster dominated the subtype in this region [5,6,10], and showed a tendency for a longer growth period compared with the other CRF01_AE clusters in China [1]. Therefore, understanding the factors associated with CRF01_AE transmission and pathogenesis is urgently required for the control and prevention of this subtype infection in China.
Several studies have demonstrated that patients infected with CRF01_AE viruses commonly had a lower baseline CD4+ cell count [11,12] and a higher frequency of CXCR4 (X4) tropic virus [13,14], and experienced a more rapid disease progression [15,16] than those infected with non-CRF01_AE viruses. These features may contribute to the quick spread of CRF01_AE strains in China. However, the underlying molecular mechanisms were largely unknown.
In the current study, we summarized the molecular epidemiology of HIV-1 infections in Jilin province of the Northeast China and found that CRF01_AE was still the dominant genotype, and specific basic amino acids and potential N-linked glycosylation sites (PNGSs) in variable regions V3 and V4 of envelope (Env) glycoprotein were closely associated with the pathogenesis of CRF01_AE strains.
Methods
The study participants and ethical approval
The information of 310 HIV-1-infected individuals was collected and analyzed in this study. Ninety-six antiviral treatment-naive individuals were first diagnosed as HIV-1 seropositive between October 2015 and May 2016 at the Changchun Infectious Disease Hospital. A written informed consent was obtained from each participant. The information of other 214 HIV-1 infected Jilin individuals was obtained from the Los Alamos National Laboratory (LANL) HIV database (http://www.hiv.lanl.gov/content/index).
HIV-1 infection status measure
Fiebig stages were determined by measurement of viral loads by real-time PCR and HIV-1 specific antibodies by EIA and Western Blot as previously described [17]. The recent and long-term infections of 96 newly identified participants were determined by the Limiting-Antigen Avidity (LAg-Avidity) assays using the HIV-1 LAg-Avidity EIA kit (Beijing Kinghawk Pharmaceutical Co., Ltd, Beijing, China) as previously described [18]. The normalized optical density (ODn) cutoff of 1.5 was used to distinguish recent from long-term HIV infection. Samples with final ODn ranging from 0.4 to 1.5 (including 1.5) were classified as recent infections, while those with values above 1.5 were classified as long-term infections. The recent infection indicated individuals acquired HIV infection within a mean time period of 130 days.
HIV-1 genomic RNA extraction, gene amplification and sequencing of the newly isolated samples
Five milliliters of whole peripheral blood was collected from each participant, and then the plasma was separated for HIV-1 genotyping. Viral genomic RNA was extracted as previously described [19] and subjected to reverse transcription–polymerase chain reaction. The gag p17-p24 (HXB2: nt836–1507) and env C2–C4 (HXB2: nt7002–7541) genes were amplified by nested PCR reactions as previously described [6,7]. The purified PCR products were subjected to direct sequencing.
Genotyping of the newly identified HIV-1 viruses
The gag and env genes were aligned separately using Clustal W. A phylogenetic tree was constructed with the HIV-1 gag or env reference sequences obtained from the LANL HIV database in Mega 6.06 software (Arizona State University, Arizona, USA) using the neighbor-joining method based on Kimura two-parameter model and 1000 bootstrap replicate test. The subtype of HIV-1 samples was determined by the distribution of both gag and env genes in the phylogenetic trees. The samples with discordant gag and env genotypes were considered as unique recombinant form viruses. The subtype of the samples which lacked the env gene sequences was determined based on the gag gene alone.
HIV-1 envelope V3–V4 amino acid sequence analysis
HIV-1 tropism was assessed using the online software Geno2pheno (coreceptor) (http://coreceptor.geno2pheno.org/index.php) [20] with 5% as the significance level (cutoff) of false positive rate (FPR) (referred as G2P5 hereinafter) [21]. The HIV-1 strain was considered as CXCR4 tropism (X4) or CCR5/CXCR4 (R5X4) dual-tropism when the FPR was lower than 5%. The numbers of PNGSs in V3, C3 and V4 regions were analyzed using the N-glycosite tool at the LANL database (http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html). The amino acid length, number of positively and negatively charged amino acid residues were analyzed using online ProtParam tool (https://web.expasy.org/protparam/).
Genbank accession numbers of nucleotide sequences
The nucleotide sequences identified in this study were submitted to GenBank with accession numbers of MH119979-MH120074 for the HIV-1 gag p17-p24 genes and MH120075-MH120165 for the env C2–C4 genes.
Statistical analysis
Data were analyzed using GraphPad software version 5.01 (GraphPad Software, Inc, La Jolla, California, USA). Difference between the means was evaluated by the Student's t test with a two-tailed 95% confidence interval. Difference in proportions or rates between two groups was assessed by the Fish's exact test. Pearson's correlation coefficient was used for linear relation determination between two parameters. A P value less than 0.05 was considered as significant.
Results
Circulating recombinant form 01_AE predominated the HIV-1 genotype in Jilin
A total of 310 samples were collected, including the 96 newly isolated samples in this study and 214 samples (one sequence per patient) obtained from the LANL database (Tables S1–S4). As shown in Table S2, 29.2% (28/96) of samples had plasma CD4+ cell counts less than 200 cells/μl, including 75.0% (21/28) of samples were at Fiebig stages III or IV of disease. With the LAg-Avidity assays, we found 34.4% (33/96) of samples were long-term infections and 65.6% (63/96) were recent infections. Among the long-term infections, 63.6% (21/33) were at Fiebig stages III or IV of disease.
Before 2007, subtype B (39/39) was the absolutely dominant HIV-1 genotype. During 2010–2012, CRF01_AE become the predominant genotype, accounting for 53.5% (92/172) of the HIV-1 infections. During 2015–2016, CRF01_AE still was the main genotype, responsible for 66.7% of infections (Fig. S1, Table S1). Among the CRF01_AE infected cases, 76.3% (119/156) were identified in the MSM population. These data indicated that CRF01_AE strain has dominated the HIV-1 genotype in Jilin, especially among the MSM population.
Circulating recombinant form 01_AE-infected individuals had a lower plasma CD4+ cell count and a higher proportion of predicted X4/R5X4 tropic virus
We compared the CD4+ cell count and the proportion of X4/C5X4-tropic virus in CRF01_AE and non-CRF01_AE samples. Because the CD4+ cell count information of the Jilin reference samples from database was lacked, the analysis on the CD4+ cell counts was conducted among the newly diagnosed samples (n = 96). No significant difference was observed between the mean CD4+ cell counts of CRF01_AE and non-CRF01_AE groups in all, recently or long-term infected samples. Among all the samples, the proportion of samples with CD4+ cell count less than 200 in CRF01_AE group (39.1%, 25/64) was significantly higher than that in non-CRF01_AE group (13.0%, 3/23, P = 0.0356). Among the recent infections or long-term infections, no difference between the two groups was observed (Fig. 1a).
Fig. 1.
Compared analyses between circulating recombinant form 01_AE and noncirculating recombinant form 01_AE infected samples.
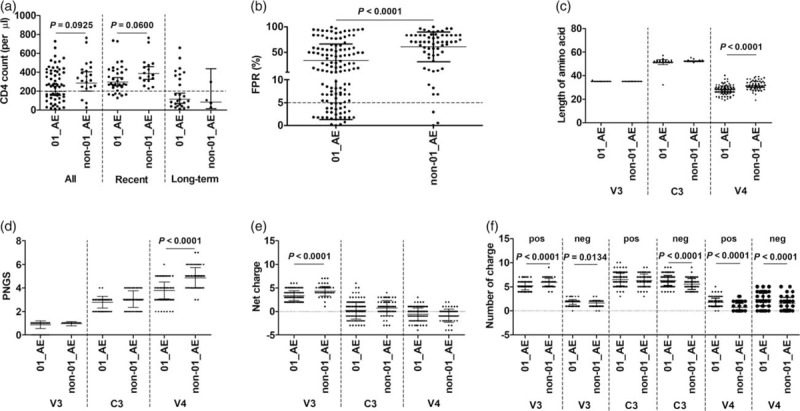
The differences in CD4+ cell count in all samples and recent and long-term infections (a), false positive rate in coreceptor usage prediction by Geno2Pheno method (b) and length of amino acids (c), number of potential N-linked glycosylation sites (d) and numbers of net, positive and negative charges in V3, C3 and V4 regions (e and f) were shown. Data were shown as mean ± SD.
A total of 187 viruses with available full-length env V3–V4 sequences, including 91 sequences identified in this study and 96 sequences from LANL HIV database, were predicted using G2P5 [21]. Thirty-one samples were predicted as X4 tropic or R5X4 dual-tropic (X4/R5X4), including 28 CRF01_AE and 3 non-CRF01_AE samples. The frequency of the predicted X4/R5X4 virus in CRF01_AE (22.4%, 28/125) was higher than that in non-CRF01_AE (4.8%, 3/62, P = 0.0016) (Fig. 1b).
Circulating recombinant form 01_AE viruses had shorter lengths, fewer potential N-linked glycosylation sites and more positively and negatively charged amino acid residues in V4 region
Considering that Env plays crucial role in viral binding to and entry into target cells, we then analyzed on the difference of Env properties between CRF01_AE and non-CRF01_AE viruses. Compared with non-CRF01_AE, CRF01_AE viruses showed a shorter length in V4 amino acid sequences, fewer PNGSs in V4, fewer net and positive charges in V3, and more negative charges in V3, more positive and negative charges in V4 (Fig. 1c–f).
The FPR value significantly negatively correlated to the numbers of net, positive charges in V3 and the numbers of positive and negative charges in V4, and positively correlated to the numbers of PNGS in V3 and V4 and the amino acid length of V4 (Fig. S2).
These findings outlined the differences between CRF01_AE and non-CRF01_AE Env sequences, especially in V3 and V4 regions, which prompted us to wonder what feature of V3 and V4 sequences contributed to CRF01_AE pathogenesis and epidemic in Northeast China.
Bigger length, more potential N-linked glycosylation site and a motif R/KNXT or NR/KT in V4 correlated to low plasma CD4+ cell counts in circulating recombinant form 01_AE samples
We divided CRF01_AE samples with available CD4+ cell count information into high CD4+ (CD4hi, CD4+ cell count ≥200, n = 34) and low CD4+ (CD4lo, CD4+ cell count <200, n = 25) groups. The amino acid length and the PNGS number between V4 regions of CD4lo and CD4hi groups were not significantly different (Fig. 2a and b). The CD4+ cell count significantly negatively correlated with the amino acid length but did not correlate with PNGS number (Fig. 2c and d).
Fig. 2.
Compared analyses between circulating recombinant form 01_AE groups with high and low CD4+ cell counts.
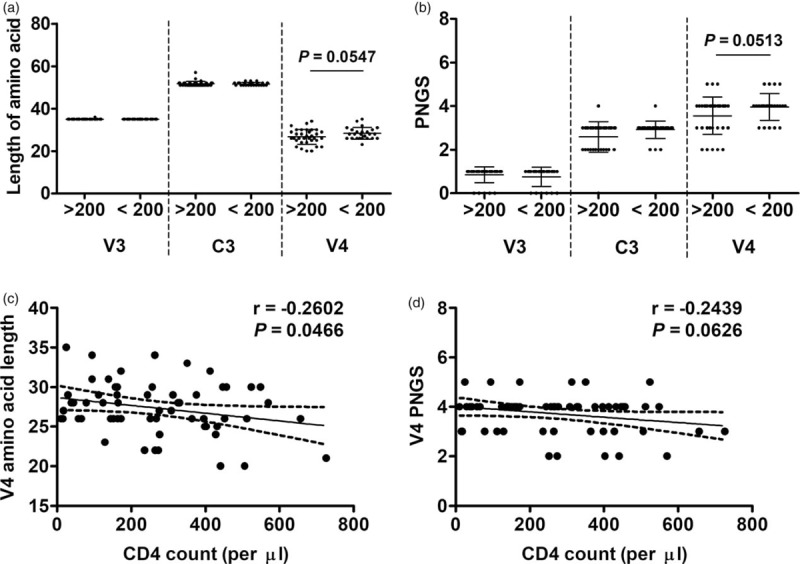
Circulating recombinant form 01_AE sequences were divided into high CD4+ (CD4hi, ≥200 cells/μl) and low CD4+ (CD4lo, <200 cells/μl) groups according to the CD4+ cell counts of the patients. The differences in length of amino acids (a) and numbers of potential N-linked glycosylation sites (b) in V3, C3 and V4 regions were analyzed. Correlation analyses between CD4+ cell count and amino acid length (c) and potential N-linked glycosylation site number (d) in V4 were also done. Data were shown as mean ± SD. r, correlation coefficient.
The V4 amino acid sequences in both CD4hi and CD4lo groups were highly conserved in N and C terminals, but highly variable from position 393–413 (according to HXB2 numbering). The diversity in sequence of this area also leaded to diversity in the V4 amino acid length. Significantly, within this highly variable area, the CD4lo group showed a higher frequency of a basic amino acid residue arginine (R) or lysine (K) just before or after the N397 residue (R/KNXT or NR/KT) (where X is any amino acid except Proline) than the CD4hi group (8/25 versus 3/34, P = 0.0404).
Basic amino acid substitution and lack of potential N-linked glycosylation site in V3 contributed to X4/R5X4 coreceptor usage of circulating recombinant form 01_AE viruses
To explore the factor associated with coreceptor usage of CRF01_AE viruses, we divided the 125 CRF01_AE Env sequences (including 62 sequences identified in this study and 63 sequences from LANL HIV database) into two sets, predicted R5-tropic (n = 97) and predicted X4/R5X4-tropic (n = 28). As expected, the predicted X4/R5X4 V3 sequences contained more net charges than the predicted R5 V3 sequences, which was mainly due to the more positively charged residues in the predicted X4/R5X4 V3 rather than the difference on the negative charge number (Fig. 3a and b). Compared with R5 V3, the predicted X4/R5X4 V3 sequence was less common to have 1–3 net positive charges, and more common to contain more than 4 of net positive charges. Two sets showed a similar proportion of sequences with 4 net positive charges.
Fig. 3.
Compared analyses between circulating recombinant form 01_AE groups harboring CCR5 and CXCR4/dual tropic viruses.
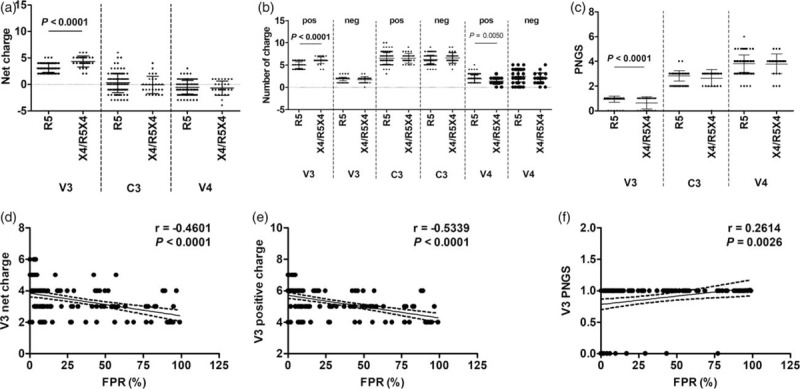
The viral tropism was predicted by the online Geno2Pheno tool with a 5% cutoff of the false positive rate. The differences in numbers of net (a), positive and negative charges (b) and potential N-linked glycosylation sites (c) in V3, C3 and V4 between CCR5 (R5, false positive rate ≥5%) and CXCR4/dual (X4/R5X4, false positive rate <5%) viruses were shown. Correlation analyses between false positive rate value and the numbers of net charges (d), positive charges (e) and potential N-linked glycosylation sites (f) in V3 region were also done. Data were shown as mean ± SD. r, correlation coefficient.
The V3 region was quite conserved in the amino acid length (35 residues) among the predicted R5 and X4/R5X4 viruses, but the sequence was highly variable. The amino acid substitution frequently occurred in V3 region, as shown in Table 1. When comparing the amino acid composition in each position between predicted R5 and X4/R5X4 V3 sequences, we found specific substitutions at several positions. The amino acid residues around the glycine-proline-glycine-glutamine crown motif exhibited a higher diversity in the two sets. Compared with R5 sequences, the predicted X4/R5X4 sequences in this area preferred to contain a higher frequency of threonine (T) and valine (V) in the position 12 and 21, respectively (Tables 1 and 2).
Table 1.
Amino acid diversity in each position in V3 region and numbers of each residue in the predicted R5 and X4/R5X4 tropic viruses.
| HXB2 | ||||||||||||||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | – | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 |
| C | T | R | P | N | N | N | T | R | K | R | I | R | I | Q | R | G | P | G | R | A | F | V | T | I | G | – | K | I | G | N | M | R | Q | A | H | C |
| R5 tropic (n = 97) | ||||||||||||||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | |
| C96 | T94 | R97 | P97 | S92 | N95 | N97 | T92 | R96 | T82 | S92 | I74 | R31 | I75 | – | – | G96 | P96 | G97 | Q57 | A34 | F89 | Y89 | R80 | T92 | G96 | D80 | I96 | I76 | G97 | D85 | I94 | R95 | Q65 | A97 | Y84 | C97 |
| S1 | I2 | F2 | T2 | V2 | I1 | K6 | G5 | T12 | S20 | M17 | A1 | L1 | R25 | T31 | Y4 | F7 | K7 | I4 | E1 | E14 | V1 | T15 | N12 | P1 | K2 | K22 | F11 | |||||||||
| S1 | Y1 | I1 | E4 | V8 | N18 | L3 | K7 | M15 | L2 | I1 | G4 | N1 | A3 | V3 | V1 | S4 | H2 | |||||||||||||||||||
| N1 | Q1 | Q3 | M2 | T14 | F2 | G3 | V15 | W1 | S4 | K2 | T1 | R3 | ||||||||||||||||||||||||
| A1 | K1 | S1 | L1 | H7 | S3 | S1 | I1 | A1 | R1 | L1 | ||||||||||||||||||||||||||
| G1 | P4 | A2 | I1 | Q1 | E1 | |||||||||||||||||||||||||||||||
| Q1 | M1 | |||||||||||||||||||||||||||||||||||
| G1 | ||||||||||||||||||||||||||||||||||||
| A1 | ||||||||||||||||||||||||||||||||||||
| X4/R5X4 tropic (n = 28) | ||||||||||||||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | |
| C27 | T26 | R28 | P28 | S17 | N25 | N27 | T20 | R27 | T21 | S25 | T11 | R18 | I24 | – | – | G28 | P28 | G26 | Q18 | V14 | F24 | Y23 | R20 | T27 | G26 | D16 | I25 | I20 | G28 | D23 | I26 | R27 | K21 | A28 | Y23 | C27 |
| S1 | I1 | F4 | S1 | T1 | I8 | G1 | K4 | G2 | I11 | S3 | T1 | V1 | R7 | M6 | L2 | H2 | K6 | R1 | E2 | E5 | T1 | T7 | N4 | L1 | G1 | Q7 | H3 | H1 | ||||||||
| A1 | Y4 | T1 | I3 | R1 | M2 | T2 | F1 | A1 | S1 | A5 | Y1 | F2 | G2 | A4 | L1 | G1 | V1 | E1 | F1 | |||||||||||||||||
| T1 | I1 | F2 | H2 | L1 | G1 | F1 | I1 | S1 | S2 | S1 | ||||||||||||||||||||||||||
| N1 | L1 | G1 | M1 | K1 | T1 | G1 | ||||||||||||||||||||||||||||||
| C1 | V1 | N1 | ||||||||||||||||||||||||||||||||||
| K1 | ||||||||||||||||||||||||||||||||||||
Amino acids were numbered according to the HXB2 V3 sequence. The number in the superscript of an amino acid residue represented the number of the residue in this position; statistically different substitutions were in bold and in box. R5, CCR5; R5X4, CCR5 and CXCR4; X4, CXCR4.
Table 2.
Specific amino acid residues in the predicted R5 and X4/R5X4 V3 regions.
| Residues in V3a | R5, n = 97 | X4/R5X4, n = 28 | P value |
| T12 | 12 (12.4%) | 11 (39.3%) | 0.0040 |
| R13 | 31 (32.0%) | 18 (64.3%) | 0.0037 |
| V21 | 15 (15.5%) | 14 (50.0%) | 0.0005 |
| K33 | 22 (22.7%) | 21 (75.0%) | <0.0001 |
| T12 + R13 | 6 (6.2%) | 11 (39.3%) | <0.0001 |
| T12 + K33 | 6 (6.2%) | 10 (35.7%) | 0.0002 |
| R13 + V21 | 6 (6.2%) | 13 (46.4%) | <0.0001 |
| R13 + K33 | 17 (17.5%) | 16 (57.1%) | 0.0001 |
| V21 + K33 | 3 (3.1%) | 13 (46.4%) | <0.0001 |
| T12 + R13 + V21 | 1 (1.0%) | 7 (25.0%) | 0.0001 |
| T12 + R13 + K33 | 5 (5.2%) | 10 (35.7%) | 0.0001 |
| T12 + V21 + K33 | 2 (2.1%) | 7 (25.0%) | 0.0004 |
| R13 + V21 + K33 | 2 (2.1%) | 12 (42.9%) | <0.0001 |
| T12 + R13 + V21 + K33 | 1 (1.0%) | 7 (25.0%) | 0.0001 |
K, lycine; R, argenine; R5, CCR5 tropic virus; R5X4, CCR5 and CXCR4 dual-tropic virus; T, threomine; V, valine; X4, CXCR4 tropic virus.
aAmino acid residues were numbered according to the HXB2 V3 sequence.
Considering that more net positive charges had been found in the predicted X4/R5X4 V3, we also analyzed the substitutions at charged amino acid residues. Indeed, the predicted X4/R5X4 V3 sequences had higher proportions of argenine (R) and lycine (K) in the position 13 and 33, respectively, compared with the predicted R5 V3 sequences. Significantly, combination substitutions including T12 and/or V21 as well as at least one basic amino acid residue (R13 or K32) seemed more common in the X4/R5X4 sequences than the R5 sequences (Tables 1 and 2). At other positions with positively or negatively charged amino acid residues, such as R3, R9, R24, R32 and asparticacid (D) at the position 30, no significant difference on the frequencies of predominant residues between the two sets was found (Table 1).
Generally, there is only one PNGS within HIV-1 Env V3 region at the position of 6–8. In this study, 12.8% (16/125) of CRF01_AE V3 sequences did not contain this PNGS, and the predicted X4/R5X4 V3 showed higher frequencies to lack this PNGS than the R5 sequences (Fig. 3c). Correlation analyses showed that the numbers of net and positive charges in V3 inversely correlated to the FPR value, and PNGS number in V3 positively correlated to the FPR value (Fig. 3d–f).
These results suggested that there were different amino acid variation patterns in the predicted R5 and X4/R5X4 tropic V3 regions, and that specific amino acid residues and the lack of the PNGS in V3 may contribute to X4/R5X4 coreceptor usage of CRF01_AE virus in Northeast China.
Discussion
In this study, we described the HIV-1 epidemic status in Jilin province, the Northeast China. The CRF01_AE strain has dominated the HIV-1 genotype since 2010 [5,22]. In the current study, we found that CRF01_AE still was the major genotype during 2015–2016, indicating CRF01_AE strain might have some epidemic advantages over non-CRF01_AE. Previous studies reported that patients infected with CRF01_AE viruses commonly harbored a lower baseline CD4+ cell count, fast CD4+ cell decline and a higher frequency of X4 tropic virus than those infected with non-CRF01_AE viruses in China and Singapore [10–16,23]. Another studies found the Chinese MSM cohort in which CRF01_AE infection accounted for 53% showed a faster CD4+ cell decline than the European MSM cohort [24] and the low baseline CD4+ cell count and fast CD4+ cell decline rate were associated with rapid progression [25]. Therefore, to better control HIV-1 infections in China, finding out and elucidating the factors contributing to the rapid transmission and high pathogenicity of CRF01_AE strains is very required.
One interesting observation in this study was that compared with non-CRF01_AE viruses, CRF01_AE viruses contained shorter amino acid lengths and fewer PNGSs in V4 region. A recent study on the HIV-1 sexual transmission pairs reported that the recipient partner tended to harbor a shorter V1–V4 region, fewer PNGSs than the transmitted partner [26], indicating that a short length and fewer PNGSs in V1–V4 may facilitate HIV-1 transmission and establishment of new infection in the recipients [27,28]. More significantly, we found the shorter lengths and fewer PNGSs in V4 were associated with the lower FPR value in Geno2Pheno prediction, indicating that these virological characteristics of V4 region may contribute to the coreceptor usage of CRF01_AE virus. This may be a new potential mechanism for the rapid transmission and high pathogenicity of CRF01_AE virus in Northeast China.
Another observation was that, the CD4+ cell count significantly negatively correlated with the V4 amino acid length in CRF01_AE samples. A retrospective investigation showed that following infection, HIV-1 subtype B viruses adapt to host immune responses through increased length and/or addition of PNGSs in V1V2 region [29]. Another study showed that CRF07_BC viruses in Xinjiang of China tended to increase the length and PNGS number in V4 region over time within IDU transmission [30]. The observations from previous studies and ours supported the notion that when the stable infection has been established, the length of variable region in HIV-1 Env begin to increase to escapes from surveillance of host humoral immunity [28,30,31]. This may be another potential mechanism for the rapid transmission and high pathogenicity of CRF01_AE in Northeast China.
It is widely accepted that the switch of coreceptor usage from CCR5 into CXCR4 or mix (R5X4 dual tropic) indicates disease progression. And the infection by CRF01_AE is commonly featured with a fast disease progression. Therefore, it is required to determine the tropism of CRF01_AE viruses for the early and correct choice of CCR5 antagonists in the treatment of HIV-1 infection. Geno2Pheno (FPR = 10%, G2P10) has been recommended to HIV-1 genotyping in Europe in 2012 [32]. In recent years, G2P10 alone [10,15] or in combination with WebPSSM x4r5 [11,14] were widely used in tropism prediction of CRF01_AE in China. However, some studies reported that G2P10 and WebPSSM seemed to overestimate CXCR4-usage for CRF01_AE [33], and G2P5 had a better concordance between genotype and phenotype tropism [21,33,34] and seemed to be a suitable algorithm to predict the coreceptor usage of CRF01_AE viruses [21]. In this study, WebPSSM x4r5 showed 100% of concordance with G2P5 in the prediction of subtype B and CRF07_BC/CRF08_BC/C, but had only 42.9% of concordance in the prediction of CRF01_AE (data not shown). Thus, G2P5 alone was utilized in the current study. The proportion of the predicted X4/R5X4 tropic CRF01_AE (22.4%) in our study was slightly lower than the observations in Shanghai (27.8%, combination of G2P10 with WebPSSM) [14] and Liaoning (30.5%, G2P10 alone) [10] of China, mainly because of the more restrict threshold of the algorithm used here.
The previous investigations and the current study demonstrated that CFR01_AE strains epidemic in China contained a higher proportion of X4 or R5X4 tropic viruses than non-CRF01_AE strains. However, the molecular mechanism associated with the switch from CCR5 to CXCR4 among Chinese CRF01_AE strains is largely unknown. In the current study, we found more than four of net charges and lack of the PNGS in V3 were independently associated with the X4/R5X4 coreceptor usage of CRF01_AE in Northeast China, in line with the recent findings in France and Japan [34,35].
It is widely recognized that the amino acid residue substitutions with arginine or lycine in the position 11, 25 or both confer the CXCR4 usage of subtype B viruses (i.e. the 11/15 rule) [36]. However, our study and the previous investigations showed the 11/25 rule did not work in the determination of CRF01_AE tropism, because these positions in CRF01_AE commonly occupied by uncharged amino acid residues [21]. Recent studies in France showed that several specific substitutions including S5Y, N7K, S11R, T12V, T12F, Q18R, I27T and S32R in V3 region were dominant among CXCR4-using CRF01_AE strains [37], and S11R was independently associated with CXCR4 switch [38]. However, we did not find these substitutions contributing to the CXCR4 usage in our study, which may be due to the diversity of CRF01_AE V3 region in different geographical areas. But, we indeed found four amino acid residues preferred to exist on predicted X4/R5X4 V3 region, including I12T, S/N13R, A21V and Q33K. A multicenter Cohort Study in America also reported that the mutations A21V and Q33K (A323V and Q336K in the article) in V3 emerged from 4.10 to 5.67 years after HIV-1 infection, and both mutations led to CXCR4 tropism switch of subtype C virus [39]. And A21V mutation (A19V in that study) leaded to a decreased affinity of HIV-1 Env gp120 for CCR5N terminus [40]. To our knowledge, our study was the first report of the amino acid substitutions for the predicted X4/R5X4 usage of CRF01_AE strains in China. Although we still did not clear the exact mechanism of such residues for viral tropism choice, our findings provide new genetic markers for monitoring the prevalence of X4/R5X4 tropic CRF01_AE strains and guiding the usage of CCR5 antagonists in Northeast China.
The limitations in this study were obvious. First, although we had made great efforts to collect samples from clinical monitoring sites and LANL database, the size of the samples in this study was small. We still believe that a large sample size will further confirm our findings. Second, we found the several factors associated with the low CD4+ cell count after CRF01_AE infection, but did not yet know the excise effects of them on viral transmission and pathogenesis. We hope this study could provide some interesting clues to understand CRF01_AE strains epidemic in Northeast China and even in the whole China. Extensive studies are still needed to explore and clear the detailed pathogenesis of CRF01_AE in future. Another weakness of this study was that the coreceptor usage of the new CRF01_AE isolates was determined only based on the genotypic prediction and phenotype confirmation assay lacked. But, we believe that this study could bring some new points to learn the pathogenesis of CRF01_AE viruses in Northeast China.
In summary, CRF01_AE has dominated HIV-1 genotype in Northeast China. Specific amino acid residues and PNGS in V3 and V4 regions as well as amino acid length of V4 region were associated with the X4/R5X4 coreceptor usage and low CD4+ cell count after CRF01_AE infection. These findings provided valuable information for monitoring epidemic situation of CRF01_AE infection in China and the correct choice of CCR5 antagonists in early treatment.
Acknowledgements
Author contributions: F.-X.W. and S.-L.L. conceived the experiments; F.-X.W. and Q.-H.L. designed the experiments; J.L., J.-Y.W, Q.-Q.H., S.-Y.L. performed the experiments; B. Shao, B. Song and Y.-L.-L. analyzed the data; F.-X.W. and S.-L.L. and Q.-H.L. wrote the article.
The current work was supported by National Megaproject on Key Infectious Diseases (2017ZX10202102) and National Natural Science Foundation of China (81672003, 81602899) and the grant of Harbin Science and Technology Bureau (2016RAXYJ059). Funders played no role in the design, analyses and conclusions of this study.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
Footnotes
Qing-Hai Li, Bing Shao and Jin Li contributed equally to the article.
References
- 1.Li X, Liu H, Liu L, Feng Y, Kalish ML, Ho SYW, et al. Tracing the epidemic history of HIV-1 CRF01_AE clusters using near-complete genome sequences. Sci Rep 2017; 7:4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He X, Xing H, Ruan Y, Hong K, Cheng C, Hu Y, et al. A comprehensive mapping of HIV-1 genotypes in various risk groups and regions across China based on a nationwide molecular epidemiologic survey. PLoS One 2012; 7:e47289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Li W, Zhong P, Fang K, Zhu K, Musa TH, et al. Nationwide trends in molecular epidemiology of HIV-1 in China. AIDS Res Hum Retroviruses 2016; 32:851–859. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Feng Y, Yang Y, Chen Y, Guo Q, Sun L, et al. Near full-length genome sequence of a novel HIV-1 recombinant form (CRF01_AE/B) detected among men who have sex with men in Jilin Province, China. AIDS Res Hum Retroviruses 2014; 30:701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Zang X, Ning C, Feng Y, Xie C, He X, et al. Molecular epidemiology of HIV-1 in Jilin province, northeastern China: emergence of a new CRF07_BC transmission cluster and intersubtype recombinants. PLoS One 2014; 9:e110738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li QH, Wang FX, Yue C, Wang JY, Jin G, Zhang CL, et al. Molecular genotyping of HIV-1 strains from newly infected men who have sex with men in Harbin, China. AIDS Res Hum Retroviruses 2016; 32:595–600. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Ma Y, Su Y, Yang L, Zhang R, Yang C, et al. HIV-1 genetic characteristics and transmitted drug resistance among men who have sex with men in Kunming, China. PLoS One 2014; 9:e87033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C, Wang Y, Kong D, Xin R, Xu W, Feng Y, et al. Characterization of a novel HIV-1 second-generation recombinant form in men who have sex with men in Beijing, China. AIDS Res Hum Retroviruses 2017; 33:1175–1179. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, He X, Hsi JH, Li F, Li X, Wang Q, et al. The rapidly expanding CRF01_AE epidemic in China is driven by multiple lineages of HIV-1 viruses introduced in the 1990s. AIDS 2013; 27:1793–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui H, Geng W, Sun H, Han X, An M, Jiang Y, et al. Rapid CD4+ T-cell decline is associated with coreceptor switch among MSM primarily infected with HIV-1 CRF01_AE in Northeast China. AIDS 2019; 33:13–22. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Xue Y, Zhou L, Lin Y, Yu X, Wang X, et al. Evidence that HIV-1 CRF01_AE is associated with low CD4+T cell count and CXCR4 co-receptor usage in recently infected young men who have sex with men (MSM) in Shanghai, China. PLoS One 2014; 9:e89462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Xue Y, Cheng H, Lin Y, Zhou L, Ning Z, et al. HIV-1 genetic diversity and its impact on baseline CD4+T cells and viral loads among recently infected men who have sex with men in Shanghai, China. PLoS One 2015; 10:e0129559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiao Y, Song Y, Kou B, Wang R, Liu Z, Huang X, et al. Primary CXCR4 co-receptor use in acute HIV infection leads to rapid disease progression in the AE subtype. Viral Immunol 2012; 25:262–267. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Zhu K, Li W, Fang K, Musa TH, Song Y, et al. Coreceptor usage of Chinese HIV-1 and impact of X4/DM transmission clusters among recently infected men who have sex with men. Medicine (Baltimore) 2016; 95:e5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Han Y, Xie J, Gu L, Li W, Wang H, et al. CRF01_AE subtype is associated with X4 tropism and fast HIV progression in Chinese patients infected through sexual transmission. AIDS 2014; 28:521–530. [DOI] [PubMed] [Google Scholar]
- 16.Chu M, Zhang W, Zhang X, Jiang W, Huan X, Meng X, et al. HIV-1 CRF01_AE strain is associated with faster HIV/AIDS progression in Jiangsu Province, China. Sci Rep 2017; 7:1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiebig EW, Wright DJ, Rawal BD, Garrett PE, Schumacher RT, Peddada L, et al. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 2003; 17:1871–1879. [DOI] [PubMed] [Google Scholar]
- 18.Duong YT, Qiu M, De AK, Jackson K, Dobbs T, Kim AA, et al. Detection of recent HIV-1 infection using a new limiting-antigen avidity assay: potential for HIV-1 incidence estimates and avidity maturation studies. PLoS One 2012; 7:e33328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JY, Chen XH, Shao B, Huo QQ, Liu SY, Li J, et al. Identification of a new HIV-1 circulating recombinant form CRF65_cpx strain in Jilin, China. AIDS Res Hum Retroviruses 2018; 34:709–713. [DOI] [PubMed] [Google Scholar]
- 20.Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R. Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol 2007; 25:1407–1410. [DOI] [PubMed] [Google Scholar]
- 21.Soulie C, Morand-Joubert L, Cottalorda J, Charpentier C, Bellecave P, Le Guen L, et al. Performance of genotypic algorithms for predicting tropism for HIV-1 CRF01_AE recombinant. J Clin Virol 2018; 99–100:57–60. [DOI] [PubMed] [Google Scholar]
- 22.Yan M, Zhao K, Du J, Li L, Wu D, Xu S, et al. HIV-1 diversity and drug-resistant mutations in infected individuals in Changchun, China. PLoS One 2014; 9:e100540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng OT, Lin L, Laeyendecker O, Quinn TC, Sun YJ, Lee CC, et al. Increased rate of CD4+ T-cell decline and faster time to antiretroviral therapy in HIV-1 subtype CRF01_AE infected seroconverters in Singapore. PLoS One 2011; 6:e15738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Lodi S, Fox Z, Li W, Phillips A, Porter K, et al. Rate of CD4 decline and HIV-RNA change following HIV seroconversion in men who have sex with men: a comparison between the Beijing PRIMO and CASCADE cohorts. J Acquir Immune Defic Syndr 2013; 62:441–446. [DOI] [PubMed] [Google Scholar]
- 25.Leelawiwat W, Pattanasin S, Sriporn A, Wasinrapee P, Kongpechsatit O, Mueanpai F, et al. Association between HIV genotype, viral load and disease progression in a cohort of Thai men who have sex with men with estimated dates of HIV infection. PLoS One 2018; 13:e0201386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi JY, Pond SLK, Anderson CM, Richman DD, Smith DM. Molecular features of the V1–V4 coding region of sexually transmitted human immunodeficiency virus type 1. J Infect Dis 2017; 215:1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 2004; 303:2019–2022. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Wan YM, Chen J, Wang J, Ren YQ, Wei Q, et al. Characterization of N-linked glycosylation sites on envelope proteins of simian/human immunodeficiency virus in peripheral blood of Chinese rhesus macaques during acute infection. Zhonghua Yu Fang Yi Xue Za Zhi 2016; 50:869–873. [DOI] [PubMed] [Google Scholar]
- 29.Curlin ME, Zioni R, Hawes SE, Liu Y, Deng W, Gottlieb GS, et al. HIV-1 envelope subregion length variation during disease progression. PLoS Pathog 2010; 6:e1001228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, Xing H, He X, Xin R, Zhang Y, Zhu J, et al. Analysis of putative N-linked glycosylation sites and variable region of envelope HIV-1 CRF07_BC recombinant in intravenous drug users in Xinjiang Autonomous Region, China. AIDS Res Hum Retroviruses 2008; 24:521–527. [DOI] [PubMed] [Google Scholar]
- 31.Bunnik EM, Euler Z, Welkers MR, Boeser-Nunnink BD, Grijsen ML, Prins JM, et al. Adaptation of HIV-1 envelope gp120 to humoral immunity at a population level. Nat Med 2010; 16:995–997. [DOI] [PubMed] [Google Scholar]
- 32.Poveda E, Paredes R, Moreno S, Alcami J, Cordoba J, Delgado R, et al. Update on clinical and methodological recommendations for genotypic determination of HIV tropism to guide the usage of CCR5 antagonists. AIDS Rev 2012; 14:208–217. [PubMed] [Google Scholar]
- 33.Mulinge M, Lemaire M, Servais JY, Rybicki A, Struck D, da Silva ES, et al. HIV-1 tropism determination using a phenotypic Env recombinant viral assay highlights overestimation of CXCR4-usage by genotypic prediction algorithms for CRF01_AE and CRF02_AG (corrected). PLoS One 2013; 8:e60566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raymond S, Delobel P, Rogez S, Encinas S, Bruel P, Pasquier C, et al. Genotypic prediction of HIV-1 CRF01-AE tropism. J Clin Microbiol 2013; 51:564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuchiya K, Ode H, Hayashida T, Kakizawa J, Sato H, Oka S, et al. Arginine insertion and loss of N-linked glycosylation site in HIV-1 envelope V3 region confer CXCR4-tropism. Sci Rep 2013; 3:2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sander O, Sing T, Sommer I, Low AJ, Cheung PK, Harrigan PR, et al. Structural descriptors of gp120 V3 loop for the prediction of HIV-1 coreceptor usage. PLoS Comput Biol 2007; 3:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shoombuatong W, Hongjaisee S, Barin F, Chaijaruwanich J, Samleerat T. HIV-1 CRF01_AE coreceptor usage prediction using kernel methods based logistic model trees. Comput Biol Med 2012; 42:885–889. [DOI] [PubMed] [Google Scholar]
- 38.Hongjaisee S, Braibant M, Barin F, Ngo-Giang-Huong N, Sirirungsi W, Samleerat T. Effect of amino acid substitutions within the V3 region of HIV-1 CRF01_AE on interaction with CCR5-coreceptor. AIDS Res Hum Retroviruses 2017; 33:946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coetzer M, Nedellec R, Salkowitz J, McLaughlin S, Liu Y, Heath L, et al. Evolution of CCR5 use before and during coreceptor switching. J Virol 2008; 82:11758–11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svicher V, Alteri C, Artese A, Zhang JM, Costa G, Mercurio F, et al. Identification and structural characterization of novel genetic elements in the HIV-1 V3 loop regulating coreceptor usage. Antivir Ther 2011; 16:1035–1045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.