

Abstract
Methylmercury (MeHg) is a toxic chemical compound naturally produced mainly in the aquatic environment through the methylation of inorganic mercury catalyzed by aquatic microorganisms. MeHg is biomagnified in the aquatic food chain and, consequently, piscivorous fish at the top of the food chain possess huge amounts of MeHg (at the ppm level). Some populations that have fish as main protein’s source can be exposed to exceedingly high levels of MeHg and develop signs of toxicity. MeHg is toxic to several organs, but the central nervous system (CNS) represents a preferential target, especially during development (prenatal and early postnatal periods). Though the biochemical events involved in MeHg-(neuro)toxicity are not yet entirely comprehended, a vast literature indicates that its pro-oxidative properties explain, at least partially, several of its neurotoxic effects. As result of its electrophilicity, MeHg interacts with (and oxidize) nucleophilic groups, such as thiols and selenols, present in proteins or low-molecular weight molecules. It is noteworthy that such interactions modify the redox state of these groups and, therefore, lead to oxidative stress and impaired function of several molecules, culminating in neurotoxicity. Among these molecules, glutathione (GSH; a major thiol antioxidant) and thiol- or selenol-containing enzymes belonging to the GSH antioxidant system represent key molecular targets involved in MeHg-neurotoxicity. In this review, we firstly present a general overview concerning the neurotoxicity of MeHg. Then, we present fundamental aspects of the GSH-antioxidant system, as well as the effects of MeHg on this system.
Keywords: Methylmercury, glutathione, antioxidant system, neurotoxicity, thiols, selenols
1. Introduction - Fundamental aspects concerning MeHg chemistry and toxicity
Mercury is a chemical element with symbol Hg, atomic number 80 and weight 200.59. Hg is not essential to living cells; conversely, it is highly toxic to animals (including humans) and plants [1]. Hg is present in the environment mainly in three chemical forms: elemental Hg vapor, inorganic Hg salts, and organic Hg compounds, such as methylmercury (MeHg) [2]. MeHg is naturally produced mainly in the aquatic environment by the methylation of inorganic mercury in a process that takes place in aquatic microorganisms [3]. In the aquatic milieu, MeHg is bioaccumulated through the aquatic food chain, reaching concentrations over 1 ppm in certain fish species [4]. Seafood ingestion is the most form of human exposure to MeHg. Accordingly, populations that have fish as main protein’s source are exposed to toxic MeHg levels [1].
Once in the body, MeHg can be toxic to several organs/systems, such as the kidney [5,6], the cardiovascular system [7,8], the immune system [9] and the reproductive system [10,11]. However, the central nervous system (CNS) represents a main target for MeHg-toxicity. In this regard, a vast epidemiological literature has reported impressive neurotoxic symptoms in humans highly exposed to MeHg, which include but are not limited to somatosensory disturbances and psychiatric symptomatology (for a review, see reference [12]). A critical event related to MeHg-neurotoxicity is its transplacental transport, allowing for the targeting of the fetus brain [13]. In line with this, epidemiological evidence shows that the exposure of pregnant women to MeHg may cause neurological deficits in their children [14,15]. Of note, developmental MeHg exposure at levels that commonly do not affect the mature (mother’s] brain is responsible for brain injury in the developing one [16].
Concerning molecular mechanisms related to MeHg-induced toxicity, it is important to mention that MeHg (CH3Hg+) has a reactive center with one positive charge in the Hg atom. Therefore, it has a relative high affinity for electrons [17], which is the cause of its relative high reactivity with nucleophilic centers, i.e. selenohydryl (-SeH or selenol) or sulfhydryl (-SH or thiol] groups. Thiols can be found in either low molecular mass molecules (i.e. glutathione (GSH]] or in proteins [18]. In eukaryotic cells, selenols are primarily present in a small number of proteins (selenoproteins] as selenocysteine [19]. In the human genome, 25 genes for selenoproteins have been identified and some of them are oxidoreductases that catalyze reactions of the detoxification of oxidants, such as glutathione peroxidase and thioredoxin reductase [20].
MeHg-induced neurotoxicity is notably linked to its interaction with either thiols or selenols groups. Such interaction changes the structure (and sometimes the function] of several proteins, as well as may change the redox state of low-molecular antioxidants, contributing to MeHg-induced oxidative stress [21]. Mechanisms other than oxidative stress are also involved in MeHg-induced neurotoxicity. In fact, changes in intracellular calcium homeostasis [22] and in proper functioning of the glutamatergic neurotransmitter system [23] also represent central events in MeHg-induced neurotoxicity. In this regard, there seems to be a relationship between the pro-oxidant properties of MeHg with its effects on calcium and glutamatergic system. In fact, it has been reported that increased levels of hydroperoxide, which can represent a consequence of MeHg-induced inhibition of glutathione peroxidase (discussed latter, [24]), are able to decrease astrocyte glutamate uptake [25], which may culminate an excitotoxicity and calcium-mediated neuronal damage [23, 26]. Accordingly, the oxidation of sulfhydryl-containing proteins may affect chemical neurotransmition through modulation of neurotransmitters release [27], as well as their binding to specific receptors [28]. In this regard, there is a significant number of nucleophilic molecules that have been reported to be targets mediating MeHg-induced toxicity (i.e., thioredoxin reductase [29,30], selenoprotein W [31], glucose-6-phosphate dehydrogenase [32,33], and 5’-deiodinase [13]). The direct interaction of the MeHg’s mercury atom with thiol or selenol groups of these target proteins has been proposed as a mechanism of toxicity [34, 35, 36]. Although any thiol or selenol-containing molecule could represent a potential target for MeHg, it is important to mention that selenols are more nucleophilic than thiols [37], consistent with the fact that selenoproteins represent primary/preferential targets compared to sulfhydryl proteins [24]. It is expected that MeHg will react preferentially with the GPx’s selenocysteine residue than with the GSH’s cysteine residue when all components (MeHg, GPx and GSH) are at equimolar concentrations. However, the intracellular levels of GSH are significantly higher (at the mM range) than those of GPx in the biological systems, which will also dictate the hierarchy of MeHg’s interaction toward components of the GSH antioxidant system. The hierarchical profile of MeHg interaction with the selenol groups of different selenoproteins will depend on their pKa values; anyway, comparative studies concerning the MeHg’s affinity for different selenoproteins (i.e. glutathione peroxidase and thioredoxin reductase) have yet to be addressed. Even though MeHg can target diverse nucleophilic molecules, this review will focus on the glutathione (GSH) antioxidant system. The choice of delving into this specific system is not only related to the fact that several toxic effects resulting from MeHg exposure are due to its effects toward the GSH and related enzymes (discussed later), but also because the GSH antioxidant system significantly affects MeHg kinetics and toxicity.
2. The glutathione antioxidant system
The term glutathione antioxidant system, used in most of the literature and in this review article, depicts the low-molecular weight glutathione (GSH) and the main enzymes involved in its metabolism. The tripeptide GSH (γ-l-glutamyl-l-cysteinylglycine) is the most important low-molecular sulfhydryl antioxidant, which is present intracellularly in millimolar concentrations. Its most important functions, which are particularly dependent on the thiol group of its cysteinyl residue, are related to the detoxification of either xenobiotics or endogenous oxidants [38]. Taking into account the great number of potentially deleterious molecules that can be detoxified in a GSH-dependent manner [39], it is predictable that impaired GSH homeostasis has been associated with human pathological conditions [40], including those related to the CNS [41].
Accordingly, prior to considerations on the role of this important antioxidant system in the detoxification and/or metabolism of pro-oxidant molecules, it is important to mention that the proper maintenance of a physiological level of oxidant challenge, currently termed oxidative eustress, is essential for governing life processes through redox signaling [42, 43]. Thus, oxidants such as hydrogen peroxide, whose levels may be increased after MeHg exposure (discussed latter), will play significant deleterious effects in oxidizable biomolecules when the physiological oxidant challenge (eustress) is surpassed, which may culminate in oxidative damage (distress). Conversely, at physiological levels, hydrogen peroxide displays essential roles in regulating cell signaling and metabolism through the oxidation of proteic nucleophilic groups, which operate as redox switches [44]. In this regard, the GSH antioxidant system may play an important role in redox signaling either via glutathionylation (direct modification of protein cysteine residues by the addition of GSH) or via metabolism of hydrogen peroxide, detailed in item 2.3.
2.1. GSH synthesis
GSH is synthesized in two ATP-consuming reactions that take place in the cytosol (Figure 1A, events a and b). Initially, the enzyme glutamate cysteine ligase (GCL) catalyzes the condensation of cysteine and glutamate to form the dipeptide γ-glutamylcysteine (γGluCys). In another reaction, glycine is added to form the tripeptide in a reaction catalyzed by GSH synthetase. The first reaction, catalyzed by GCL, represents a regulated step that controls GSH synthesis. Chemical inhibition of GCL with the buthionine sulfoximine (BSO) has been reported to significantly decrease GSH levels in both in vitro [45] and in vivo [46] conditions. Of note, both the GCL and GSH synthase genes are regulated by the nuclear transcription factor erythroid-2-related factor 2 (Nrf2) [47], which translocates into the nucleous during oxidative stress, mainly via oxidation of an associated protein (the Kelch-like ECH-associated protein 1; KEAP1) [48]. Nuclear translocation of Nrf2 stimulates the transcription of Nfr2-related genes, such as phase 2 detoxifying and antioxidant enzymes (i.e., GCL). Not only oxidative stress, but also additional events (i.e. PKC and AMPK-dependent phosphorylation) can stimulate Nrf2 translocation to the nucleus [49,50], increasing antioxidant and xenobiotic-metabolizing capacity.
Figure 1: General overview of glutathione synthesis and most important intracellular roles.
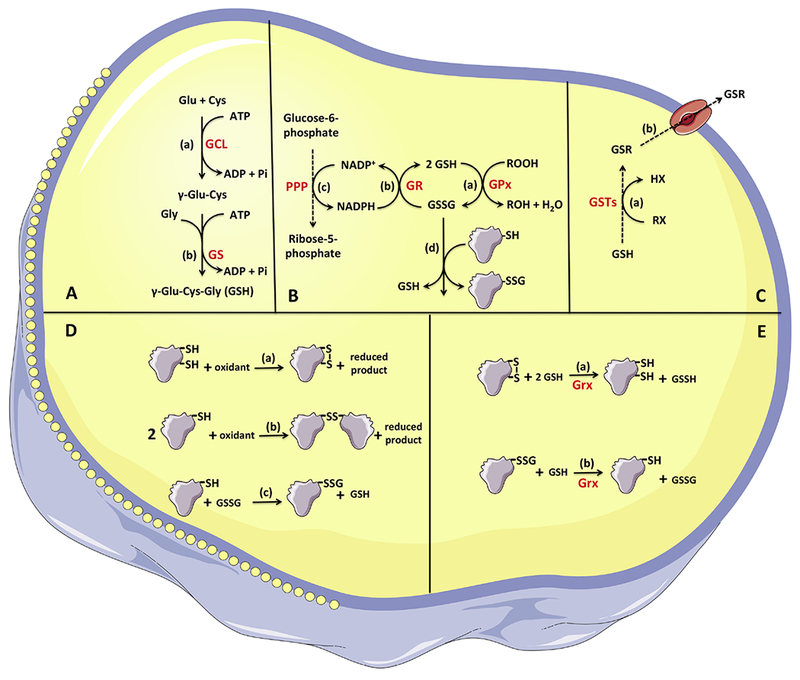
A) Glutathione synthesis - The tripeptide glutathione is synthesized in two ATP-consuming reactions. Event (a) represents the condensation of cysteine and glutamate to form the dipeptide γ-glutamylcysteine in a reaction catalyzed by glutamate cysteine ligase. Event (b) represents the addition of glycine to form the tripeptide in a reaction catalyzed by GSH synthetase. B) GSH-GSSG redox cycling - Glutathione peroxidases catalyze the reduction of H2O2 and organic peroxides at the expenses of electrons from GSH (event a). The oxidized glutathione generated by the GPx-catalyzed reaction can be reduced by glutathione reductase (event b), which uses NADPH generated mainly by the oxidative phase of the pentose phosphate pathway (event c). C) GSH complexation with xenobiotics - The conjugation of glutathione with diverse xenobiotics is catalyzed by glutathione-S-transferases (event a), forming a xenobiotic-GSH conjugate that can be exported from the intra- to the extracellular environment via multidrug resistance-associated proteins (event b). D) Thiol oxidation - The formation of protein disulfides can occur via either the oxidation of two protein thiols (from a single or different proteins, events a and b, respectively) or when a single sulfhydryl protein forms a disulfide with GSH via glutathionylation, forming a mixed disulfide (event c). E) Glutaredoxins - Glutaredoxins use electrons from glutathione to reduce oxidized target proteins, restoring the reduced state in cysteine(s) from proteins (event a) or from a mixed disulfide (event b).
GSH (reduced glutathione = γ-l-glutamyl-l-cysteinylglycine); GSSG (oxidized glutathione); Glu (glutamate); Cys (cysteine); γGluCys (dipeptide γ-glutamylcysteine); GCL (glutamate cysteine ligase); GS (GSH synthetase); GPx (glutathione peroxidase); GR (glutathione reductase); GSTs (glutethione-S-transferases); RX (xenobiotic); ROOH (peroxide = hydrogen or organic peroxide); Grx (glutaredoxin).
With respect to the regulation of GSH synthesis, is important to mention that the maintenance of proper intracellular GSH concentrations is achieved through tightly regulated events, which occur at both transcriptional and post-transcriptional (either mRNA stabilization/destabilization or post-translational modification) levels. The major determinants of GSH synthesis are the availability of precursor (cysteine is a main limiting amino acid), as well as the activity/levels of GCL and GS [47]. The expression of GSH synthetic enzymes is increased during oxidative stress and the main transcription factors involved in this process include Nrf2/Nrf1 via the antioxidant response element (ARE), activator protein-1 (AP-1) and nuclear factor κ B (NFκB) [47]. Nrf2 seems to play a pivotal role in regulating GSH levels.
Nrf2 is a transcription factor of the cap ‘n’ collar-basic leucine zipper proteins (CNC-bZIP) [51–53]. It is a key regulator of the antioxidant cell response, modulating the expression of more than a thousand genes involved in the antioxidant defenses, metabolism, cell proliferation, and immune and detoxifying responses, including GCL and GS. When activated, Nrf2 translocates to the nucleus and forms heterodimers with other transcripts (i.e. c-Jun and small Maf proteins), binding to the antioxidant response element (ARE), favoring the transcription of specific genes [54]. Keap1 (Kelch-like ECH-associated protein 1) is the most important regulator of Nrf2 activity. Under basal conditions, Keap-1 interacts with Nrf2, stimulating its ubiquitination and consequent proteolysis. A well-known mechanism associated with the disruption of the Keap-1/Nrf2 association is the oxidation of specific thiol groups from Keap-1 (Cysl51, Cys273, Cys288 and Cys297], which leads to Nrf2 translocation to the nucleus, stimulating the expression of several genes [55]. Additional mechanisms modulating Nrf2 nuclear translocation and/or degradation involve protein phosphorylation. GSK-3β, by stimulating Nrf2 phosphorylation, creates a degradation domain that, once recognized by ubiquitin ligase, triggers Nrf2 proteasomal degradation [56]. Additionally, GSK-3β phosphorylates Fyn (a member of the src-kinase family], which can phosphorylate Nrf2, leading to nuclear export ubiquitination, and degradation of Nrf2 [57]. In this scenario, PI3K/Akt activation inhibits (by phosphorylation] GSK-3p, thus preventing Nrf2 degradation. Additionally, PI3K/Akt induces extracellular-signal related kinase (Erk1/2] activation, which favors Nrf2 translocation to the nucleus [58].
Because Nrf2 activation results in the upregulation of several antioxidant enzymes and cy to protective genes, several lines of evidence have pointed it as an attractive therapeutic target for oxidative stress-related pathological conditions [59,60], including neurodegenerative diseases [61]. As result of the stimulation of Nrf2 translocation after the oxidation of critical cysteine residues, several electrophiles have been reported to display protective effects in a Nrf2-dependent manner (for review, see [61]]. In addition to GCL and GS (involved in GSH synthesis], further proteins linked to the GSH system are also upregulated via Nrf2, such as GR [62] and some members of the GST [63] and GPx [64] families. Of note, the neurotoxicant MeHg has been reported to stimulate Nrf2 activation and, in turn, Nrf2 has been reported to protect against MeHg toxicity (discussed latter in Section 3.6).
2.2. Glutathione S-transferase: GSH complexation with xenobiotics
Glutathione S-transferase (GST), which are present in bacteria, plants, and animals, is also part of the GSH system and represents a large family of proteins that act in detoxication pathways by catalyzing the conjugation of GSH with diverse xenobiotics (Figure 1C, event a), usually resulting in more efficient transport and elimination [65]. They also participate in a number of anabolic and catabolic pathways and play an important role in defense against oxidative stress by reducing peroxides and dehydroascorbates [66]. Human GSTs are divided into eight classes (Alpha, Mu, Pi, Theta, Kappa, Zeta, Omega, and Sigma), which are based on protein structure similarity, as well as kinetic properties [67, 68].
From a biochemical point of view, it is noteworthy that the basis for all GST catalytic activities relies on their capability of lowering the pKa of the thiol group of GSH from 9.0 (in aqueous solution) to approximately 6.5, when GSH is bound in the enzyme’s catalytic site [69]. With the reduction of the thiol’s pKa, GSH will predominantly be as the thiolate (GS-) anion and, consequently, will be more reactive to electrophilic xenobiotics. The binding of these compounds to the hydrophobic motif of GSTs also favors the occurrence of the reaction [70]. The conjugation of GSH with the diverse xenobiotics leads to the formation of a conjugate/complex (xenobiotic-GSH) that is generally more hydrophilic than the xenobiotic alone, allowing for export and/or excretion. The export of the xenobiotic-GSH conjugate from the intra- to the extracellular environment generally occurs via a transport protein from the multidrug resistance-associated proteins family [71], (Figure 1C, event b). GSTs are involved in the detoxification of several xenobiotics (i.e., polychlorinated biphenyls, triazines, thiocarbamates, sulfonylureas, chloracetanilides, just to name a few), but also catalyze the conjugation of GSH with endogenous compounds (i.e., 4-hydroxinonenal, formed as a product of lipid peroxidation) [72]. Of particular importance, some lines of evidence suggest that GSTs can moderate MeHg transport and toxicity [73], suggesting its participation in the catalysis of the complexation of GSH with MeHg; this is discussed in item 3. Even though GSH conjugation has been identified as an important detoxication process, several GST-dependent bioactivation reactions have been identified [74].
2.3. Glutathione peroxidase and glutathione reductase: GSH-GSSG redox cycling
As already mentioned, GSH contributes to the detoxification of endogenous oxidants. In this regard, GSH is able to directly (non-enzymatically) react with hydroxyl (−OH) and superoxide (O2−) radicals. Moreover, GSH donates electrons for the detoxification (reduction) of H2O2 and organic peroxides in a reaction catalyzed by GSH-dependent oxidoreductases - enzymes of the glutathione peroxidase (GPx) family. Glutathione peroxidases (GPxs) represent an important family of antioxidant enzymes that catalyze the reduction of H2O2 and organic peroxides at the expenses of electrons from GSH (Figure 1B, event a) [75]. In humans, 8 isoforms (GPx1 - GPx8) have been so far identified; among then, the cytosolic (GPx1), the gastrointestinal-specific (GPx2), the plasma (GPx3) and the phospholipid hydroperoxide (GPx4) contain selenocysteine in the catalytic site. GPx6, which is expressed in the olfactory epithelium, is a seleneprotein just in humans, but not in rodents and other species [76]. GPx5, GPx7 and GPx8 contain cysteine in the active site [75]. The presence of critical nucleophilic groups (selenols or thiols) at their active sites implies that all GPx isoforms can be target of electrophiles, such as MeHg (discussed latter).
In addition to alcohols or water, derived from the reduction of organic peroxides or H2O2, respectively, the usual GPx-catalyzed reaction also produces oxidized glutathione (GSSG), which is a disulfide formed from the oxidation of two GSH molecules. The GSSG generated by the GPx-catalyzed reaction can be reduced by the flavoenzyme glutathione reductase (GR) (Figure 1B, event b), which uses NADPH as reducing agent [77]. The reduction of GSSG to GSH (2 molecules) is needed to the maintenance of the proper GSH-GSSG redox cycling; the availability of NADPH, generated mainly by the oxidative phase of the pentose phosphate pathway (PPP) (Figure 1B, event c) represents a crucial event [78]. During oxidative stress, GSSG can oxidize critical protein thiols through the formation of mixed protein-GSH disulfides (Figure 1B, event d), affecting protein function. This process can be mitigated by the export of GSSG from the cell by multidrug resistance protein 1 [79], but it results in decreased intracellular GSH levels.
As already mentioned, the detoxification of peroxides and the maintenance of the proper GSH-GSSG redox cycling are crucial events to avoid extreme oxidative events toward lipids peroxidation, protein and nucleic acids. In this scenario, GPx and GR are two important enzymes of the GSH antioxidant system that, with the reducing support from GSH, help to maintain appropriate thiol redox state, preventing such oxidative events. Figure 1B depicts the main events related to the GSH-GSSG redox cycling.
2.4. Glutaredoxins
In sulfhydryl-containing proteins, their thiol groups (PSH] may display crucial roles in modulating their respective functions [80]. As consequence, pro-oxidative conditions that cause excessive PSH oxidation may affect protein functions and, consequently, lead to pathological conditions. Particularly in the CNS, oxidation of PSH may affect neurotransmitter release [27] and their binding to specific receptors [28], which importantly affects chemical neurotransmission.
Depending on the oxidative stimulus, PSH oxidation can lead to the formation of disulfide linkages (–S–S–), cysteinyl radical (P-S−], sulfenic (PSOH), sulfinic (PSO2H), sulfonic (PSO3H) acids, etc. [81]. Of note, PSH oxidation to disulfides is frequently reversible in cells under normal physiological conditions [82]; disulfides can be formed by either the oxidation of two PSHs (from a single or different proteins, Figure 1D, events a and b, respectively] or when a single PSH form a disulfide with GSH (or free cysteine] via glutathionylation (or cysteinylation], respectively, forming a mixed disulfide (Figure 1D, event c]. The reduction of disulfides (in proteins or mixed disulfides] is done at the expense of electrons initially derived from NADPH in events that may have glutaredoxins or thioredoxins as intermediary reducing agents [83]. Because thioredoxin and thioredoxin reductase are not part of the GSH system, these proteins will be not discussed in this review (for details about the role of thioredoxin and thioredoxin reductase in reducing oxidized sulfhydryl proteins, see Lu and Holmgren [84]).
Glutaredoxins (Grxs) represent a group of sulfhydryl-containing proteins that can be considered part of the GSH system. Depending on the number of active site cysteine residues, they are divided into dithiol and monothiol Grxs [85]. In dithiol Grxs, cysteine residues at the active centre may be in either a reduced (thiol) or an oxidized (disulfide) form. In the oxidized form (disulfide), these Grxs can be non-enzymatically reduced by GSH. Once reduced, Grxs act in a system in which electrons are transferred to oxidized target proteins, restoring the reduced state in cysteine(s) from proteins (Figure 1E, event a) or from a mixed disulfide (Figure 1E, event b). Consequently, dithiol Grxs display a crucial role in post-translational redox modification, which have been implied in the modulation of numerous physiological and disease-related conditions, such as immune defense, cardiac hypertrophy, hypoxia-reoxygenation insult, neurodegeneration and cancer [86]. Figure 1E depicts the main events related to the reduction of oxidized sulfhydryl proteins and mixed disulfides by dithiol Grxs. In contrast to dithiol Grxs, monothiol Grxs function mainly in iron homeostasis and in the biosynthesis of FeS proteins [87].
3. The complex interplay between MeHg and the GSH antioxidant system: protective and toxic consequences
3.1. MeHg and GSH interaction
As already mentioned, the interaction of MeHg with nucleophilic groups (i.e., thiols) has been reported as a key event mediating its toxicity. In this scenario, more than 4 decades have passed since the direct chemical interaction between MeHg and GSH was reported in the literature. Such interaction displays a crucial role in decreasing MeHg deposition in tissues [88] and increasing Hg excretion in the bile of MeHg-exposed rats [89], indicating a protective role of GSH against MeHg-toxicity. From the molecular point of view, it is noteworthy MeHg can interact with the thiol group of GSH forming a complex (MeHg-GSH) (Figure 2, event a), which can be exported from the cells (Figure 2, event b). In line with this, in vitro experiments with cultured cells have shown that (i) increased GSH levels are protective against MeHg-induced neurotoxicity [90,91] and (ii) GSH monoethyl ester (a GSH precursor) protected against MeHg-neurotoxicity in a manner dependent on MRP1-mediated efflux [92], highlighting the important role of this family of proteins (multidrug resistance-associated proteins) in exporting the MeHg-GSH complex from cells. Under in vivo conditions, treatment with N-acetylcysteine (NAC, a precursor of cysteine, essential for GSH synthesis) reduced MeHg levels in the developing embryo in a rodent model [93]. The authors observed that NAC increased the urinary excretion of MeHg, highlighting its potential role as antidote agent in MeHg poisonings. Consistent with this, Dutczak and Ballatori [94] reported that the MeHg-GSH complex represents a major form by which MeHg is transported across liver canalicular membranes into bile, a major route of excretion. According to these observations, GSH depletion has been reported as a consequence of MeHg exposure in rodent models [95,96]. Even though the formation of the MeHg-GSH complex tend to decrease MeHg toxicity, it is important to mention that MeHg can be exchanged between different thiols, allowing for the transference of MeHg from the MeHg-GSH complex to another thiol (i.e., sulfhydryl protein) [97], leading to PSH oxidation and potential changes in protein function (Figure 2, event c). Thus, even though MeHg is much more reactive toward thiols than MeHg-GSH, one may hypothesize that this complex could also contribute to thiol oxidation because, according to the studies of Rabenstein and collaborators [97], MeHg can be exchanged between different thiols. It is noteworthy that MeHg can also interact directly with free cysteine [98], forming a cysteine-MeHg complex, which can be taken up by cells via a neutral amino acid carrier transport System L [99]. Even though the formation of this cysteine-MeHg complex can theoretically decrease cysteine availability for GSH synthesis, to the best of our knowledge, there are no studies concerning the actual toxicological relevance of this phenomenon.
Figure 2: Glutathione complexation with methylmercury.
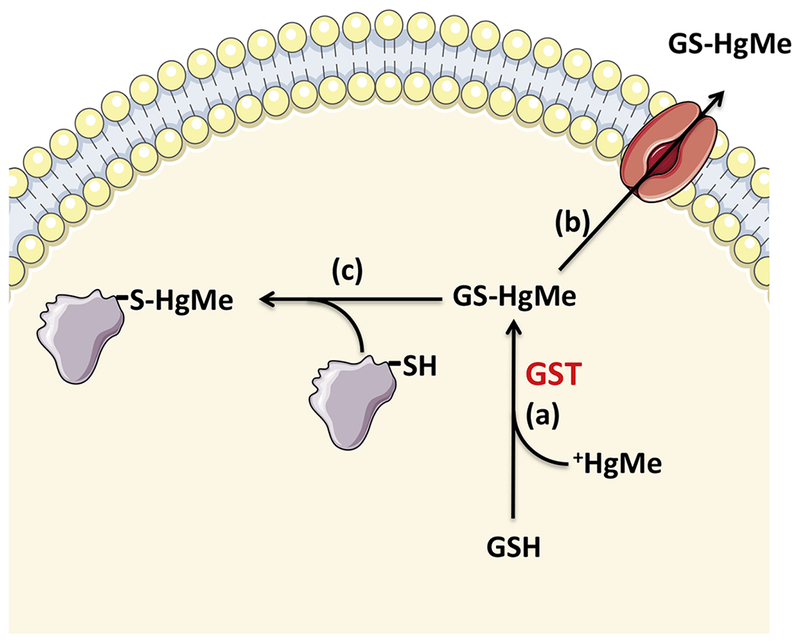
The conjugation of glutathione with methylmercury is catalyzed by glutathione-S-transferases (event a), forming a methylmercury-glutathione conjugate that can be exported from the intra- to the extracellular environment via multidrug resistance-associated proteins (event b). Event (c) represents the transference of methylmercury from the methylmercury-glutathione complex to another thiol (i.e. sulfhydryl protein), leading to protein thiol oxidation.
GSH (reduced glutathione = γ-l-glutamyl-l-cysteinylglycine); GSTs (glutathione-S-transferases); +HgMe (methylmercury); GS-HgMe (methylmercury-glutathione complex).
Based on the direct chemical interaction between MeHg and GSH, MeHg-induced GSH depletion is an expected event. Nevertheless, it is noteworthy that intracellular GSH levels are commonly in the millimolar (mM) range in several organs’tissues (liver, brain, kidney, etc.). Taking into account that GSH depletion has been observed in the brain of MeHg-exposed rodents whose brain mercury levels were in the low micromolar range [95,96], it is reasonable to suppose that MeHg-GSH interaction is not enough to completely explain MeHg-induced GSH oxidation.
3.2. MeHg decreases intracellular levels of GSH precursors
Peripheral cysteine delivered to the CNS is oxidized extracellularly to cystine [100], a disulfide formed by the oxidation of two cysteine molecules. Taking into account that cysteine is a limiting precursor for GSH synthesis, its intracellular levels are dependent on the availability of cystine [101]. With particular emphasis on the CNS, in vitro studies with cultured cells have shown that cultured astrocytes and hippocampal neurons can accomplish the uptake of both cysteine and cystine (for a review, see [102]). With respect to cystine, treatment of these cells (cultured astrocytes or hippocampal neurons) with MeHg causing decreased cystine uptake in astrocytes, but not in neurons [103]. With respect to cysteine, MeHg treatment also did not change cysteine uptake in neurons, although decreasing cysteine uptake in astrocytes [104]. Although MeHg-induced inhibition of cysteine and cystine uptake has not been observed in neurons, it is important to mention that MeHg could cause an indirect decrease of neuronal GSH synthesis as consequence of MeHg-induced changes in the astrocytic uptake of GSH; this is believed because astrocytes provide GSH precursors to neurons [105].
3.3. MeHg modulates enzymes involved in the GSH-GSSG redox cycle
A remarkable process related to the effects of MeHg on the GSH-GSSG redox cycle is its effect on GPx. Most of the GPx isoforms are selenoproteins, which present crucial selenocysteine residues (in the form of selenol group or selenolate: -SeH or -Se−) at their catalytic sites [75]. On the other hand, it is well known that mercurials, including MeHg, present a great affinity for selenols (greater than that for thiols) [106]. In fact, MeHg is a soft electrophile (or soft acid) that preferentially interacts with nucleophilic groups (mainly thiols and selenols) from proteins and low-molecular-weight molecules. On the other hand, these thiols and selenols (present in several molecules of the GSH antioxidant system: GSH, GPx, Grx) are soft nucleophilic groups (soft bases), which have high affinities for soft acids, such as MeHg [107]. Of note, selenium is a softer nucleophile compared with sulfur thiols and, therefore, selenols are more prone to be target by MeHg compared to thiols [108]. Accordingly, both in vitro and in vivo data indicate that the activities of different GPx isoforms, such as GPx1, GPx 3 and GPx 4, are decreased after MeHg exposure [24, 29,109–112]. With respect to mechanisms, in an in vitro study with cultured neurons, Farina and collaborators [24] reported that MeHg is a direct inhibitor of GPx1 likely due to the direct interaction of MeHg with selenocysteine, located at the enzymes active site. On the other hand, it seems that post-transcriptional and pre-translational phenomena related to mercury-selenium interaction are also linked to the decreased activity of GPx and other selenoproteins [113,114]. In this regard, it seems that mercurials can cause a depletion of selenium necessary for de novo synthesis of selenoproteins [115]. In fact, the biosynthesis of protein-incorporated selenocysteine occurs on its tRNA, and the pathway begins with the attachment of the amino acid serine to tRNA, which is further converted to selenocysteine by the addition of selenide (HSe−) [116]. Of note, MeHg is known to interact with HSe− [117,118], decreasing its availability to be incorporated into selenoproteins [113].
With respects to GR, in vivo data with rodents indicate that the effects of MeHg toward GR are dependent on different variables, such as tissue and developmental life stage. In a study where adult male C57/BL6 mice were orally administered 5 mg/kg/day MeHg for 28 days, Kirkpatrick and collaborators [119] observed that GR activity decreased in liver and increased in brain tissue of MeHg-treated animals. An increase in brain GR activity was also observed in three independent studies [110, 120, 121], where adult Swiss mice were orally exposed to MeHg during 2-3 weeks. In line with these studies, it has been proposed that the increase in GR may represent a compensatory response to preserve GSH homeostasis [121]. In sharp contrast to the increased GR activity observed in the brain of MeHg exposed adult mice, studies with developmental (in utero or early post natal) exposures to MeHg have shown decreased GR activity in the CNS of mice pups [96, 122].
Based on the effects of MeHg on the enzymes involved in GSH-GSSG redox cycle (especially GPx), one might expect increased levels of peroxide and lipid peroxidation after MeHg exposures. In line with this, increased peroxide levels [123,124] and enhanced lipid peroxidation [120,125] have been vastly reported in the literature as a consequence of MeHg exposures. From a mechanistic point of view, an in vitro study with cultured mouse cerebellar neurons showed that MeHg causes significant inhibition of GPx activity, leading to a decreased ability to deal with exogenously peroxides, increased lipid peroxidation, and cell death [24]. On the other hand, evidence shows that MeHg has the capacity to increase the generation of hydrogen peroxide (or its precursor superoxide anion) in mitochondria [123,124]. Based on these evidences, it seems that the increased levels of hydrogen peroxide observed in MeHg exposed animals [126] represent a consequence of both increased generation and decreased detoxification.
The effect of MeHg on the GSH-GSSG redox cycle seems to be also important in MeHg-induced glutamatergic dyshomeostasis (for a review, see [23]), which is a significant phenomenon related to its neurotoxicity. In an in vitro study with cultured astrocytes, Allen and collaborators [25] showed that MeHg is able to inhibit astrocytic glutamate transporters, leading to increased glutamate concentrations in the extracellular fluid, and that this event was prevented by adding catalase. These results indicate that hydrogen peroxide, generated after MeHg exposure, caused the observed decrease in glutamate uptake in astrocytes. These in vitro results [25] are in line with in vivo evidences showing MeHg exposure caused changes in glutamate uptake by rodent cerebral cortex slices [127,128] and increased the levels of extracellular glutamate in the cortices of rats [129]. The most important events related to the effects of MeHg in the enzymes involved in GSH-GSSG redox cycle (with focus in the CNS), as well as related consequences, are depicted in Figure 3.
Figure 3: Methylmercury inhibits glutathione peroxidase and leads to increased levels of hydrogen peroxide, glutamate dyshomeostasis and lipoperoxidation.
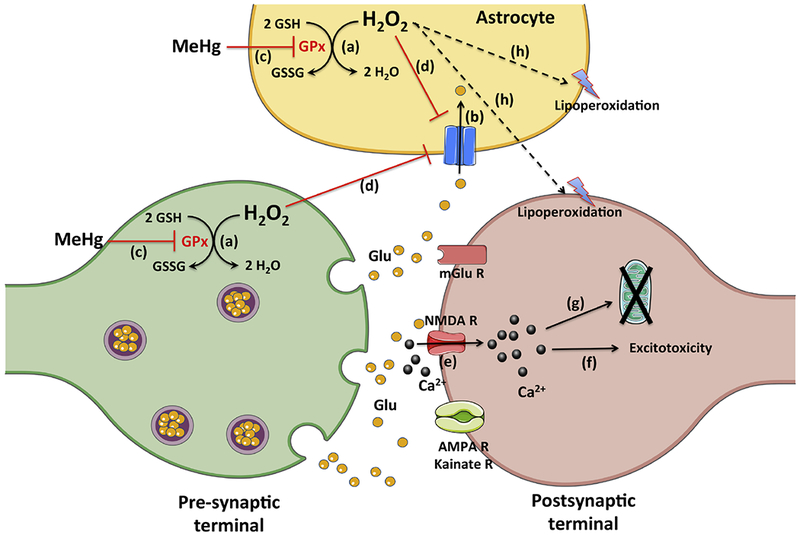
Event (a) represents the glutathione peroxidase-catalyzed reduction of hydrogen peroxide to water with the simultaneous oxidation of glutathione to its disulfide form. Event (b) represents the astrocyte glutamate uptake, which is crucial to remove glutamate from the synaptic cleft, preventing excitotoxicity. As consequence of methylmercury-induced inhibition of glutathione peroxidase (event c), increased levels of hydrogen peroxide may lead to decreased glial glutamate uptake (event d) and increased lipoperoxidation (event h). As result of decreased astrocyte glutamate uptake, the increased levels of extracellular glutamate may lead to hyperactivation of its receptors (i.e. N-methyl-D-aspartate receptor) and increase in calcium influx (event e), which may culminate in excitotoxicity (event f) and mitochondrial dyshomeostasis (event g).
GSH (reduced glutathione = γ-l-glutamyl-l-cysteinylglycine); GSSG (oxidized glutathione); Glu (glutamate); GPx (glutathione peroxidase); H2O2 (hydrogen peroxide); NMDA R (N-methyl-D-aspartate receptor); mGlu R (metabotropic glutamate receptors).
3.4. MeHg and GSTs
Studies concerning the effects of MeHg in GSTs are scarce [110]; however, there is a significant amount of data (including in humans) reporting that GSTs affect MeHg elimination by catalyzing MeHg-GSH conjugation (Figure 2, event a). In experiments with rats, the treatment with pharmacological inhibitors of GSTs (benziodarone or azathioprine) significantly decreased the biliary transport of MeHg [130]. In a search for MeHg tolerance genes in Drosophila, Mahapatra and collaborators [131] identified three GST genes (GSTD3, GSTE3, and GSTE7) with higher expression in strains of flies that were more resistant to MeHg. In another study with Drosophila, Vorojeikina et al. [73] investigated a possible functional role for GSTs in modulating MeHg toxicity during development. The authors observed that RNAi knockdown of endogenous GSTD1, GSTE1, or GSTS1 increased susceptibility to MeHg in developing pupa, leading to decreased rate of adult eclosion. On the other hand, the exogenous expression of human GSTP1 in developing flies resulted in increased MeHg resistance. In addition, the authors observed a trend whereby Hg body burden was inversely related to the levels of GST activity [73].
The potential association between polymorphic variants of GSTs and MeHg levels in humans has also been reported. Concerning the GSTs from classes Mu (GSTM) and Theta (GSTT), whose deletions seem to impair catalytic activity leading to greater sensitivity to toxic compounds [132, 133], Barcelos and collaborators [134] investigated the genotype frequency of the GSTM1 and GSTT1 polymorphisms and examine the influence of the GSTM1*0 and GSTT1*0 polymorphisms (homozygous deletions] on hair and blood levels of Hg in a group of riparian individuals exposed to MeHg in the Amazon region of Brazil. The authors observed that GSTT1 genotype carriers presented lower levels of blood and hair Hg when compared to other genotypes carriers. Moreover, GSTM1*0/GSTT1*0 individuals presented higher Hg levels in blood and hair than subjects presenting any other genotypes.
Concerning the GSTs from class Pi (GSTP], two common polymorphisms in the coding region of the human GSTP1, Ile105Val [GSTP1105] and Ala114Val [GSTP1114], lead to lower catalytic activity compared to the wild-type GSTP1wt. [135, 136] In studies with humans, some evidence has shown that carriers of the GSTP1114 allele are more prone to present increased Hg levels in blood or hair [133, 137]. On the other hand, other studies reported that GSTP1105 and GSTP1114 carriers showed association with lower Hg retention [138, 139]. Although there are some contradictory results in the literature (mainly related to human studies), findings from experimental and population studies indicate a potential role for GSTs in moderating Hg body burden in MeHg-exposed subjects. The most important role of GSTs in MeHg metabolism is the catalysis of MeHg and GSH conjugation (Figure 2).
3.5. MeHg and glutaredoxins:
Studies on the effects of MeHg on glutaredoxins are scarce. In an in vitro study with CCF-STTG1 human astrocytoma cells, Robitaille and coworkers [140] observed that exposure to a low MeHg concentration (1 μM), which did not significantly change GSH levels, increased GSSG levels by approximately 12-fold, leading to reduced GSH/GSSG ratio. Moreover, the authors observed that the enzyme glutaredoxin-1, which is involved in reversing protein S-glutathionylation, was inhibited by approximately 50% after MeHg exposure. Consistent with this effect, the authors also observed that MeHg treatment increased the S-glutathionylation of high molecular weight proteins. This study [140] seems to be the first to show that treatment with low MeHg concentrations directly interferes with glutaredoxin-1 activity, thus prolonging protein S-glutathionylation. In another in vitro study with cultured cells, Branco and coworkers [141] observed that MeHg exposure (1 μM) decreased the activity of Grx activity (< 20%; p<0.05) in neuroblastoma cells (SH-SY5Y), although no change in the expression of either Grx1 or Grx2 was observed. In this same study [141] , experiments with primary cerebellar neurons from mice depleted of mitochondrial Grx2 (mGrx2D) showed that such protein is important in protecting thioredoxin 2 from oxidation caused by another organic mercurial (ethylmercury). In this regard, a potential link between the effects of mercurials on the GSH system along with its effects on the thioredoxin system may have synergistic/additive toxicological relevance. To the best of our knowledge, there are no in vivo studies on the effects of MeHg on Grxs.
3.6. MeHg and Nrf2
The oxidation of critical cysteine residues from Keap-1 (Cys151, Cys273, Cys288 and Cys297) (see section 2.1) has been reported as an important event that activates the disruption of the Keap-1/Nrf2 association, favoring Nrf2 nuclear translocation [55]. In line with the electrophilic properties of MeHg, in vitro studies have reported the direct interaction of MeHg with the cysteine residues in Keap-1 structure [48, 142]. Accordingly, in vitro [143, 144] and in vivo [145] studies have shown that MeHg increases in the Nrf2 nuclear translocation. In accordance, MeHg has been reported to increase the expression of Nrf2-related genes, such as ho-1, cglc, nqo1 and xCT [143, 145]. In general, the aforementioned studies indicate that MeHg is able to interact with (and oxidize) critical cysteine residues of Keap-1, stimulating Nrf2 nuclear translocation and, consequently, favoring the expression of Nrf2-related genes. In addition to the oxidation of critical cysteine residues of Keap-1, phosphorylation events have also been reported as modulators of MeHg-induced Nrf2 nuclear translocation. In this regard, MeHg seems to stimulate Nrf2 activity by downregulating the Src family kinase Fyn [146] and by modulating phosphatidylinositol 3 (PI3) kinase [144]. For mechanistic details concerning phosphorylation events and signaling pathways modulating MeHg-induced Nrf2 nuclear translocation, see Antunes dos Santos et al. [147] and Unoki et al. [148].
The relationship between MeHg and Nrf2 is not only related to the stimulatory effects of MeHg toward Nrf2 nuclear translocation, but also to the protective effects of Nrf2 activation against MeHg toxicity. In this context, several compounds that are known to stimulate Nrf2 nuclear translocation, increasing the expression of Nrf2-related genes (i.e., sulforaphane [145], isothiocyanates 6-methylsulfinylhexyl isothiocyanate [149], hydroxytyrosol [150]) have been reported to ameliorate some toxic effects induced by MeHg. In this scenario, it seems that the activation of Nrf2 induced by MeHg is linked to increased protection against this compound [48].
4. Concluding remarks and perspectives
As previously discussed, MeHg is an electrophilic compound that interacts with thiol and selenol groups of several biomolecules. Even though MeHg can form a relatively stable chemical interaction with thiols and selenols, it can be exchanged between different nucleophilic groups [97], allowing for its transfer between different molecules. In this regard, there seem to be preferential targets for MeHg, which present nucleophilic groups with high affinity for its electrophilic core (-Hg+). Because GSH and some GSH-related enzymes (i.e. GPx, Grxs) represent some of these targets, MeHg affects multiple branches of GSH homeostasis, which has been extensively reported to have toxicological relevance [21].
Because of the low-dose and chronic profile of most cases of human exposure to MeHg, clear signs of toxicity are commonly detected only when important neurotoxic damage is achieved, at which point it is commonly irreversible. In this regard, it is important to identify and validate biomarkers that are predictive of early adverse effects prior to adverse health outcomes becoming irreversible [151]. Based on the relative high affinity of MeHg for GSH, one might propose this tripeptide as potential biomarker of MeHg exposures. However, many other compounds (i.e., paraquat, arsenic, benzo(a)pyrene, and acetaminophen) may also decrease GSH levels [151]. Consequently, in a multi-contaminant or therapeutic context GSH does not permit distinction of MeHg-related toxicity from that produced by other compounds. With respect to the use of GPx as a potential biomarker for MeHg exposure/poisoning, Usuki and Fujimura [111] have pointed out the plasma selenium-dependent glutathione peroxidase (GPx3) as a potential biomarker of ongoing MeHg cytotoxicity. Nevertheless, it is important to note that the activity of most of the GPx isoforms depends on the selenium status, which may be significantly affected by nutritional and regional characteristics. The measurement of MeHg-targeted GPxs (i.e., plasma GPx3) could represent a perspective related to the topic of “potential biomarkers” for MeHg exposure. However, studies are necessary to elucidate temporal aspects concerning the fate of MeHg after interaction with GPx3
Although the use of pharmacological strategies to recover patients from neuropathological conditions resulting from exposure to MeHg is impractical/ineffective, some lines of evidence point to the benefit of using antidote strategies to eliminate the toxicant from the body. In this context sulfhydryl-containing molecules display significant effects in increasing MeHg excretion from the body, at least in cases of acute exposure [152]. Even though GSH presents relative high affinity for MeHg, its use as antidote is not useful because GSH does not effectively enter the cells. On the other hand, N-acetylcysteine (a GSH precursor), as well as D-penicillamine and 2,3-dimercaptopropane sulfonate (DMPS), enhance MeHg excretion from the body [93,152]. Considering that MeHg has a higher affinity for selenols compared to thiols, the use of selenocompounds to remove MeHg from the body represents a perspective. In line with this, an in vivo study showed that diphenyl diselenide, an organic selenium compound, decreased Hg levels in several organs of MeHg-treated mice [153]. To the best of our knowledge, studies concerning the use of selenocompounds to increase MeHg excretion in humans have never been performed and represent a perspective.
An interesting aspect on the interaction of MeHg with GSH (or with GSH-related proteins, such as GPx) is that from a toxicological point of view, it represents a double-edged sword. In fact, on one hand the chemical interaction of MeHg with either GSH or GSH-related enzymes triggers toxicity [109, 121], while on the other hand such interaction is important in protecting against toxicity, favoring MeHg excretion [93]. In fact, the chemical interaction of MeHg with the GSH’s thiol group and/or with GPx’s selenol group decreases the cellular capacity to detoxify oxidants, favoring toxicity [24, 91]. On the other hand, MeHg excretion is significantly favored by its interaction with GSH [154]. This reciprocal relationship, which modulates toxic and protective pathways, highlights the intriguing interplay between MeHg and the GSH antioxidant system.
Highlights.
Methylmercury (MeHg) is toxic to several organs, but the central nervous system represents a preferential target especially during development (prenatal and early postnatal periods).
MeHg interacts with (and oxidize) nucleophilic groups present in proteins or low-molecular weight molecules and such interactions modify the redox state of these groups, leading to oxidative stress, impaired function and neurotoxicity.
Glutathione (GSH) and thiol- or selenol-containing enzymes belonging to the GSH antioxidant system represent key molecular targets involved in MeHg-neurotoxicity.
The chemical interaction of MeHg with the GSH’s thiol group and/or with GPx’s selenol group decreases the cellular capacity to detoxify oxidants, favoring toxicity.
MeHg excretion is significantly favored by its interaction with GSH
Acknowledgments:
The author would like to thank the colleagues/co-authors who have contributed to several studies referenced in this review. These studies were funded in part by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) and National Institute of Environmental Health Sciences, NIEHS R01ES07331, NIEHS R01ES10563 and NIEHS R01ES020852. The author would like to thank Dr. João Batista Teixeira da Rocha for the critical reading of this article and for significant suggestions that substantially improves the text. Figures were made using Servier Medical Art (https://smart.servier.com).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Clarkson TW, Magos L, & Myers GJ (2003). The Toxicology of Mercury-Current Exposures and Clinical Manifestations. New England Journal of Medicine. 10.1056/NEJMra022471 [DOI] [PubMed] [Google Scholar]
- [2].Clarkson TW (2002). The three modern faces of mercury. Environmental Health Perspectives. 10.1289/ehp.02110s111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Compeau GC, & Bartha R (1985). Sulfate-reducing bacteria: Principal methylators of mercury in anoxic estuarine sediment. Applied and Environmental Microbiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hintelmann H (2010). Organomercurials. Their formation and pathways in the environment. Met Ions Life Sci. 10.1039/BK9781847551771-00365. [DOI] [PubMed] [Google Scholar]
- [5].Jin X, Lok E, Caldwell D, Mueller R, Kapal K, Liston V, Kubow S, Chan HM, Mehta R. (2009). Dietary fats altered nephrotoxicity profile of methylmercury in rats. J Appl Toxicol. 10.1002/jat.1389. [DOI] [PubMed] [Google Scholar]
- [6].Shi JZ, Kang F, Wu Q, Lu YF, Liu J, Kang YJ. (2011) Nephrotoxicity of mercuric chloride, methylmercury and cinnabar-containing Zhu-Sha-An-Shen-Wan in rats. 10.1016/j.toxlet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- [7].Moreira EL, de Oliveira J, Dutra MF, Santos DB, Gonçalves CA, Goldfeder EM, de Bem AF, Prediger RD, Aschner M, Farina M. (2012) Does methylmercury-induced hypercholesterolemia play a causal role in its neurotoxicity and cardiovascular disease? Toxicol Sci. 10.1093/toxsci/kfs252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wells EM, Herbstman JB, Lin YH, Hibbeln JR, Halden RU, Witter FR, Goldman LR. (2017), Methyl mercury, but not inorganic mercury, associated with higher blood pressure during pregnancy. Environ Res. https://doi.org/0.1016/j.envres.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tonk EC, de Groot DM, Penninks AH, Waalkens-Berendsen ID, Wolterbeek AP, Slob W, Piersma AH, van Loveren H. (2010) Developmental immunotoxicity of methylmercury: the relative sensitivity of developmental and immune parameters.Toxicol Sci. 10.1093/toxsci/kfq223. [DOI] [PubMed] [Google Scholar]
- [10].Frenedoso da Silva R, Missassi G, dos Santos Borges C, Silva de Paula E, Hornos Carneiro MF, Grotto D, Barbosa Junior F, De Grava Kempinas W. (2014). Phytoremediation potential of Maná-Cubiu (Solanum sessiliflorum Dunal) for the deleterious effects of methylmercury on the reproductive system of rats. Biomed Res Int. 10.1155/2014/309631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen N, Lin M, Liu N, Wang S, Xiao X. (2018). Methylmercury-induced testis damage is associated with activation of oxidative stress and germ cell autophagy. J Inorg Biochem. 10.1016/j.jinorgbio.2018.10.007. [DOI] [PubMed] [Google Scholar]
- [12].Ekino S, Susa M, Ninomiya T, Imamura K, Kitamura T. (2007). Minamata disease revisited: an update on the acute and chronic manifestations of methyl mercury poisoning. J Neurol Sci. 2007; 262(1-2):131–44. [DOI] [PubMed] [Google Scholar]
- [13].Watanabe C, Yoshida K, Kasanuma Y, Kun Y, & Satoh H (1999). In utero methylmercury exposure differentially affects the activities of selenoenzymes in the fetal mouse brain. Environmental Research. 10.1006/enrs.1998.3889 [DOI] [PubMed] [Google Scholar]
- [14].Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, … Jørgensen PJ (1997). Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicology and Teratology. 10.1016/S0892-0362(97)00097-4 [DOI] [PubMed] [Google Scholar]
- [15].Murata K, Weihe P, Budtz-Jørgensen E, Jørgensen PJ, & Grandjean P (2004). Delayed brainstem auditory evoked potential latencies in 14-year-old children exposed to methylmercury. Journal of Pediatrics. 10.1016/j.jpeds.2003.10.059 [DOI] [PubMed] [Google Scholar]
- [16].Grandjean P, & Landrigan P (2006). Developmental neurotoxicity of industrial chemicals. Lancet. 10.1016/S0140-6736(06)69665-7 [DOI] [PubMed] [Google Scholar]
- [17].Pearson RG, & Songstad J (1967). Application of the Principle of Hard and Soft Acids and Bases to Organic Chemistry. Journal of the American Chemical Society. 10.1021/ja00984a014 [DOI] [Google Scholar]
- [18].Go YM, & Jones DP (2013). The redox proteome. Journal of Biological Chemistry. 10.1074/jbc.R113.464131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hatfield DL, Tsuji PA, Carlson BA, & Gladyshev VN (2014). Selenium and selenocysteine: Roles in cancer, health, and development. Trends in Biochemical Sciences. 10.1016/j.tibs.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lu J, & Holmgren A (2009). Selenoproteins. The Journal of Biological Chemistry. 10.1074/jbc.R800045200 [DOI] [PubMed] [Google Scholar]
- [21].Farina M, Aschner M, & Rocha JBT (2011). Oxidative stress in MeHg-induced neurotoxicity. Toxicology and Applied Pharmacology. 10.1016/j.taap.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Atchison WD. (2005). Is chemical neurotransmission altered specifically during methylmercury-induced cerebellar dysfunction? Trends Pharmacol Sci. 10.1016/j.tips.2005.09.008 [DOI] [PubMed] [Google Scholar]
- [23].Aschner M, Syversen T, Souza DO, Rocha JBT, & Farina M (2007). Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Brazilian Journal of Medical and Biological Research. 10.1590/S0100-879X2007000300001 [DOI] [PubMed] [Google Scholar]
- [24].Farina M, Campos F, Vendrell I, Berenguer J, Barzi M, Pons S, & Suñol C (2009). Probucol increases glutathione peroxidase-1 activity and displays long-lasting protection against methylmercury toxicity in cerebellar granule cells. Toxicological Sciences. 10.1093/toxsci/kfp219 [DOI] [PubMed] [Google Scholar]
- [25].Allen JW, Mutkus LA, Aschner M. (2001). Methylmercury-mediated inhibition of 3H-D-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res. 2001; 902(1): 92–100. [DOI] [PubMed] [Google Scholar]
- [26].Hidalgo C, Donoso P. (2008). Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal. 10.1089/ars.2007.1886. [DOI] [PubMed] [Google Scholar]
- [27].LoPachin RM, & Barber DS (2006). Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Toxicological Sciences. 10.1093/toxsci/kfl066 [DOI] [PubMed] [Google Scholar]
- [28].Soares FA, Farina M, Santos FW, Souza D, Rocha JBT, & Nogueira CW (2003). Interaction between Metals and Chelating Agents Affects Glutamate Binding on Brain Synaptic Membranes. Neurochemical Research. 10.1023/A:1026175825871 [DOI] [PubMed] [Google Scholar]
- [29].Branco V, Canário J, Lu J, Holmgren A, & Carvalho C (2012). Mercury and selenium interaction in vivo: Effects on thioredoxin reductase and glutathione peroxidase. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2011.12.002 [DOI] [PubMed] [Google Scholar]
- [30].Wagner C, Sudati JH, Nogueira CW, & Rocha JBT (2010). In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. BioMetals. 10.1007/s10534-010-9367-4 [DOI] [PubMed] [Google Scholar]
- [31].Kim YJ, Chai YG, & Ryu JC (2005). Selenoprotein W as molecular target of methylmercury in human neuronal cells is down-regulated by GSH depletion. Biochemical and Biophysical Research Communications. 10.1016/j.bbrc.2005.03.080 [DOI] [PubMed] [Google Scholar]
- [32].Wolf MB, & Baynes JW (2007). Cadmium and mercury cause an oxidative stress-induced endothelial dysfunction. BioMetals. 10.1007/s10534-006-9016-0 [DOI] [PubMed] [Google Scholar]
- [33].Kenow KP, Hoffman DJ, Hines RK, Meyer MW, Bickham JW, Matson CW, Elfessi A (2008). Effects of methylmercury exposure on glutathione metabolism, oxidative stress, and chromosomal damage in captive-reared common loon (Gavia immer) chicks. Environmental Pollution. 10.1016/j.envpol.2008.06.009 [DOI] [PubMed] [Google Scholar]
- [34].Mori K, Yoshida K, Tani J, Hoshikawa S, Ito S, Watanabe C. (2006). Methylmercury inhibits type II 5’-deiodinase activity in NB41A3 neuroblastoma cells. Toxicol Lett. 2006; 161(2): 96–101. [DOI] [PubMed] [Google Scholar]
- [35].Wagner C, Sudati JH, Nogueira CW, Rocha JB. (2010). In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. Biometals. 10.1007/s10534-010-9367-4. [DOI] [PubMed] [Google Scholar]
- [36].Rodrigues J, Branco V, Lu J, Holmgren A, Carvalho C. (2015). Toxicological effects of thiomersal and ethylmercury: Inhibition of the thioredoxin system and NADP(+)-dependent dehydrogenases of the pentose phosphate pathway. Toxicol Appl Pharmacol. 10.1016/j.taap.2015.05.002. [DOI] [PubMed] [Google Scholar]
- [37].Johansson L, Gafvelin G, Arnér ES. (2005). Selenocysteine in proteins-properties and biotechnological use. Biochim Biophys Acta. 10.1016/j.bbagen.2005.05.010 [DOI] [PubMed] [Google Scholar]
- [38].Deponte M (2013). Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochimica et Biophysica Acta - General Subjects. 10.1016/j.bbagen.2012.09.018 [DOI] [PubMed] [Google Scholar]
- [39].Dringen R, Brandmann M, Hohnholt MC, & Blumrich EM (2015). Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochemical Research. 10.1007/s11064-014-1481-1 [DOI] [PubMed] [Google Scholar]
- [40].Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, & Hammond CL (2009). Glutathione dysregulation and the etiology and progression of human diseases. Biological Chemistry. 10.1515/BC.2009.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Aoyama K, & Nakaki T (2013). Impaired glutathione synthesis in neurodegeneration. International Journal of Molecular Sciences. 10.3390/ijms141021021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sies H, Berndt C, Jones DP. (2017). Oxidative Stress. Annu Rev Biochem. 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- [43].Ren et al. , (2017). Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid Redox Signal. 27(13): 989–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sies H (2014). Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Devesa A, O’Connor J, Garciá C, Puertes IR, & Viña JR (1993). Glutathione metabolism in primary astrocyte cultures: flow cytometric evidence of heterogeneous distribution of GSH content. Brain Research. 10.1016/0006-8993(93)91264-S [DOI] [PubMed] [Google Scholar]
- [46].Griffith OW, & Meister A (1979). Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). Journal of Biological Chemistry. [PubMed] [Google Scholar]
- [47].Lu SC (2009). Regulation of glutathione synthesis. Molecular Aspects of Medicine. 10.1016/j.mam.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Toyama T, Sumi D, Shinkai Y, Yasutake A, Taguchi K, Tong KI, … Kumagai Y. (2007). Cytoprotective role of Nrf2/Keap1 system in methylmercury toxicity. Biochemical and Biophysical Research Communications. 10.1016/j.bbrc.2007.09.017 [DOI] [PubMed] [Google Scholar]
- [49].Huang HC, Nguyen T, & Pickett CB (2002). Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. Journal of Biological Chemistry. 10.1074/jbc.M206911200 [DOI] [PubMed] [Google Scholar]
- [50].Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, & Kim SG (2016). AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Molecular and Cellular Biology. 10.1128/MCB.00118-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jaiswal AK (2004). Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2004.02.074 [DOI] [PubMed] [Google Scholar]
- [52].Kwong M, Kan YW, & Chan JY (1999). The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stress-inducing agents. Role for Nrf1 in γ-gcs(L) and gss expression in mouse fibroblasts. Journal of Biological Chemistry. 10.1074/jbc.274.52.37491 [DOI] [PubMed] [Google Scholar]
- [53].Chan JY, & Kwong M (2000). Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochimica et Biophysica Acta - Gene Structure and Expression. 10.1016/S0167-4781(00)00238-4 [DOI] [PubMed] [Google Scholar]
- [54].Silva-Islas CA, & Maldonado PD (2018). Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacological Research. 10.1016/j.phrs.2018.06.013 [DOI] [PubMed] [Google Scholar]
- [55].Canning P, Sorrell FJ, & Bullock AN (2015). Structural basis of Keap1 interactions with Nrf2. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2015.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cuadrado A (2015). Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2015.04.029 [DOI] [PubMed] [Google Scholar]
- [57].Jain AK, & Jaiswal AK (2007). GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. Journal of Biological Chemistry. 10.1074/jbc.M611336200 [DOI] [PubMed] [Google Scholar]
- [58].Culbreth M, Aschner M (2018). GSK-3β, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000Res. 10.12688/f1000research.15239.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tan SM, & de Haan JB (2014). Combating oxidative stress in diabetic complications with Nrf2 activators: How much is too much? Redox Report. 10.1179/1351000214Y.0000000087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ramprasath T, Vasudevan V, Sasikumar S, Puhari SS, Saso L, & Selvam GS (2015). Regression of oxidative stress by targeting eNOS and Nrf2/ARE signaling: a guided drug target for cardiovascular diseases. Curr Top Med Chem. 10.2174/1568026615666150220114417 [DOI] [PubMed] [Google Scholar]
- [61].Joshi G, & Johnson A, J. (2012). The Nrf2-ARE Pathway: A Valuable Therapeutic Target for the Treatment of Neurodegenerative Diseases. Recent Patents on CNS Drug Discovery. 10.2174/157488912803252023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N, Biswal S (2009). Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2008.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD (2002). Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 10.1042/bj20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Banning A, Deubel S, Kluth D, Zhou Z, & Brigelius-Flohe R (2005). The GI-GPx Gene Is a Target for Nrf2. Molecular and Cellular Biology. 10.1128/MCB.25.12.4914-4923.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hayes JD, Flanagan JU, & Jowsey IR (2005). Glutathione Transferases. Annual Review of Pharmacology and Toxicology. 10.1146/annurev.pharmtox.45.120403.095857 [DOI] [PubMed] [Google Scholar]
- [66].Whitbread AK, Masoumi A, Tetlow N, Schmuck E, Coggan M, & Board PG (2005). Characterization of the omega class of glutathione transferases. Methods in Enzymology. 10.1016/S0076-6879(05)01005-0 [DOI] [PubMed] [Google Scholar]
- [67].Josephy PD (2010). Genetic variations in human glutathione transferase enzymes: significance for pharmacology and toxicology. Human Genomics and Proteomics : HGP. 10.4061/2010/876940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mannervik B, Board PG, Hayes JD, Listowsky I, & Pearson WR (2005). Nomenclature for mammalian soluble glutathione transferases. Methods in Enzymology. 10.1016/S0076-6879(05)01001-3 [DOI] [PubMed] [Google Scholar]
- [69].Armstrong RN (2006). Glutathione S-Transferases: Structure and Mechanism of an Archetypical Detoxication Enzyme. In Advances in Enzymology and Related Areas of Molecular Biology. 10.1002/9780470123157.ch1 [DOI] [PubMed] [Google Scholar]
- [70].Hayes JD, & Pulford DJ (1995). The Glutathione-S-Transferase Supergene Family - Regulation of Gst and the Contribution of the Isoenzymes to Cancer Chemoprotection and Drug-Resistance. Crit Rev Biochem Molec Biol. 10.3109/10409239509083491 [DOI] [PubMed] [Google Scholar]
- [71].Homolya L, Váradi A, & Sarkadi B (2003). Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. In BioFactors. 10.1002/biof.5520170111 [DOI] [PubMed] [Google Scholar]
- [72].Balogh LM, & Atkins WM (2011). Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metabolism Reviews. 10.3109/03602532.2011.558092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Vorojeikina D, Broberg K, Love TM, Davidson PW, van Wijngaarden E, & Rand MD (2017). Glutathione S-transferase activity moderates methylmercury toxicity during development in Drosophila. Toxicological Sciences. 10.1093/toxsci/kfx033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dekant W, & Vamvakas S (1993). Glutathione-dependent bioactivation of xenobiotics. Xenobiotica. 10.3109/00498259309059415 [DOI] [PubMed] [Google Scholar]
- [75].Brigelius-Flohé R, & Maiorino M (2013). Review: Glutathione peroxidases. BBA - General Subjects. 10.1016/j.bbagen.2012.11.020 [DOI] [PubMed] [Google Scholar]
- [76].Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, & Gladyshev VN (2003). Characterization of mammalian selenoproteomes. Science 10.1126/science.1083516 [DOI] [PubMed] [Google Scholar]
- [77].Dringen R, & Gutterer JM (2002). Glutathione reductase from bovine brain. Methods in Enzymology. 10.1016/S0076-6879(02)48646-6 [DOI] [PubMed] [Google Scholar]
- [78].Wamelink MMC, Struys EA, & Jakobs C (2008). The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: A review. Journal of Inherited Metabolic Disease. 10.1007/s10545-008-1015-6 [DOI] [PubMed] [Google Scholar]
- [79].König J, Nies AT, Cui Y, Leier I, & Keppler D (1999). Conjugate export pumps of the multidrug resistance protein (MRP) family: Localization, substrate specificity, and MRP2-mediated drug resistance. Biochimica et Biophysica Acta - Biomembranes. 10.1016/S0005-2736(99)00169-8 [DOI] [PubMed] [Google Scholar]
- [80].Ghezzi P (2005). Oxidoreduction of protein thiols in redox regulation. Biochemical Society Transactions. 10.1042/BST20051378 [DOI] [PubMed] [Google Scholar]
- [81].Dalle-Donne I, Rossi R, Colombo G, Giustarini D, & Milzani A (2009). Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends in Biochemical Sciences. 10.1016/j.tibs.2008.11.002 [DOI] [PubMed] [Google Scholar]
- [82].Seres T, Ravichandran V, Moriguchi T, Rokutan K, Thomas JA, & Johnston RB (1996). Protein S-thiolation and dethiolation during the respiratory burst in human monocytes. A reversible post-translational modification with potential for buffering the effects of oxidant stress. Journal of Immunology (Baltimore, Md. : 1950). [PubMed] [Google Scholar]
- [83].Mustacich D, & Powis G (2000). Thioredoxin reductase. The Biochemical Journal. 10.1042/0264-6021:3460001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lu J, & Holmgren A (2014). The thioredoxin antioxidant system. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2013.07.036 [DOI] [PubMed] [Google Scholar]
- [85].Hanschmann E-M, Godoy JR, Berndt C, Hudemann C, & Lillig CH (2013). Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: from Cofactors to Antioxidants to Redox Signaling. Antioxidants & Redox Signaling. 10.1089/ars.2012.4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lillig CH, Berndt C, & Holmgren A (2008). Glutaredoxin systems. Biochimica et Biophysica Acta - General Subjects. 10.1016/j.bbagen.2008.06.003 [DOI] [PubMed] [Google Scholar]
- [87].Rodriguez-Manzaneque MT (2002). Grx5 Is a Mitochondrial Glutaredoxin Required for the Activity of Iron/Sulfur Enzymes. Molecular Biology of the Cell. 10.1091/mbc.01-10-0517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Richardson RJ, & Murphy SD (1975). Effect of glutathione depletion on tissue deposition of methylmercury in rats. Toxicology and Applied Pharmacology. 10.1016/0041-008X(75)90274-4 [DOI] [PubMed] [Google Scholar]
- [89].Osawa M, & Magos L (1974). The chemical form of the methylmercury complex in the bile of the rat. Biochem Pharmacol. 10.1016/0006-2952(74)90199-3 [DOI] [PubMed] [Google Scholar]
- [90].Kaur P, Aschner M, & Syversen T (2011). Biochemical factors modulating cellular neurotoxicity of methylmercury. Journal of Toxicology. 10.1155/2011/721987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Shanker G, Syversen T, Aschner JL, & Aschner M (2005). Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Molecular Brain Research. 10.1016/j.molbrainres.2005.02.006 [DOI] [PubMed] [Google Scholar]
- [92].Rush T, Liu XQ, Nowakowski AB, Petering DH, & Lobner D (2012). Glutathione-mediated neuroprotection against methylmercury neurotoxicity in cortical culture is dependent on MRP1. NeuroToxicology. 10.1016/j.neuro.2012.03.004 [DOI] [PubMed] [Google Scholar]
- [93].Aremu DA, Madejczyk MS, & Ballatori N (2008). N-acetylcysteine as a potential antidote and biomonitoring agent of methylmercury exposure. Environmental Health Perspectives. 10.1289/ehp.10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Dutczak WJ, & Ballatori N (1994). Transport of the glutathione-methylmercury complex across liver canalicular membranes on reduced glutathione carriers. Journal of Biological Chemistry. J Biol Chem. 1994; 269(13):9746–51. [PubMed] [Google Scholar]
- [95].Franco JL, Teixeira A, Meotti FC, Ribas CM, Stringari J, Garcia Pomblum SC, Farina M (2006). Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environmental Research. 10.1016/j.envres.2006.02.003 [DOI] [PubMed] [Google Scholar]
- [96].Stringari J, Nunes AKC, Franco JL, Bohrer D, Garcia SC, Dafre AL, … Farina M. (2008). Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicology and Applied Pharmacology. 10.1016/j.taap.2007.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rabenstein DL, Isab AA, & Reid RS (1982). A proton nuclear magnetic resonance study of the binding of methylmercury in human erythrocytes. Biochim. Biophys. Acta. 10.1016/0167-4889(82)90038-6 [DOI] [PubMed] [Google Scholar]
- [98].Kerper LE, Ballatori N, Clarkson TW (1992). Methylmercury transport across the blood–brain barrier by an amino acid carrier Am J Physiol, 262 (1992), pp. R761–R765 [DOI] [PubMed] [Google Scholar]
- [99].Aschner M, Eberle NB, Goderie S, & Kimelberg HK (1990). Methylmercury uptake in rat primary astrocyte cultures: the role of the neutral amino acid transport system. Brain Research. 10.1016/0006-8993(90)91546-S [DOI] [PubMed] [Google Scholar]
- [100].Wade LA, & Brady HM (1982). Cysteine and Cystine Transport at the Blood-Brain Barrier. Journal of Neurochemistry. 10.1111/j.1471-4159.1982.tb12548.x [DOI] [PubMed] [Google Scholar]
- [101].Bannai S, Takada A, Kasuga H, Tateishi N (1986). Induction of cystine transport activity in isolated rat hepatocytes by sulfobromophthalein and other electrophilic agents. Hepatology 10.1002/hep.1840060624 [DOI] [PubMed] [Google Scholar]
- [102].Shanker G, & Aschner M (2001). Identification and characterization of uptake systems for cystine and cysteine in cultured astrocytes and neurons: Evidence for methylmercury-targeted disruption of astrocyte transport. In Journal of Neuroscience Research. 10.1002/jnr.10066 [DOI] [PubMed] [Google Scholar]
- [103].Allen JW, Shanker G, & Aschner M (2001). Methylmercury inhibits the in vitro uptake of the glutathione precursor, cystine, in astrocytes, but not in neurons. Brain Research. 10.1016/S0006-8993(01)01988-6 [DOI] [PubMed] [Google Scholar]
- [104].Shanker G, Allen JW, Mutkus L. a, & Aschner M. (2001). Methylmercury inhibits cysteine uptake in cultured primary astrocytes, but not in neurons. Brain Research 10.1016/S0006-8993(01)02791-3 [DOI] [PubMed] [Google Scholar]
- [105].Dringen R, & Hirrlinger J (2003). Glutathione pathways in the brain. Biological Chemistry. 10.1515/BC.2003.059 [DOI] [PubMed] [Google Scholar]
- [106].Arnold AP, Tan KS, & Rabenstein DL (1986). Nuclear Magnetic Resonance Studies of the Solution Chemistry of Metal Complexes. 23. Complexation of Methylmercury by Selenohydryl-Containing Amino Acids and Related Molecules. Inorganic Chemistry. 10.1021/ic00234a030 [DOI] [Google Scholar]
- [107].Ouyang Y, Peng Y, Li J, Holmgren A, Lu J. (2018). Modulation of thiol-dependent redox system by metal ions via thioredoxin and glutaredoxin systems . Metallomics 10.1039/c7mt00327g [DOI] [PubMed] [Google Scholar]
- [108].Reich and Hondal, (2016). ReicWhy Nature Chose Selenium. ACS Chem Biol. 11(4): 821–41. [DOI] [PubMed] [Google Scholar]
- [109].Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R, Bainy AC, Marques MR, Dafre AL, Farina M (2009). Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic Biol Med. 10.1016/j.freeradbiomed.2009.05.013 [DOI] [PubMed] [Google Scholar]
- [110].Zemolin APP, Meinerz DF, de Paula MT, Mariano DOC, Rocha JBT, Pereira AB, … Franco JL. (2012). Evidences for a role of glutathione peroxidase 4 (GPx4) in methylmercury induced neurotoxicity in vivo. Toxicology 10.1016/j.tox.2012.07.013 [DOI] [PubMed] [Google Scholar]
- [111].Usuki F, & Fujimura M (2016). Decreased plasma thiol antioxidant barrier and selenoproteins as potential biomarkers for ongoing methylmercury intoxication and an individual protective capacity. Archives of Toxicology. 10.1007/s00204-015-1528-3 [DOI] [PubMed] [Google Scholar]
- [112].Meinerz DF, Branco V, Aschner M, Carvalho C, & Rocha JBT (2017). Diphenyl diselenide protects against methylmercury-induced inhibition of thioredoxin reductase and glutathione peroxidase in human neuroblastoma cells: a comparison with ebselen. Journal of Applied Toxicology. 10.1002/jat.3458 [DOI] [PubMed] [Google Scholar]
- [113].Usuki F, Yamashita A, & Fujimura M (2011). Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. Journal of Biological Chemistry. 10.1074/jbc.M110.168872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Penglase S, Hamre K, & Ellingsen S (2014). Selenium prevents downregulation of antioxidant selenoprotein genes by methylmercury. Free Radical Biology and Medicine. 10.1016/j.freeradbiomed.2014.07.019 [DOI] [PubMed] [Google Scholar]
- [115].Spiller HA (2018). Rethinking mercury: the role of selenium in the pathophysiology of mercury toxicity . Clinical Toxicology. 10.1080/15563650.2017.1400555 [DOI] [PubMed] [Google Scholar]
- [116].Allmang C, Krol A. (2006). Selenoprotein synthesis: UGA does not end the story. Biochimie. 2006; 88(11):1561–71. [DOI] [PubMed] [Google Scholar]
- [117].Magos L, Webb M, Hudson AR. (1979). Complex formation between selenium and methylmercury. Chem Biol Interact. 1979; 28(2-3):359–62. [DOI] [PubMed] [Google Scholar]
- [118].Khan MA, Wang F. (2009) Mercury-selenium compounds and their toxicological significance: toward a molecular understanding of the mercury-selenium antagonism. Environ Toxicol Chem. 28(8): 1567–77. doi: 10.1897/08-375.1. [DOI] [PubMed] [Google Scholar]
- [119].Kirkpatrick M, Benoit J, Everett W, Gibson J, Rist M, & Fredette N (2015). The effects of methylmercury exposure on behavior and biomarkers of oxidative stress in adult mice. NeuroToxicology. 10.1016/j.neuro.2015.07.001 [DOI] [PubMed] [Google Scholar]
- [120].Farina M, Franco JL, Ribas CM, Meotti FC, Missau FC, Pizzolatti MG, … Santos ARS (2005). Protective effects of Polygala paniculata extract against methylmercury-induced neurotoxicity in mice. The Journal of Pharmacy and Pharmacology. 10.1211/jpp.57.11.0017 [DOI] [PubMed] [Google Scholar]
- [121].Stringari J, Meotti FC, Souza DO, Santos ARS, & Farina M (2006). Postnatal methylmercury exposure induces hyperlocomotor activity and cerebellar oxidative stress in mice: Dependence on the neurodevelopmental period. Neurochemical Research. 10.1007/s11064-006-9051-9 [DOI] [PubMed] [Google Scholar]
- [122].Sumathi T, & Christinal J (2016). Neuroprotective Effect of Portulaca oleraceae Ethanolic Extract Ameliorates Methylmercury Induced Cognitive Dysfunction and Oxidative Stress in Cerebellum and Cortex of Rat Brain. Biological Trace Element Research. 10.1007/s12011-015-0546-6 [DOI] [PubMed] [Google Scholar]
- [123].Franco JL, Braga HC, Stringari J, Missau FC, Posser T, Mendes BG, … Farina M (2007). Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: Protective effects of quercetin. Chemical Research in Toxicology. 10.1021/tx7002323 [DOI] [PubMed] [Google Scholar]
- [124].Mori N, Yasutake A, & Hirayama K (2007). Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Archives of Toxicology. 10.1007/s00204-007-0209-2 [DOI] [PubMed] [Google Scholar]
- [125].Carvalho MC, Franco JL, Ghizoni H, Kobus K, Nazari EM, Rocha JBT, Farina M (2007). Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology. 10.1016/j.tox.2007.07.009 [DOI] [PubMed] [Google Scholar]
- [126].Yee S, Choi BH. (1994) Methylmercury poisoning induces oxidative stress in the mouse brain. Exp Mol Pathol. 1994; 60(3):188–96. [DOI] [PubMed] [Google Scholar]
- [127].Farina M, Dahm KCS, Schwalm FD, Brusque AM, Frizzo MES, Zeni G, … Rocha JBT. (2003). Methylmercury increases glutamate release from brain synaptosomes and glutamate uptake by cortical slices from suckling rat pups: Modulatory effect of ebselen. Toxicological Sciences. 10.1093/toxsci/kfg058 [DOI] [PubMed] [Google Scholar]
- [128].Manfroi CB, Schwalm FD, Cereser V, Abreu F, Oliveira A, Bizarro L, Farina M (2004). Maternal milk as methylmercury source for suckling mice: Neurotoxic effects involved with the cerebellar glutamatergic system. Toxicological Sciences. 10.1093/toxsci/kfh201 [DOI] [PubMed] [Google Scholar]
- [129].Juárez BI, Martínez ML, Montante M, Dufour L, García E, & Jiménez-Capdeville ME (2002). Methylmercury increases glutamate extracellular levels in frontal cortex of awake rats. Neurotoxicology and Teratology. 10.1016/S0892-0362(02)00270-2 [DOI] [PubMed] [Google Scholar]
- [130].Magos L, Cikrt M, & Snowden R (1985). The dependence of biliary methylmercury secretion on liver GSH and ligandin. Biochemical Pharmacology. 10.1016/0006-2952(85)90035-8 [DOI] [PubMed] [Google Scholar]
- [131].Mahapatra CT, Bond J, Rand DM, & Rand MD (2010). Identification of methylmercury tolerance gene candidates in drosophila. Toxicological Sciences. 10.1093/toxsci/kfq097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ginsberg G, Smolenski S, Hattis D, Guyton KZ, Johns DO, & Sonawane B (2009). Genetic polymorphism in glutathione transferases (GST): Population distribution of GSTM1, T1, and P1 conjugating activity. Journal of Toxicology and Environmental Health - Part B: Critical Reviews. 10.1080/10937400903158375 [DOI] [PubMed] [Google Scholar]
- [133].Gundacker C, Komarnicki G, Jagiello P, Gencikova A, Dahmen N, Wittmann KJ, & Gencik M (2007). Glutathione-S-transferase polymorphism, metallothionein expression, and mercury levels among students in Austria. Science of the Total Environment. 10.1016/j.scitotenv.2007.07.033 [DOI] [PubMed] [Google Scholar]
- [134].Barcelos GRM, Grotto D, de Marco KC, Valentini J, Lengert A van H, Oliveira, A. Á. S. de, … Barbosa F. (2013). Polymorphisms in glutathione - related genes modify mercury concentrations and antioxidant status in subjects environmentally exposed to methylmercury. Science of the Total Environment. 10.1016/j.scitotenv.2013.06.029 [DOI] [PubMed] [Google Scholar]
- [135].Ali-Osman F, Akande O, Antoun G, Mao JX, & Buolamwini J (1997). Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants: Evidence for differential catalytic activity of the encoded proteins. Journal of Biological Chemistry. 10.1074/jbc.272.15.10004 [DOI] [PubMed] [Google Scholar]
- [136].Goodrich JM, & Basu N (2012). Variants of glutathione s-transferase pi 1 exhibit differential enzymatic activity and inhibition by heavy metals. Toxicology in Vitro. 10.1016/j.tiv.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Custodio HM, Broberg K, Wennberg M, Jansson JH, Vessby B, Hallmans G, … Skerfving S. (2004). Polymorphisms in Glutathione -Related Genes Affect Methylmercury Retention. Archives of Environmental Health. 10.1080/00039890409603438 [DOI] [PubMed] [Google Scholar]
- [138].Goodrich JM, Wang Y, Gillespie B, Werner R, Franzblau A, & Basu N (2011). Glutathione enzyme and selenoprotein polymorphisms associate with mercury biomarker levels in Michigan dental professionals. Toxicology and Applied Pharmacology. 10.1016/j.taap.2011.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Engström KS, Strömberg U, Lundh T, Johanson I, Vessby B, Hallmans G, … Broberg K. (2008). Genetic variation in glutathione -related genes and body burden of methylmercury. Environmental Health Perspectives. 10.1289/ehp.10804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Robitaille S, Mailloux RJ, & Chan HM (2016). Methylmercury alters glutathione homeostasis by inhibiting glutaredoxin 1 and enhancing glutathione biosynthesis in cultured human astrocytoma cells. Toxicology Letters. 10.1016/j.toxlet.2016.05.013 [DOI] [PubMed] [Google Scholar]
- [141].Branco V, Coppo L, Solá S, Lu J, Rodrigues CMP, Holmgren A, & Carvalho C (2017). Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury. Redox Biology. 10.1016/j.redox.2017.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Toyama T, Shinkai Y, Kaji T, & Kumagai Y (2013). A convenient method to assess chemical modification of protein thiols by electrophilic metals. The Journal of Toxicological Sciences. 10.2131/jts.38.477 [DOI] [PubMed] [Google Scholar]
- [143].Ni M, Li X, Yin Z, Sidoryk-Weogonekgrzynowicz M, Jiang H, Farina M, … Aschner M (2011). Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. GLIA. 10.1002/glia.21153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Wang L, Jiang H, Yin Z, Aschner M, & Cai J (2009). Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicological Sciences. 10.1093/toxsci/kfn201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Feng S, Xu Z, Wang F, Yang T, Liu W, Deng Y, & Xu B (2017). Sulforaphane Prevents Methylmercury-Induced Oxidative Damage and Excitotoxicity Through Activation of the Nrf2-ARE Pathway. Molecular Neurobiology. 10.1007/s12035-015-9643-y [DOI] [PubMed] [Google Scholar]
- [146].Culbreth M, Zhang Z, & Aschner M (2017). Methylmercury augments Nrf2 activity by downregulation of the Src family kinase Fyn. NeuroToxicology. 10.1016/j.neuro.2017.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Antunes dos Santos A, Ferrer B, Marques Gonçalves F, Tsatsakis A, Renieri E, Skalny A, … Aschner M. (2018). Oxidative Stress in Methylmercury-Induced Cell Toxicity. Toxics. 10.3390/toxics6030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Unoki T, Akiyama M, Kumagai Y, Gonçalves FM, Farina M, Rocha JB, & Aschner M (2018). Molecular pathways associated with methylmercury-induced Nrf2 modulation. Front. Genet. 10.3389/fgene.2018.00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Toyama T, Shinkai Y, Yasutake A, Uchida K, Yamamoto M, & Kumagai Y (2011). Isothiocyanates reduce mercury accumulation via an Nrf2-dependent mechanism during exposure of mice to methylmercury. Environmental Health Perspectives. 10.1289/ehp.1003123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Mohan V, Das S, & Rao SBS (2016). Hydroxytyrosol, a dietary phenolic compound forestalls the toxic effects of methylmercury-induced toxicity in IMR-32 human neuroblastoma cells. Environmental Toxicology. 10.1002/tox.22134 [DOI] [PubMed] [Google Scholar]
- [151].Branco V, Caito S, Farina M, Teixeira da Rocha J, Aschner M, & Carvalho C (2017). Biomarkers of mercury toxicity: Past, present, and future trends. Journal of Toxicology and Environmental Health - Part B: Critical Reviews. 10.1080/10937404.2017.1289834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Lund ME, Banner W Jr, Clarkson TW, Berlin M. (1984). Treatment of acute methylmercury ingestion by hemodialysis with N-acetylcysteine (Mucomyst) infusion and 2,3-dimercaptopropane sulfonate. J Toxicol Clin Toxicol. 22(1): 31–49. [DOI] [PubMed] [Google Scholar]
- [153].de Freitas AS, Funck VR, Rotta Mdos S, Bohrer D, Mörschbächer V, Puntel RL, Nogueira CW, Farina M, Aschner M, Rocha JB. (2009). Diphenyl diselenide, a simple organoselenium compound, decreases methylmercury-induced cerebral, hepatic and renal oxidative stress and mercury deposition in adult mice. Brain Res Bull. 10.1016/j.brainresbull.2008.11.001. [DOI] [PubMed] [Google Scholar]
- [154].Yasutake A, Hirayama K, Inoue M. (1989). Mechanism of urinary excretion of methylmercury in mice. Arch Toxicol. 63(6): 479–83. [DOI] [PubMed] [Google Scholar]