

Supplemental Digital Content is available in the text.
Abstract
Background:
We hypothesized that crosstalk between noncoding RNAs, including microRNA (miRNA), lncRNA, and circRNA, might play a critical role in keloids development and physiology. To reveal the molecular mechanisms involved in the pathogenesis of keloids, we compared their gene expression profiles and differential expressions in keloid and normal skin tissues.
Methods:
Expression profiles of mRNAs and lncRNAs and circRNAs in 2 pairs (identification set) of keloid and matched normal skin tissues were analyzed through sequencing. Real-time quantitative PCR was performed to validate the sequencing results using 5 pairs (validation set) of keloid and matched normal skin tissues. Presumed targets of differentially expressed lncRNAs and circRNAs were functionally annotated by bioinformatics approaches.
Results:
The differential expression of mRNAs in keloid and normal skin by high-throughput sequencing was 2,528, of which 1,271 were downregulated, whereas 1,257 were upregulated. In the meantime, sequencing identified 2,227 differentially expressed lncRNAs, including 1,224 upregulated and 1,003 downregulated in keloid tissue compared with normal skin tissue. Additionally, 154 differentially expressed circRNAs were identified, including 81 upregulated and 73 downregulated in keloid tissue compared with normal skin tissue. Functional annotations of differentially expressed circRNA targets revealed their enrichment in several signaling pathways important for scar wound healing.
Conclusions:
Expression profiles of mRNAs, lncRNAs, and circRNAs were altered in keloid tissue, which may partly contribute to the etiology of keloids by affecting several signaling pathways relevant to scar wound healing. A better understanding of keloids pathogenesis may identify new therapeutic targets for keloids.
INTRODUCTION
Keloid is a benign fibrous dermal tumor, characterized by abnormal proliferation of excessive fibroblasts and the lack of fibroblast apoptosis in response to skin injuries.1 In keloid, skin grows beyond the boundaries of the wound, causing pain, itching, and contracture, which becomes a cosmetic and psychological burden for patients.2 Some treatment methods are described in the literature, although there is still no consensus on recurrence rate. Keloid remains a therapeutic challenge because its molecular mechanism is unclear. At present, a general optimal treatment method has not been formulated. So far, most studies have focused on the establishment of keloid lesions and the surrounding extracellular matrix.3
Recently, the discovery of lncRNAs was introduced as a new catalog of diagnostic and therapeutic opportunities for cancer biology.4 lncRNAs, a subtype of RNA transcripts containing more than 200 nucleotides,5 are previously considered as a genetic byproduct because they lack capability of encoding proteins or exhibit limited physiological potential.6 Another type of noncoding RNAs, circRNAs, is formed by back-splice events joined with a junction of the 5′ and 3′ ends.7 It belongs to a novel class of noncoding genome with distinct properties and diverse biological functions. Although circRNAs were reported a few decades ago,8 they were misinterpreted as errors of splicing processes.9 A major turning point of understanding the complex roles of circRNAs came with the development of sequencing technology. Thousands of well-expressed, diverse, stable circRNAs have been discovered recently. Sequence analysis has indicated crucial regulatory roles for circRNAs, in a tissue-specific manner.
Surprisingly, despite the fact that more than 90% of human genome is actively transcribed, protein-coding genes account for less than 2%, which suggests that noncoding RNAs represent most of the human transcriptome.10–12 The circRNAs contribute significantly to total RNAs: although typically circular isoforms account for 5%–10% of total transcripts of corresponding coding genes, certain circRNAs are up to 200 times more abundant than their linear counterparts.7,13–16 Accumulating evidence strongly supports that noncoding RNAs, including miRNAs, lncRNAs, and circRNAs, are key elements of tumorigenesis and progression.17,18 Furthermore, lncRNAs and circRNAs could be mediated by microRNA-binding sites in 3′ untranslated regions (3′ UTRs), suggesting that mRNAs act as competitive endogenous RNAs (ceRNAs).19 Previous studies on cancer genomes and transcriptomes have identified profound alterations in multiple genes and proteins.20 RNA-RNA crosstalk and competing endogenous networks are also important for tumor development and progression.21–23
In this study, we compared the expression profiles of mRNAs, lncRNAs, and circRNAs in keloid tissue with normal skin, to develop an ever-deepening understanding of the potential molecular mechanisms involved in the pathogenesis of keloids.
MATERIALS AND METHODS
Keloid and Normal Tissue Samples
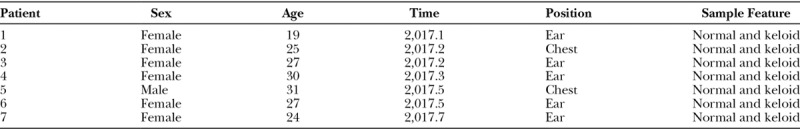
From January 2017 to July 2017, a total of 7 keloid patients were included in the criteria according to the following criteria, of which 2 were used for gene sequencing and 5 for qRT-PCR validation: (1) the mass protruded on the skin surface, hard, smooth, and bright surface, irregular boundary, no sign of retreat within 1 year; (2) the lesion exceeded the edge of the original injury, infiltrated into the surrounding normal tissue, and grew like a crab foot; (3) sustained growth, redness, itching, and other clinical symptoms, no self-healing tendency, cannot self-subsidize; (4) final pathological examination confirmed keloid; (5) the focus has no infection; (6) none of the patients received adjuvant treatment before operation; (7) there were no severe systemic diseases of heart, brain, kidney, liver, and other important organs; (8) nonpregnant and lactating women; (9) no history of glucocorticoid allergy; (10) keloid weight > 100 mg; (11) normal skin came from the side of keloid of the same patient; (12) keloid resection (incisal margin > 0.5 mm); (13) informed consent was signed by all patients. Clinical data: age 18–60 years, mean age (31.83 ± 12.75) years; male 1, female 6; ear keloid 5, chest keloid 2; and diameter between 1.0 and 5.0 cm (Table 1). The participants were H.W. and J.W and during or after date collection, authors looked at the information used to identify participants. All the patients agreed to donate biospecimens for scientific research and publication and signed the informed consent form before surgery. The sample tissue was freshly frozen using liquid nitrogen and stored at −80°C. All studies were approved by the ethics committee of Harbin Medical University.
Table 1.
Summary.
mRNA, lncRNA, and circRNA High-throughput Sequence Analysis
Total RNA from tissues were isolated from the whole tissue layer using Hipure Total RNA Mini Kit (Magen) according to the protocol. The total RNA sample were eluted with 50 µl of RNase-free water and repurified; then, the concentration and integrity of the extracted total RNA were estimated by Qubit 3.0 Fluorometer (Invitrogen, Carlsbad, Calif.) and Agilent 2100 Bioanalyzer (Applied Biosystems, Carlsbad, Calif.), respectively. RNA samples with a RIN value of at least 6.6 or higher were used for further processing.
RNA-seq library was prepared with approximately 1 µg of total RNA using KAPA Stranded RNA-Seq Kit with RiboErase (HMR) for Illumina Platforms (Kapa Biosystems, Inc., Woburn, Mass.). Briefly, ribosomal RNA was removed from the total RNA. Next, first strand and directional second strand synthesis were performed. Then the RNA tailing and adapter ligation were performed with the purified cDNA. Finally, the purified, adapter-ligated DNA was amplified. The library quality and concentration was assessed by utilizing a DNA 1000 chip on an Agilent 2100 Bioanalyzer. Accurate quantification for sequencing applications was determined using the qPCR-based KAPA Biosystems Library Quantification kit (Kapa Biosystems, Inc., Woburn, Mass.). Each library was diluted to a final concentration of 10 nM and pooled equimolar before clustering. Paired-end sequencing was performed on all samples. Quality analysis of gene coverage graph and thermal graph. All samples meet the requirements.
The reads were first mapped to the latest UCSC transcript set using Bowtie2 version 2.1.047 and gene expression level was estimated using RSEM v1.2.15.48 For lncRNA expression analysis, we used transcripts set form Lncipedia (http://www.lncipedia.org). Trimmed mean of M values was used to normalize gene expression. Differentially expressed genes were identified using edgeR program.49 Genes with altered levels (with a P < 0.05 and more than 1.5-fold changes) were considered differentially expressed. The pathway and network analyses were performed using IPA. IPA computed a score for each network according to the fit of the set of supplied focus genes. These scores indicated the likelihood of focus genes belonging to a specific network instead of being obtained by chance. A score >2 indicated ≥99% confidence that a focus gene network was not generated by chance. The canonical pathways generated by IPA were the most significant for the uploaded dataset. Fischer’s exact test with FDR option was used to calculate the significance of canonical pathways.
For circRNA expression analysis, the reads were mapped to genome using the STAR,50 and DCC51 was used to identify specific circRNAs and to estimate the expression of circRNAs. Trimmed mean of M values was used to normalize gene expression. Differentially expressed genes were identified using edgeR program. miRanda52 was used to predict the corresponding miRNA targets of specific circRNAs.
Enrichment Analysis
GO (http://www.geneontology.org) and KEGG (http://www.genome.jp/kegg) databases were used to predict the possible functions of differentially expressed mRNAs and to explore the pathways in which they participate. GO and KEGG analyses for differentially expressed lncRNAs and circRNAs identified downstream genes regulated by RNAs. A P value of <0.05 and |log2 (fold change)| >1 were defined as statistically significant. The calculation of a P value for GO analysis was performed using the following equation:
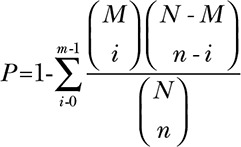 |
Quantitative Real-time PCR
To confirm the reproducibility of the data obtained from the lncRNA and circRNA sequencing, 1 downregulated circRNA was analyzed. qRT-PCR was performed using the ABI Prism 7500 Sequence Detection System (Applied Biosystems). Trizol Reagent (Invitrogen) was applied to extract total RNA from tissues. Geneseed II First Strand cDNA Synthesis Kit (Geneseed, Guangzhou, China) was used to reverse transcription RNA into cDNA. cDNA samples were loaded for qRT-PCR detection. Human β-actin was used as an internal control. The primer sequences were as follows: hsa-circ-0000994—5′-TCGAGGAAATGTTATCGTTCCA-3′ (forward); 5′-TCCAACTGTCACAACCTAACAAT-3′ (reverse); and human β-actin—5′-CATGTACGTTGCTATCCAGGC-3′ (forward); 5′-CTCCTTAATGTCACGCACGAT-3′ (reverse). The PCR conditions were as follows: 5 minutes at 95°C, 40 cycles of 10 seconds at 95°C, and 34 seconds at 60°C. Melt curve analysis was performed to determine the specificity of the reaction. Each sample was tested in triplicate. Relative expression level changes were evaluated using 2-ΔΔCt method. Each circRNA from keloid tissue was compared with the corresponding normal skin tissue by using Student’s t test.
Statistical Analysis
All data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA) unless where indicated Student's t test was used to compare aberrant levels of circRNAs expression between distinct groups. Fisher’s exact tests were performed in GO and KEGG pathway analyses. The data were expressed as mean ± SEM. All experiments were performed in triplicate, and a P value of <0.05 was considered statistically significant.
RESULTS
Expression Profiles of mRNAs in Keloids Analyzed with GO and KEGG
Differential expression profiles were described in SDC 1 (see figure, Supplemental Digital Content 1, which displays analysis of differentially expressed mRNAs. A and B, Volcano plot and clustered heat map of differentially expressed circRNAs depicting up- and downregulated circRNAs in keloids, compared with matched normal skin tissue. Control 1 and control 2 represented matched normal skin tissue, whereas tumor 1 and tumor 2 represented keloids tissue, http://links.lww.com/PRSGO/B101). Expression levels of all biuniquely annotatable and reliably quantifiable mRNAs in 2 matched normal and keloids samples were summarized in SDC 1B. Among these differentially expressed mRNAs, 1,271 were downregulated and 1,257 were upregulated. Furthermore, differentially expressed mRNAs were presented using Volcano Plot filtering (SDC 1A). Gene ontology (GO) analysis of genes downstream from differentially expressed mRNAs was conducted (SDC 2, see figure, Supplemental Digital Content 2, which displays GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. A, GO enrichment of differentially expressed mRNAs. B, Significant terms in KEGG pathways, http://links.lww.com/PRSGO/B102). The 2 most common biological processes were skeletal system development (GO:0001501) and blood vessel morphogenesis (GO:0045514). The most enriched cellular component was glycosaminoglycan catabolic process (GO:0005578). The most enriched molecular functions were cell adhesion molecule binding (GO:0098742) and glycosaminoglycan binding (GO:0005539). KEGG pathway analysis (SDC 2) revealed that, among them, the most enrichment factor is focal adhesion and the most reliable enrichment factor is ECM-receptor interaction.
Ingenuity Pathway Analyses
Ingenuity pathway analyses (IPA) contain canonical pathway, function and disease, upstream regulators, and network analyses (SDC 3 and 4, see figure, Supplemental Digital Content 3, which displays function and diseases analyses of chronic inflammatory disorder and there are 239 potential regulatory target genes, http://links.lww.com/PRSGO/B103; see figure, Supplemental Digital Content 4, which displays (A) upstream regulator predicted by IPA to regulate those molecules that have changed in the submitted dataset. The predicted activation of HIF1α upstream regulator is activated and the z score is 2.405. A total of 131 molecules were predicted as target molecules in dataset. B, The predicted activation of TGF-β upstream regulator is activated and the z score is 3.044. A total of 142 molecules were predicted as target molecules in the dataset. (c) The molecules in network. The red arrow identifies the upregulated molecules in the dataset, whereas the green one means the downregulated molecules. However, the molecules of imaginary lines were potential molecules, which were predicted by IPA, http://links.lww.com/PRSGO/B104).
For canonical pathway analyses, we give priority to adopt-log (P value) for sorting and analyses, the value represents the enrichment degree of pathway in the dataset. Our result shows that the value of 103 canonical pathways are greater than 1.3, which generally means the results for the possibility of random match are less than 5% (SDC 3).
Function and diseases analyses of chronic inflammatory disorder is presented in SDC 4A, and there are 239 potential regulatory target genes. The P value of it is 0.00000246, and z score is 2.2 of which the absolute value is greater than 2. In general, it means the predicted activation state is increased.
Upstream regulator is predicted by IPA to regulate those molecules that have changed in the submitted dataset. The predicted activation of HIF1α upstream regulator is stimulated and the z score is 2.405. Activation z score measures the probability of occurrence of this phenomenon. The higher the absolute value, the higher the probability, which means that the downstream gene expression changes and the possible regulatory relationship of this regulator are more consistent. We predicted 131 target molecules in dataset as shown in SDC 4. The predicted activation of TGF-β upstream regulator is activated of which the z score is 3.044. A total of 142 molecules were predicted as target molecules in dataset (SDC 4).
The molecules in network is presented in SDC 4B, in which the red arrow identifies the upregulated molecules in dataset, whereas the green one means the downregulated molecules. However, the molecules of imaginary lines were potential molecules which were predicted by IPA. Scores demonstrate the enrichment degree of the network, generally more than 20 points means the network is reliable.
Expression Profiles of lncRNAs in Keloids Analyzed with GO and KEGG
To explore potential biological functions of lncRNAs and circRNAs in keloids, RNA sequencing was performed to demonstrate lncRNA and circRNA expression profiles in human keloids and matched normal skin tissues. A total of 2,227 lncRNAs were identified differentially expressed between keloids and normal skin tissues. The volcano plot for lncRNAs was shown in SDC 5A. Of these lncRNAs, 1,224 were upregulated, whereas 1,033 were downregulated in keloids compared with normal skin tissue (SDC 5, see figure, Supplemental Digital Content 5, which displays analysis of differentially expressed lncRNAs. A and B, Volcano plot and clustered heat map of differentially expressed lncRNAs depicting up- and downregulated lncRNAs in keloids, compared with matched normal skin tissue. Control 1 and control 2 represented matched normal skin tissue, whereas tumor 1 and tumor 2 represented keloids tissue, http://links.lww.com/PRSGO/B105).
Potential functions of these noncoding RNAs were analyzed using GO (http://www.geneontology.org) and KEGG (http://www.genome.jp/kegg) databases (SDC 6, see figure, Supplemental Digital Content 6, which displays GO and KEGG analyses. A, GO enrichment of differentially expressed lncRNAs. B and C, Significant terms in KEGG pathways, http://links.lww.com/PRSGO/B106). Differentially expressed lncRNAs were classified as biological processes, cellular components, and molecular functions with GO (SDC 6A). The most enriched and meaningful biological processes terms were related to spermatogenesis (GO:0007283), blood vessel morphogenesis (GO:0048514), protein secretion (GO:0009306), signal release (GO:0023061), ossification (GO:0001503), and kidney development (GO:0001822). The most enriched term under cellular components was transcription factor complex (GO:0005667). The most enriched term under the molecular functions was transcription factor activity, RNA polymerase II core promoter proximal region sequence-specific binding (GO:0000982). The lncRNAs analyzed with KEGG identified specific genes downregulated and involved in RAS signaling pathway (SDC 6). Those upregulated genes were involved in pathways responsible for cancer and metabolic transcriptional misregulation.
Expression Profiles of circRNAs in Keloids Analyzed with GO and KEGG
circRNA length and chromosome distribution were detected and differential expression profiles in SDC 7. Expression levels of all biuniquely annotatable and reliably quantifiable circRNAs in 2 matched normal and keloids samples were summarized in SDC 8B. Among these differentially expressed circRNAs, 81 were downregulated, whereas 73 were upregulated. Furthermore, differentially expressed circRNAs were presented using Volcano Plot filtering (SDC 7, see figure, Supplemental Digital Content 7, which displays analysis of differentially expressed circRNAs. A and B, Volcano plot and clustered heat map of differentially expressed circRNAs depicting up- and downregulated circRNAs in keloids, compared with matched normal skin tissue. Control 1 and control 2 represented matched normal skin tissue, whereas tumor 1 and tumor 2 represented keloids tissue, http://links.lww.com/PRSGO/B107).
GO analysis of genes downstream from differentially expressed circRNAs was conducted (SDC 8, see figure, Supplemental Digital Content 8, which displays (A) GO enrichment of differentially expressed circRNAs. B and C, Significant terms in KEGG pathways, http://links.lww.com/PRSGO/B108). The 2 most common biological processes were extracellular matrix organization (GO:0030198) and extracellular structure organization (GO:0043062). The most enriched cellular component was adherens junction (GO:0005912). The most enriched molecular functions were dioxygenase activity (GO:0051213) and nucleotidyltransferase activity (GO:0016779). KEGG pathway analysis (SDC 8) revealed those downregulated circRNAs in keloids related to PI3K-Akt signaling pathway, whereas upregulated circRNAs involved in regulation of actin cytoskeleton and focal adhesion.
Construction of a circRNAs-miRNA Coexpression Network
To systematically explore the influence of dynamic changes in regulation of circRNAs and miRNAs on gene expression in keloids, a network was constructed by integrating matched expression profiles of circRNAs, miRNAs, and mRNAs. Targetscan 7.0 (http://www.targetscan.org/) and miRanda (http://www.microrna.org/microrna/home.do) were used to identify circRNA-miRNA interactions. Target genes were selected according to GO or KEGG pathway analysis or previous reports of keloid-associated genes, and then global circRNAs-miRNA network was constructed (SDC 9, see figure, Supplemental Digital Content 9, which displays construction of a circRNAs-miRNA coexpression network. The network contained 1,000 miRNAs with the highest total scores. The green, red, and blue nodes represent downregulated circRNAs, upregulated circRNAs, and miRNAs, respectively, http://links.lww.com/PRSGO/B109). Those differentially expressed circRNAs were identified as interacting with 8,271 miRNAs. This network contained 1,000 miRNAs with the highest total scores (SDC 9). Based on this coexpression network, circRNAs are involved in keloids formation, and many miRNAs or genes are regulated by circRNAs.
Validation of the Sequence Data by qRT-PCR
To validate the results from sequencing, quantitative real-time PCR (qRT-PCR) was employed to measure the abundance of circRNAs, including downregulated has-circ-0000994 [SDC 10, see figure, Supplemental Digital Content 10, which displays validation of sequencing data by qRT-PCR. qRT-PCR was employed to measure the abundance of has-circ-0000994 (n = 3, **P < 0.01, ***P < 0.001) http://links.lww.com/PRSGO/B110]. The transcriptional level of has-circ-0000994 coincided perfectly with sequencing data. The expression level of downregulated has-circ-0000994 was decreased by 3.0773648-fold.
DISCUSSION
Keloids are common diseases in the skin for plastic surgery. Keloids are resistant to treatment, typically characterized by gradual invasion of scar tissue into surrounding normal skin and original wound area.2 At present, the molecular mechanism of keloid remains unclear. Therefore, it is important to explore the molecules involved in scar formation and keloid development. Recent research suggests abnormal gene expression at transcriptional and/or translation level in keloids. Our previous studies have demonstrated that miRNA-21 plays a role in regulating apoptosis in scar tissue.24 circRNAs act as a miRNA molecular sponge to regulate the functions of miRNAs. In addition, in the study by Onoufriadis et al.,25 gene association network analysis was also performed to further classify the differentially expressed genes from the RNA-Seq dataset according to reactome pathway terms and correlated expression values among them. It demonstrated a divergent average expression profile of cytokine signaling genes between keloid-prone and healthy individuals during wound healing.25 In consequence, it is key and feasible to better understand circRNAs and to construct lncRNA-miRNA-mRNA network, to explore the specific pathogenesis of keloid.
CircRNAs which belong to noncoding RNAs are different from the traditional linear RNAs. They are widely present in paleontology, flies, mouse, and human.13,16,26–28 circRNAs participate in the regulation of gene expression after transformation29 and have the potential to encode proteins. Hansen et al.15 found that testicular-specific circRNAs produced on Y chromosome act as miR-138 “sponge” and negatively regulate the expression of miR-138. circRNAs are stable and widely expressed in many tissues.30 Some circRNAs have important biological functions and can be used as biomarkers for disease diagnosis.31 Gene regulation based on space and time is critical for normal development of tissues and organs, and for pathogenesis of diseases. circRNAs are inherently stable by virtue of closed covalent structure and exonuclease resistance, even in exosomes.9,27,32–34 This observation opens up an interesting possibility that circRNAs, like miRNAs, may participate in paracrine signaling or cell-to-cell crosstalk. Nevertheless, many functions of circRNAs remain unknown. circRNAs are emerging as potentially essential regulators of cellular physiology and biomarkers for the diagnosis of diseases.
lncRNAs are noncoding transcripts with lengths of more than 200 nucleotides.35,36 lncRNAs are different from mRNAs, although they share common structural characteristics for 5 ‘methylation and polyadenylated tails. lncRNAs were once thought to be merely by-products of RNA polymer type 2 transcription, “noise” and “garbage” in gene transcription, not exerting specific biological functions, and even “dark matter”. However, with the development of research on lncRNAs, lncRNAs and short-chain RNAs have the ability to produce splicing variants and exert similar regulatory functions in the course of tumor evolution. lncRNAs are involved in the biological processes of various cancer cells, including cell proliferation, apoptosis, and metastasis.37–39 Recently, lncRNAs have become key molecules in regulating multiple aspects of coding gene expression. In addition, circRNAs are indicated to function at every stage of tumor progression.40 For example, XIST is involved in progression of glioblastoma, gastric cancer, and hepatocellular carcinoma.41–45 So as a benign tumor, keloid formation may be closely related to lncRNAs and circRNAs. We analyzed the difference between keloids and normal skin tissue through high-throughput sequencing. Various possible regulatory pathways and interaction networks were established with bioinformatics. It is inferred that lncRNAs may be related to the development and progression of keloids, which require further research. In addition, the establishment of lncRNA-miRNA-mRNA mutual control network will be the focus of our future work.
In this study, we used high-throughput sequencing and bioinformatics to predict the changes and functions of noncoding RNAs and to construct gene networks in keloids. The up- or downregulation of circRNAs was validated. Expression of miRNAs in human hypertrophic scar tissue was shown to be significantly different from that in normal skin, which might stimulate the growth and proliferation of tumor cells. Compared with normal skin, 81 circRNAs were upregulated, whereas 73 circRNAs downregulated; and 1,224 lncRNAs were upregulated whereas 1,003 lncRNAs downregulated in keloids. Our results verified that has-circ-0000994 was significantly differentially expressed in scar tissue compared with normal skin tissue, and miR-21 was regulated by has-circ-0000994 in keloids. We speculate that has-circ-0000994/miR-21 axis may be a potential therapeutic target for keloids. miRNA-21 is one of the 23 upregulated miRNAs in keloid tissues, which is closely related to cell proliferation and antiapoptosis. miRNA-21 inhibits apoptosis of tumor cells by promoting proliferation and migration. Previous studies have shown that miRNA-21 inhibits mitochondria-mediated apoptosis pathway.24,46 Key media including miRNA-circRNA networks and cellular signaling pathways were also discovered. Our finding explains a significant portion of thousands of unrepresented lncRNA and circRNAs in keloids. There are some limitations in our study. We use mostly earlobe keloids, the mechanism of action for these may differ from chest keloids. At this stage, it is difficult to elucidate different distribution and functions of circRNAs, lncRNAs, and miRNAs in different parts of the body. It is challenging to prove the corresponding network for signal transduction, which will be our next research focus. The expression of noncoding RNAs in keloid tissues is different from that in normal skin tissues. However, this study does not fully explain the underlying causes. Therefore, further research on molecular networks connected with miRNA-circRNA-mRNA is guaranteed.
CONCLUSIONS
Here, we demonstrate the expression profiles of circRNAs, lncRNAs, and mRNAs in keloids and normal skin tissues detected by high-throughput sequencing. GO and KEGG path analyses enable us to annotate the potential functions of differentially expressed circRNAs and lncRNAs. circRNA-miRNA interaction gene coexpression networks were constructed. Complicated crosstalk among circRNAs, lncRNAs, and miRNAs may affect many signal pathways associated with pathology of keloids. This study may help us understand keloid pathogenesis and even identify therapeutic targets. Besides, as a new field that has not yet been explored, further researches are needed to clarify the role of noncoding RNAs in keloids.
Supplementary Material
Footnotes
Published online 20 June 2019.
These authors contributed equally to this work.
Supported by National Natural Science of China (Grant 81471796) and Heilongjiang Province Science Foundation for Excellent Youth Scholars (Grant JC2017019).
Disclosure: The authors have no financial interest to declare in relation to the content of this article. All authors have given consent for the publication of the article.
Supplemental digital content is available for this article. Clickable URL citations appear in the text.
REFERENCES
- 1.Luo S, Benathan M, Raffoul W, et al. Abnormal balance between proliferation and apoptotic cell death in fibroblasts derived from keloid lesions. Plast Reconstr Surg. 2001;107:87–96. [DOI] [PubMed] [Google Scholar]
- 2.Robles DT, Berg D. Abnormal wound healing: keloids. Clin Dermatol. 2007;25:26–32. [DOI] [PubMed] [Google Scholar]
- 3.He Y, Deng Z, Alghamdi M, et al. From genetics to epigenetics:new insights into keloid scarring. Cell Prolif. 2017;50:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo X, Qiu Y, Jiang Y, et al. Long non-coding RNA implicated in the invasion and metastasis of head and neck cancer: possible function and mechanisms. Mol Cancer. 2018;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol. 2015;22:5–7. [DOI] [PubMed] [Google Scholar]
- 6.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007;14:103–105. [DOI] [PubMed] [Google Scholar]
- 7.Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. [DOI] [PubMed] [Google Scholar]
- 8.Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280:339–340. [DOI] [PubMed] [Google Scholar]
- 9.Cocquerelle C, Mascrez B, Hétuin D, et al. Mis-splicing yields circular RNA molecules. FASEB J. 1993;7:155–160. [DOI] [PubMed] [Google Scholar]
- 10.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature. 2007;447:799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katayama S, Tomaru Y, Kasukawa T, et al. ; RIKEN Genome Exploration Research Group; Genome Science Group (Genome Network Project Core Group); FANTOM Consortium. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. [DOI] [PubMed] [Google Scholar]
- 13.Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. [DOI] [PubMed] [Google Scholar]
- 16.Salzman J, Chen RE, Olsen MN, et al. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li JH, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JY, Lee JE, Park JB, et al. Roles of long non-coding RNAs on tumorigenesis and glioma development. Brain Tumor Res Treat. 2014;2:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatesh T, Suresh PS, Tsutsumi R. Non-coding RNAs: functions and applications in endocrine-related cancer. Mol Cell Endocrinol. 2015;416:88–96. [DOI] [PubMed] [Google Scholar]
- 21.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272–283. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Zheng Q, Bao C, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25:981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu H, Wang J, Ma H, et al. microRNA-21 inhibits mitochondria-mediated apoptosis in keloid. Oncotarget. 2017;8:92914–92925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Onoufriadis A, Hsu CK, Ainali C, et al. Time series integrative analysis of RNA sequencing and microRNA expression data reveals key biologic wound healing pathways in keloid-prone individuals. J Invest Dermatol. 2018;138:2690–2693. [DOI] [PubMed] [Google Scholar]
- 26.Danan M, Schwartz S, Edelheit S, et al. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012;40:3131–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nigro JM, Cho KR, Fearon ER, et al. Scrambled exons. Cell. 1991;64:607–613. [DOI] [PubMed] [Google Scholar]
- 28.Capel B, Swain A, Nicolis S, et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993;73:1019–1030. [DOI] [PubMed] [Google Scholar]
- 29.Kulcheski FR, Christoff AP, Margis R. Circular RNAs are miRNA sponges and can be used as a new class of biomarker. J Biotechnol. 2016;238:42–51. [DOI] [PubMed] [Google Scholar]
- 30.Qu S, Zhong Y, Shang R, et al. The emerging landscape of circular RNA in life processes. RNA Biol. 2017;14:992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Werfel S, Nothjunge S, Schwarzmayr T, et al. Characterization of circular RNAs in human, mouse and rat hearts. J Mol Cell Cardiol. 2016;98:103–107. [DOI] [PubMed] [Google Scholar]
- 32.Lan PH, Liu ZH, Pei YJ, et al. Landscape of RNAs in human lumbar disc degeneration. Oncotarget. 2016;7:63166–63176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwanhäusser B, Busse D, Li N, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. [DOI] [PubMed] [Google Scholar]
- 34.Lasda E, Parker R. Circular RNAs co-precipitate with extracellular vesicles: a possible mechanism for circRNA clearance. PLoS One. 2016;11:e0148407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boon RA, Jaé N, Holdt L, et al. Long noncoding RNAs: from clinical genetics to therapeutic targets? J Am Coll Cardiol. 2016;67:1214–1226. [DOI] [PubMed] [Google Scholar]
- 36.Qiu MT, Hu JW, Yin R, et al. Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biol. 2013;34:613–620. [DOI] [PubMed] [Google Scholar]
- 37.Jiao F, Hu H, Yuan C, et al. Elevated expression level of long noncoding RNA MALAT-1 facilitates cell growth, migration and invasion in pancreatic cancer. Oncol Rep. 2014;32:2485–2492. [DOI] [PubMed] [Google Scholar]
- 38.Lian Y, Wang J, Feng J, et al. Long non-coding RNA IRAIN suppresses apoptosis and promotes proliferation by binding to LSD1 and EZH2 in pancreatic cancer. Tumour Biol. 2016;37:14929–14937. [DOI] [PubMed] [Google Scholar]
- 39.Zheng S, Chen H, Wang Y, et al. Long non-coding RNA LOC389641 promotes progression of pancreatic ductal adenocarcinoma and increases cell invasion by regulating E-cadherin in a TNFRSF10A-related manner. Cancer Lett. 2016;371:354–365. [DOI] [PubMed] [Google Scholar]
- 40.Spizzo R, Almeida MI, Colombatti A, et al. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31:4577–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tantai J, Hu D, Yang Y, et al. Combined identification of long non-coding RNA XIST and HIF1A-AS1 in serum as an effective screening for non-small cell lung cancer. Int J Clin Exp Pathol. 2015;8:7887–7895. [PMC free article] [PubMed] [Google Scholar]
- 42.Yao Y, Ma J, Xue Y, et al. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 2015;359:75–86. [DOI] [PubMed] [Google Scholar]
- 43.Chen DL, Ju HQ, Lu YX, et al. Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J Exp Clin Cancer Res. 2016;35:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang J, Sun CC, Gong C. Long noncoding RNA XIST acts as an oncogene in non-small cell lung cancer by epigenetically repressing KLF2 expression. Biochem Biophys Res Commun. 2016;478:811–817. [DOI] [PubMed] [Google Scholar]
- 45.Zhuang LK, Yang YT, Ma X, et al. microRNA-92b promotes hepatocellular carcinoma progression by targeting Smad7 and is mediated by long non-coding RNA XIST. Cell Death Dis. 2016;7:e2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Yang D, Xiao Z, et al. miRNA expression profiles in keloid tissue and corresponding normal skin tissue. Aesthetic Plast Surg. 2012;36:193–201. [DOI] [PubMed] [Google Scholar]
- 47.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32:1094–1096. [DOI] [PubMed] [Google Scholar]
- 52.Enright AJ, John B, Gaul U, et al. microRNA targets in drosophila. Genome Biol. 2003;5:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]