

Abstract
Background and Objectives:
Pfu DNA polymerase is an enzyme that exhibits the lowest error rate in the 3′ to 5′ exonuclease (proofreading) activity during DNA synthesis in Polymerase Chain Reactions (PCRs). This study was aimed to express and purify Pfu DNA polymerase in a bacterial expression system under a simple purification method.
Materials and Methods:
Pfu polymerase gene sequence, derived from Pyrocuccus furiosus (Pfu) genomic DNA, was cloned and overexpressed in E. coli BL21 (DE3) pLysS. Upon overexpression, bacterial lysate containing the Pfu DNA polymerase was heated at 94°C for 5 minutes. Pfu DNA polymerase having high thermal stability was retained while the other bacterial proteins were denatured. The resulting thermo stable Pfu DNA polymerase was separated from the other debris of the denatured proteins by simple centrifugation.
Results:
The enzymatic activity of the resulting Pfu DNA polymerase was estimated by comparing with the commercial Pfu DNA Polymerases. An estimated 50000 units of functional Pfu DNA polymerase was produced from a 400 ml culture.
Conclusion:
The in-house produced Pfu DNA Polymerase could be used for routine amplification that requires high-fidelity such as cloning and DNA sequencing.
Keywords: Protein expression, Pyrococcus furiosus, DNA polymerase, Polymerase chain reaction
INTRODUCTION
DNA amplification using Polymerase Chain Reaction (PCR) has long emerged as one of the fundamental technologies of molecular biology. Generic PCR consists of three main thermal steps: 1) DNA template denaturation where the double stranded DNA is denatured into two single stranded DNA; 2) primer annealing where the primers anneal to the complementary sequences in the template strands 3) and elongation - DNA polymerase synthesizes new copies of DNA from the template (1). Commonly, Taq polymerase is used as the enzyme of choice in the PCR amplification. This polymerase has no proof-reading capacity (3′-5′ exonuclease), with an error rate of 8×10−6 mutations/bp/duplication (2). In applications that require high fidelity such as cloning, a polymerase capable of proof-reading is highly desirable and one such DNA polymerase is the DNA Polymerase from Pyrococcus furiosus (Pfu).
The Pfu DNA polymerase gene encodes a polypeptide of 775 amino acids with a predicted molecular weight of 90, 109 Da (3, 4). The polymerase possesses a 3′-5′ exonuclease activity that allows for removal of mis-incorporated bases (“proof-reading”) and has been reported to exhibit a low error rate in PCR (1.3×10−6 mutations/bp/duplication) which is approximately 8-fold lower as compared to Taq Polymerase (2, 5). Various researchers have highlight on the expression and purification strategy of the polymerase with a focus on the biophysical characteristic of the protein and/or recombinant DNA technology such as the immobilized metal affinity-tagged purification, weak cation exchange resins, polyethylenemine precipitation, ammonium sulfate precipitation and ion exchange chromatography (6–11). In this study, we reported an in-house expression and purification method of Pyrococcus furiosus (Pfu) DNA polymerase in E. coli BL21 (DE3) pLysS which capitalized on the thermal stability of the Pfu DNA polymerase as the key strategy for the purification which averts the usage of other tedious, specialized and expensive strategies. The overexpressed Pfu DNA polymerase containing lysate was subjected to heating which denatures the rest of the less thermally stable protein, forming debris which is subsequently removed. As a test of functionality, the protein was used to amplify a template plasmid.
The work aims to obtain purified Pfu DNA polymerase at a low cost using a simple in-house purification method that averts specialized techniques and equipment for research and teaching use.
MATERIALS AND METHODS
Plasmid.
Pyrococcus furiosus chromosomal DNA was kindly provided by Dr. Boris at the University of Muenster, Germany. Pfu DNA polymerase gene sequence was amplified and cloned into pSE420 plasmid (Invitrogen, MA, USA). The forward and reverse primers used were Fpfu 5′-AATATTGAATTccatggTTTTAGATGTGGATTAC-3′, which contains the NcoI Restriction site and Rpfu 5′-AATATTGAATTCactagtCTAGGATTTTTTAATGTTAAGC-3′ that contains the SpeI restriction site (restriction sites are represented with lower case and underlined). Following restriction digestion of the both, plasmid and the PCR product, ligation was carried out for 16 hours at 4°C. The ligation mixture was electro-transformed into E. coli DH5α and the recombinant plasmid named pSEPfu was confirmed with sequencing.
Protein expression.
The pSEPfu plasmid was extracted and electro-transformed into E. coli BL21 (DE3) pLysS (Merck, MA, USA). A single positive colony was picked and inoculated into a 10 ml LB broth (containing 100 μg/ml ampicillin and 35 μg/ml chloramphenicol) and cultured for 16 hours at 37°C, 200 rpm. The starter culture was inoculated at a ratio of 1:100 to LB broth (containing 100 μg/ml ampicillin and 35 μg/ml chloramphenicol). The culture was incubated at 37°C and was shaken at 200 rpm. When the OD600 reached 0.5, induction with 0.5 mM IPTG was carried out for a total of 6 hours. One mL of culture was taken at each hour and subjected to protein profiling by 12% SDS PAGE analysis. The intensities of the bands that correspond to the expected size of the Pfu Polymerase were compared between the different hours of induction period. The band of expected size with the highest intensity constituted the most optimal hour of induction.
Protein purification.
The culture was pelleted at 10,000 × g for 10 minutes and the cells were lysed with BugBuster® Protein Extraction Reagent (Merck, MA, USA) as per manufacturer protocol. The lysate was centrifuged at 15,000 × g for 15 minutes and the supernatant was heated at 94°C for 5 minutes. The insoluble fraction was separated from the soluble fraction by centrifugation at 10, 000 × g for 5 minutes at 4°C. The soluble fraction was dialyzed against a buffer containing 20 mM Tris-Cl pH 7.5, 100 mM KCl, 0.1 mM EDTA, 1 mM DTT and 50% glycerol (Pfu Buffer). Dialysis was repeated for 4 times, 8 hours each. The dialyzed fraction was subjected to protein purity analysis via 12% SDS PAGE.
Functional assay of the Pfu Polymerase based on PCR amplification.
To assess the functionality of the purified Pfu Polymerase, PCR amplification was carried out using the template p600 plasmid (recombinant plasmid containing a 600 bp insert of ICR mouse c-myc gene). The PCR reactions carried out were in 20 μl volume containing 20 mM Tris-Cl pH 8.8, 10 mM KCl, 10 mM (NH4)2 SO4, 2 mM MgSO4, 0.1% Triton® X-100 and 0.1 mg/ml nuclease-free BSA, 0.2 mM dNTPs, 0.6 μM p600 primers, and 150 ng of the template. To estimate the enzymatic activity of the Pfu Polymerase, the band intensity of the PCR amplification was compared to that of the commercial Pfu Polymerases from Promega (3U/μl) (Promega, WI, USA) and Thermo Scientific Fisher (2.5U/μl) (Thermo Scientific, MA, USA). Different titration stocks of the Pfu Polymerase were made accordingly by dilution with the Pfu buffer (2×, 5×, 10×, 20×). The PCR parameters were: Initial denaturation: 94°C for 5 min, 35 cycles of denaturation: 94°C for 40 seconds, annealing: 68°C for 40 seconds, extension: 72°C for 40 seconds, and final extension: 72°C for 5 minutes. Band intensity was analyzed via ImageJ software using plot profile and 3D surface plot (12).
RESULTS
Optimum time for overexpression of Pfu polymerase.
Overexpression of the Pfu DNA polymerase was carried out in E. coli BL21 (DE3) pLysS. The time course expression was performed by IPTG-based induction with sample collection at different time points up to 6 hrs. SDS-PAGE analysis was carried out with the collected samples to get an overview on the expression level of Pfu polymerase at the different induction periods (Fig. 1). A band of an expected size of 90 kDa was seen, which represent the overexpressed Pfu DNA polymerase across the period of 6 hours. The optimum time for overexpression was determined to be 5 hours as at this time point, the highest band intensity was reflected which corresponded to the highest level Pfu DNA Polymerase expression. Subsequently, to extract the overexpressed protein, we chemically lysed the cell using the BugBuster® Protein Extraction Reagent.
Fig. 1.

Time course induction of Pfu protein expression. SDS PAGE analysis to gain insight on the time course analysis of Pfu DNA Polymerase expression following IPTG induction after 1, 2, 3, 4, 5 & 6 hours. Lane 1: Protein size marker, Lane 2: 0 hour (before IPTG was added), Lane 3: 1 hour, Lane 4: 2 hours, Lane 5: 3 hours, Lane 6: 4 hours, Lane 7: 5 hours, Lane 8: 6 hours. Pfu Polymerase (arrow) band with the size of 90 kDa was seen to be expressed. 5 hours was chosen as the optimal induction period.
Heat-based purification and centrifugation of Pfu DNA Polymerase.
Following the separation of the soluble from the insoluble fraction by centrifugation at 10,000 × g for 5 min, the soluble proteins were heated at 94°C for 5 minutes. This heating step denatured most of the E. coli proteins while sustaining the thermostable Pfu polymerase (11). The lysate was centrifuged to obtain the soluble fraction. The soluble fraction, which comprises the purified protein was dialyzed extensively with the Pfu Buffer. SDS PAGE analysis of the dialyzed protein shows that presence of the band, which corresponds to the estimated size of the 90 kDa protein Pfu DNA Polymerase (Fig. 2).
Fig. 2.
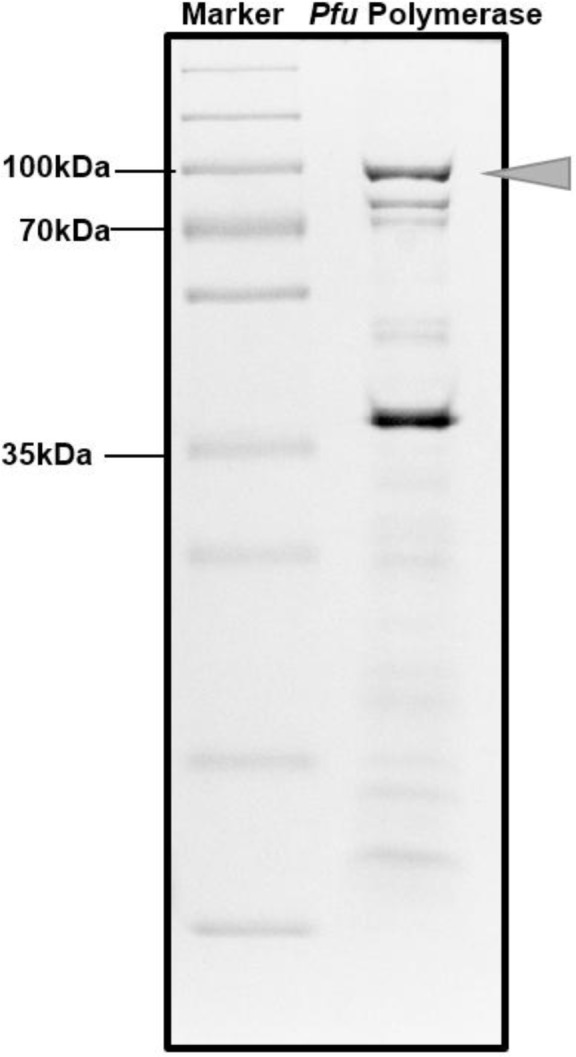
SDS PAGE of Purified Pfu Polymerase. Arrow shows the purified Pfu Polymerase after dialysis.
Titration of enzymatic activity.
Titration stocks were made to assess the activity of the polymerase. The enzymatic activity of Pfu DNA polymerase was estimated based on the band intensity matching of the PCR amplicon to that of the commercial Thermo Scientific Fisher (TSF) and Promega (P) Pfu DNA Polymerase. One microliter of each diluted fractions as well as commercial polymerases were used in the PCR. The relative intensity of the PCR band as measured using ImageJ produced using 1 μL of the 5× diluted in-house Pfu DNA polymerase was estimated to be similar to that of the 2.5U Thermo Scientific Pfu Polymerase (TSF) (Fig. 3). Thus, the enzymatic activity for the current batch of Pfu DNA polymerase was estimated at 12.5 U/μL.
Fig. 3.

Estimation of enzymatic activity for purified Pfu DNA Polymerase. (a) Gel electrophoresis of PCR product with Pfu Polymerase titration with the relative intensity of the amplicons. Pfu Polymerase from the stock was diluted in Pfu buffer (2×, 5×, 10×, 20×) accordingly. The PCR amplification was run in parallel with 1 μL of Pfu DNA Polymerase from Promega (3U/μL) and 1 μL of Pfu DNA Polymerase from Thermo Scientific (2.5U/μL). Lane 1: your marker, Lane 2: 2× dilution, Lane 3: 5× dilution, Lane 4: 10× dilution, Lane 5: 10× dilution, Lane 6: Negative Control, Lane 7: Promega (P) Pfu Polymerase, Lane 8: Thermo Scientific (TSF) Pfu Polymerase. (b) Plot Profile analysis of the bands.
Furthermore, we also tested the activity of 1U from our 5× diluted Pfu polymerase with 1U of both commercial polymerases. We found the band intensity of the PCR amplicon using the purified Pfu DNA polymerase was similar to the commercial polymerases, Promega and Thermo Scientific Fisher (Fig. 4). The 3D surface plot analysis corroborated the analysis, which showed that the peaks and areas to be similar for all three bands. Collectively, an estimated 50000 units of Pfu DNA polymerase was obtained from a 400 ml of bacterial starter culture.
Fig. 4.

Analysis of Pfu DNA polymerase performance compared with commercial polymerases. (a) Gel electrophoresis of the PCR product to assess the functionality of the purified Pfu DNA polymerase in 1U. PCR amplification was carried out using p600 plasmid as the template and 1U of the purified Pfu DNA Polymerase, Promega Pfu DNA Polymerase and Thermo Scientific Pfu DNA Polymerase, respectively. Lane M: DNA size marker, Lane 1: 1U Purified Pfu Polymerase, Lane 2: 1U Promega Pfu Polymerase, Lane 3: 1U Thermo Scientific Pfu Polymerase. (b) 3D Surface Plot analysis on the intensity profile of the bands (axis units in pixels).
DISCUSSION
The PCR method has changed the molecular biology field in a multitude of applications such as sequencing and diagnostics. In a DNA amplification process, one pivotal point that must be thoroughly considered is the accuracy of the copied DNA sequences. Common DNA polymerases have errors in their activity, limiting their applications in high fidelity PCR such as in amplification of long sequences. In applications DNA like sequencing, polymerases with proof-reading activities such as the Pfu is often the main choice (5). Subsequently, various modified polymerases were developed with a prime focus on Pfu to cater for a faster polymerization rate using fusions such as DNA binding proteins that essentially highlights the superiority of this proof reading enzyme (13). Alternatively, researchers have also opted for a combinational usage of Pfu and Taq DNA polymerase for high fidelity PCR with a high polymerization rate (2).
In this study, we cloned the Pfu DNA polymerase gene into the pSE420 plasmid. After successfully cloning the gene, we proceeded to overexpress the native Pfu DNA polymerase with standard IPTG induction protocol in the E. coli BL21 (DE3) pLysS for a tighter expression regulation as toxicity was reported in Pfu DNA polymerase overexpression on the E. coli BL21 (DE3) strain even when IPTG induction was not performed (11). Our study demonstrated an induction time of 5 hours for optimal protein expression. Previous publications using the standard shake-flask protein expression have collectively denoted Pfu polymerase induction time no longer than 5 hrs under IPTG concentration of 0.5–1 mM (7, 10, 14). We heated the lysate to denature and precipitate most of the host proteins. SDS PAGE analysis showed that the dialyzed soluble fraction lysate contains predominantly Pfu polymerase, which is retained due to its high thermostability while most of the bacterial proteins are denatured. Similar to our findings, other investigators have shown that the polymerase activity is not affected by host proteins (7, 15). Additionally, the removal of possible E. coli DNA contaminants can be applied depending on the research application by the use of Benzonase or DNAse treatment (16).
The functionality of the protein was assessed by standard PCR amplification using a plasmid as template for amplifying a 600 bp sequence. Dialysis of the soluble fraction which contains the in-house overexpressed Pfu DNA polymerase, removes small molecules and exchanges the buffer composition with that of the stabilizing buffer. The enzymatic activity of the purified Pfu DNA polymerase activity was estimated to be at 12.5U as determined by comparison with the known activity of the commercial Pfu DNA polymerase. This was carried out by diluting the protein to the common working concentration of 1U/μL and compared with the same concentration of the commercial Pfu DNA polymerases. Collectively, we have accumulated Pfu DNA polymerase with a total enzymatic activity of 50000 units from this method.
CONCLUSION
The Pfu DNA polymerase was successfully overexpressed and purified with a simple heating step with minimal resources. We showed that the Pfu DNA polymerase is functioning and an estimated 50000 units of enzymatic activity was obtained from a 400 ml culture in our experimental setting. The purified Pfu DNA polymerase could be applicable for cloning and DNA sequencing applications.
ACKNOWLEDGEMENTS
SS Prabu is funded by the Vice Chancellor’s Award. Citartan M and Tang TH were supported by E-science fund (305/CIPPT/613235), FRGS (203/CIPPT/6711441), FRGS grant (203/CIPPT/6711510) and RU grant (1001/CIPPT/811319).
The authors have no financial conflicts of interest.
REFERENCES
- 1.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988;239:487–491. [DOI] [PubMed] [Google Scholar]
- 2.Cline J, Braman J, Hogrefe H. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 1996;24:3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uemori T, Sato Y, Kato I, Doi H, Ishino Y. A novel DNA polymerase in the hyperthermophilic archaeon, Pyrococcus furiosus: gene cloning, expression, and characterization. Genes Cells 1997;2:499–512. [DOI] [PubMed] [Google Scholar]
- 4.Uemori T, Ishino Y, Toh H, Asada K, Kato I. Organization and nucleotide sequence of the DNA polymerase gene from the archaeon Pyrococcus furiosus. Nucleic Acids Res 1993;21:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McInerney P, Adams P, Hadi MZ. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol Biol Int 2014;2014:287430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bing LI, Cheng GU, Jindong Z. Molecular cloning and expression of Pfu DNA polymerase gene and its application in long-distance PCR. Chin Sci Bull 1998;43:863–867. [Google Scholar]
- 7.Chae YKJW, Kyoung Suk CHO. Rapid and simple method to prepare functional Pfu DNA polymerase expressed in Escherichia coli periplasm. J Microbiol Biotechnol 2002;12:841–843. [Google Scholar]
- 8.Dabrowski S, Kur J. Cloning and expression in Escherichia coli of the recombinant his-tagged DNA polymerases from Pyrococcus furiosus and Pyrococcus woesei. Protein Expr Purif 1998;14:131–138. [DOI] [PubMed] [Google Scholar]
- 9.Hu JH, Wang F, Liu CZ. Development of an efficient process intensification strategy for enhancing Pfu DNA polymerase production in recombinant Escherichia coli. Bioprocess Biosyst Eng 2015;38:651–659. [DOI] [PubMed] [Google Scholar]
- 10.Sun Z, Cai J. Purification of recombinant Pfu DNA polymerase using a new JK110 resin. Korean J Chem Eng 2006;23:607–609. [Google Scholar]
- 11.Lu C, Erickson HP. Expression in Escherichia coli of the Thermostable DNA Polymerase from Pyrococcus furiosus. Protein Expr Purif 1997;11:179–184. [DOI] [PubMed] [Google Scholar]
- 12.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Śpibida M, Krawczyk B, Olszewski M, Kur J. Modified DNA polymerases for PCR troubleshooting. J Appl Genet 2017;58:133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SW, Kim D-UU, Kim JK, Kang L-WW, Cho H-SS. Crystal structure of Pfu, the high fidelity DNA polymerase from Pyrococcus furiosus. Int J Biol Macromol 2008;42:356–361. [DOI] [PubMed] [Google Scholar]
- 15.Ding Y, Liu S, Qi Q. Preparation of Taq DNA polymerase by thermal purification. Anhui Agric Sci 2011;12:375–378. [Google Scholar]
- 16.Chen S, Zheng X, Cao H, Jiang L, Liu F, Sun X. A simple and efficient method for extraction of Taq DNA polymerase. Electron J Biotechn 2015:8–11. [Google Scholar]