

Abstract
Background
Chrysin is a strong inhibitor of breast cancer resistance protein (BCRP) but it is practically insoluble in water. Effective solubilization of chrysin is critical for its pharmaceutical application as an absorption enhancer via inhibition of BCRP-mediated drug efflux.
Objective
This study aimed to develop an effective oral formulation of chrysin to improve its in vivo effect as an absorption enhancer.
Method
Solid dispersions (SDs) of chrysin were prepared with hydrophilic carriers having surface acting properties and a pH modulator. In vitro and in vivo characterizations were performed to select the optimal SDs of chrysin.
Results
SDs with Brij®L4 and aminoclay was most effective in increasing the solubility of chrysin by 13-53 fold at varying drug-carrier ratios. Furthermore, SDs significantly improved the dissolution rate and extent of drug release. SDs (chrysin: Brij®L4: aminoclay=1:3:5) achieved approximately 60% and 83% drug release within 1 h and 8 h, respectively, in aqueous medium, while the dissolution of the untreated chrysin was less than 13%. XRD patterns indicated the amorphous state of chrysin in SDs. The SD formulation was effective in improving the bioavailability of topotecan, a BCRP substrate in rats. Following oral administration of topotecan with the SDs of chrysin, the Cmax and AUC of topotecan was enhanced by approximately 2.6- and 2-fold, respectively, while the untreated chrysin had no effect.
Conclusion
The SD formulation of chrysin with Brij®L4 and aminoclay appeared to be promising in improving the dissolution of chrysin and enhancing its in vivo effect as an absorption enhancer.
Keywords: Chrysin, solid dispersion, BCRP inhibitor, dissolution, topotecan, ATP-binding cassette (ABC)
1. INTRODUCTION
Breast Cancer Resistance Protein (BCRP), an ATP-Binding Cassette (ABC) transporter, involves in tumor cell resistance against diverse chemotherapeutic drugs by actively extruding the drugs from cells [1]. Moreover, it is highly expressed in the apical membrane of the intestinal epithelium and often leads to the limited oral absorption of anticancer drugs including topotecan [2, 3]. The pharmaceutical application of BCRP inhibitors may therefore lead to more effective oral chemotherapy with anticancer drugs belonging to the BCRP substrates. Consequently, a significant need exists for more effective and safer inhibitors to enhance the bioavailability and in vivo efficacy of BCRP substrate drugs.
Flavonoids are common dietary components with a long history of human consumption [4] and BCRP inhibitors derived from flavonoids are therefore expected to have the advantage of low toxicity. Chrysin (5,7-dihydroxyflavone) is a natural flavone present in several plants and honeycombs [5]. It exhibits various pharmacological activities including antioxidant, anti-inflammatory, antihypertensive, antidiabetic, and anticancer effects [5-10]. In addition, chrysin is one of the most potent BCRP inhibitors with an IC50 of 0.39 ± 0.13 μM against mitoxantrone efflux [11]. Therefore, chrysin was expected to improve the cellular accumulation of BCRP substrates by reducing BCRP-mediated drug efflux. However, the in vivo effects of chrysin were found to be very limited despite its in vitro potency as a BCRP inhibitor [11, 12]. A possible reason for this discrepancy may be the low aqueous solubility and low bioavailability of chrysin, although additional causes cannot be excluded [13-15]. Chrysin is soluble in some organic solvents but is practically insoluble in water [14-17]. Therefore, effective solubilization of chrysin is critical for its pharmaceutical application as an absorption enhancer via inhibition of BCRP-mediated drug efflux.
Among the various formulation techniques, solid dispersion (SD) formulation is one of the effective approaches for the enhancement of the solubility and dissolution rate of many poorly water soluble drugs [18-21]. In SDs, hydrophobic drugs may exist in amorphous forms driving improved dissolution due to their highly disordered structure and higher Gibbs free energy compared with crystalline materials [22-25]. In addition, agglomeration of drug particles is prevented by interactions between the drug and the carrier, and wettability of drug molecules is improved by water absorption or dissolution of hydrophilic carriers surrounding the drug particles, leading to rapid drug absorption as released in a supersaturated state [21]. In particular, SDs prepared using surfactants may stabilize solid dispersions, avoiding drug recrystallization [26, 27]. Furthermore, pH modifiers can alter pH of the internal and external microenvironment of the SDs and enhance drug solubility. Therefore, in the present study, ternary SDs of chrysin were prepared with hydrophilic carriers having surface acting properties and also with a pH modulator. In vitro characterization was performed to select the optimal SD formulation of chrysin. Next, the in vivo effectiveness of the SD formulation as an absorption enhancer was evaluated in rats using topotecan as a typical BCRP substrate drug.
2. MATERIALS AND METHODS
2.1. Materials
Chrysin, poloxamer 407 (Pluronic® F-127), D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS), polyoxyethylene (4) lauryl ether (Brij®L4 (formerly Brij 30)) and 3-aminopropyltriethoxysaline (ATPES, 99%) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Macrogol 15 Hydroxystearate (Kolliphor® HS 15) was obtained from BASF (Ludwigshafen, Germany). Magnesium chloride hexahydrate (98.0%) and other inorganic salts were supplied by Junsei Chemical Co., Ltd. (Tokyo, Japan). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and all other reagents used in cell culture studies were obtained from GE Healthcare Life Sciences (South Logan, UT, USA). All other chemicals were of analytical grade and all solvents were of HPLC grade.
2.2. Cells
HT29 cells were obtained from the Korean Cell Line Bank (Seoul, Korea). Cells were grown in DMEM containing 10% FBS and 1% antibiotics, and incubated at 37 ºC in an atmosphere of 5% CO2 and 90% relative humidity.
2.3. Preparation of Solid Dispersions (SDs) and Physical Mixtures (PMs)
For the selection of optimal carriers, SDs of chrysin were prepared with different surface active carriers at a weight ratio of 1:5 by using the solvent evaporation method. In addition, SDs were prepared with or without aminoclay, a pH modulator, at different weight ratios. Aminoclay was prepared following the method reported by Yang et al. [28]. In brief, 3-aminopropyltriethoxysilane (2.6 mL, 11.7 mmol) was added dropwise with stirring to magnesium chloride (1.68 g, 7.24 mmol) in ethanol (40 g). A white precipitate formed immediately and was stirred overnight. After centrifugation, the resulting product was washed three times in ethanol, and dried in oven at 40°C.
For the preparation of SDs, chrysin was dissolved in acetone and each carrier dissolved in acetone or ethanol was added to the drug solution. After vigorous mixing, all the solvents were eliminated under vacuum at room temperature. The obtained product was milled and sieved through 50-mesh screen. PMs were also prepared by mixing chrysin, carrier and aminoclay in a mortar and pestle. To determine the optimal composition, SDs and PMs were prepared at various weight ratios of each component.
2.4. Solubility Studies
Each formulation (drug amount equivalent to 1 mg of chrysin) was dissolved in 1 mL of distilled water and stirred at 600 rpm for 24 h at room temperature. After the centrifugation at 13,000 rpm for 10 min, the supernatant was filtered through 0.45 µm pore-sized cellulose syringe filter (Target®, National scientific, USA). The filtrates were diluted with ethanol and the drug concentration was measured using HPLC.
2.5. Differential Scanning Calorimetry (DSC), X-Ray Diffraction (XRD), and Scanning Electron Microscopy
Thermal transition properties of samples were determined using a DSC Q2000 (TA Instruments, Ghent, Belgium) equipped with an intercooler. Indium was used to calibrate the temperature scale and enthalpy response. Samples were placed in aluminum pans and heated at a scanning rate of 10°C/min from 20°C to 300°C.
XRD patterns were examined at room temperature using an X-ray diffractometer (X’Pert APD, PHILIPS, Amsterdam, Netherlands). The diffraction pattern was obtained with CuKα radiation at 40 kV and 30 mA. Scans were recorded over a 2θ range of 3°-40° using a step size of 0.05° at a scanning speed of 2°/min. The XRD study was conducted at the Korea Basic Science Institute (Daegu Center, Korea).
The morphology of SD formulation was examined using scanning electron microscopy (SEM). Samples were spread on specimen stub using double-sided sticky tape, coated with platinum, and examined by an analytical high resolution scanning electron microscope (SU-70; Hitachi, Tokyo, Japan).
2.6. Dissolution Studies
Dissolution tests were performed in a dissolution tester DT 1420 (ERWEKA, Heusenstamm, Germany) at a paddle rotation speed of 50 rpm in 900 mL of aqueous media containing 0.25% Tween 80 at 37°C. Each formulation (drug amount equivalent to 50 mg of chrysin) was filled into a hard gelatin capsule with 30% Ac-Di-Sol and 1% talc and exposed to dissolution medium. One milliliter of each sample was withdrawn at the predetermined time points (0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, and 8 h) and an equal volume of fresh medium was added into the vessel to maintain a constant volume of the dissolution medium. The collected samples were filtered through 0.2 µm pore-sized cellulose syringe filters. The filtrates were diluted with the mobile phase and the drug concentration was determined using HPLC assay.
2.7. In vitro Cytotoxicity Study
The cytotoxicity of chrysin and chrysin SD was evaluated in HT29 cells by using MTT assay. Cells were seeded at 1×105 cells per well of 96-well plates. After incubation for 24 h, the cells were treated with chrysin or chrysin SD at the concentrations equivalent to 5-160 μM of chrysin and incubated for 48 h. At the end of incubation, thiazolyl blue tetrazolium bromide was added to each well and incubated for 4 h. The medium was removed, and DMSO (100 μL) was added to dissolve the formazan crystals. The absorbance of each sample was determined by a microplate reader at 550 nm.
2.8. Pharmacokinetic Studies in Rats
Animal studies were carried out in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (USA) and the study protocol was approved by the review committee of Dongguk University (IACUC-2017-016-1). Male Sprague-Dawley rats (250-290 g) were supplied by Orient bio Co., Ltd. (Seongnam, Korea). All rats were given free access to tap water and a normal standard chow diet (Superfeed Company, Wonju, Korea). SD rats were fasted for 12 h before the experiments, and divided into three groups (n=3 per group). The untreated drug or SD (equivalent to 100 mg/kg of chrysin) was suspended in 0.5% aqueous methylcellulose with 5% PEG and topotecan (10 mg/kg) was dissolved in saline. Each drug solution was given orally to the three groups of rats (group 1, topotecan only; group 2, topotecan + untreated chrysin; group 3, topotecan + chrysin SD). Blood samples were obtained from the femoral artery at the predetermined time points. Blood samples were centrifuged at 13,000 rpm for 5 min and the obtained plasma samples were kept frozen at -80 °C until analyzed by HPLC.
2.9. HPLC Assay
2.9.1. In Vitro Samples
The concentration of chrysin was determined using a high performance liquid chromatography (HPLC) system (Perkin Elmer series 200), consisting of a UV detector, a pump and an automatic injector. A reversed-phase C18 column (Gemini C18, 4.6x250 mm, 5 µm; Phenomenex, Torrance, CA, USA) was eluted with a mobile phase consisting of acetonitrile and 0.1% formic acid (60:40, v/v) at a flow rate of 1.0 mL/min at 30°C. The UV wavelength was set at 267 nm. Nimodipine was used as an internal standard and the calibration curve of chrysin was linear (r2 = 0.99) within the concentration range of 0.5-20 µg/mL.
2.9.2. In Vivo Plasma Samples
The concentration of topotecan in rat plasma was determined using HPLC assay. Briefly, 40 μL of samples were spiked with 4 μL of mesalazine (internal standard), and then 120 μL of acetonitrile and 100 μL of 1% HCl were added with vigorous mixing. After centrifugation at 13,000 rpm for 5 min, aliquots of the supernatants were injected into the HPLC. The mobile phase consisted of acetonitrile and 10 mM sodium phosphate buffer (16:84, v/v at pH 3.7) and the flow rate was 1.0 mL/min at 30°C. The fluorescence detector was set at an excitation wavelength of 350 nm and emission wavelength of 527 nm. The calibration curve of topotecan in rat plasma was linear (r2 = 0.99) within the concentration range of 10-1000 ng/mL.
2.10. Pharmacokinetic Analysis and Statistical Analysis
Based on noncompartmental analysis, the area under the plasma concentration-time curve (AUC) was calculated using the linear trapezoidal method. The peak plasma concentration (Cmax) and the time to reach the peak plasma concentration (Tmax) were values recorded from the experimental data.
Data were represented as mean values with standard deviation (SD). Statistical analysis was performed using one-way ANOVA followed by Dunnett’s test. A p value <0.05 was considered statistically significant.
3. RESULTS AND DISCUSSION
3.1. Carrier Selection and Preparation of Solid Dispersion
Because the solubilization of chrysin is a critical factor to ensure the adequate in vivo performance of chrysin as a BCRP inhibitor for enhancing oral absorption of BCRP substrates, SDs of chrysin were prepared with various hydrophilic carriers. In particular, given that surfactants or self-emulsifying agents are effective in stabilizing solid dispersions by avoiding drug recrystallization, the four surface active carriers such as TPGS, Kolliphor® HS 15, Brij® L4, and poloxamer 407 were selected to prepare the chrysin SDs, and their effects on improving the solubility of chrysin were determined in water. As shown in Table 1, all SD formulations enhanced significantly the aqueous solubility of chrysin. Among the tested carriers, Brij®L4 (Brij 30) was the most effective in enhancing the solubility of the chrysin. Given that Brij®L4, a nonionic surfactant, has amphiphilic properties consisting of hydrophobic and hydrophilic regions, it could form micelles and distribute hydrophobic drugs into the core of micelles, resulting in improved drug solubility [29, 30]. In addition, previous studies have reported a strong inhibitory effect of Brij®L4 against BCRP-mediated drug efflux [31] and thereby, the presence of Brij®L4 in SD formulation of chrysin may have a synergistic effect on BCRP inhibition. Therefore, in the present study, Brij®L4 was selected as a hydrophilic carrier to prepare chrysin SDs.
Table 1. Solubility of chrysin in various formulations (Mean±SD, n=3).
| Formulation(w/w) | Solubility (μg/mL) |
|---|---|
| chrysin (untreated drug) | 3.038 ± 0.19 |
| chrysin: TPGS = 1:5 | 27.39 ± 0.33 |
| chrysin: Kolliphor® HS 15= 1:5 | 25.52 ± 0.43 |
| chrysin: Brij®L4 = 1:5 | 37.81 ± 0.59 |
| chrysin: Poloxamer 407 = 1:5 | 10.14 ± 0.17 |
| chrysin: Brij®L4:aminoclay = 1:5:1 | 38.08 ± 0.58 |
| chrysin: Brij®L4:aminoclay = 1:5:2 | 50.49 ± 0.55 |
| chrysin: Brij®L4:aminoclay = 1:5:5 | 97.55 ± 0.89 |
For the adsorption of semisolid SDs and modulation of microenvironmental pH, aminoclay (3-aminopropyl functionalized magnesium phyllosilicate) was also added to the SD formulations. After the amine group of aminoclay is protonated in water, aminoclay is delaminated to cationic and water soluble nanosheets [32]. Aminoclay can drive the pH of the internal and external microenvironment of the SDs to a more basic state [28]. As the pKa1 of chrysin is 6.72, chrysin could be deprotonated at a high pH and be more soluble in basic environments [17]. Accordingly, the drug solubilizing effect of SDs increased as the amount of aminoclay increased in SDs (Table 1). Finally, the combination of Brij® L4 and aminoclay was chosen to prepare chrysin SDs and PMs in subsequent studies.
The solubility of chrysin increased in both PMs and SDs as the drug-carrier ratio was increased (Table 2 and Fig. 1). Given that the concentration of Brij®L4 used in the SD formulation was greater than its critical micelle concentration (CMC) (0.00097%, w/v) [33], drug distribution into the micelles might have been increased with the increase in Brij®L4 concentration, leading to enhanced drug solubility. Consequently, the SDs increased the solubility of chrysin by 13-53 fold depending on the drug-carrier ratios. However, the SDs appeared to be more effective in improving the solubility of chrysin than the corresponding PMs did, implying that there might have been the additional factors affecting the drug solubility other than micellar solubilization, such as the change of drug crystallinity to an amorphous form in the SD formulations. Therefore, their structural characteristics were examined using DSC and XRD.
Table 2. Solubility of chrysin in SDs at the different drug-carrier ratios (Mean±SD, n=3).
| Formulation(w/w) | Solubility (μg/mL) |
|---|---|
| chrysin: Brij®L4:aminoclay = 1:1:5 | 42.92 ± 0.60 |
| chrysin: Brij®L4:aminoclay = 1:3:5 | 92.72 ± 0.38 |
| chrysin: Brij®L4:aminoclay = 1:5:5 | 97.55 ± 0.89 |
| chrysin: Brij®L4:aminoclay = 1:10:5 | 126.0 ± 0.43 |
| chrysin: Brij®L4:aminoclay = 1:20:5 | 159.7 ± 0.27 |
Fig. (1).
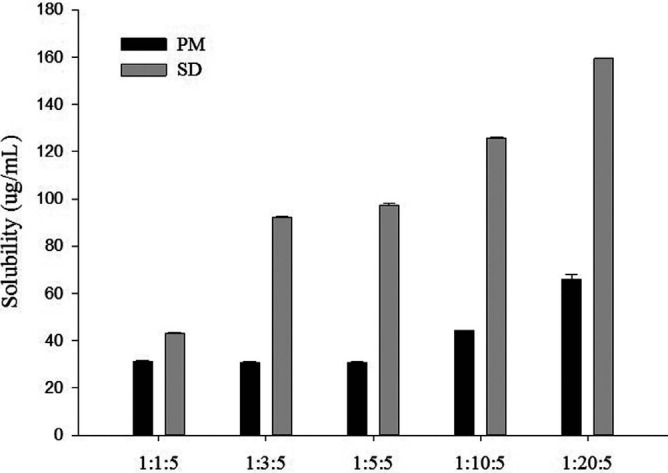
Solubility of chrysin in SDs and PMs with Brij®L4 and aminoclay at various weight ratios (Mean±SD, n=3).
3.2. Structural and Morphological Characteristics
As illustrated in Fig. (2A), the thermograms of untreated drug, aminoclay, PMs, and SDs were analyzed using differential scanning calorimetry (DSC). The untreated chrysin (conventional powder formulation) clearly showed a sharp and single endothermic peak corresponding to its melting point at 288°C, which is consistent with a previous report [34]. However, this peak disappeared in the PMs and SDs, which may be because of drug dissolution in the melted carriers. In addition, X-ray diffraction was used to examine the crystallinity of chrysin in each formulation. XRD patterns of untreated drug, aminoclay, PMs, and SDs are shown in Fig. (2B). The untreated chrysin exhibited the multiple distinct peaks at 7.5°, 12.7°, 14.9°, 17.8°, and 27.9°, which were also observed in the PMs. However, these distinct peaks of crystalline chrysin disappeared in the SD formulations, implying that drug crystallinity was changed to the amorphous form. Therefore, in addition to the micelle-solubilizing effect of the hydrophilic carriers, the amorphous state of chrysin may also have contributed to the enhanced solubility of chrysin in the SD formulations.
Fig. (2).
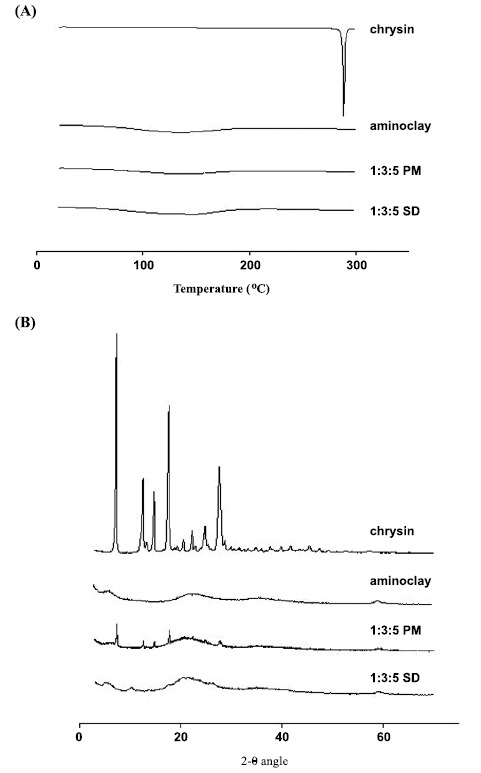
DSC thermograms (A) and X-ray diffraction patterns (B) of chrysin, aminoclay, PMs and SDs.
The morphology of the SD formulation was observed by SEM (Fig. 3). The SDs exhibited a homogeneous blend of all ternary components in irregular-shaped particles, indicating the amorphous state of drug.
Fig. (3).

SEM image of the SD formulation.
3.3. In vitro Dissolution Study
The dissolution profiles of the SD formulations were evaluated in comparison with those of the untreated drug and the PMs. As shown in Fig. (4A), the dissolution of untreated drug over a period of 8 h was less than 13%. Compared with the untreated drug, the SDs and PMs significantly increased the dissolution rate and extent of drug release. In particular, SDs (chrysin: Brij®L4: aminoclay=1:3:5) achieved approximately 60% and 83% drug release in water within 1 h and 8 h, respectively. This may be explained by several factors including improved drug wetting and micellar solubilization by the hydrophilic carriers, increased microenvironmental pH in the presence of aminoclay, and the amorphous state of the drug in the SD formulations. As explained in section 3.1., chrysin is known to be more soluble in alkaline solutions, thus, the dissolution of chrysin in the present study also exhibited pH-dependency. All the tested formulations showed a low extent of drug release at acidic conditions (pH 1.2 and pH 4.0), but the SD formulation could still achieve significantly higher dissolution than untreated chrysin (Fig. 4B and 4C).
Fig. (4).
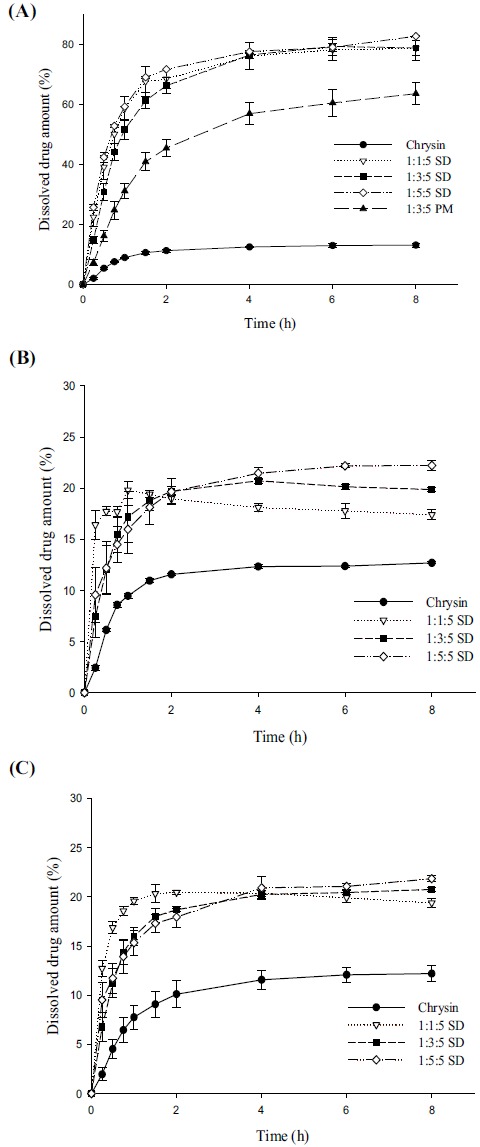
Dissolution profiles of chrysin in different formulations in water (A), in pH 1.2 buffer (B), and in pH 4.0 buffer (C) (Mean±SD, n=3).
3.4. In vitro Cytotoxicity
The cytotoxicity of chrysin and chrysin SD was evaluated in HT29 cells, the human colorectal carcinoma cells. As shown in Fig. (5), chrysin SD exhibited higher cytotoxicity than chrysin, where the CC50 value of chrysin SD was 26.3 μM but the cytotoxicity of untreated chrysin was low even at 160 μM. This result may be explained by the enhanced solubilization of chrysin by SD formulation. Therefore, chrysin SD may have a synergistic effect in the combination therapy with other anticancer drugs.
Fig. (5).
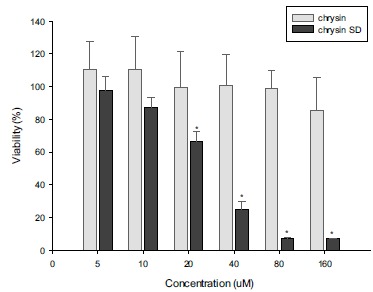
Cytotoxicity of chrysin and chrysin SD in HT29 cells (Mean±SD, n=6). *p<0.05.
3.5. In vivo Pharmacokinetics
The in vivo effectiveness of the SD formulations as an absorption enhancer was evaluated in rats using topotecan as a typical BCRP substrate drug. Topotecan was administered orally in the presence and the absence of chrysin in different formulations and then the plasma concentration-time profiles of topotecan were determined as summarized in Table 3 and Fig. (6).
Table 3. Plasma concentration-time profiles of topotecan after an oral administration of topotecan (10mg/kg) in the presence and the absence of chrysin in different formulations to rats (Mean±SD, n=3).
| Control (Topotecan Only) | Topotecan + Untreated Chrysin | Topotecan + Chrysin SD | |
|---|---|---|---|
| Cmax (ng/mL) | 109.4 ± 44.55 | 115.9 ± 40.63 | 278.7 ± 114.2 |
| Tmax (h) | 1.3 ± 0.66 | 0.83 ± 0.14 | 0.50 ± 0.00 |
| AUC (ng* h/mL) | 577.8 ± 37.21 | 522.3 ± 153.9 | 1113 ± 141.6* |
*p < 0.05, compared to the control.
Fig. (6).
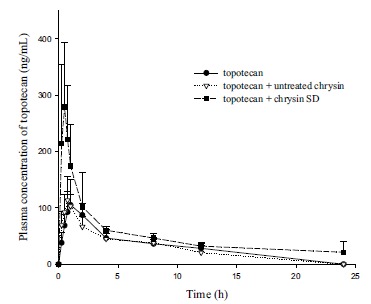
Plasma concentration-time profiles of topotecan after an oral administration of topotecan (10 mg/kg) to rats in the presence or absence of chrysin in different formulations (Mean±SD, n=3).
The SD formulations of chrysin significantly improved oral exposure of topotecan in rats, while the untreated chrysin had no effect. The Cmax and AUC of topotecan was enhanced by approximately 2.6- and 2-fold, respectively, via the current use of chrysin in the SD formulations. These results may be attributed to the effective inhibition of drug efflux by chrysin in SD formulations during intestinal absorption. Since topotecan undergoes very limited metabolism
[35], the interaction of chrysin with drug-metabolizing enzymes can be excluded at this point. Considering that (i) BCRP is highly expressed in the apical membrane of intestines [2], (ii) topotecan is a BCRP substrate limiting oral absorption [3], and (iii) the affinity of topotecan for P-gp is low [36], it is most likely that the enhanced solubility and dissolution of chrysin by the SD formulation could increase the luminal concentration of chrysin in the GI tract and led to an improved in vivo effect of chrysin against BCRP-mediated drug efflux during the intestinal absorption of topotecan.
Given that the cytotoxicity of chrysin was significantly enhanced by SD formulation (Fig. 5), the concurrent use of chrysin SD may have a synergistic effect in chemotherapy via the inhibition of BCRP-mediated efflux of anticancer drugs and also the cytotoxic effect of chrysin.
CONCLUSION
The SD formulation of chrysin with Brij®L4 and aminoclay appeared to be promising in improving the dissolution of chrysin and enhancing its in vivo effects as an absorption enhancer.
ACKNOWLEDGEMENTS
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2016R1A2B2010097 and No. 2018R1A5A2023127), and by the research program of Dongguk University in 2017.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study protocol was approved by the review committee of Dongguk University (IACUC-2017-016-1).
HUMAN AND ANIMAL RIGHTS
No Humans were used for studies that are basis of this Research. The Animal studies were carried out in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (USA).
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Mao Q. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005;7:E118–E133. doi: 10.1208/aapsj070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taipalensuu J., Tornblon H., Lindberg G., Einarsson C., Sjoqvist F., Melhus H., Garberg P., Sjostrom B., Lundgren B., Artursson P. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial CaCo-2 cell monolayers. J. Pharmacol. Exp. Ther. 2001;299:164–170. [PubMed] [Google Scholar]
- 3.Jonker J.W., Smit J.W., Brinkhuis R.F., Maliepaard M., Beijnen J.H., Schellens J.H., Schinkel A.H. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J. Natl. Cancer Inst. 2000;92:1651–1656. doi: 10.1093/jnci/92.20.1651. [DOI] [PubMed] [Google Scholar]
- 4.Havsteen B.H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002;96:67–202. doi: 10.1016/s0163-7258(02)00298-x. [DOI] [PubMed] [Google Scholar]
- 5.Mani R., Natesan V. Chrysin: Sources, beneficial pharmacological activities, and molecular mechanism of action. Phytochemistry. 2018;145:187–196. doi: 10.1016/j.phytochem.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y.H., Yang Z.S., Wen C.C., Chang Y.S., Wang B.C., Hsiao C.A., Shih T.L. Evaluation of the structure-activity relationship of flavonoids as antioxidants and toxicants of zebrafish larvae. Food Chem. 2012;134:717–724. doi: 10.1016/j.foodchem.2012.02.166. [DOI] [PubMed] [Google Scholar]
- 7.Gresa‐Arribas N., Serratosa J., Saura J., Solà C. Inhibition of CCAAT/enhancer binding protein δ expression by chrysin in microglial cells results in anti‐inflammatory and neuroprotective effects. J. Neurochem. 2010;115:526–536. doi: 10.1111/j.1471-4159.2010.06952.x. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto Y. Effects of dietary chrysin supplementation on blood pressure and oxidative status of rats fed a high-fat high-sucrose diet. Food Sci. Technol. Res. 2014;20:295–300. [Google Scholar]
- 9.Samarghandian S., Azimin-Nezhad M., Samini F., Farkhondeh T. Chrysin treatment improves diabetes and its complications in streptozotocin-induced diabetic rat. Can. J. Physiol. Pharmacol. 2016;94:388–393. doi: 10.1139/cjpp-2014-0412. [DOI] [PubMed] [Google Scholar]
- 10.Khoo B.Y., Chua S.L., Balaram P. Apoptotic effects of chrysin in human cancer cell lines. Int. J. Mol. Sci. 2010;11:2188–2199. doi: 10.3390/ijms11052188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S., Yang X., Morris M.E. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol. Pharmacol. 2004;65:1208–1216. doi: 10.1124/mol.65.5.1208. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S., Wang X., Sagawa K., Morris M.E. Flavonoids chrysin and benzoflavone, potent breast cancer resistance protein inhibitors, have no significant effect on topotecan pharmacokinetics in rats or mdr1a/1b (-/-) mice. Drug Metab. Dispos. 2005;33:341–348. doi: 10.1124/dmd.104.002501. [DOI] [PubMed] [Google Scholar]
- 13.Walle T., Otake Y., Brubaker J.A., Walle U.K., Halushka P.V. Disposition and metabolism of the flavonoid chrysin in normal volunteers. Br. J. Clin. Pharmacol. 2001;51:143–146. doi: 10.1111/j.1365-2125.2001.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou L., Zhang P., Yang G., Lin R., Wang W., Liu T., Zhang L., Zhang J. solubility of chrysin in ethanol and water mixtures. J. Chem. Eng. Data. 2014;59:2215–2220. [Google Scholar]
- 15.Kwon Y.E., Kim H.M., Park S.Y., Jung S.H. Enhancement of solubility and antioxidant activity of some flavonoids based on the inclusion complexation with sulfobutylether β-cyclodextrin. Bull. Korean Chem. Soc. 2010;31:3035–3037. [Google Scholar]
- 16.Sassa-Deepaeng T., Pikulkaew S., Okonogi S. Development of chrysin loaded poloxamer micelles and toxicity evaluation in fish embryos. Drug Discov. Ther. 2016;10:150–155. doi: 10.5582/ddt.2016.01039. [DOI] [PubMed] [Google Scholar]
- 17.Castro G.T., Ferretti F.H., Blanco S.E. Determination of the overlapping pK(a) values of chrysin using UV-vis spectroscopy and ab initio methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005;62:657–665. doi: 10.1016/j.saa.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen M.N.U., Vo T.V., Tran P.H.L., Tran T.T.D. Zein-based solid dispersion for potential application in targeted delivery. J. Pharm. Investig. 2017;47:357–364. [Google Scholar]
- 19.Singh J., Walia M., Harikumar S. Solubility enhancement by solid dispersion method: A review. J. Drug Deliv. Sci. Technol. 2013;3:148–155. [Google Scholar]
- 20.Yamashita S., Fukunishi A., Higashino H., Kataoka M., Wada K. Design of supersaturable formulation of telmisartan with pH modifier: In vitro study on dissolution and precipitation. J. Pharm. Investig. 2017;47:163–171. [Google Scholar]
- 21.Vo C.L.N., Park C., Lee B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013;85:799–813. doi: 10.1016/j.ejpb.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Liu X., Feng X., Williams R.O., III, Zhang F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2018;48:19–41. [Google Scholar]
- 23.Ziaee A., Albadarin A.B., Padrela L., Faucher A., O’Reilly E., Walker G. Spray drying ternary amorphous solid dispersions of ibuprofen - An investigation into critical formulation and processing parameters. Eur. J. Pharm. Biopharm. 2017;120:43–51. doi: 10.1016/j.ejpb.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Hwang I., Kang C-Y., Park J-B. Advances in hot-melt extrusion technology toward pharmaceutical objectives. J. Pharm. Investig. 2017;47:123–132. [Google Scholar]
- 25.Prasad D., Chauhan H., Atef E. Amorphous stabilization and dissolution enhancement of amorphous ternary solid dispersions: combination of polymers showing drug-polymer interaction for synergistic effects. J. Pharm. Sci. 2014;103:3511–3523. doi: 10.1002/jps.24137. [DOI] [PubMed] [Google Scholar]
- 26.Seo S.W., Han H.K., Chun M.K., Choi H.K. Preparation and pharmacokinetic evaluation of curcumin solid dispersion using Solutol® HS15 as a carrier. Int. J. Pharm. 2012;424:18–25. doi: 10.1016/j.ijpharm.2011.12.051. [DOI] [PubMed] [Google Scholar]
- 27.Karata A., Yuksel N., Baykara T. Improved solubility and dissolution rate of piroxicam using gelucire 44/14 and labrasol. Farmaco. 2005;60:777–782. doi: 10.1016/j.farmac.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 28.Yang L., Shao Y., Han H.K. Improved pH-dependent drug release and oral exposure of telmisartan, a poorly soluble drug through the formation of drug-aminoclay complex. Int. J. Pharm. 2014;471:258–263. doi: 10.1016/j.ijpharm.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Edwards D.A., Luthy R.G., Liu Z. Solubilization of polycyclic aromatic hydrocarbons in micellar nonionic surfactant solutions. Environ. Sci. Technol. 1991;25:127–133. [Google Scholar]
- 30.Jafvert C.T., Van Hoof P.L., Heath J.K. Solubilization of non-polar compounds by non-ionic surfactant micelles. Water Res. 1994;28:1009–1017. [Google Scholar]
- 31.Yamagata T., Kusuhara H., Morishita M., Takayama K., Benameur H., Sugiyama Y. Effect of excipients on breast cancer resistance protein substrate uptake activity. J. Control. Release. 2007;124:1–5. doi: 10.1016/j.jconrel.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 32.Datta K.K.R. Achari, A.; Eswaramoorthy, M. Aminoclay: A functional layered material with multifaceted applications. J. Mater. Chem. A Mater. Energy Sustain. 2013;1:6707–6718. [Google Scholar]
- 33.Kim I.S., Park J-S., Kim K-W. Enhanced biodegradation of polycyclic aromatic hydrocarbons using nonionic surfactants in soil slurry. Appl. Geochem. 2001;16:1419–1428. [Google Scholar]
- 34.Chadha R., Bhalla Y., Nandan A., Chadha K., Karan M. Chrysin cocrystals: Characterization and evaluation. J. Pharm. Biomed. Anal. 2017;134:361–371. doi: 10.1016/j.jpba.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Platzer P., Schaden S., Thalhammer T., Hamilton G., Rosenberg B., Silgoner I., Jager W. Biotransformation of topotecan in the isolated perfused rat liver: Identification of three novel metabolites. Anticancer Res. 1998;18:2695–2700. [PubMed] [Google Scholar]
- 36.Kruijtzer C.M.F., Beijnen J.H., Rosing H., ten Bokkel Huinink W.W., Schot M., Jewell R.C., Paul E.M., Schellens J.H.M. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-Glycoprotein inhibitor GF120918. J. Clin. Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]