

Abstract
Work in yeast models has benefitted tremendously from the insertion of epitope or fluorescence tags at the native gene locus to study protein function and behavior under physiological conditions. In contrast, work in mammalian cells largely relies on overexpression of tagged proteins because high-quality antibodies are only available for a fraction of the mammalian proteome. CRISPR/Cas9-mediated genome editing has recently emerged as a powerful genome-modifying tool that can also be exploited to insert various tags and fluorophores at gene loci to study the physiological behavior of proteins in most organisms, including mammals. Here we describe a versatile toolset for rapid tagging of endogenous proteins. The strategy utilizes CRISPR/Cas9 and microhomology-mediated end joining repair for efficient tagging. We provide tools to insert 3×HA, His6FLAG, His6-Biotin-TEV-RGSHis6, mCherry, GFP, and the auxin-inducible degron tag for compound-induced protein depletion. This approach and the developed tools should greatly facilitate functional analysis of proteins in their native environment.
Keywords: CRISPR/Cas, auxin, protein engineering, gene expression, protein purification, protein degradation, AID, epitope tagging, microhomology
Introduction
The precision and simplicity of CRISPR-based gene modification has brought unprecedented advances in gene knockout and functional genomic studies (1–3). The Cas9 nuclease generates a DNA double-strand break (DSB)2 at sites selected by the co-expressed guide RNA (gRNA). The DSB is then repaired by either nonhomologous end joining (NHEJ) or homologous recombination (HR)–mediated repair. For gene knockouts, NHEJ randomly joins the two processed ends of DNA from the DSB site, which is efficient in generating insertion- or deletion-based gene knockouts. In contrast, HR is a favored method for gene knockins and tagging because it is a precise repair mechanism and allows in-frame modification. However, HR is extremely inefficient compared with NHEJ and requires a large fragment of a homologous DNA sequence corresponding to the genomic DNA surrounding the DSB site (4, 5). In contrast, microhomology-mediated end joining (MMEJ), a recently discovered high-fidelity alternative to HR, only requires about 20 bp of (micro)homology flanking the Cas9-induced DSB site (6, 7). An MMEJ-based mechanism has been shown previously to be effective in tagging endogenous proteins in human cells (7). Currently, four mammalian CRISPR tagging systems are available (4, 7, 8, 10). However, most of the available methods require labor-intensive cloning for large homologous sequences (200–700 bp) (4, 8, 10), and only one of them provides protein N terminus tagging, albeit with very low efficiency (2%) because of a lack of antibiotic selection markers (4). To address the need for an easy, efficient, and versatile N-terminal tagging method, we expanded the tagging concept from Yamamoto and co-workers (6, 7) by redesigning and optimizing MMEJ-based tools to allow N-terminal insertion. We provide a set of plasmids with a variety of tags that help to fully exploit the potential of MMEJ-mediated CRISPR protein tagging.
Conceptually, our system is based on a similar two-plasmid precise integration into target chromosome (PITCh) system (Fig. 1A) as described by Yamamoto and co-workers (7). Plasmid 1 (Fig. 1A, pX330-YFG-PITCH) is the Cas9-expressing vector, which also expresses two gRNAs. The target gRNA directs the cut at the genomic locus that is modified, and the PITCh gRNA targets the second plasmid (Fig. 1A, pN-PITCh-HA) to release the repair fragment that contains the tags flanked by microhomologies to the target locus. The inserted DNA fragment contains the tag, a puromycin-resistant gene for selection of successful integration, and GFP for visual assessment of tagging efficiency. These features are all expressed as one polycistronic mRNA separated by the “self-cleaving” 2A peptide, which leads to ribosome skipping (11–13) to produce the three polypeptides: GFP, PuroR, and tagged target protein. The tags available in our collection include 3×HA, His6FLAG, and HBTH (14, 15) for protein purification under native or denaturing conditions as well as immunoblotting, mCherry and GFP for protein intracellular localization, and the AID degron for auxin-induced conditional protein depletion (16) (Fig. 1B). The system is easily expandable with other tags and will be a useful tool set for scientists.
Figure 1.

Experimental schematic of MMEJ mediated N-terminal CRISPR knockin tagging. A, experimental schematic of MMEJ-mediated tagging. The PX330 vector expresses Cas9 and two gRNAs, one targeting the genomic locus of choice and the other one targeting the pN-PITCh vector to release the repair fragment containing the tag. The pN-PITCh vector contains GFP, the puromycin resistant marker, and the designated tag, all separated by self-cleaving T2A and P2A peptides. Two 20-bp microhomology regions corresponding to the regions adjoining the Cas9 cleavage site are flanking the insertion cassette. The pN-PITCh and PX330 vectors are co-transfected, and cells are selected with puromycin to identify the desired clones. B, pN-PITCh versions available for various tags. C, schematic for reconstitution of the native N-terminal sequences. For gRNAs that select the Cas9 cut site inside of the ORF, reconstitution is required and is achieved by including the removed 5′ ORF fragment into the pN-PITCh construct during cloning of the microhomology sequences. To prevent Cas9 cleavage of the repair fragment, either the gRNA targeting sequence or the PAM domain should be changed during the repair process.
Results and discussion
N terminus tagging of PP2AC with MMEJ-mediated repair
The MMEJ-mediated PITCh tagging system utilizes the pX330 vector, which expresses the Cas9 nuclease and two gRNAs. We rearranged the U6 promoter and PITCh gRNA directly after the target gRNA (YFG) cloning site that is flanked by BbsI to bypass the golden gate assembly step required for the parent plasmid (7). One of the gRNAs targets the genome to generate a DSB, whereas the other targets the PITCh vector to release the tagging cassette. The linearized DNA fragment from the PITCh vector is then integrated into the genome through short microhomologies via MMEJ-mediated repair (Fig. 1A) (7). MMEJ repair can occur during G1 and early S phase compared with HR repair, which is limited to the G2/M phase (17–19). This makes the PITCh system more efficient than HR-based approaches. In addition, the microhomology sequence required for MMEJ is extremely short (∼20 bp), which allows the sequence to be changed for different targets with a simple PCR procedure (Fig. 1A).
MMEJ-mediated PITCh tagging is very efficient, but currently available systems are limited to modifications with GFP at the C terminus of endogenous proteins (6, 7). Many proteins do not tolerate C-terminal tags, either because of structural distortion or because of modification sites. We therefore redesigned and optimized the MMEJ-based tagging approach for N-terminal tagging and expanded the repertoire of tags (Fig. 1B). This required rearranging the location of the GFP and the flanking microhomology sequence and addition of an additional 2A peptide. Compared with the Yamamoto system, which fuses GFP to the C terminus of the target protein, GFP expression in our system solely serves as a reporter for successful integration, which greatly reduces the number of samples for PCR and Western blot confirmation.
The need for N-terminal tagging tools became evident when we needed to tag the catalytic subunit of the protein phosphatase 2A (PP2AC). The C terminus of the essential PP2AC subunit is highly regulated by posttranslational modifications (20). The 309-amino-acid-long PP2AC is phosphorylated at Thr-304 and Tyr-307 and carboxylmethylated at Leu-309 (21), making C-terminal tagging impossible. We therefore re-engineered the C-terminal PITCh tagging system for N-terminal tagging (Fig. 1B). The N-terminal PITCh tagging cassette (pN-PITCh) starts with GFP, followed by the puromycin resistance gene and one of several available tags, all divided by self-cleaving 2A sequences for separation (Fig. 1B). The PITCh tagging cassette is flanked by 20-bp microhomology sequences to direct insertion at the desired genomic site by MMEJ (Fig. 1A). To test the efficiency of our approach, we designed a gRNA that targets the N terminus of the PP2AC ORF to insert the PITCh cassette containing GFP, PuroR, and 3×HA tag, all separated by 2A self-cleaving peptides.
Note that, depending on the exact Cas9 cleavage position, part of the nucleotide sequence encoding the N-terminal amino acids needs to be artificially added back into the PITCh cassette to prevent the loss of the amino acid sequence (Fig. 1C, the blue sequence on the repair template). We transiently transfected 293T cells with pX330X2-PP2Ac-PITCh, which expresses the Cas9 nuclease and the two gRNAs that target the PITCh cassette for release of the repair fragment and the genomic sequence encoding the PP2Ac N terminus, respectively (Fig. 1A).
Furthermore, we cloned the U6 promoter and PITCh gRNA directly after the target gRNA (YFG) cloning site that is flanked by BbsI to bypass the golden gate assembly step. Cells were co-transfected with the second plasmid, pN-PITCh-HA, which contains the repair fragment (Fig. 1B).
Three days after transfection, cells were selected using puromycin. Because the PuroR gene is driven by the endogenous promoter of the target gene (here PP2AC), only correctly integrated cassettes will be puromycin resistant, show a GFP signal, and express 3×HA-PP2AC. Depending on the target gene, the GFP expression driven by the endogenous promoter is typically weaker compared with overexpressed CMV promoter-driven GFP (Fig. 2A) but, nevertheless, is a useful visual indicator of successful integration. Single clones were selected and tested by immunoblotting with anti-PP2AC and anti-HA antibodies. Ten of 12 clones (83%) were CRISPR modified in both alleles and produced homozygous 3×HA-tagged PP2AC (Fig. 2B). One clone had no tagged PP2AC (#4), and another clone was heterozygous for tagged PP2AC (#10) (Fig. 2B). Note that there is a trace amount of lower molecular species of PP2Ac visible when protein extracts are probed with anti-PP2Ac antibodies (Fig. 2B). These species are not detected with anti-HA antibodies, suggesting nonspecific N-terminal degradation. This appears to be a problem caused by the HA tag only because no such degradation products were seen when other tags were fused to the PP2Ac N terminus (Fig. 2C).
Figure 2.

MMEJ-mediated N-terminal tagging of PP2Ac with HA and HBTH. A, GFP expression 2 days after puromycin selection of pN-PITCh-HA–transfected 293T cells. Exposure time for GFP was 12 s. B, HA-tagged PP2Ac was detected in single clones using HA antibodies and PP2Ac antibodies. 10 of 12 clones were homozygous knockins. Clone #10 is heterozygous for the tag. C, HBTH-PP2Ac was detected in single cell clones using RGS6H antibodies to recognize the RGSHis6 epitope, which is part of the tag. All analyzed clones were tagged on both alleles.
Expanding the tagging repertoire for protein purification and visualization
To broaden the application of our method, we generated a selection of different tags in the pN-PITCh system, including His6FLAG, mCherry, HBTH, and GFP (Fig. 1B). The N-terminal HBTH tagging construct was also tested on PP2AC, and six of six (100%) tested single colonies showed homozygous expression of HBTH-PP2AC (Fig. 2C). The HBTH tag is a combination of two copies of the His6 tag (HBTH) flanking an autonomous biotinylation peptide (HBTH) and a tobacco etch virus protease cleavage site (HBTH). This tag is useful for cross-linking MS, tandem purification under fully denaturing conditions, and general applications that benefit from highly stringent purification conditions (14, 15, 22). The HBTH and mCherry tags were also tested on the proteasome component RPN1 to evaluate application at a different genomic locus (Fig. 3A). Three of three (100%) tested single colonies were homozygous for HBTH-RPN1 (Fig. 3A). RPN1 is critical for 26S proteasome activity, as it coordinates substrate recruitment, deubiquitination, and movement toward the catalytic core (23). To test whether the N-terminal HBTH-tagged RPN1 is completely functional, we performed proteasome activity assays using fluorogenic substrate peptides. Proteasome activity was unaffected in cells expressing HBTH-tagged RPN1 (Fig. 3B). Leveraging the high affinity of the biotinylated HBTH tag for streptavidin beads, we purified the proteasome complex and analyzed the affinity-purified complex by SDS-PAGE and MS. Subunits of the catalytic 20S core particle and the 19S regulatory particle were readily visible by protein staining after separation by SDS-PAGE (Fig. 3C) (24). Mass spectrometry confirmed that all 26S subunits were purified and detected with high peptide coverage (Table S2).
Figure 3.

N-terminal tagging of the proteasome subunit RPN1 with HBTH and mCherry. A, HBTH-tagged RPN1 was detected using HRP-conjugated streptavidin and RPN1 antibodies. B, cells expressing HBTH-tagged RPN1 were lysed for proteasome activity assay using fluorogenic peptide as substrates. Fluorescent signals were normalized to total protein loaded to the assay. C, affinity purification of the proteasome using cells expressing HBTH-RPN1 and streptavidin beads. 19S and 20S proteasome subunits are visualized on a PVDF membrane with Amido Black stain. Mass spectrometry analysis confirmed that the pulldown had all proteasome subunits (Table S2). D, mCherry-RPN1 expression 3 days after puromycin selection.
Both mCherry and GFP tagging allow intracellular protein tracing. Although mCherry-tagged RPN1 expressed at the endogenous level only produces a weak fluorescent signal, it was enough to visualize intracellular RPN1 with extended exposure time (∼10-s exposure) (Fig. 3D). The N-terminal GFP tagging construct pN-PITCh-GFP was tested by tagging catalase. Catalase is a key antioxidant enzyme with a C-terminal peroxisome targeting sequence (25–27). GFP-catalase was readily detected and, as expected, colocalized with peroxisomes, which were visualized with red fluorescent protein (RFP) (Fig. 4).
Figure 4.
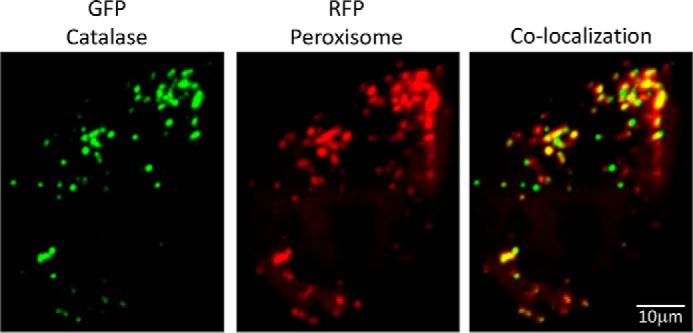
MMEJ-mediated GFP tagging and visualization of endogenous catalase. 293T cells selected after pN-PITCh-GFP tagging of catalase were transfected with RFP fused to a peroxisome targeting sequence to visualize peroxisomes. Cells were imaged 24 h after transfection.
Notably, when we attempted to tag the N terminus of PP2AC with GFP or mCherry, we were unable to retrieve viable cell clones with correct GFP or mCherry-fused PP2AC. This might be the result of spatial hindrance from bulky tags, such as GFP or mCherry, which abrogates essential protein interactions and thereby prevents formation of a functional PP2A holoenzyme complex. Because PP2AC is essential for cell viability, failure to obtain GFP- and mCherry-tagged PP2AC is a good indication that the tags interfere with protein function. Such negative effects on the functionality of proteins is less obvious for nonessential proteins, and, as with all tagging approaches, careful evaluation of protein function after tagging is important.
During gRNA design, we noticed that not all genes have gRNA target sites with good scores (>0.5 from a scale of 0 to 1, using the web tool developed by Doench et al. (28)). This situation requires placing the Cas9 cut site upstream of the start codon. To test the efficiency of using gRNA positions upstream of the start codon, we compared two gRNAs that target mRNA guanine-N7 methyltransferase (RNMT). gRNA1 targets the coding region, and placement of the microhomology region allows seamless repair. In contrast, gRNA2 targets more than 20 bp upstream of the start codon, and the microhomology regions needed to be designed so that the 20-bp noncoding fragment is removed by the repair machinery during MMEJ, producing the tag directly fused to the beginning of the ORF (Fig. 5A). In addition to providing the flexibility to choose optimal gRNAs, this approach also avoids the need to reinsert the lost ORF fragments when gRNAs target inside the ORF (Figs. 1C and 5A, gRNA1).
Figure 5.

Comparison of Cas9 cleavage site positions for N-terminal HBTH tagging of mRNA RNMT. A, schematic layout of the RNMT ORF region surrounding the start codon. Two gRNAs were selected using gRNA Designer (28). gRNA1 directs Cas9 cleavage 3′ of the start codon, and microhomology regions can be located next to the cut site to direct insertion. gRNA2 guides Cas9 cleavage to the 5′ UTR. Therefore, microhomology sequences need to be chosen so that part of the 5′ UTR is removed during the repair process and the tag is fused seamlessly to the RNMT1 ORF. B, HBTH-RNMT was detected using RGS6H and RNMT antibodies. gRNA2, which mediates intra-ORF cleavage, yielded more homozygous knockins.
However, the upstream gRNA2 placement is less efficient than the intra-ORF gRNA1, with only one of seven (14%) homozygous tag insertions for gRNA2. Most of the resulting clones carried heterozygous HBTH-RNMT. In contrast, consistent with other tagging approaches, three of three (100%) clones were homozygous for HBTH-RNMT when gRNA1 was used (Fig. 5B). This is most likely due to the additional trimming required to insert the repair template (Fig. 5A). To overcome this inefficiency, a potential solution would be to employ two identical tagging cassettes that carry different selection markers. This will increase the frequency of homozygous insertions because of the resistance to both antibiotics.
Harnessing the AID system for essential gene study and the immediate protein depletion phenotype
There are about 2,000 essential genes in human cell lines (29, 30), and conditional depletion of these essential genes is important to study their functions. Furthermore, even for nonessential proteins, rapid conditional degradation of the protein can be important to detect the immediate phenotype after target protein depletion, before compensatory effects can obscure the phenotype. Therefore, compared with CRISPR-mediated gene knockout, conditional protein depletion offers flexibility to study essential genes for cell viability and avoids adaptation to knockout of nonessential genes. Various approaches for conditional protein depletion have been described (31–34), but the recent development of the auxin-inducible degron (AID) strategy seems universally efficient (10, 35, 36) and can be incorporated into the PITCh system. AID is a plant-specific pathway controlled by the phytohormone auxin. The F-box protein TIR1 assembles with Skp1 and cullin-1 to form the CRL1TIR1 ubiquitin ligase. TIR1 is the substrate receptor in this ubiquitin ligase and recognizes protein substrates containing AID. However, binding of CRL1TIR1 to AID only occurs in the presence of the plant hormone auxin (37), which can be viewed as a molecular glue that strengthens the TIR1–AID interaction (Fig. 6A). When CRL1TIR1 binds AID, the AID-containing protein is ubiquitylated and subsequently degraded by the 26S proteasome (36, 38). To harness the AID system for protein depletion, we generated the H3F-AID (RGS6H + 3×FLAG + AID) and F-AID (1×FLAG + AID) constructs, which can be used for N-terminal tagging of endogenous proteins. Cells also need to express TIR1 to complete the system for auxin-inducible protein depletion. We therefore inserted CMV-OsTIR1 (derived from Oryza sativa) to the adeno-associated virus integration site (AAVS) safe harbor locus as described previously (10). After hygromycin selection, OsTIR1 expression was confirmed by immunoblotting (Fig. 6B). OsTIR1 assembles with endogenous human Skp1 and cullin-1 to form the CRL1TIR1 ubiquitin ligase. The H3F-AID tag (Fig. 1C) was fused to PP2AC via MMEJ-mediated knockin as described for other tags. Degradation of H3F-AID-PP2Ac in OsTIR1-expressing cells was tested with 500 μm of synthetic (1-naphthaleneacetic acid) and natural (indole-3-acetic acid) auxin. H3F-AID-PP2Ac protein levels were dramatically reduced to about ∼5% after addition of either natural or synthesized auxin for 4 h (Fig. 6B). This versatile method for inducible protein depletion is applicable to any protein.
Figure 6.

Auxin-induced degradation of H3F-AID–tagged PP2Ac. PP2Ac was tagged on the N terminus with pN-PITCh-H3F-AID in 293T cells expressing CMV-driven OsTIR1 integrated into the AAVS safe harbor. PP2Ac was detected using FLAG antibodies during auxin-induced conditional protein depletion. Synthetic (1-naphthaleneacetic acid, NAA) as well as natural (indole-3-acetic acid, IAA) auxin were tested.
Recent developments of various CRISPR-based technologies have simplified genomic engineering. Our study further enriches the CRISPR gene editing toolbox by a number of useful vectors for tagging with epitopes, purification handles, fluorophores, and AID for inducible protein depletion.
Experimental procedures
Generation of plasmids
The pX330 vector that harbors gRNAs that target the YFG and pN-PITCh vectors was generated as described previously (7). Parental pX330 and PITCh plasmids were developed by the Sakuma laboratory (Addgene 63670, 63671, and 63672) (7). Parental vectors for OsTIR1 and the AID sequence were gifts from the Kanemaki laboratory (Addgene 72828, 72833, and 72834) (10). gRNA sequences were designed using the website platforms developed by Zhang lab (3, 28).3 pN-PITCH tagging vectors with PP2Ac microhomologies were generated by gene synthesis of the 2A-3×HA fragment (Fig. 1C), which is a fusion of the self-cleaving peptide P2A (GSGATNFSLLKQAGDVEENPGP) and three copies of the HA epitope. The 2A-3×HA fragment was then fused to the 3′ end of the parental GFP-2A-Puro cassette by Gibson assembly (38). pX330–2-PITCh, which carries PITCH gRNA and a BbsI site for further gRNA insertion, was generated through golden gate assembly with pX330-S-PITCh. All the primers used in this study are listed in Table S1.
Construction of pX330–2-PITCh that carries YFG gRNA
Guide RNAs were designed using the tool provided at https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design.3 Oligos were synthesized at Eurofins Scientific with a 20-bp gRNA sequence containing a 5′ overhang CACC and 3′overhang CAAA (Fig. S1A) to facilitate cloning into the Bbs1 site of pX330–2-PITCh. To anneal oligos, 10 μl of each oligo (100 μm) was mixed with 80 μl of annealing buffer (10 mm Tris (pH 7.5), 50 mm NaCl, and 1 mm EDTA), boiled for 5 min, and cooled at room temperature. The pX330–2-PITCH plasmid was digested with BbsI at 37 °C overnight, and 50 ng of digested vector was used to ligate with annealed oligos (Fig. S2A). 1 μl of the ligation reaction was transformed with into electro-competent cells and plated on Luria broth ampicillin plates.
Changing microhomologies on pN-PITCh tagging vectors
The 20-bp microhomologies to direct MMEJ are based on the genomic sequences adjacent to the gRNA cut site (3 bp prior to the protospacer adjacent motif (PAM) domain, Fig. S1B). Microhomologies on the pN-PITCh tagging vector were changed by PCR and cloned into the vector backbone with Gibson assembly (Fig. S2B). The PITCh tagging cassette and backbone vector were amplified by PCR. The cloning strategy is illustrated in Fig. S2. Briefly, to amplify the tagging cassette, a 10-μl reaction with 1 μl of 50 ng pN-PITCh tagging vector, 10 μm primer, and 5 μl of 2× PrimeSTAR Phusion MAX mixture (Takara) was assembled and amplified as follows: 98 °C for 2 min; three times 98 °C for 10 s, 58 °C for 15 s, and 72 °C for 1 min; fifteen times 98 °C for 10 s and 72 °C for 1 min; and once 72 °C for 2 min. To amplify the vector backbone, a 10-μl reaction with 1 μl of 50 ng pN-PITCh tagging vector, 10 μm primer (5′, ccaaacacgtacgcgtacgatgctctagaatg; 3′, tgctatgtaacgcggaactccatatatggg), and 5 μl of 2× PrimeSTAR MAX mixture (Takara) was assembled and amplified as follows: 98 °C for 2 min; 18 times 98 °C for 10 s and 72 °C for 4 min; and once 72 °C for 2 min.
To remove the parental vector, 1 μl of DpnI was added, followed by 37 °C incubation for 30 min. Note that vector and cassette primers were designed to have 20-bp overlap for efficient Gibson assembly (Fig. S2B). PCR products were gel purified after DpnI digestion and then fused by Gibson assembly. For Gibson assembly, 7.5 μl of Gibson assembly mixture (100 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 0.8 mm dNTP mixture, 10 mm DTT, 5% PEG-8000, 1 mm NAD, 5.3 units/ml T5 exonuclease, 33.3 units/ml Phusion polymerase, and 5.3 units/ml Taq ligase) were used with 50 ng of vector backbone and 50 ng of the PITCh cassette fragment in a 10-μl reaction. The assembly reaction was incubated at 50 °C for 30 min before 1 μl was used for transformation.
Generation of cell pools expressing tagged proteins
5 μg of pX330 and 2.5 μg of pN-PITCh tagging vector were used to transfect cells. DNA and 22.5 μl of BioT were each mixed separately with 400 μl of Opti-MEM. The solutions were combined, incubated for 5 min at room temperature, and then added dropwise to the target cells. 72 h after transfection, cells were divided to three 15-cm plates and selected with 0.25, 0.5, or 1 μg/ml puromycin. Different concentrations should be tested in parallel because puromycin resistance is driven by the target gene promoter, and, depending on promoter strength, resistance strength may vary. Visual confirmation of GFP expression in surviving cells can typically be observed after 5 days of selection.
Generation of single cell clones
One to two weeks after the initial selection, plates with the right selection pressure should yield about 95% killing, and surviving cells will have started to form single colonies. These colonies are typically visible by eye. A sterilized clonal ring and vacuum grease were used to circle the clone for trypsinization and extraction. Alternatively, limited dilution, which aims to have 0.5 cell/well on 96-well plates, can also be used to select individual clones. Clonal rings were dabbed with vacuum grease before circling the clone. 100 μl of trypsin-EDTA (0.05%) was added to the rings, followed by incubation for 10 min at 37 °C. Cells were suspended by repeated pipetting before moving the solution to 6-well plates containing 2 ml of growth medium per well with the appropriate antibiotic. When cells reached more than 60% confluence, uniform GFP expression was confirmed using a microscope before part of the cells was harvested for immunoblot analyses.
Cell culture
HEK293T cells were obtained from the ATCC and maintained at 37 °C with 5% CO2 in DMEM containing 10% FBS and 1% penicillin–streptomycin–amphotericin.
Immunoblotting
Cells were harvested and lysed in 8 m urea buffer (8 m urea, 200 mm NaCl, 100 mm Tris (pH 7.5), 0.2% SDS, 1 mm Na-pyrophosphate, 0.5 mm EDTA, 0.5 mm EGTA, 5 mm NaF, and 10 μm Na-orthovanadate), and protein concentration was quantified by A280 nm. Equal amounts of lysates were separated on a 10% SDS-PAGE gel, and proteins were transferred to a PVDF membrane. Blots were blocked in 5% milk/TBST (TBS with 0.1% Tween 20) and incubated with the primary antibody overnight. Blots were washed for 4 min twice with TBST before incubation with secondary antibody at room temperature for 1 h. After secondary antibody incubation, blots were washed twice for 4 min with TBST and once with TBS before incubation for 4 min with Super Signal West Dura (Thermo Fisher) and imaging with Fuji Imager Las4000.
Antibodies used in this study were as follows: anti-methyl-PP2Ac (Abcam, ab66597), anti-PP2A (Thermo Fisher, MA5-18060), anti-RNMT (Fisher Scientific, 061355MI), anti-HA-HRP (Roche, 12CA5), and anti-RGSH (Qiagen, 34610). Antibodies were used at 1:1000 dilution in 5% milk/TBST.
Proteasome activity assay
The proteasome substrates SUC-LLVY-AMC, SUC-LLE-AMC, and SUC-ARR-AMC were purchased from Boston Biochem. In-solution proteolytic activity assays for the lysates were performed with the fluorogenic peptide substrates SUC-LLVY-AMC, SUC-LLE-AMC, and SUC-ARR-AMC, as described previously (9). The activity readings were normalized to lysate protein concentrations. Briefly, 10 μl of each sample was incubated with 100 μm substrate for 30 min at 37 °C. The reaction was quenched by 1% SDS, and the fluorescence was measured at an excitation of 380 nm and emission of 460 nm. Total protein concentrations were determined by a Bradford assay and used to normalize the proteasome activities. Three biological replicates were performed.
Affinity purification of the human 26S proteasome and sample preparation
Stable 293 cell lines expressing HBTH-Rpn1 were grown to confluence in DMEM containing 10% FBS and 1% penicillin/streptomycin, trypsinized, and washed three times with PBS buffer. The cell pellets from two 15-cm plates were collected and lysed in buffer A (100 mm sodium chloride, 50 mm sodium phosphate, 10% glycerol, 5 mm ATP, 1 mm DTT, 5 mm MgCl2, 1× protease inhibiter (Roche), 1× phosphatase inhibitor, and 0.5% NP-40 (pH 7.5)). The lysates were centrifuged at 13,000 rpm for 15 min to remove cell debris, and the supernatant was incubated with streptavidin resin for 2 h at 4 °C. The streptavidin beads were then washed with 50 bed volumes of the lysis buffer, followed by a final wash with 20 bed volumes of TEB buffer (50 mm Tris-HCl (pH 7.5)) containing 10% glycerol. The purified proteins were then reduced with 2 mm tris(2-carboxyethyl)phosphine at 37 °C for 15 min, alkylated with 25 mm iodoacetamide in the dark at room temperature for 30 min, and digested with endopeptidase Lys-C at 37 °C for 4 h in urea buffer (8 m urea and 25 mm ammonia bicarbonate). Finally, the urea concentration was adjusted to 1.5 m for subsequent trypsin digestion at 37 °C overnight. The samples were desalted using a C18 tip (Agilent Technologies) prior to mass spectrometry analysis.
Mass spectrometry analysis: LC-MS/MS and database searching for protein identification
Liquid chromatography and tandem MS (LC-MS/MS) were carried out using an Orbitrap Fusion Lumos MS (Thermo Fisher Scientific) coupled online with an Ultimate 3000 HPLC system (Thermo Fisher Scientific). MS1 and MS2 scans were acquired in the Orbitrap. MS1 scans were measured with a scan range of 375 to 1500 m/z, resolution set to 120,000, and the automatic gain control (AGC) target set to 4 × 105. MS1 acquisition was performed in top speed mode with a cycle time of 5 s. For MS2 scans, the resolution was set to 30,000, the AGC target was 5e4, the precursor isolation width was 1.6 m/z, and the maximum injection time was 100 ms for collision-induced dissociation (CID). The CID-MS2 normalized collision energy was 25%. For MS2_MS2 analysis, 3+ ions were chosen from the MS1 scan and submitted for sequential CID_EThcD MS2 acquisitions. MS2 scans were acquired in the Orbitrap at 30,000 resolution with an isolation window of 1.6 m/z and AGC target 5e4. For CID analysis, 25% normalized collision energy was used with a maximum injection time of 100 ms.
To identify proteins through database searching, monoisotopic masses of parent ions and corresponding fragment ions, parent ion charge states, and ion intensities from LC-MS/MS spectra were first extracted based on the Raw Extract script from Xcalibur v2.4. The data were searched using the Batch-Tag in the developmental version (v5.10.0) of Protein Prospector against a decoy database consisting of a normal SwissProt database concatenated with its randomized version (SwissProt.2014.12.4.random.concat with total of 20,196 protein entries searched). Homo sapiens was selected as the species. The mass accuracy for parent ions and fragment ions was set at ±20 ppm and 0.6 Da, respectively. Trypsin was set as the enzyme, and a maximum of two missed cleavages was allowed. Protein N-terminal acetylation, methionine oxidation, and N-terminal conversion of glutamine to pyroglutamic acid were selected as variable modifications. The proteins were identified by at least two peptides with a false positive rate of 0.5% or less.
Author contributions
D.-W. L., B. P. C., J.-W. H., X. W., and L. H. data curation; D.-W. L., L. H., and P. K. formal analysis; D.-W. L. and P. K. validation; D.-W. L., B. P. C., J.-W. H., and P. K. investigation; D.-W. L. and P. K. methodology; D.-W. L. writing-original draft; L. H. and P. K. conceptualization; L. H. and P. K. resources; P. K. funding acquisition; P. K. project administration; P. K. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the Sakuma and Kanemaki laboratories for generously providing plasmids and Dr. Phang-Lang Chen for OsTIR1 antibodies and helpful discussions.
This work was supported by the National Institutes of Health (R01GM-066164 and R01GM128432 to P. K. and R01GM074830 to L. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figures S1 and S2 and Tables S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- DSB
- double-strand break
- gRNA
- guide RNA
- PITCh
- precise integration into target chromosome
- NHEJ
- nonhomologous end joining
- HR
- homologous recombination
- MMEJ
- microhomology-mediated end joining
- RHMT
- guanine-N7 methyltransferase
- AID
- auxin-inducible degron
- HBTH
- His6-Biotin-TEV-RGSHis6
- CID
- collision-induced dissociation
- YFG
- your favorite gene.
References
- 1. Esvelt K. M., Mali P., Braff J. L., Moosburner M., Yaung S. J., and Church G. M. (2013) Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 10, 1116–1121 10.1038/nmeth.2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dalvai M., Loehr J., Jacquet K., Huard C. C., Roques C., Herst P., Côté J., and Doyon Y. (2015) A scalable genome-editing-based approach for mapping multiprotein complexes in human cells. Cell Rep. 13, 621–633 10.1016/j.celrep.2015.09.009 [DOI] [PubMed] [Google Scholar]
- 5. Mao Z., Bozzella M., Seluanov A., and Gorbunova V. (2008) Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 7, 1765–1771 10.1016/j.dnarep.2008.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakade S., Tsubota T., Sakane Y., Kume S., Sakamoto N., Obara M., Daimon T., Sezutsu H., Yamamoto T., Sakuma T., and Suzuki K. T. (2014) Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 5, 5560 10.1038/ncomms6560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sakuma T., Nakade S., Sakane Y., Suzuki K. T., and Yamamoto T. (2016) MMEJ-Assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 11, 118–133 10.1038/nprot.2015.140 [DOI] [PubMed] [Google Scholar]
- 8. Savic D., Partridge E. C., Newberry K. M., Smith S. B., Meadows S. K., Roberts B. S., Mackiewicz M., Mendenhall E. M., and Myers R. M. (2015) CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Res. 25, 1581–1589 10.1101/gr.193540.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elsasser S., Schmidt M., and Finley D. (2005) Characterization of the Proteasome Using Native Gel Electrophoresis. Methods Enzymol. 10.1016/S0076-6879(05)98029-4 [DOI] [PubMed] [Google Scholar]
- 10. Natsume T., Kiyomitsu T., Saga Y., and Kanemaki M. T. (2016) Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218 10.1016/j.celrep.2016.03.001 [DOI] [PubMed] [Google Scholar]
- 11. de Felipe P., Hughes L. E., Ryan M. D., and Brown J. D. (2003) Co-translational, intraribosomal cleavage of polypeptides by the foot-and-mouth disease virus 2A peptide. J. Biol. Chem. 278, 11441–11448 10.1074/jbc.M211644200 [DOI] [PubMed] [Google Scholar]
- 12. Ryan M. D., Donnelly M., Lewis A., Mehrotra A. P., Wilkie J., and Gani D. (1999) A model for nonstoichiometric, cotranslational protein scission in eukaryotic ribosomes. Bioorg. Chem. 27, 55–79 10.1006/bioo.1998.1119 [DOI] [Google Scholar]
- 13. Donnelly M. L., Luke G., Mehrotra A., Hughes L. E., Gani D., and Ryan M. D. (2001) Analysis of the aphthovirus 2A/2B polyprotein “cleavage” mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal “skip.” J. Gen. Virol. 82, 1013–1025 [DOI] [PubMed] [Google Scholar]
- 14. Guerrero C., Tagwerker C., Kaiser P., and Huang L. (2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell. Proteomics 5, 366–378 10.1074/mcp.M500303-MCP200 [DOI] [PubMed] [Google Scholar]
- 15. Tagwerker C., Flick K., Cui M., Guerrero C., Dou Y., Auer B., Baldi P., Huang L., and Kaiser P. (2006) A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol. Cell. Proteomics 5, 737–748 10.1074/mcp.M500368-MCP200 [DOI] [PubMed] [Google Scholar]
- 16. Morawska M., and Ulrich H. D. (2013) An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30, 341–351 10.1002/yea.2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pardo B., Gómez-González B., and Aguilera A. (2009) DNA repair in mammalian cells. Cell. Mol. Life Sci. 66, 1039–1056 10.1007/s00018-009-8740-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paix A., Wang Y., Smith H. E., Lee C.-Y., Calidas D., Lu T., Smith J., Schmidt H., Krause M. W., and Seydoux G. (2014) Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 198, 1347–1356 10.1534/genetics.114.170423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taleei R., and Nikjoo H. (2013) Biochemical DSB-repair model for mammalian cells in G1 and early S phases of the cell cycle. Mutat. Res. 756, 206–212 10.1016/j.mrgentox.2013.06.004 [DOI] [PubMed] [Google Scholar]
- 20. Janssens V., Longin S., and Goris J. (2008) PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem. Sci. 33, 113–121 10.1016/j.tibs.2007.12.004 [DOI] [PubMed] [Google Scholar]
- 21. Wlodarchak N., and Xing Y. (2016) PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 51, 162–184 10.3109/10409238.2016.1143913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tagwerker C., Zhang H., Wang X., Larsen L. S., Lathrop R. H., Hatfield G. W., Auer B., Huang L., and Kaiser P. (2006) HB tag modules for PCR-based gene tagging and tandem affinity purification in Saccharomyces cerevisiae. Yeast 23, 623–632 10.1002/yea.1380 [DOI] [PubMed] [Google Scholar]
- 23. Rosenzweig R., Bronner V., Zhang D., Fushman D., and Glickman M. H. (2012) Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. J. Biol. Chem. 287, 14659–14671 10.1074/jbc.M111.316323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X., Chen C. F., Baker P. R., Chen P. L., Kaiser P., and Huang L. (2007) Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 10.1021/BI061994U [DOI] [PubMed] [Google Scholar]
- 25. Murakami K., Ichinohe Y., Koike M., Sasaoka N., Iemura S., Natsume T., and Kakizuka A. (2013) VCP Is an integral component of a novel feedback mechanism that controls intracellular localization of catalase and H2O2 levels. PLoS ONE 8, e56012 10.1371/journal.pone.0056012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Z., and Kang Y. J. (2000) Cellular and subcellular localization of catalase in the heart of transgenic mice. J. Histochem. Cytochem. 48, 585–594 10.1177/002215540004800502 [DOI] [PubMed] [Google Scholar]
- 27. Brocard C., and Hartig A. (2006) Peroxisome targeting signal 1: is it really a simple tripeptide? Biochim. Biophys. Acta 1763, 1565–1573 10.1016/j.bbamcr.2006.08.022 [DOI] [PubMed] [Google Scholar]
- 28. Doench J. G., Fusi N., Sullender M., Hegde M., Vaimberg E. W., Donovan K. F., Smith I., Tothova Z., Wilen C., Orchard R., Virgin H. W., Listgarten J., and Root D. E. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K. R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S., Mero P., Dirks P., Sidhu S., Roth F. P., Rissland O. S., et al. (2015) High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 10.1016/j.cell.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 30. Wang T., Birsoy K., Hughes N. W., Krupczak K. M., Post Y., Wei J. J., Lander E. S., and Sabatini D. M. (2015) Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 10.1126/science.aac7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banaszynski L. A., Chen L. C., Maynard-Smith L. A., Ooi A. G., and Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 10.1016/j.cell.2006.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonger K. M., Chen L. C., Liu C. W., and Wandless T. J. (2011) Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 7, 531–537 10.1038/nchembio.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neklesa T. K., Tae H. S., Schneekloth A. R., Stulberg M. J., Corson T. W., Sundberg T. B., Raina K., Holley S. A., and Crews C. M. (2011) Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 7, 538–543 10.1038/nchembio.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chung H. K., Jacobs C. L., Huo Y., Yang J., Krumm S. A., Plemper R. K., Tsien R. Y., and Lin M. Z. (2015) Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 11, 713–720 10.1038/nchembio.1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nishimura K., and Kanemaki M. T. (2014) Rapid depletion of budding yeast proteins via the fusion of an auxin-inducible degron (AID). Curr. Protoc. Cell Biol. 64, 20.9.1–20.9.16 10.1002/0471143030.cb2009s64 [DOI] [PubMed] [Google Scholar]
- 36. Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., and Kanemaki M. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 10.1038/nmeth.1401 [DOI] [PubMed] [Google Scholar]
- 37. Tan X., Calderon-Villalobos L. I., Sharon M., Zheng C., Robinson C. V., Estelle M., and Zheng N. (2007) Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645 10.1038/nature05731 [DOI] [PubMed] [Google Scholar]
- 38. Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.