

Abstract
During infection, the fungal pathogen Aspergillus fumigatus forms biofilms that enhance its resistance to antimicrobials and host defenses. An integral component of the biofilm matrix is galactosaminogalactan (GAG), a cationic polymer of α-1,4-linked galactose and partially deacetylated N-acetylgalactosamine (GalNAc). Recent studies have shown that recombinant hydrolase domains from Sph3, an A. fumigatus glycoside hydrolase involved in GAG synthesis, and PelA, a multifunctional protein from Pseudomonas aeruginosa involved in Pel polysaccharide biosynthesis, can degrade GAG, disrupt A. fumigatus biofilms, and attenuate fungal virulence in a mouse model of invasive aspergillosis. The molecular mechanisms by which these enzymes disrupt biofilms have not been defined. We hypothesized that the hydrolase domains of Sph3 and PelA (Sph3h and PelAh, respectively) share structural and functional similarities given their ability to degrade GAG and disrupt A. fumigatus biofilms. MALDI-TOF enzymatic fingerprinting and NMR experiments revealed that both proteins are retaining endo-α-1,4-N-acetylgalactosaminidases with a minimal substrate size of seven residues. The crystal structure of PelAh was solved to 1.54 Å and structure alignment to Sph3h revealed that the enzymes share similar catalytic site residues. However, differences in the substrate-binding clefts result in distinct enzyme-substrate interactions. PelAh hydrolyzed partially deacetylated substrates better than Sph3h, a finding that agrees well with PelAh's highly electronegative binding cleft versus the neutral surface present in Sph3h. Our insight into PelAh's structure and function necessitate the creation of a new glycoside hydrolase family, GH166, whose structural and mechanistic features, along with those of GH135 (Sph3), are reported here.
Keywords: Aspergillus, biofilm, glycoside hydrolase, extracellular matrix, protein structure, virulence factor, substrate specificity, galactosaminogalactan, lung infection
Introduction
Aspergillus fumigatus is a ubiquitous filamentous fungus that causes invasive pulmonary infections in patients who are immunosuppressed due to cytotoxic chemotherapy, organ and stem cell transplantation, or biological therapies such as tumor necrosis factor inhibitors (1–3). During pulmonary infection, filamentous hyphae of A. fumigatus grow within biofilms: multicellular communities of organisms embedded in a self-produced extracellular matrix (4). This biofilm extracellular matrix plays several roles in the pathogenesis of invasive aspergillosis including mediating the adherence of hyphae to host tissues and enhancing resistance to antifungal drugs and host immune defenses (5–8).
Solid-state NMR spectroscopy analyses have revealed that polysaccharides are the most abundant component of the A. fumigatus extracellular matrix (9). Immunohistochemical and EM studies of Aspergillus biofilms have identified α-1,3-glucan, galactomannan, and galactosaminogalactan (GAG)6 as key polysaccharide components of the matrix (10). GAG is a linear cationic polymer of α-1,4-d-galactose and partially deacetylated α-1,4-N-acetyl-d-galactosamine (GalNAc) that is produced by activity of the products of a five-gene cluster located on chromosome 3 (11). GAG synthesis is thought to be initiated by the synthesis of UDP-GalNAc and UDP-Gal by the glucose-4-epimerase Uge3 (12). These sugars are then linked and exported through the action of the predicted glycosyltransferase Gtb3, and the resulting polymer is partially deacetylated in the extracellular space by secreted Agd3 (13). Cleavage of the emerging polymer is thought to be mediated by two glycoside hydrolases encoded within this gene cluster, Sph3 and Ega3 (11). GAG, plays a central role in biofilm formation and virulence (14). GAG-deficient strains are markedly impaired in their ability to form biofilms and adhere to pulmonary epithelial cells in vitro (15). Hyphae-associated GAG enhances resistance to neutrophil extracellular traps (16) and conceals pathogen-associated molecular patterns such as β-glucan from immune detection (17). Secreted GAG also promotes infection by inducing neutrophil apoptosis and the production of the anti-inflammatory cytokine IL-1 receptor antagonist (18). Consistent with these observations, GAG-deficient strains of A. fumigatus exhibit attenuated virulence in mouse models of invasive aspergillosis (17).
The importance of GAG in biofilm formation and virulence suggest that this exopolysaccharide is a promising therapeutic target. We recently reported that the recombinant glycoside hydrolase domains from two microbial proteins, A. fumigatus Sph3 and Pseudomonas aeruginosa PelA, degrade GAG and disrupt A. fumigatus biofilms (19). PelA is a multidomain protein with both deacetylase and glycoside hydrolase activity that is required for production of the P. aeruginosa Pel polysaccharide (20–22). In addition to its degrading A. fumigatus biofilms, the recombinant hydrolase domain of PelA (PelAh) also disrupts Pel-dependent biofilms of P. aeruginosa (21). Although, the structure and anomeric configuration of the Pel polysaccharide remains to be determined, it has been found to contain partially deacetylated 1,4-linked GalNAc and N-acetylglucosamine (GlcNAc) in a ratio of 5:1 (20, 23). The presence of GalNAc-rich regions in both GAG and Pel polysaccharides suggests that PelAh may also be specific for GalNAc.
Structural and functional characterization of the glycoside hydrolase domain of Sph3 (Sph3h) revealed that this enzyme is essential for GAG biosynthesis and belongs to the glycoside hydrolase (GH) family 135 (24). Sph3h has an (β/α)8-fold with a shallow conserved active site groove. Co-crystallization of Sph3h with GalNAc and mutagenesis of residues within the active site groove suggest that this enzyme hydrolyzes GAG within GalNAc-rich regions of the polymer (24).
Less is known about structure and function of the glycoside hydrolase domain of PelA, which is predicted by Phyre2 to contain a (β/α)-barrel fold (20) and by the conserved domain database to belong to the GH114 superfamily (www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). However, a BLAST search against all CAZy, GH114 members, and a search of the sequence against all Hidden Markov Models of CAZy families, failed to find any significant sequence identity with classified glycoside hydrolases, suggesting that PelAh belongs to a new CAZy family.
Herein, we characterize the molecular mechanisms underlying the cross-kingdom GAG activity of the glycoside hydrolases, Sph3h and PelAh. Mass spectrometry enzymatic fingerprinting and NMR studies revealed that both enzymes share retaining endo-α-1,4-N-acetylgalactosaminidase activity and require a minimal substrate length of seven GalNAc residues. The substrate interacts with the binding cleft differently in the two enzymes leading Sph3h to cleave substrates closer to the nonreducing end, whereas PelAh cleaves proximal to the reducing end of oligosaccharides. The enzymes also differ in their ability to cleave partially deacetylated substrates. Although neither enzyme is active against fully deacetylated oligosaccharides, PelAh has a higher propensity to cleave GalNAc linkages within regions of partially deacetylated GAG. The structure of PelAh was determined, and comparison with Sph3h revealed a high degree of structural similarity within the catalytic site of these enzymes. The presence of a deeper, more electronegative groove in PelAh likely underlies its ability to bind and cleave cationic partially deacetylated substrates more effectively than Sph3h.
Results
Sph3h and PelAh are α-1,4-N-acetylgalactosaminidases
To elucidate the molecular mechanisms by which Sph3h and PelAh mediate biofilm disruption, these enzymes were incubated with pre-grown A. fumigatus biofilms and the profile of oligosaccharides that were released was analyzed by matrix-assisted laser desorption and ionization time of flight mass spectrometry (MALDI-TOF MS) enzymatic fingerprinting. Consistent with our previous studies, nanomolar concentrations of Sph3h or PelAh disrupted A. fumigatus biofilms (Fig. 1, A and B) (19). Analysis of the oligosaccharides released by treatment with either enzyme revealed similar MS fingerprints, with spectra displaying ions of m/z ratio corresponding to N-acetylhexosamine (HexNAc) oligomers. Treatment of biofilms with 100 nm Sph3h produced ions with a m/z ratio from 1056.5684 to 2478.2696 with a repetitive occurrence of 203.1002 ± 0.0059 matching with the HexNAc m/z ratio (25). This profile is consistent with the release of HexNAc oligomers ranging in size from pentamers to dodecamers (12-mers) (Fig. 1A). Treatment of fungal biofilms with 100 nm PelAh generated a similar spectrum of ions ranging from m/z 1056.6345 to 3290.8078, with an interval m/z difference of 203.1107 ± 0.0053 between ions, suggesting that PelAh treatment released HexNAc oligosaccharides ranging in size from pentamers to hexadecamer (16-mers) (Fig. 1B). MS-MS fragmentation analysis confirmed these ions were composed uniquely of m/z ratio 203.10 units, suggesting that these oligosaccharides are HexNAc homopolymers (Fig. 1C). GC coupled to MS (GC-MS) analysis of the monosaccharide composition of these oligosaccharides confirmed they are composed solely of GalNAc, supporting the hypothesis that these enzymes cleave regions of homo-GalNAc within GAG (Fig. 1D).
Figure 1.

A. fumigatus biofilm degradation by Sph3h and PelAh release α-1,4-GalNAc oligosaccharides. A, crystal violet staining of pre-grown A. fumigatus biofilm treated with the indicated concentration of Sph3h and the associated MALDI-TOF MS spectra of oligosaccharides released from the biofilm treated with 100 nm Sph3h (circled concentration). B, crystal violet staining of pre-grown A. fumigatus biofilms treated with the indicated concentration of PelAh and the associated MALDI-TOF MS spectra of oligosaccharides released from the biofilm treated with 100 nm PelAh (circled concentration). C, MS-MS fragmentation spectra of m/z = 1665.7 ion issued of the Sph3h treatment and matching with HexNAc8 oligosacaccharide. This structure is a homopolymer of units with an average m/z of 203.1086, corresponding to the m/z ratio of a N-acetylhexosamine (white square), consistent with a HexNAc8 oligosaccharide. D, GC-MS extracted ion chromatogram m/z = 173.0 of HexNAc monosaccharide standards mix. E, GC-MS extracted ion chromatogram m/z = 173.0 of the biofilm released HexNAc oligosaccharides.
To further confirm the specificity of Sph3h and PelAh for the α-1,4-GalNAc linkages within GAG, the ability of these enzymes to cleave synthetic oligo-α-1,4-galactose, and oligo-α-1,4-galactosamine were tested. Neither Sph3h nor PelAh exhibited activity against these components of GAG, even at concentrations as high as 5 μm (Fig. 2, A and B). Similarly, neither enzyme was able to degrade chitin (β-1,4–linked GlcNAc), the only other major hexosamine-containing polysaccharide within the fungal cell wall (Fig. 2C). These data suggest that biofilm disruption by Sph3h and PelAh is a consequence of cleavage of α-1,4-GalNAc homopolymeric regions within GAG.
Figure 2.

Sph3h and PelAh are specific for α-1,4-GalNAc. MALDI-TOF MS analysis of the products released by the incubation of Sph3h and PelAh with (A) a fraction enriched in α-1,4-galactose 9-mers, (B) a fraction enriched in α-1,4-galactosamine 9-mers, and (C) chemically partially hydrolyzed chitin. The initial sample is represented in white, following Sph3h treatment in gray, and PelAh treatment in black. MALDI-TOF MS spectra of (D) a sample enriched in α-1,4-GalNAc 4-mer to 15-mer. MALDI-TOF MS spectra of the enzymatic products released by (E) Sph3h and (F) PelAh from the sample presented in D.
Sph3h and PelAh are α-1,4-GalNAc endo-acting hydrolases with a minimal substrate length of 7 GalNAc units
To confirm the α-1,4-N-acetylgalactosaminidase activity of Sph3h and PelAh, MALDI-TOF MS enzymatic fingerprinting was performed using a purified fraction of α-1,4-GalNAc oligosaccharides obtained by partial Sph3h digestion of A. fumigatus biofilms (Fig. 2D). Treatment of these predominately decameric to dodecameric oligosaccharides with either 1 μm Sph3h (Fig. 2E) or 1 μm PelAh (Fig. 2F) for 1 h resulted in a shift of the spectra consistent with final product sizes of penta-, hexa-, and heptamers, confirming the ability of these enzymes to cleave α-1,4-GalNAc linkages.
To determine the minimum substrate size that can be cleaved by Sph3h and PelAh, α-1,4-GalNAc hexamers and heptamers purified from partial Sph3h digestion of A. fumigatus biofilms were treated with each enzyme and the degradation products were analyzed by MALDI-TOF MS fingerprinting. GalNAc heptasaccharides but not hexasaccharides were rapidly hydrolyzed by both enzymes (Fig. 3, A–D). Hydrolysis of GalNAc heptasaccharides by Sph3h resulted in the accumulation of pentasaccharides (Fig. 3B), whereas PelAh hydrolysis produced predominantly both tetra- and pentasaccharides (Fig. 3D), suggesting that these enzymes function as endo-acting glycoside hydrolases. The ability of PelAh to degrade the heptamers into two sets of products suggests some flexibility in the positioning of the oligosaccharide in the PelAh-binding site.
Figure 3.

Sph3h and PelAh are endo-N-acetylgalactosaminidases with a minimum substrate size of seven residues. Degradation time course of pure oligosaccharides monitored over 30 min by MALDI-TOF MS. Sph3h degradation kinetic of: A, α-1,4-GalNAc 6-mers and B, α-1,4-GalNAc 7-mers. PelAh degradation kinetic of: C, α-1,4-GalNAc 6-mers and D, α-1,4-GalNAc 7-mers. Relative proportion of each ions was calculated and reported here under the color purple for 7-mer; green for 6-mer, blue for 5-mer, red for 4-mer, and yellow for 3-mer.
Sph3h and PelAh are retaining endoglycoside hydrolases
To investigate the molecular mechanism of the two glycoside hydrolases, the stereochemical outcome of oligosaccharide cleavage by these enzymes was evaluated by 1H NMR spectroscopy using synthetic GalNAc octamers as a substrate. Spectral analysis of the pre-reaction substrate demonstrated the presence of four doublets with coupling constants between 3.5 and 4.0 Hz in the anomeric region of the spectra, characteristic of α-glycosidic linkages. The addition of Sph3h or PelAh led to the appearance of a new peak at 5.29 ppm with a coupling constant of 4.0 Hz characteristic of an α-anomer (Fig. 4, A and B). Acquisition of 1D spectra at 25 and 37 °C (Fig. 4, A and B), and of 2D COSY 1H-1H spectra (Fig. S1) after 24 h of reaction revealed the appearance of a β-anomer signal at 4.73 ppm on the 1D spectra consistent with secondary mutarotation at the new reducing end. The addition of purified monomeric GalNAc to samples at the end of the experiment resulted in the appearance of anomeric signals at 4.66 and 5.25 ppm. These signals were distinct from the experimental products, demonstrating that neither Sph3h nor PelAh released monosaccharides from GalNAc octamers (Fig. S2). Collectively, these data suggest that Sph3h and PelAh are retaining endoglycosidases.
Figure 4.

Sph3h and PelAh are acting as retaining glycoside hydrolases. Degradation time course of α-1,4-GalNAc octamers by (A) Sph3h and (B) PelAh monitored by NMR spectroscopy highlighting the region between 4.6 and 5.4 ppm. All spectra were recorded at 25 °C except the final point at 48 h, which was also acquired at 37 °C as indicated by the asterisk.
PelAh has a (β/α)-barrel with a deep substrate-binding groove
To shed light on the mechanisms by which PelAh and Sph3h hydrolyze α-1,4-GalNAc polymers, and to complement the available structure of Sph3h, we pursued the structure determination of PelAh. Crystals of PelAh formed readily in a variety of different crystallization conditions and the structure was subsequently determined using selenomethionine incorporation and the single-wavelength anomalous diffraction method to 1.54 Å and refined to a final Rwork and Rfree of 16.9 and 19.2%, respectively. Unambiguous electron density allowed the modeling of residues 48 to 300. No interpretable density was observed for residues 47, 301–303, or the hexa-histidine purification tag. The structure of PelAh revealed a β8/α7-barrel–fold with a C-terminal region lacking the secondary structure (Fig. 5A). This region, residues 290–300, packs against the core β-barrel and makes multiple contacts to neighboring helices suggesting that the lack of helical structure is not an artifact of the construct design. Furthermore, secondary structure prediction using Jpred4 (26) predicts that residues 297–310 form a β-strand supporting the lack of an 8 helix.
Figure 5.

The structure of PelAh reveals a (β/α)-barrel. A, tertiary structure of PelAh with the (β/α)-fold colored in orange and yellow, respectively. The βHL and the insertion after β3 (Loop3) are colored in blue. B, the active-site groove is shown with a transparent surface allowing for visualization of the conserved residues residing in the cleft based on Consurf analysis. In orange are the three residues (denoted with single letter abbreviations) identified as highly conserved in Sph3 homologues. C, Cα alignment of PelAh (yellow and blue) to hypothetical protein TM1410 (PDB 2AAM, gray and black) shows similarity in tertiary structure topology. The insertions of PelAh are colored as in A showing that TM1410 also contains these additions including a loop after β3 (Loop3TM1410, black). An unknown ligand (red) and glycerol (gray) were found in the groove of the TM1410 structure. D, visualization of PelAh structure B-factors, colored blue to red for relative low to high values.
In addition to the core (β/α)-fold, PelAh contains two structural insertions. The first is a 38-amino acid loop between β3 and α3 containing a two strand anti-parallel β-sheet and two α-helices (Loop3 in Fig. 5A). The second is a β-hairpin loop (βHL) between β6 and α6. These insertions contribute to the creation of a 15-Å deep, ∼36 Å long, active site groove on the “top” face of the barrel at the C termini of the β-strands (Fig. 5A). This groove is composed of several aromatics that could potentially participate in polysaccharide-protein interactions (Fig. 5B).
When the structure of PelAh was submitted to the tertiary structure similarity server, DALI (27), the most similar structures found were: (i) a hypothetical protein TM1410 from Thermatoga maritima, which has sequence similarity to GH114 family members (PDB 2AAM, 2.5 Å root mean square deviation (RMSD) over 203 residues); (ii) a cycloisomaltooligosaccharide glucotransferase from GH66 (PDB 3WNK, 2.9 Å RMSD over 165 residues); (iii) Cwp19, a peptidoglycan hydrolase reported as a GH-like 10 family member (PDB 5OQ2, 2.9 Å RMSD over 193 residues), and (iv) a dextranase from GH66 (PDB 3VMN, 2.9 RMSD over 155 residues). TM1410 has the highest similarity in structure and highest sequence identity to PelAh (15.7%). TM1410 also contains an insertion after β3, consisting of a three-stranded β-sheet and a small α-helix (Loop3TM1410). Loop3TM1410 folds further over the putative active site than the equivalent Loop3 in PelAh (Fig. 5C). Electron density corresponding to a ring containing ligand, as well as multiple glycerol molecules, was found in the deep groove of the TM1410 structure. It is possible that Loop3TM1410 has some flexibility and the presence of a ligand in TM1410 causes the loop to cap the groove. Loop3PelA and the βHL have the highest B-factors of the PelAh structure, reaching 134 Å2 in Loop3 as compared with an average protein B-factor of 31.6 Å2, suggesting conformational heterogeneity in the crystal, further supporting the flexibility of these regions (Fig. 5D).
PelAh is most closely related to GH114 family as previously noted. According to the automated carbohydrate active enzymes annotation server dbCAN2, PelAh has very low overlap with the GH114 HMM profile and has insufficient sequence identity to be assigned to this family (28, 29). These results suggest that PelAh represents a new GH family related to GH114. Our experimental evidence that PelAh exhibits glycoside hydrolase activity allows for the creation of the GH166 family. Although no GH114 structure has been solved to date, the similarity in the predicted structure suggests that GH166 and GH114 may create a new GH clan (30).
Structural comparison of GAG hydrolyzing PelAh and Sph3h reveals architectural differences but conserved active-site residues
A superimposition of PelAh and Sph3h (RMSD of 3.05 Å over 167 Cα atoms) aligned the active site of Sph3h (24) with that of PelAh (19) and revealed that the insertions in the PelAh structure are unique to this protein (Fig. 6A). Sph3h has a much shallower binding cleft compared with PelAh (Fig. 6B). Active sites situated in clefts and tunnels suggest endo-activity and correlates with increased processivity (31–33). Shallow substrate grooves correlate with low processivity but have been found in enzymes that cleave crystalline, recalcitrant substrates (31–33). The deeper cleft found in PelAh is indicative of a more processive activity as compared with Sph3h.
Figure 6.

PelAh and Sph3h differ in their substrate-binding cleft architecture but share catalytic motifs. A, tertiary structure alignment of Sph3h (PDB 5D5G, purple) with PelAh. B, transparent surface representation of PelAh (yellow and blue) and Sph3h (purple) in the same orientation shows the relative depths of the active site groove. C, alignment of the active site residues of PelAh (yellow) and Sph3h (purple) based on the Sph3h active site motifs shows high identity between the hydrolases around the GalNAc (gray)-binding site of Sph3h (PDB 5D6T). D, primary sequence alignment of P. aeruginosa PelAh (PelAPa) with homologues from Geobacter metallireducens (PelAGm), Ralstonia solanacearum (PelARs), as well as TM1410 and Sph3 (Aspergillus clavatus) done by MUSCLE. Sequence identity to PelAh is listed based on MUSCLE alignment for the two homologues and Sph3. Sequence identity to TM1410 is based on structural alignment.
Despite differences in cleft architecture, examination of the putative active sites (RMSD 0.25 Å over active site motif backbone atoms) revealed a high degree of conservation, with residues Asp-166, Asn-202, and Glu-222 of Sph3h that interact with GalNAc in our co-crystal structure (24), superimposing with residues Asp-160, Asn-199, and Glu-218 of PelAh (Fig. 5, C and D). Sph3h activity was previously shown to be dependent on Asp-166 as conservative mutation of this residue abolished Sph3h degradation of GAG (24). Mutation of Glu-222 to alanine also abolished Sph3h activity but replacement with glutamine had a lesser affect. Biofilm disruption by PelAh was shown to involve Glu-218, which aligns to Glu-222 of Sph3h, suggesting this residue is involved in catalysis (19). The similarity of the residues within the catalytic site of Sph3h and PelAh suggests that they share a common catalytic mechanism. The distance between carboxyl side chains of the catalytic residues Asp-166 and Glu-222 in Sph3h and the homologous residues Asp-160 and Glu-218 in PelAh are 5.2–6.7 and 4.8–6.8 Å, respectively. This distance is consistent with a retaining (∼5.5 Å) rather than inverting mechanism of cleavage (∼10 Å) (32).
Sph3h and PelAh have different distribution of substrate-binding subsites
To determine whether Sph3h and PelAh cleave GalNAc oligosaccharides closer to the reducing or nonreducing end, an oligosaccharide preparation enriched in (α-1,4-GalNAc)9 was reduced by sodium borohydride treatment, conferring an additional m/z ratio of 2 to the reducing end. These reduced oligosaccharides were then incubated with each enzyme and the resulting products analyzed by MALDI-TOF MS. Both enzymes produced multiple oligosaccharide products ranging in length from tetramers to heptamers. However, oligosaccharides produced by Sph3h treatment were consistent with cleavage of (α-1,4-GalNAc)9 near the reducing end of the substrate, whereas PelAh treatment released reduced GalNAc4–6 consistent with cleavage near the nonreducing end (Fig. 7).
Figure 7.
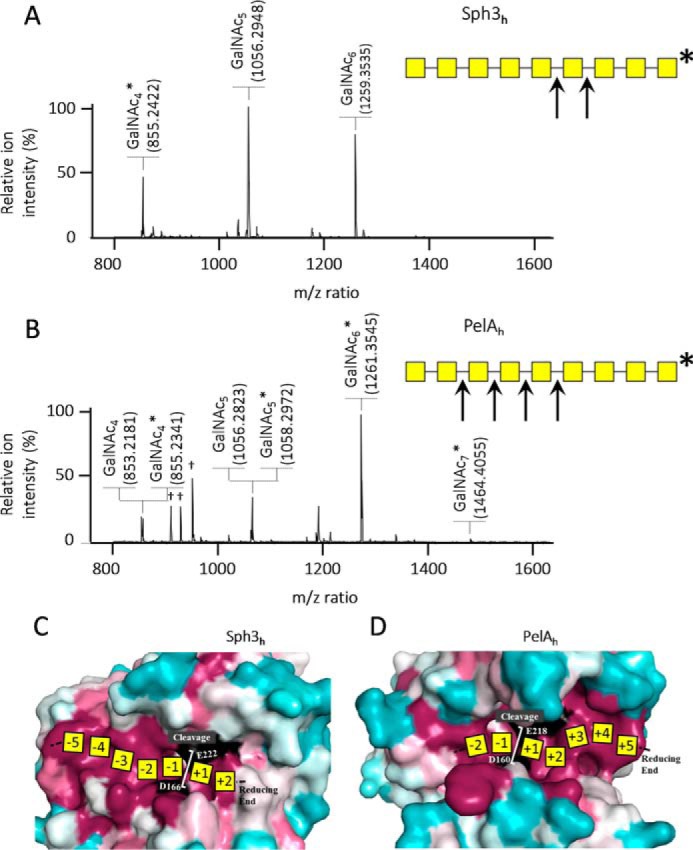
Sph3h and PelAh do not hydrolyze the oligosaccharide at the same location. A, MALDI-TOF MS spectra of the enzymatic product of Sph3h and B, PelAh cleavage of reduced α-1,4-GalNAc 9-mers and their associated schematic view of the cleavage sites. Yellow squares represent GalNAc; arrows indicate enzymatic cleavage sites; * indicates the reducing end of the oligosaccharides; † indicates matrix ion signals. C, surface representation colored by conservations (conserved in magenta and variable in teal, catalytic residues in black) showing a proposed map of a heptamer substrate and product subsites on Sph3h, and D, PelAh.
Mapping of sequence conservation based on alignments of Sph3h and PelAh to respective homologous proteins revealed high degrees of sequence conservation in the active-site groove of each enzymes (Fig. 7, C and D). However, closer examination of the patterns of conservation shows differences between the two enzymes (Fig. 7, C and D). For Sph3h, surface conservation extends further on the nonreducing side of +1/−1 cleavage site suggesting approximately five conserved substrate-binding subsites (−5 to −1, Fig. 7C). This observation is consistent with our data that shows that this enzyme produces a minimum length pentasaccharide from the nonreducing end (Fig. 7A). In contrast, PelAh has surface conservation that extends on the reducing end side of cleavage and at least five subsites could be mapped (+1 to +5, Fig. 7B). Surface residue conservation thus correlates well with the results of the GalNAc9 hydrolysis experiments.
PelAh cleaves deacetylated-rich regions within GAG
Comparison of the structures of Sph3h and PelAh revealed that the PelAh active site groove is more electronegative than that of Sph3h (Fig. 8A), suggesting that PelAh may be able to bind and potentially cleave cationic oligosaccharides, such as partially deacetylated oligo-GalNAc. Although neither enzyme was able to cleave α-1,4 GalN homopolymers, it is possible that these enzymes may be able to cleave α-1,4 GalNAc linkages within GalN-rich regions of GAG. Although GalN-containing oligosaccharides were not detected during the biofilm disruption assay (Fig. 1), these cationic degradation products could have remained adherent to the negatively charged hyphal cell wall. To test this hypothesis, a cell-free GAG degradation assay was performed using purified secreted GAG. Sph3h treatment of secreted GAG resulted in the release of predominately homo-GalNAc oligosaccharides (86%) with a smaller amount of mono-deacetylated GalNAc oligosaccharides (14%, Fig. 8B). In contrast, treatment of secreted GAG with PelAh produced almost exclusively partially deacetylated GalNAc oligosaccharides (98%, Fig. 8B) suggesting that PelAh preferentially binds and degrades GalN-containing regions of GAG. Consistent with our prior results, no galactose homo- or heteropolymers, nor GalN homopolymers were found, confirming that Sph3h and PelAh specifically cleave α-1,4 GalNAc linkages (Fig. 2B).
Figure 8.
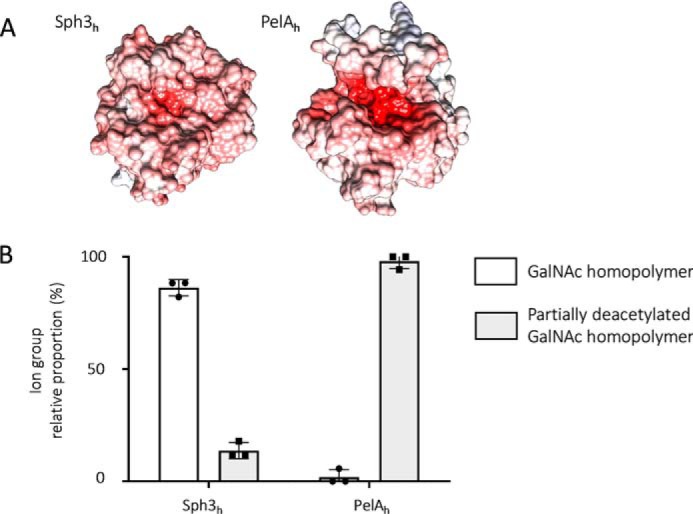
Difference in electrostatic charge surface predict PelAh is preferentially able to cleave oligosaccharides containing deacetylated GalNAc. A, electrostatic surface representation of Sph3h and PelAh generated using APBS in Chimera. Quantitative electrostatics are colored from red (−15 kT) to blue (+15 kT). B, relative proportions of oligosaccharides obtained from digestion of purified, secreted GAG degradation products with 1 μm Sph3h or PelAh. Oligosaccharide products were detected by MALDI-TOF MS. Ions were categorized according to their composition as indicated in the legend. No galactose-homopolymers were detected.
Discussion
We previously described the structure of Sph3h and found that this enzyme constituted the first member of a new glycoside hydrolase family, GH135, with activity against GAG and Aspergillus biofilms (24). More recently, the glycoside hydrolase domain of PelA from P. aeruginosa has also been reported to disrupt A. fumigatus biofilms (19). However, the molecular mechanism and specificity these enzymes had not been elucidated.
Herein, the mechanism by which these enzymes disrupt A. fumigatus biofilms was studied using a combination of structural biology, and mass and NMR spectroscopy. These studies revealed that Sph3h and PelAh share a conserved active site and cleave α-1,4-GalNAc glycosidic linkages using a retaining enzyme mechanism. Despite these similarities of specificity and mechanism, the interaction between the enzymes and the substrate differ. The Sph3h has a neutral electrostatic cleft in contrast to the deep electronegative groove of the PelAh. These observations correlate with the nature of the products observed after cleavage of secreted GAG. The electronegative PelAh was able to bind and release cationic GalN-containing oligosaccharide more efficiently, whereas the neutral Sph3h released mostly neutral GalNAc oligosaccharides. Furthermore, despite interacting with the same substrate, the two glycoside hydrolases have extended conserved substrate-binding sites on opposites sides of the catalytic −1/+1 site. This difference leads to Sph3h cleaving closer to the reducing end, whereas PelAh cleaves nearer to the nonreducing end of oligosaccharides.
Sph3h is structurally similar to GH27 family members, which rely on two acidic residues to cleave the glycosidic bond (24). GH27 catalytic residues align structurally with Asp-166 and Glu-222 in Sph3h. Previously, mutagenesis studies on Sph3h identified Asp-166 as essential for catalysis (24). Glu-222 was not essential in vitro as mutation to glutamine retained some activity (24). Replacement of the catalytic acid/base residue with glutamine does not abolish activity in some retaining hydrolases but slows the rate of reaction (34, 35). Mutation of the acid/base glutamate in the Sulfolobus solfataricus β-glycosidase reduced activity 10–60–fold depending on the substrate (36). In Sph3h, Glu-222 is coordinated by Tyr-88 and Asn-202, and these residues may aid in activation of the glutamine in the E222Q mutant. PelAh has equivalent catalytic residues that align structurally to those of Sph3h. NMR analysis found that both enzymes utilize a retaining mechanism. Analysis of the structure of Sph3h and PelAh revealed that the distances between the carboxyl side chains of the catalytic residues Glu-222 and Asp-166 in Sph3h and Glu-218 and Asp-160 in PelAh correspond to the average distance between the catalytic residues of retaining hydrolases (32). Thus, the structures of Sph3h and PelAh and the NMR results are consistent with these enzymes utilizing a retaining mechanism (Fig. 4).
Our findings demonstrate that PelAh and Sph3h share a similar (β/α)-barrel–fold with central grooves containing a highly conserved active sites, underlying their endo-α-1,4-N-acetylgalactosaminidase activity. The high identity between the catalytic residues of the active site suggests that the difference in the ability of these enzymes to hydrolyze oligosaccharides containing deacetylated residues is a reflection of differences in polysaccharide binding affinity. This hypothesis is supported by the fact that, unlike Sph3h, the binding groove of PelAh is highly electronegative, suggesting an affinity for cationic substrates as seen in the soluble GAG-degradation assay. PelA is a multidomain protein with both hydrolase and carbohydrate deacetylase activity. The deacetylase activity has been shown to be required for in vivo Pel-dependent biofilm formation (20). Our results suggest that the PelA deacetylase domain likely acts on Pel first, rendering the polysaccharide cationic prior to hydrolysis of the polymer. Similar findings have been reported for the poly-β1–6-N-acetyl-d-glucosamine modifying enzyme, PgaB, which contains both a deacetylase domain and a GH153 hydrolase domain (37). Studies of PgaB demonstrated that the GH153 domain requires a specific pattern of partially deacetylated poly-β1–6-N-acetyl-d-glucosamine as a substrate (37). PelAh appears to be more promiscuous in its substrate specificity, as the enzyme can hydrolyze pure acetylated oligomers.
Another difference between Sph3h and PelAh was revealed by their activity on reduced GalNAc nonamers (Fig. 7). Sph3h hydrolyzed oligosaccharides proximal to the reducing end, whereas PelAh cleaved near the nonreducing end. This observation suggests differences in the roles of the substrate-binding residues on either side of the catalytic site in each enzyme. This hypothesis is substantiated by the differences in surface residue conservation in the putative binding grooves of Sph3h and PelAh. Sph3h contains a longer stretch of conserved residues on the nonreducing side of the −1/+1 cleavage site. The conserved residues in Sph3h span the −5 to +2 binding subsites. In contrast, the conserved surface of PelAh aligns with −2 to +5 subsites.
Previously, we reported that PelAh was able to disrupt both GAG- and Pel-dependent biofilms. In contrast, Sph3h was active only against GAG-dependent fungal biofilms, and was unable to cleave Pel-dependent biofilms despite being able to bind Pel polysaccharide (19, 24). The results of our structure-function studies reveal important differences in the substrate specificity and structure of these enzymes that may explain these observations. PelAh was found to have a deeper, more electronegative substrate-binding groove and exhibit preferential activity against partially deacetylated substrates compared with Sph3h. Although Sph3h treatment of soluble GAG releases predominately acetylated oligomers, treatment with PelAh releases 98% partially deacetylated products. Although the detailed structure of Pel has not been determined, compositional studies suggest it contains both GlcNAc and GalNAc (23). The degree of deacetylation, and the identity of the sugars deacetylated in the Pel polysaccharide have not been determined. Our results suggest the inactivity of Sph3h against Pel may reflect the fact that Pel is more extensively deacetylated than GAG. Alternately, whereas the activity of PelA against α-linked GalNAc suggests that these α-linkages are present within Pel, it is possible that the presence of GlcNAc sugars within the Pel polymer may interfere with Sph3h ability to cleave the Pel polymer. Distinguishing among these possibilities, and confirming the presence of α-linked GalNAc in Pel will require a complete structural analysis of this polysaccharide. The results of these studies assign enzymatic function to two new glycoside hydrolases. Sph3h is the only GH135 family member that has been functionally characterized. Although the structure of Sph3h had been previously determined (24), the current study expands our understanding of the activity of this enzyme and demonstrates that Sph3h functions as a retaining endo-α-1,4-N-acetylgalactosaminidase with specificity for fungal GAG. We also found that the bacterial enzyme PelAh is a retaining endo-α-1,4-N-acetylgalactosaminidase. As the sequence identity between PelAh and current glycoside hydrolase families is low, our structural and functional characterization of this protein enables us to classify PelAh as the first member of a new family, GH166.
Materials and methods
Protein expression and purification of PelAh for structural studies
PelAh encompassing residues 47–303 of the mature PelAh protein was expressed and purified as previously described (21). Selenomethionine (SeMet)-labeled protein was produced as previously described (38) with B834 Met Escherichia coli cells (Novagen) and purified as described for the native protein.
Biofilm disruption assay
104 A. fumigatus conidia were grown in Brian media in polystyrene, nontissue culture treated 96-well-plates for 21 h at 37 °C and then treated with the indicated concentration of glycoside hydrolase in 1× PBS for 1 h at room temperature under gentle agitation. Biofilms were then washed, stained with 0.1% (w/v) crystal violet and destained with 100% ethanol for 10 min. The optical density of the destain solution was measured at 600 nm.
MALDI-TOF MS enzymatic fingerprint
Products of enzymatic digestions were diluted in 0.2% trifluoroacetic acid (TFA) before being spotted on the MALDI-TOF plate in a ratio 1:1 (v/v) with 5 mg/ml of dihydroxybenzoic acid matrix reconstituted in acetonitrile (ACN), 0.2% TFA (70:30, v/v). Spectra were recorded on a Bruker UltrafleXtreme in positive reflector mode and represent an accumulation of 5000 laser shots. MALDI-TOF MS/MS experiments were performed using the same mass spectrometer.
GC-mass spectrometry monosaccharide composition
Oligosaccharides enzymatically extracted from biofilm were hydrolyzed with 6 m hydrochloric acid (HCl) for 4 h at 100 °C. After drying, samples were derivatized and analyzed as previously described (39). Briefly, samples were then converted in methyl glycosides by heating in 1 m methanol-HCl (Supelco) for 16 h at 80 °C. Samples were dried and washed twice with methanol prior re-N-acetylating hexosamine residues. Re-N-acetylation was performed by incubation with a mix of methanol:pyridine:anhydride acetic (10:2:3) for 1 h at room temperature. Samples were then treated with hexamethyldisilazane:trimethylchlorosilane:pyridine solution (3:1:9, Supelco) for 20 min at 80 °C. The resulting TMS methyl glycosides were dried, resuspended in 1 ml of cyclohexane, and injected in the Trace1300 GC-MS system equipped with a CP-Sil5-CB capillary column (Agilent Technologies). Elution was performed with the following temperature gradient: 120 to 160 °C at a rate of 10 °C/min, 160 to 220 °C at a rate of 1.5 °C/min, 220 to 280 °C at a rate of 20 °C/min. Identification and quantification of each monosaccharide were carried out using standards and response factors determined for each monosaccharide.
Production of oligosaccharide of chitin, α-1,4-Gal, and α-1,4-GalN and specificity study
Oligosaccharides of chitin/chitosan were produced by acidic partial hydrolysis of chitin from shrimp shell (Sigma). Briefly, chitin was incubated in 0.1 m HCl for 2 h at 100 °C. Solubilized oligosaccharides were then purified on a Hypersep Hypercarb SPE cartridge (ThermoFisher) conditioned as per the manufacturer's instructions. After loading the sample, the cartridge was washed with water, 5% (v/v) ACN, and oligosaccharides were eluted with 50% (v/v) ACN.
The α-1,4-Gal and α-1,4-GalN oligosaccharides were chemically synthetized based on the use of di-tert-butylsilylene group-protected building blocks. These extremely powerful cis-galactosylating agents only produced α-isomer products. After glycosylation reactions, the di-tert-butylsilylene group was removed with HF-pyridine. The resulting free 6-hydroxyl group was protected with the benzoyl group selectively to afford the acceptor. The final compounds were obtained after deprotection by saponification, debenzylation, azide reduction, and acetylation of amine groups.
All oligosaccharides were incubated with 5 μm Sph3h or PelAh for 1 h at room temperature and analyzed by MALDI-TOF MS. Controls of enzyme functionality were performed using as substrate α-1,4-GalNAc oligosaccharides produced as described below.
Enzymatic mechanism determination by 1H NMR
Proton NMR spectra were recorded on an AVANCE III HD 600 NMR spectrometer (AscendTM 600 magnet, Bruker Biospin Ltd.) operating at a frequency of 600.17 MHz for 1 h and equipped with a quadruple resonance CryoProbe (CPQCI 1H-31P/13C/15N) and a SampleJETTM autosampler. For each sample, 0.45 mg of α-1,4-GalNAc octamer was resuspended in 160 μl of 0.1× PBS in D2O containing 0.5 mm trimethylsilylpropanoic acid and transferred in a 3-mm NMR tube. The 1H NMR spectra were continuously acquired at 25 °C for 4 h with lock and shim performed on every 10 experiments. A new spectrum at 25 °C was recorded after 24 h to observe the mutarotation and confirmation of this event was validated with the acquisition of 1H NMR spectra at 37 °C and a 2D 1H-1H correlation spectroscopy (COSY) spectra at 25 °C. The 1H spectra were acquired using the pulse sequence noesypr1d (Bruker Biospin Ltd.) to achieve good suppression of the water signal. Each 1H spectrum was acquired with 32 scans, a 1H 90° pulse length of 7.8 μs, a mixing time of 10 ms, a spectral width of 12 kHz, and a recycle delay of 4 s for a total of 66,000 data points. The 2D 1H-1H COSY spectra were acquired using 16 scans with a 1H 90° pulse length of 8 μs, a spectral width of 3 kHz in both dimensions, and a repetition delay of 1.8 s for a total of 2048 data points in F2 and 128 increments in F1. All spectra were processed using TOPSPIN software (version 3.5 pl 7, Bruker Biospin Ltd.).
Production of pure α-1,4-GalNAc oligosaccharides
A. fumigatus biofilm were incubated with 5 nm Sph3h for 1 h at room temperature, solubilized oligosaccharides were then further purified on a Sep-Pak C18 cartridge. In brief, cartridges were conditioned using absolute ethanol followed by water. Samples were then loaded onto the cartridge before washing and eluting using a 0.25% (v/v) step gradient of methanol from 0 to 4% (v/v) followed by a 1% (v/v) step gradient of ACN from 1 to 4%.
Reduction of oligosaccharides
Reduction of the α-1,4-GalNAc 9-mer was performed resuspending the oligosaccharides in 1 m ammonium hydroxide containing 10 mg/ml of sodium borohydride and incubating overnight at room temperature. Reaction was quenched adding dropwise 30% (v/v) acetic acid and samples were purified using a Hypersep Hypercarb SPE cartridge (ThermoFisher) as per chitin oligosaccharide purification.
Crystallization, data collection, and structure solution
Purified PelAh was concentrated to ∼20 mg/ml and crystallization trials were performed using MCSG1–4 sparse-matrix screens (Microlytic) in 48-well hanging-drop VDX plates (Hampton Research) using a 2-μl drop with a 1:1 protein:precipitant ratio at 20 °C. Initial crystallization hits were obtained in several conditions. Diffraction quality native PelAh crystals were grown using 0.1 m BisTris, pH 7.5, 25% (w/v) PEG MME 5000 at a 1:2 ratio of protein to crystallization solution at 20 °C. SeMet-labeled PelAh was crystallized in a similar condition with 26% (w/v) PEG MME 5000. Both crystals were cryoprotected for 10 s in mother liquor supplemented with 15% (v/v) ethylene glycol prior to vitrification in liquid nitrogen.
Diffraction data were collected at −173 °C with wavelengths of 0.9791 and 1.075 Å on beamline X29, National Synchrotron Light Source (NSLS) (Table 1) for the SeMet labeled and native crystal, respectively. A high redundancy dataset was generated for the SeMet-labeled PelAh by collecting 90 images with 2° oscillation at 90% beam attenuation and with an exposure time of 0.3 s/image and 360 images with 1° oscillation with 50% beam attenuation with an exposure time of 0.4 s/image on an ADSC Quantum-315 detector with a 260-mm crystal-to-detector distance. The native PelAh dataset was collected using the same strategy as described above for the SeMet crystal but with a 180-mm crystal-to-detector distance. Autosol (40) was used to determine initial phases and generate a density-modified map. The resulting electron density map was of high quality and enabled PHENIX AutoBuild to build >95% of the protein. The remaining residues were built manually in COOT (41, 42) and the structure refined using PHENIX.REFINE (43). Translation/Libration/Screw (TLS) groups were added to the refinement in PHENIX through the use of the TLSMD server (44, 45).
Table 1.
Summary of data collection and refinement statistics
Values in parentheses correspond to the highest resolution shell.
| SeMet PelAh | PelAh | |
|---|---|---|
| Data collection | ||
| Beamline | NSLS X29 | NSLS X29 |
| Wavelength (Å) | 0.979 | 1.075 |
| Space group | P21212 | P21212 |
| Cell dimensions | ||
| a, b, c (Å) | 61.3, 85.2, 47.2 | 65.0, 84.0, 47.2 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50.0–1.90 (1.97–1.90) | 50.00–1.54 (1.58–1.54) |
| No. reflections | 304,767 | 665,419 |
| No. of unique reflections | 19,863 | 39,193 |
| I/σI | 21.7 (4.5) | 18.6 (6.6) |
| Completeness (%) | 99.8 (99.7) | 99.9 (99.5) |
| Rmerge (%)a | 12.2 (62.5) | 18.1 (44.3) |
| Refinement | ||
| Rworkb/Rfreec | 16.9/19.2 | |
| No. of atoms | ||
| Protein | 1,935 | |
| Water | 193 | |
| Average B-factors (Å2) | ||
| Protein | 31.6 | |
| Water | 38.3 | |
| RMS deviations | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 0.94 | |
| Ramachandran plotd | ||
| Total favored (%) | 96.8 | |
| Total allowed (%) | 100 | |
| Coordinate error (Å)e | 0.13 | |
| PDB code | 5TCB |
a Rmerge = ∑∑|I (k) − 〈I〉|/∑ I (k), where I (k) and 〈I〉 represent the diffraction intensity values of the individual measurements and the corresponding mean values. The summation is over all unique measurements.
b Rwork = ∑‖Fobs| − k|Fcalc‖/|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
c Rfree is the sum extended over a subset of reflections (5%) excluded from all stages of the refinement.
d As calculated using MolProbity (51).
e Maximum-Likelihood Based Coordinate Error, as determined by PHENIX (42).
All structure figures were generated using the PyMOL molecular graphics system (DeLano Scientific) (46), or Chimera (47) for electrostatics using APBS (48). Structural similarity to deposited structures in the Protein Data Bank (PDB) was determined using DALI and structure alignment was performed in COOT (27, 41). Amino acid conservation was calculated using the Consurf server (49) aligned to 113 proteins using the default settings. Programs used for crystallographic data processing and analysis were accessed through SBGrid (50).
Secreted GAG purification and digest
Secreted purified GAG was prepared as previously reported (13). Briefly, culture supernatant of a 3-day-old Af293 culture was filtered on Miracloth prior to being ethanol precipitated. Precipitate was then successively washed with 70% (v/v) ethanol twice, 150 mm NaCl, and water. The remaining gel was then freeze dried. The dried purified GAG was incubated with the 1 μm glycoside hydrolase for 1 h in 0.1× PBS. The released soluble oligosaccharides were then analyzed using the MALDI-TOF MS enzymatic fingerprint technique.
Author contributions
F. L. M., N. C. B., and P. L. H. conceptualization; F. L. M. and N. C. B. formal analysis; F. L. M., N. C. B., N. A., P. B., and H. R. investigation; F. L. M. and N. C. B. visualization; F. L. M., N. C. B., and P. L. H. methodology; F. L. M., N. C. B., P. L. H., and D. C. S. writing-original draft; F. L. M., N. C. B., N. A., Y. Z., P. B., H. R., J. D. C., P. L. H., and D. C. S. writing-review and editing; Y. Z. and J. D. C. resources; P. L. H. and D. C. S. supervision; P. L. H. and D. C. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Todd Lowary, Department of Chemistry, University of Alberta, for assistance in the design and interpretation of the NMR spectra. We thank the Drug Discovery platform of the Research Institute of the McGill Health Center for use of the MALDI-TOF mass spectrometer and NMR equipment. Beam Line X29 at the National Synchrotron Light Source is supported by the United States Department of Energy Office and National Center for Research Resources Grant P41RR012408 and NIGMS Grant P41GM103473 from the National Institutes of Health.
This work was supported by Canadian Institutes of Health Research (CIHR) Operating Grants 81361 (to P. L. H. and D. C. S.), FDN-154327 and 43998 (to P. L. H.), and 123306 and FDN-159902 (to D. C. S.), Cystic Fibrosis Canada Grant 558692 (to D. C. S. and P. L. H.), in part by a graduate scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC) including Vanier and CGS-M, Mary H. Beatty, and Dr. James A. and Connie P. Dickson Scholarships from the University of Toronto, Cystic Fibrosis Canada, and The Hospital for Sick Children (to N. C. B.), and European Research Council (ERC) Grant ERC-CoG-726072-GLYCONTROL (to J. D. C. C.). P. L. H. is the recipient of a Tier I Canada Research Chair. D. C. S. is supported by a Chercheur-Boursier Award from Fonds de Recherche Quebec Santé (FRQS) Grant 34961. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- GAG
- galactosaminogalactan
- GH
- glycoside hydrolase
- CAZy
- carbohydrate active enzyme
- HexNAc
- N-acetylhexosamine
- RMSD
- root mean square deviation
- ACN
- acetonitrile
- SPE
- solid phase extraction
- Sph3h
- hydrolase domain of Sph3
- PelAh
- hydrolase domain of PelA
- βHL
- β-hairpin loop
- SeMet
- selenomethionine
- PDB
- Protein Data Bank
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Latgé J.-P. (2001) The pathobiology of Aspergillus fumigatus. Trends Microbiol. 9, 382–389 10.1016/S0966-842X(01)02104-7 [DOI] [PubMed] [Google Scholar]
- 2. Taccone F. S., Van den Abeele A. M., Bulpa P., Misset B., Meersseman W., Cardoso T., Paiva J. A., Blasco-Navalpotro M., De Laere E., Dimopoulos G., Rello J., Vogelaers D., Blot S. I., and AspICU Study Investigators (2015) Epidemiology of invasive aspergillosis in critically ill patients: clinical presentation, underlying conditions, and outcomes. Crit. Care (London, England) 19, 7 10.1186/s13054-014-0722-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mehrad B., Strieter R. M., and Standiford T. J. (1999) Role of TNF-α in pulmonary host defense in murine invasive Aspergillosis. J. Immunol. 162, 1633–1640 [PubMed] [Google Scholar]
- 4. Müller F.-M., Seidler M., and Beauvais A. (2011) Aspergillus fumigatus biofilms in the clinical setting. Med. Mycology 49, S96–S100 10.3109/13693786.2010.502190 [DOI] [PubMed] [Google Scholar]
- 5. Pierce C. G., Srinivasan A., Uppuluri P., Ramasubramanian A. K., and López-Ribot J. L. (2013) Antifungal therapy with an emphasis on biofilms. Curr. Opin. Pharmacol. 13, 726–730 10.1016/j.coph.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seidler M. J., Salvenmoser S., and Müller F.-M. (2008) Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob. Agents Chemother. 52, 4130–4136 10.1128/AAC.00234-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kernien J. F., Snarr B. D., Sheppard D. C., and Nett J. E. (2017) The interface between fungal biofilms and innate immunity. Front. Immunol. 8, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramage G., Rajendran R., Sherry L., and Williams C. (2012) Fungal biofilm resistance. Int. J. Microbiol. 2012, 528521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reichhardt C., Ferreira J. A., Joubert L.-M., Clemons K. V., Stevens D. A., and Cegelski L. (2015) Analysis of the Aspergillus fumigatus biofilm extracellular matrix by solid-state nuclear magnetic resonance spectroscopy. Eukaryot. Cell 14, 1064–1072 10.1128/EC.00050-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loussert C., Schmitt C., Prevost M. C., Balloy V., Fadel E., Philippe B., Kauffmann-Lacroix C., Latgé J. P., and Beauvais A. (2010) In vivo biofilm composition of Aspergillus fumigatus. Cell. Microbiol. 12, 405–410 10.1111/j.1462-5822.2009.01409.x [DOI] [PubMed] [Google Scholar]
- 11. Sheppard D. C., and Howell P. L. (2016) Biofilm exopolysaccharides of pathogenic fungi: lessons from bacteria. J. Biol. Chem. 291, 12529–12537 10.1074/jbc.R116.720995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee M. J., Gravelat F. N., Cerone R. P., Baptista S. D., Campoli P. V., Choe S. I., Kravtsov I., Vinogradov E., Creuzenet C., Liu H., Berghuis A. M., Latgé J. P., Filler S. G., Fontaine T., and Sheppard D. C. (2014) Overlapping and distinct roles of Aspergillus fumigatus UDP-glucose 4-epimerases in galactose metabolism and the synthesis of galactose-containing cell wall polysaccharides. J. Biol. Chem. 289, 1243–1256 10.1074/jbc.M113.522516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee M. J., Geller A. M., Bamford N. C., Liu H., Gravelat F. N., Snarr B. D., Le Mauff F., Chabot J., Ralph B., Ostapska H., et al. (2016) Deacetylation of fungal exopolysaccharide mediates adhesion and biofilm formation. MBio 7, e00252–00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Briard B., Muszkieta L., Latgé J.-P., and Fontaine T. (2016) Galactosaminogalactan of Aspergillus fumigatus, a bioactive fungal polymer. Mycologia 108, 572–580 10.3852/15-312 [DOI] [PubMed] [Google Scholar]
- 15. Sheppard D. C. (2011) Molecular mechanism of Aspergillus fumigatus adherence to host constituents. Curr. Opin. Microbiol. 14, 375–379 10.1016/j.mib.2011.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee M. J., Liu H., Barker B. M., Snarr B. D., Gravelat F. N., Al Abdallah Q., Gavino C., Baistrocchi S. R., Ostapska H., Xiao T., et al. (2015) The fungal exopolysaccharide galactosaminogalactan mediates virulence by enhancing resistance to neutrophil extracellular traps. PLoS Pathog. 11, e1005187 10.1371/journal.ppat.1005187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gravelat F. N., Beauvais A., Liu H., Lee M. J., Snarr B. D., Chen D., Xu W., Kravtsov I., Hoareau C. M., Vanier G., et al. (2013) Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal β-glucan from the immune system. PLoS Pathog. 9, e1003575 10.1371/journal.ppat.1003575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van de Veerdonk F. L., Gresnigt M. S., Romani L., Netea M. G., and Latgé J.-P. (2017) Aspergillus fumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 15, 661–674 10.1038/nrmicro.2017.90 [DOI] [PubMed] [Google Scholar]
- 19. Snarr B. D., Baker P., Bamford N. C., Sato Y., Liu H., Lehoux M., Gravelat F. N., Ostapska H., Baistrocchi S. R., and Cerone R. P. (2017) Microbial glycoside hydrolases as antibiofilm agents with cross-kingdom activity. Proc. Natl. Acad. Sci. U.S.A. 114, 7124–7129 10.1073/pnas.1702798114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Colvin K. M., Alnabelseya N., Baker P., Whitney J. C., Howell P. L., and Parsek M. R. (2013) PelA deacetylase activity is required for Pel polysaccharide synthesis in Pseudomonas aeruginosa. J. Bacteriol. 195, 2329–2339 10.1128/JB.02150-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baker P., Hill P. J., Snarr B. D., Alnabelseya N., Pestrak M. J., Lee M. J., Jennings L. K., Tam J., Melnyk R. A., Parsek M. R., Sheppard D. C., Wozniak D. J., and Howell P. L. (2016) Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci. Adv. 2, e1501632 10.1126/sciadv.1501632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Franklin M. J., Nivens D. E., Weadge J. T., and Howell P. L. (2011) Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, Alginate, Pel, and Psl. Front. Microbiol. 2, 167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jennings L. K., Storek K. M., Ledvina H. E., Coulon C., Marmont L. S., Sadovskaya I., Secor P. R., Tseng B. S., Scian M., and Filloux A. (2015) Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl. Acad. Sci. U.S.A. 112, 11353–11358 10.1073/pnas.1503058112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bamford N. C., Snarr B. D., Gravelat F. N., Little D. J., Lee M. J., Zacharias C. A., Chabot J. C., Geller A. M., Baptista S. D., and Baker P. (2015) Sph3 is a glycoside hydrolase required for the biosynthesis of galactosaminogalactan in Aspergillus fumigatus. J. Biol. Chem. 290, 27438–27450 10.1074/jbc.M115.679050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang S., Wang M., Chen L., Yin B., Song G., Turko I. V., Phinney K. W., Betenbaugh M. J., Zhang H., and Li S. (2015) QUANTITY: an isobaric tag for quantitative glycomics. Sci. Rep. 5, 17585 10.1038/srep17585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–394 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holm L., and Laakso L. M. (2016) Dali server update. 44, W351–W355 10.1093/nar/gkw357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yin Y., Mao X., Yang J., Chen X., Mao F., and Xu Y. (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, 445–W451 10.1093/nar/gks479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., Busk P. K., Xu Y., and Yin Y. (2018) dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101 10.1093/nar/gky418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henrissat B., and Bairoch A. (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316, 695–696 10.1042/bj3160695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Payne C. M., Baban J., Horn S. J., Backe P. H., Arvai A. S., Dalhus B., Bjørås M., Eijsink V. G., Sørlie M., Beckham G. T., and Vaaje-Kolstad G. (2012) Hallmarks of processivity in glycoside hydrolases from crystallographic and computational studies of the Serratia marcescens chitinases. J. Biol. Chem. 287, 36322–36330 10.1074/jbc.M112.402149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davies G., and Henrissat B. (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859 10.1016/S0969-2126(01)00220-9 [DOI] [PubMed] [Google Scholar]
- 33. Kleywegt G. J., Zou J.-Y., Divne C., Davies G. J., Sinning I., Stâhlberg J., Reinikainen T., Srisodsuk M., Teeri T. T., and Jones T. A. (1997) The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 Å resolution, and a comparison with related enzymes1. J. Mol. Biol. 272, 383–397 10.1006/jmbi.1997.1243 [DOI] [PubMed] [Google Scholar]
- 34. Cecchini D. A., Fauré R., Laville E., and Potocki-Veronese G. (2015) Biochemical identification of the catalytic residues of a glycoside hydrolase family 120 β-xylosidase, involved in xylooligosaccharide metabolisation by gut bacteria. FEBS Lett. 589, 3098–3106 10.1016/j.febslet.2015.08.012 [DOI] [PubMed] [Google Scholar]
- 35. Zechel D. L., and Withers S. G. (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res. 33, 11–18 10.1021/ar970172 [DOI] [PubMed] [Google Scholar]
- 36. Moracci M., Capalbo L., Ciaramella M., and Rossi M. (1996) Identification of two glutamic acid residues essential for catalysis in the β-glycosidase from the thermoacidophilic archaeon Sulfolobus solfataricus. Protein Eng. 9, 1191–1195 10.1093/protein/9.12.1191 [DOI] [PubMed] [Google Scholar]
- 37. Little D. J., Pfoh R., Le Mauff F., Bamford N. C., Notte C., Baker P., Guragain M., Robinson H., Pier G. B., Nitz M., Deora R., Sheppard D. C., and Howell P. L. (2018) PgaB orthologues contain a glycoside hydrolase domain that cleaves deacetylated poly-β-(1,6)-N-acetylglucosamine and can disrupt bacterial biofilms. PLoS Pathog. 14, e1006998 10.1371/journal.ppat.1006998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee J. E., Cornell K. A., Riscoe M. K., and Howell P. L. (2001) Structure of E. coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure 9, 941–953 10.1016/S0969-2126(01)00656-6 [DOI] [PubMed] [Google Scholar]
- 39. Le Mauff F., Loutelier-Bourhis C., Bardor M., Berard C., Doucet A., D'Aoust M. A., Vezina L. P., Driouich A., Couture M. M., and Lerouge P. (2017) Cell wall biochemical alterations during Agrobacterium-mediated expression of haemagglutinin-based influenza virus-like vaccine particles in tobacco. Plant Biotechnol. J. 15, 285–296 10.1111/pbi.12607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terwilliger T. C., and Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 849–861 10.1107/S0907444999000839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., and Grosse-Kunstleve R. W. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Afonine P. V., Mustyakimov M., Grosse-Kunstleve R. W., Moriarty N. W., Langan P., and Adams P. D. (2010) Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 66, 1153–1163 10.1107/S0907444910026582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 10.1107/S0907444906005270 [DOI] [PubMed] [Google Scholar]
- 45. Painter J., and Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 10.1107/S0021889805038987 [DOI] [Google Scholar]
- 46. DeLano W. L. (2002) The PyMOL Molecular Graphics System. Schroedinger, LLC, New York [Google Scholar]
- 47. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 48. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ashkenazy H., Erez E., Martz E., Pupko T., and Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 10.1093/nar/gkq399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Cutting edge: collaboration gets the most out of software. elife 2, e01456 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.