

Abstract
Human IgG2 antibody displays distinct therapeutically-useful properties compared with the IgG1, IgG3, and IgG4 antibody subclasses. IgG2 is the second most abundant IgG subclass, being able to bind human FcγRII/FcγRIII but not to FcγRI or complement C1q. Structural information on IgG2 is limited by the absence of a full-length crystal structure for this. To this end, we determined the solution structure of human myeloma IgG2 by atomistic X-ray and neutron-scattering modeling. Analytical ultracentrifugation disclosed that IgG2 is monomeric with a sedimentation coefficient (s20, w0) of 7.2 S. IgG2 dimer formation was ≤5% and independent of the buffer conditions. Small-angle X-ray scattering in a range of NaCl concentrations and in light and heavy water revealed that the X-ray radius of gyration (Rg) is 5.2–5.4 nm, after allowing for radiation damage at higher concentrations, and that the neutron Rg value of 5.0 nm remained unchanged in all conditions. The X-ray and neutron distance distribution curves (P(r)) revealed two peaks, M1 and M2, that were unchanged in different buffers. The creation of >123,000 physically-realistic atomistic models by Monte Carlo simulations for joint X-ray and neutron-scattering curve fits, constrained by the requirement of correct disulfide bridges in the hinge, resulted in the determination of symmetric Y-shaped IgG2 structures. These molecular structures were distinct from those for asymmetric IgG1 and asymmetric and symmetric IgG4 and were attributable to the four hinge disulfides. Our IgG2 structures rationalize the existence of the human IgG1, IgG2, and IgG4 subclasses and explain the receptor-binding functions of IgG2.
Keywords: antibody, analytical ultracentrifugation, molecular modeling, neutron-scattering, small-angle X-ray scattering (SAXS), immunoglobulin G (IgG)
Introduction
Up to 75% of the total antibody content in serum is composed of the IgG class. The IgG class is divided into four subclasses, IgG1, IgG2, IgG3, and IgG4. The differences between these arise due to their variable regions, upper CH2 domains, and the hinge (Fig. 1). The hinge consists of the upper, middle, and lower hinge (Fig. 2). The core hinge (upper and middle) contains 15, 12, 62, and 12 residues for IgG1, IgG2, IgG3, and IgG4, respectively. The IgG subclasses present much variety in structure and function, even though the constant domains possess over 95% sequence homology (1). IgG2 is the second most abundant subclass with an average concentration of 3 mg/ml in adult serum (1). IgG2 has a serum half-life of 21 days as for the IgG1 and IgG4 subclasses. IgG2 has a unique role as this is the predominant IgG subclass that binds to bacterial capsular polysaccharide antigens (2, 3). Also, IgG2 shows an increased resistance to microbial proteases for reasons of the unique sequence of the lower hinge (4, 5). IgG2 binds complement C1q weakly, and thus IgG2 predominantly activates the complement cascade through the alternative pathway (6). Of the three classes of human FcγR receptors, IgG2 binds to only FcγRII and FcγRIII and not to FcγRI. Through its ligand binding and the formation of antigen–antibody immune complexes, IgG2 activates antibody-dependent cell-mediated cytotoxicity through macrophages and polymorphonuclear leukocytes, in order that antibody-dependent cell-mediated phagocytosis will clear any pathogens such as bacteria. In biotechnology, IgG2 is regarded as the antibody with the least overall effector function, and thus it makes a perfect scaffold for designing therapeutic antibodies with lack of function, similar to IgG4. Several Food and Drug Administration–approved drugs are based on IgG2, including evolocumab, denosumab, panitumumab, brodalumab, and erenumab (Antibody Society).
Figure 1.
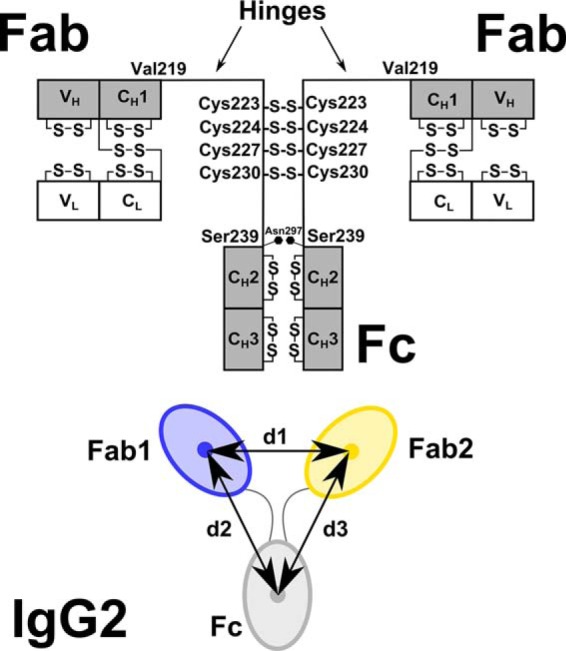
Human IgG2 domain structure. The two heavy chains each possess VH, CH1, CH2, and CH3 domains, and the two light chains each possess VL and CL domains. The heavy chains are connected by four Cys–Cys disulfide bridges at Cys-223, Cys-224, Cys-227, and Cys-230. There is one N-linked oligosaccharide site at Asn-297 on each of the CH2 domains. The hinge region between the Fab and Fc fragments is composed of 19 residues (ERKCCVECPPCPAPPVAGP) between Val-219 and Ser-239 (EU numbering). Below the black diagram, the distance between the centers of mass of the two Fab regions (blue, yellow) was denoted as d1. Those between the two Fab and Fc regions were denoted as d2 and d3. The antibody is shown as a 2-fold symmetric structure with d2 = d3. In general, d2 and d3 are unequal. In the text, the smaller of the two values is denoted as min(d2,d3), and the larger of the two is denoted as max(d2,d3).
Figure 2.

Sequence alignment of IgG2 with human IgG1 6a and IgG1 19a and IgG4. The IgG2 sequence was taken from IgG2 Li33 (13). The IgG1 6a and 19a sequences were taken from Ref. 27. A and B, VL, and CL domains. C–E, VH and CH1 domains and the hinge. F and G, CH2 and CH3 domains. H, comparison of hinge sequences from human IgG1, IgG2, and IgG4 subclasses. E and F, yellow indicates the contact residues involved in the IgG1–Fc complex with the C1q globular head, and blue indicates the contact residues required for interacting with FcγRI, and green indicates the contact residues that interact with both C1q and FcγRI.
IgG2 is ∼150 kDa and has the typical IgG structure consisting of two heavy chains (H)3 and two light chains (L) that are divided into variable (V) and constant (C) domains (Fig. 1). The heavy chains are connected via four disulfide bonds that form between cysteine residues in the hinge. Structurally, human IgG2 from myeloma forms covalent dimers through inter-protein disulfide bonds arising from the hinge cysteine residues (7). IgG2 possesses three different isoforms termed IgG2A, IgG2A/B, and IgG2B with different hinge disulfide bonds (8–10). IgG2A is regarded as the “classical” or “canonical” IgG2 structure with four intact disulfide bonds in the hinge (Fig. 1). The IgG2A/B isoform contains one Fab region disulfide-linked to the hinge, and the IgG2B isoform contains both Fab regions disulfide-linked to the hinge. The significance of these isoforms on the structure and function is currently unknown.
Structural information on the IgG subclasses is lacking because only two crystal structures for full-length human antibodies are available, namely IgG1 b12 and IgG4 (PDB codes 1HZH and 5DK3) (11, 12). This is attributed to the inherent flexibility in the antibody hinge in the order of IgG3 > IgG1 > IgG4 > IgG2 that makes crystallizations difficult (13). The crystal structures only offer a single “snapshot” of a potential broad range of IgG structures in physiological conditions (11). Electron micrographs of human–mouse chimeric IgG2 in vacuo show the existence of different shapes (14). Although no full-length IgG2 crystal structure is yet available, crystal structures for the Fab and Fc regions of human IgG2 are available (PBD codes 3KYM, 4HAF, 4HAG, and 4L4J) (15–17). Myeloma IgG2 has been studied by EM, differential scanning microcalorimetry, and fluorescence to reveal an asymmetric structure with one Fab region closer to the Fc region than the other Fab region, similar to that seen for IgG1 and IgG4 (18). Human monoclonal and polyclonal IgG2, human myeloma IgG2, human–mouse chimeric human IgG2, and humanized IgG2 have been previously studied using X-ray or neutron solution scattering or analytical ultracentrifugation (8, 14, 15, 19–27). The recent studies of human monoclonal IgG1 and IgG4 utilized modeling to fit the scattering curves in terms of molecular structures (28, 29). More accurate modeling for human IgG1 and IgG4 based on joint X-ray and neutron-scattering data sets with Monte Carlo simulations has been performed using a newly developed workflow termed SASSIE (30).4 The outputted structures are atomistic in their nature, because they are physically-realistic models with correctly-joined amino acid and glycan residues. These outputs revealed asymmetric solution structures that resembled the IgG1 and (in part) the IgG4 crystal structures. Here, we used joint small-angle X-ray and neutron-scattering (SAXS and SANS), analytical ultracentrifugation (AUC), and Monte Carlo modeling to analyze 123,371 physically-realistic IgG2 structures. The resulting best-fit atomistic models revealed that classical IgG2 possesses a Y-shaped symmetric conformation in solution. This outcome explained in structural terms for the first time the different IgG2 isoforms and the ligand-binding functions of IgG2 to C1q and the three human FcγR receptors.
Results
Purification and characterization of IgG2
Human IgG2 from myeloma plasma was subjected to Superose 6 gel filtration to ensure that this was monodisperse immediately prior to AUC, SAXS, and SANS experiments. It was eluted as a large main peak at ∼16 ml, with a minor peak at 14.5 ml that was discarded (Fig. 3). Nonreducing and reducing SDS-polyacrylamide gels were run for IgG2, IgG1 6a, IgG1 19a, and IgG4 B72.3. A single band in Fig. 3, lane 2, between 200 and 116 kDa in nonreducing SDS-PAGE corresponds to the expected mass of 147.4 kDa for intact IgG2. Under reducing conditions in Fig. 3, lane 3, the heavy chains were observed at an apparent molecular mass of 55.4 kDa, and the light chains were observed between 21.5 and 31 kDa, both as expected (Fig. 3). The corresponding nonreducing and reducing samples for the other antibodies IgG1 6a, IgG1 19a and IgG4 B72.3 were also consistent with previous studies, noting that IgG4 forms trace amount of a half-molecule (28, 29).
Figure 3.

Purification of IgG2. The IgG2 elution peak from a Superose 6 10/300 gel-filtration column is shown on the left (mAU, milli-absorbance units). The nonreduced and reduced SDS-PAGE analysis of the IgG subclasses is shown on the right with H2L2 representing the intact antibody molecule, H the heavy chain, and L the light chain. Lanes 1 and 10 contain Mark 12 molecular mass markers labeled in kDa. Lanes 2- 3, 4–5, 6–7, and 8–9 contain nonreduced and reduced IgG2, nonreduced and reduced IgG1 6a, nonreduced and reduced IgG1 19a, and nonreduced and reduced IgG4 B72.3, respectively.
Native and deglycosylated myeloma IgG2 was subjected to native MS to determine its molecular mass size range. The mass spectra show that native and deglycosylated IgG2 existed as three main populations. For native IgG2 (Fig. 4A), the masses for the three populations were 154,527 ± 52, 156,392 ± 139, and 157,988 ± 62 Da. The observed molecular masses were higher than the calculated molecular mass of 147.4 kDa from the sequence of IgG2 anti-LINGO1 Li33 (Fig. 2), suggesting polydispersity arising from variable protein and glycan contents, but as this spanned 3,461 Da (2.2%) in mass, this polydispersity was comparatively low. The amounts of the three species were 8.9% for 154.5 kDa, 48% for 156.4 kDa, and 43.1% for 158.0 kDa. The high mass error range was attributed to the different glycoforms present in native IgG2. For deglycosylated IgG2 (Fig. 4B), the signals were clearer with reduced error ranges. The masses and corresponding distributions for the three populations were decreased by 3,199 to 151,405 ± 4 Da (37.2%), 153,101 ± 3 Da (43.7%), and 154,805 ± 10 Da (19%), again spanning 3,400 Da in mass. The reduction was attributed to the removal of two biantennary glycan chains at Asn-297 (each with an approximate mass of 2,200 Da) through PNGase digests. Compared with IgG2 anti-LINGO1 Li33 as a baseline, the protein molecular masses of the three species were increased by up to 4.0–10.6 kDa (2.7–7.2%).
Figure 4.
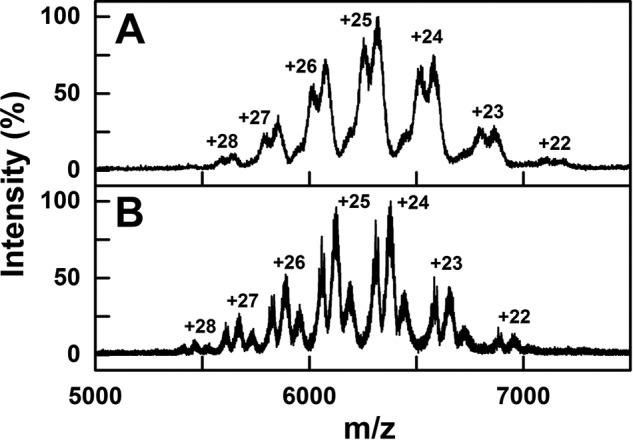
Native MS of glycosylated and deglycosylated IgG2. Native mass spectra of myeloma IgG2 are shown at an m/z between 5,000 and 7,500. The glycosylated and deglycosylated IgG2 mass spectra are shown in A and B, respectively. The theoretical charge states were generated using Amphitrite software and labeled.
Analytical ultracentrifugation of IgG2
The size and shape of IgG2 were examined using sedimentation velocity runs in AUC experiments. The SEDFIT analyses involved fits of up to 300 scans. Excellent agreement between the experimental boundary scans and fitted lines was seen (left panels, Fig. 5). The size distribution analyses c(s) for IgG2 showed a major monomeric species in solution and accompanied by a negligible dimer peak (right panels, Fig. 5). The monomer peak was observed at mean s20, w0 values of 7.33 ± 0.07 S for IgG2 in H2O and 7.07 ± 0.20 S in 2H2O, within error of each other (Fig. 6A). These s20, w0 values were comparable with values of 6.4–7.0 S previously reported for IgG2 (8, 14, 15, 19, 20) but not with the value of 5.4 S reported in one study (21). These previous studies did not state the protein partial specific volume, and the buffer density and viscosity in use, thus explaining small differences with earlier s20, w0 values. Some variation is attributed to instrumental effects; the s20, w0 values from 67 different laboratories showed a ±4.4% deviation in an AUC study of reproducibility (30). Given that the IgG2A/B and IgG2B isoforms possess different hinge disulfide bonds (8–10), the appearance of a single c(s) peak indicated that, if present, these two other isoforms showed similar shapes to the classic IgG2A structure.
Figure 5.
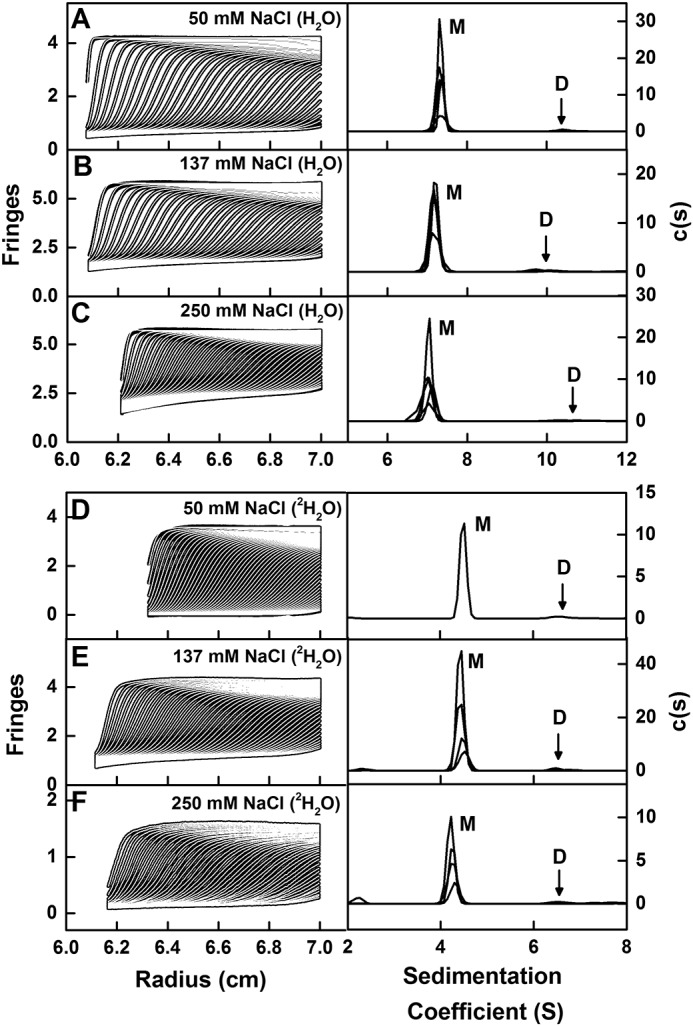
Sedimentation velocity analyses of IgG2. The experimentally observed sedimentation boundaries for IgG2 in the left panels in PBS-50 (A), PBS-137 (B), PBS-250 in H2O buffers (C), and in PBS-50 (D), PBS-137 (E), and PBS-250 (F) in 100% 2H2O buffers were recorded at a rotor speed of 40,000 rpm and 20 °C. Approximately 50 boundaries (black outlines) are shown from up to 300 scans for every sixth scan for clarity, and they were fitted using SEDFIT as shown (white lines). In the right panels, the corresponding size-distribution analyses c(s) are shown to reveal a major monomer (M) peak and a minor dimer (D) peak. The observed c(s) peaks are shifted to lower s values in 2H2O buffers.
Figure 6.
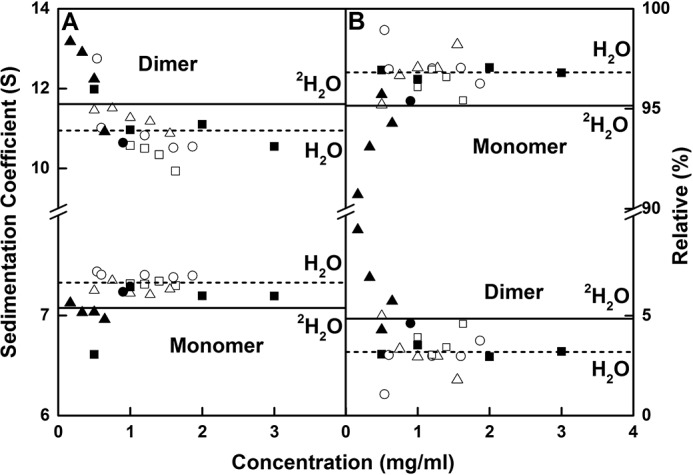
Sedimentation coefficient values and amounts of IgG2. A, s20, w values for the monomer and dimer peaks are shown as a function of IgG2 concentration in the six buffers of this study. B, percentages of monomer and dimer from the c(s) integrations. IgG2 is shown in PBS-50 (○), PBS-137 (□), and PBS-250 (▵) buffers in H2O at 20 °C and in PBS-50 (●), PBS-137 (■), and PBS-250 (▴) buffers at 20 °C in 100% 2H2O. The average s20, w values of monomer and dimer from the integration of the c(s) analyses for IgG2 are shown for H2O (- -) and 2H2O (—) buffers at 20 °C.
Given the scatter in s20, w0 values (Fig. 6A), the sedimentation rates of IgG2 did not display a clear dependence on sample concentration or buffer, indicating that the overall shape remained unchanged (Fig. 6A). Thus, the mean s20, w0 values in H2O were only slightly reduced with an increase in NaCl concentration, being 7.40 ± 0.02, 7.32 ± 0.02, and 7.26 ± 0.06 S for PBS-50, PBS-137, and PBS-250, respectively. The corresponding mean s20, w0 values in 2H2O were 7.24, 7.07 ± 0.31, and 7.04 ± 0.07 S for PBS-50, PBS-137, and PBS-250, respectively. The mean IgG2 dimer s20, w0 values in H2O were 11.1 ± 0.9, 10.3 ± 0.3, and 11.3 ± 0.3 S for PBS-50, PBS-137, and PBS-250, respectively. The mean s20, w0 values observed in 2H2O were 10.6, 11.2 ± 0.6, and 12.3 ± 1.0 S for PBS-50, PBS-137, and PBS-250, respectively (Fig. 6A). Given that protein partial specific volumes (v̄) are affected by the hydration shell (33, 34) and that the hydration shell for 2H2O has a higher mass than that for H2O, the v̄ values will be reduced in 2H2O. The heavy water v̄ value of 0.70 ml/g in place of 0.73 ml/g for light water proved sufficient to account for the s20, w0 values. Thus for H2O, the overall average s20, w0 values were 7.33 ± 0.07 and 10.95 ± 0.69 S for IgG2 monomer and dimer, respectively. In 2H2O, the overall average s20, w0 values were similar at 7.07 ± 0.20 and 11.61 ± 0.99 S for IgG2 monomer and dimer, respectively.
Heavy water is a promoter of dimer formation and aggregates, and small increases in IgG2 dimer were seen for the 2H2O samples. To examine this for IgG2, the mean proportions of the monomer for H2O were 97.2 ± 1.0, 96.3 ± 0.7, and 96.8 ± 1.1% for PBS-50, PBS-137, and PBS-250, respectively (Fig. 6B). For 2H2O, the mean amounts of monomer were 95.4, 96.8 ± 0.3, and 93.4 ± 2.1% for PBS-50, PBS-137, and PBS-250, respectively. In H2O, the mean value of 96.8 ± 0.5% decreased to 95.2 ± 2.1% for the IgG2 monomer in 2H2O. The mean proportion of dimers were 2.8, 3.7 ± 0.7, and 3.2 ± 1.1% for PBS-50, PBS-137, and PBS-250, respectively, in H2O (Fig. 6B). These showed small increases in 2H2O to 4.6, 3.2 ± 0.3, and 6.6 ± 2.1% for PBS-50, PBS-137, and PBS-250, respectively. For the IgG2 dimer in H2O buffer, the mean value of 3.2 ± 0.5% increased to 4.8 ± 2.1% in 2H2O buffer. High salt may have promoted dimer formation in PBS-250 in 2H2O.
The masses from the c(s) analyses compared well with the composition-calculated and MS values. The c(s) masses were 155 ± 18, 163 ± 6, and 147 ± 13 kDa for PBS-50, PBS-137, and PBS-250, respectively, in H2O. Those for 2H2O were 141 and 145 ± 17 kDa for PBS-50 and PBS-137 (not available in PBS-250). These values agreed with the expected molecular mass of 147.4 kDa calculated from the IgG2 composition and those of 151–158 kDa determined by native MS. For the dimer, the mean masses were doubled as expected at 282 ± 6, 274 ± 6, and 284 ± 19 kDa for PBS-50, PBS-137, and PBS-250 respectively, in H2O. In 2H2O, whereas the errors were greater for reason of low intensities, the mean masses were similar at 251, 285 ± 13, and 219 ± 27 kDa for PBS-50, PBS-137, and PBS-250, respectively, and were consistent with the expected composition-calculated mass of 297.4 kDa for the dimer. These agreements validated the use of myeloma IgG2 in the SAXS and SANS analyses.
X-ray and neutron-scattering data for IgG2
The IgG2 solution structure was jointly analyzed by both SAXS and SANS as complementary approaches (Table 1). SAXS monitored the shape of the hydration shell surrounding IgG2 as well as its overall antibody structure, whereas SANS using heavy water buffer monitored the overall shape of the unhydrated structure because the hydration shell was largely invisible in this buffer (32).
Table 1.
Experimental X-ray and neutron scattering data and sedimentation coefficients for human myeloma IgG2
| Sample | Rga | Rxs-1 | Rxs-2 | Dmax | s20, w0 |
|---|---|---|---|---|---|
| nm | nm | nm | S | ||
| X-ray scattering | |||||
| 0.5 mg/ml, PBS-50 | 5.24; 5.40 | 2.61 | 1.35 | 18 | 7.44b |
| 1.0 mg/ml, PBS-137 | 5.23; 5.36 | 2.61 | 1.37 | 18 | 7.32 |
| 1.5 mg/ml, PBS-250 | 5.38; 5.52 | 2.61 | 1.37 | 18 | 7.26b |
| Neutron scattering | |||||
| .45 mg/ml, PBS-50 in 100% 2H2O | 5.04; 5.20 | 2.39 | 1.12 | 17 | 7.24b |
| 1.0 mg/ml, PBS-137 in 100% 2H2O | 4.97; 5.00 | 2.45 | 1.15 | 17 | 7.28 |
| 1.99 mg/ml, PBS-250 in 100% 2H2O | 4.95; 5.03 | 2.39 | 1.12 | 17 | 6.96b |
SAXS was used to examine IgG2 at 0.5–4.0 mg/ml at 20 °C in PBS-50, PBS-137, and PBS-250, using time-frame analyses to ensure the absence of radiation damage effects. Overall, cross-sectional Guinier analyses resulted in high-quality linear plots in three distinct regions of the I(Q) curves, as expected for antibodies, from which the Rg, Rxs-1, and Rxs-2 values were obtained within satisfactory Q.Rg and Q.Rxs limits (Fig. 7, A–C), as in our previous studies (28, 29). The lowest Q values were not used in the Guinier Rg fits to minimize any potential effect of trace aggregates in the samples. The X-ray Rg values showed an apparent concentration dependence. These increased with concentration from 5.24 to 5.71 nm for PBS-50, 5.02 to 5.41 nm for PBS-137, and 5.16 to 5.38 nm for PBS-250 (Fig. 8A). This increase was attributed to X-ray radiation-induced damage of IgG2, because a small increase in intensities was seen in I(Q) at low Q values, and because this concentration effect was not seen in the AUC and neutron data (Figs. 6A and 8B). SAXS data above 1.5 mg/ml were thus discarded for reasons of radiation damage. The Rxs-1 and Rxs-2 values were unchanged (Fig. 8A). The average Rxs-1 values were 2.64 ± 0.03, 2.59 ± 0.04, and 2.61 ± 0.03 nm, and the average Rxs-2 values were 1.41 ± 0.06, 1.34 ± 0.08, and 1.34 ± 0.04 nm for PBS-50, PBS-137, and PBS-250 buffers, respectively. The Rg, Rxs-1, and Rxs-2 values in the three buffers were within error of each other. The Rg values of 5.0–5.2 nm here agreed with the earlier Rg values of 5.0 to 5.8 nm for humanized IgG2 (26), and they were slightly larger than those for humanized IgG2 of 4.76 ± 0.048 nm (20, 25), panitumumab of 5.1 nm (22), and polyclonal human IgG2 of 4.8 nm (27).
Figure 7.

X-ray and neutron Guinier Rg and Rxs analyses for IgG2. The X-ray scattering curves of IgG2 are shown for three buffers: A, PBS-50; B, PBS-137; and C, PBS-250 at 20 °C. The concentrations were ∼0.5, 1.0, and 1.5 mg/ml for PBS-50 and PBS-137 and 1.0 and 1.5 mg/ml for PBS-250 from bottom to top. The filled circles between the arrowed data points represent the Q.Rg and Q.Rxs ranges used to determine the Rg and Rxs values. The Q-ranges used for the Rg, Rxs-1, and Rxs-2 values were 0.15–0.28, 0.31–0.47, and 0.65–1.04 nm−1, respectively. The neutron-scattering curves of IgG2 are shown for three buffers: D, PBS-50; E, PBS-137; and F, PBS-250 at 20 °C in 100% heavy water. The concentrations were ∼0.30, 0.59, 1.19, and 2.38 mg/ml for PBS-50; 0.5, 1.0, 2.0, and 3.0 mg/ml for PBS-137; and 0.33, 1.99, and 2.66 mg/ml for PBS-250 from bottom to top. The filled circles between the arrowed data points represent the Q.Rg and Q.Rxs ranges used to determine the Rg and Rxs values. The Q-ranges used for the Rg, Rxs-1, and Rxs-2 values were 0.15–0.28, 0.31–0.47, and 0.65–1.04 nm−1, respectively. Two neutron curves (4 mg/ml in PBS-137 and 0.45 mg/ml in PBS-50) were omitted for clarity.
Figure 8.
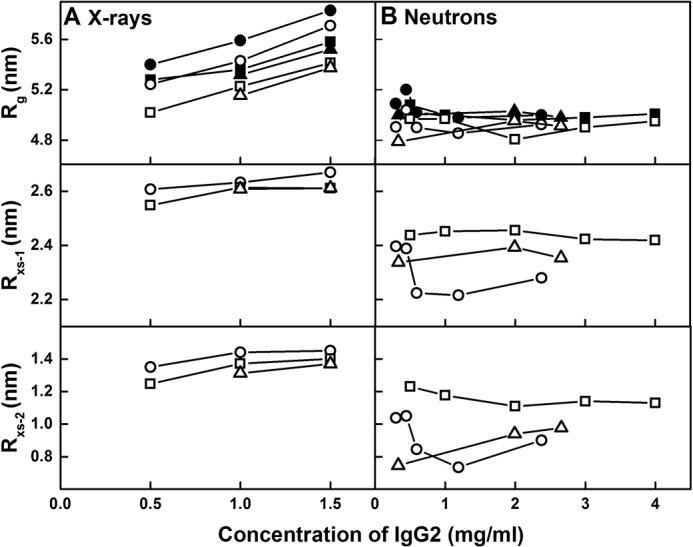
Concentration dependence of the X-ray and neutron Guinier parameters for IgG2. The Rg, Rxs-1, and Rxs-2 values are displayed from top to bottom for each buffer. A, X-ray Guinier values for IgG2 are shown for PBS-50 (○), PBS-137 (□), and PBS-250 (▵) buffers at 20 °C. The P(r) Rg values are shown for PBS-50 (●), PBS-137 (■), and PBS-250 (▴) at 20 °C. B, neutron Guinier values for IgG2 are shown for PBS-50 (○), PBS-137 (□), and PBS-250 (▵) buffers at 20 °C in 100% heavy water. The P(r) Rg values are shown for PBS-50 (●), PBS-137 (■), and PBS-250 (▴) at 20 °C in 100% heavy water.
SANS was also used to examine IgG2 at 0.3–4.0 mg/ml at 20 °C in PBS-50, PBS-137, and PBS-250 in 2H2O. Likewise, the neutron Guinier analyses also revealed high-quality linear plots in three distinct regions of the I(Q) curves from which the Rg, Rxs-1, and Rxs-2 values were obtained within satisfactory Q.Rg and Q.Rxs limits (Fig. 7, D–F). The Rg, Rxs-1, and Rxs-2 values were consistent within error (Fig. 8B). The mean Rg values were 4.93 ± 0.07, 4.97 ± 0.07, and 4.88 ± 0.09 nm, the average Rxs-1 values were 2.30 ± 0.09, 2.44 ± 0.02, and 2.36 ± 0.03 nm, and the average Rxs-2 values were 0.91 ± 0.13, 1.16 ± 0.05, and 0.93 ± 0.12 nm, for PBS-50, PBS-137, and PBS-250 respectively. These neutron Rg, Rxs-1, and Rxs-2 values were lower than those for X-rays, with this being attributed to the near invisibility of the surface hydration shell in heavy water, as well as the high-negative solute-solvent contrast difference compared with that of IgG2 (34). The Rg values reported here were slightly larger than that of 4.76 ± 0.06 nm for human anti-streptavidin IgG2 in 10 mm sodium acetate (pH 5.2) in 2H2O (23).
The distance distribution function P(r) provided structural information on full-length human IgG2 in real space, with this being equivalent to a histogram of all the inter-atom distances within IgG2. The X-ray P(r) analyses gave Rg values similar to those from the X-ray Guinier analyses, showing that the two analyses were self-consistent (filled and open symbols in Fig. 8A). The maximum length (L) of IgG2 was determined to be 18 nm from the value of r when the P(r) curve intersected 0 at large r (Fig. 9A). This L value agreed with the maximum dimension (Dmax) values of 17.2 ± 0.7 and 17.3 ± 0.4 nm for humanized IgG2 (25). The maxima in the P(r) curves corresponded to the most frequently occurring inter-atomic distances within the structure. Two peaks, M1 and M2, were identified in all the P(r) curves at r values of 4.8 ± 0.3 and 7.6 ± 0.3 nm, respectively. The M1 peak corresponds to distances within each Fab and Fc region, and the M2 peak corresponds to distances between the Fab–Fab and Fab–Fc regions. No concentration dependence in the M1 and M2 positions was observed (Fig. 9C).
Figure 9.

X-ray and neutron distance distribution analyses P(r) for IgG2. The peak maxima M1 and M2 and maximum length at L are indicated by arrows. A, X-ray P(r) curves for IgG2 in PBS-50, PBS-137, and PBS-250 (H2O) are shown for concentrations at ∼0.5, 1.0, and 1.5 mg/ml for PBS-50 and PBS-137 and 1.0 and 1.5 mg/ml for PBS-250 from bottom to top. B, neutron P(r) curves for IgG2 in PBS-50, PBS-137, and PBS-250 at 20 °C in 100% heavy water. Concentrations were ∼0.30, 0.45, 0.59, 1.19, and 2.38 mg/ml for PBS-50, 0.5, 1.0, 2.0, 3.0, and 4.0 mg/ml for PBS-137, and 0.33, 1.99, and 2.66 mg/ml for PBS-250 from bottom to top. C, X-ray M1 and M2 values are shown for PBS-50 (○), PBS-137 (□), and PBS-250 (▵) buffers. D, neutron M1 and M2 values are shown for PBS-50 (○), PBS-137 (□), and PBS-250 (▵) in 100% heavy water buffer. The lines are the mean values for M1 (—) and M2 (- - -).
The neutron P(r) analyses of IgG2 in heavy water revealed similar Rg values compared with the Guinier analyses (filled and open symbols in Fig. 8B). The majority of L values was 17 nm except for 0.59 and 2.38 mg/ml IgG2 in PBS-50 (average L of 16.9 ± 0.4 nm) (Fig. 9B). The M1 and M2 peaks were observed for most of the P(r) curves, except for 0.33 mg/ml IgG2 in PBS-250, and showed r values of 4.8 ± 0.5 and 7.6 ± 0.3 nm, respectively. The neutron and X-ray M1 and M2 values were in excellent agreement, and the reduced neutron L values compared with the X-ray L values were attributed to the hydration shell being not visible in neutron scattering.
Scattering models for IgG2
The IgG2 starting model was generated using the crystal structures of the human IgG2 Fab and Fc regions (see “Experimental procedures”). Residues missing in the Fc region were replaced with the corresponding residues from the other heavy chain (see “Experimental procedures”). The starting structure was Y-shaped with the Fab arms crossed over one another. This starting structure was energy-minimized using NAMD (see “Experimental procedures”).
In the Monte Carlo simulations based on backbone dihedral angles, the 19 IgG2 residues (Fig. 2E) represented the full-length hinge, which was assigned to be flexible (Fig. 1) and was varied in four different simulations (“Experimental procedures”). The first search involved 200,000 simulations to yield 106,799 sterically-accepted models that included asymmetric as well as symmetric IgG2 models. The second, third, and fourth searches reduced the maximum rotation angle per step from 30 to 15° to include smaller movements of the IgG2 hinge. These involved distance constraints of 1 or 0.75 nm between the α-carbons of each of the four cysteine residue pairs that form hinge disulfide bonds (Fig. 1). A total of 100,000 simulations were carried out for each of these two distance constraints using five different starting structures (20,000 simulations for each structure). This resulted in 12,597 and 3,975 accepted models for distance constraints of 1 and 0.75 nm, respectively. Different asymmetric and symmetric IgG2 starting structures with or without crossover Fab regions explored four types of Fab arrangements, limited any biased structures that favor certain conformations, and allowed the sampling of the maximum conformational space. Overall, 123,371 models were accepted for evaluation from a total of 400,000 simulated ones.
X-ray scattering modeling fits for IgG2
The 123,371 models were converted to their hydrated small-sphere representations for comparison with the SAXS curves. Their Rg values ranged between 3.85 and 6.21 nm (Fig. 10, A and B; Table 2). The Rxs-1 and Rxs-2 ranges were 1.34–3.36 and 0.02–2.30 nm, respectively. The models thus covered a broad range of conformational space as desired (gray, Fig. 11A). Following an examination of the R-factors, the experimental X-ray curves used for the fits were taken to be those for 0.5, 1.0, and 1.5 mg/ml IgG2 in PBS-50, PBS-137, and PBS-250, respectively, for which radiation damage was seen to be minimal. The scattering curve fits gave rise to a wide range of R-factors from 3.5 to 22.9% in a V-shaped distribution with its minimum close to the experimental Rg value (gray, Fig. 10, A and B). The lowest R-factors for the three X-ray scattering curves in PBS-50, PBS-137, and PBS-250 were 4.0, 3.5, and 3.6%, respectively, showing that these structures were improved compared with the starting IgG2 structure R-factors of 5.1, 4.8, and 5.1%. The use of R-factor cutoffs of 5.5, 5.0, and 5.7% as filters resulted in the selection of 35,141, 30,088, and 42,292 good-fit models for the three scattering curves in PBS-50, PBS-137, and PBS-250 buffers, respectively (orange, Fig. 10, A and B; Table 2), and reduced the number of accepted models by two-thirds. Views of the 30,088 models for 1 mg/ml IgG2 in PBS-137 showed a broad conformational distribution (gold, Fig. 11B).
Figure 10.
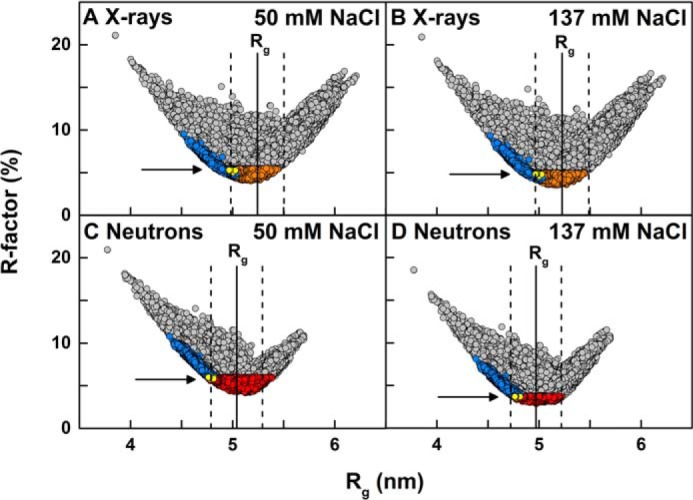
Modeling analyses for IgG2. The 123,371 goodness-of-fit R-factors were compared with the X-ray and neutron Rg values calculated for the IgG2 models. All 123,371 models are shown in gray. The 5,242 models filtered using an α-carbon separation of 0.75 nm for each of the four pairs of cysteine residues in the hinge (Fig. 1) are shown as blue circles. The 13 best-fit models that were accepted for each X-ray and neutron pair according to three filters (X-ray and neutron R-factor cutoffs and disulfide separations) are shown as yellow circles and arrowed. The experimentally observed Guinier Rg values are shown by vertical solid lines with error ranges of ± 5% shown by dashed lines. A, hydrated X-ray models were compared with the experimental X-ray curve of 0.5 mg/ml IgG2 in PBS-50 where the orange circles show 35,141 models with the R-factor below 5.5%. B, hydrated X-ray models were compared with experimental X-ray curve of 1 mg/ml IgG2 in PBS-137, where the orange circles show 30,088 models with the R-factor below 5%. C, unhydrated neutron models were compared with the experimental neutron curve of 0.45 mg/ml IgG2 in PBS-50 in 100% 2H2O, where the red circles show 44,835 models with the R-factor below 6%. D, unhydrated neutron models were compared with the experimental neutron curve of 1 mg/ml IgG2 in PBS-137 in 100% 2H2O, where the red circles show 10,731 models with the R-factor below 3.75%.
Table 2.
Modelling of the X-ray (upper) and neutron (lower) scattering data for human myeloma IgG2
| Sample | Filter | Models | Rg | Rxs-1 | Rxs-2 | R-factor | d1 | Min(d2,d3) | Max(d2,d3) |
|---|---|---|---|---|---|---|---|---|---|
| nm | nm | nm | % | nm | nm | nm | |||
| All IgG2 models | None | 123,371 | 3.85–6.21 | 1.34–3.36 | 0.02–2.30 | NA | 3.48–13.97 | 3.33–9.70 | 4.55–10.34 |
| X-ray fit, 0.5 mg/ml, PBS-50 | R-factor ≤5.5% | 35,141 | 4.83–5.45 | 2.23–2.96 | 0.71–1.78 | 4.04–5.50 | 4.95–12.70 | 4.91–9.66 | 6.27–10.21 |
| R-factor ≤5.5% and four disulfides | 1,474 | 4.83–5.04 | 2.56–2.79 | 1.26–1.51 | 4.61–5.50 | 5.55–7.54 | 6.80–8.39 | 7.62–8.92 | |
| X-ray fit, 1 mg/ml, PBS-137 | R-factor ≤5% | 30,088 | 4.84–5.45 | 2.23–2.94 | 0.71–1.78 | 3.53–5.00 | 4.95–12.70 | 4.91–9.66 | 6.27–10.21 |
| R-factor ≤5% and four disulfides | 1,247 | 4.84–5.04 | 2.56–2.79 | 1.26–1.51 | 4.15–5.00 | 5.55–7.54 | 7.15–8.39 | 7.62–8.92 | |
| X-ray fit, 1.5 mg/ml, PBS-250 | R-factor ≤5.7% | 42,292 | 4.85–5.52 | 2.23–3.01 | 0.61–1.82 | 3.62–5.70 | 4.90–12.86 | 4.91–9.69 | 6.19–10.23 |
| R-factor ≤5.7% and four disulfides | 1,100 | 4.86–5.04 | 2.56–2.79 | 1.26–1.51 | 4.72–5.70 | 5.55–7.54 | 6.97–8.39 | 7.65–8.92 | |
| All IgG2 models | None | 123,371 | 3.77–5.70 | 1.55–2.96 | 0.05–2.14 | NA | 3.48–13.97 | 3.33–9.70 | 4.55–10.34 |
| Neutron fit, 0.45 mg/ml, PBS-50 in 100% 2H2O | R-factor ≤6% | 44,835 | 4.75–5.39 | 2.03–2.79 | 0.64–1.56 | 4.15–6.00 | 5.21–13.87 | 5.17–9.70 | 6.53–10.23 |
| R-factor ≤6% and four disulfides | 13 | 4.76–4.82 | 2.44–2.51 | 1.28–1.36 | 5.77–6.00 | 6.84–7.37 | 7.81–8.14 | 7.89–8.39 | |
| Neutron fit, 1 mg/ml, PBS-137 in 100% 2H2O | R-factor ≤3.75% | 10,731 | 4.75–5.22 | 2.16–2.65 | 0.87–1.48 | 2.85–3.75 | 5.69–13.39 | 5.51–9.70 | 6.65–10.23 |
| R-factor ≤3.75% and 4 disulfides | 13 | 4.76–4.81 | 2.44–2.48 | 1.28–1.33 | 3.54–3.75 | 6.84–7.26 | 7.47–8.01 | 8.00–8.39 | |
| Neutron fit, 1.99 mg/ml, PBS-250 in 100% 2H2O | R-factor ≤8.2% | 35,213 | 4.70–5.48 | 1.92–2.75 | 0.06–1.62 | 6.13–8.20 | 5.16–13.97 | 4.99–9.70 | 6.53–10.23 |
| R-factor ≤8.2% and four disulfides | 19 | 4.74–4.81 | 2.41–2.48 | 1.26–1.36 | 7.89–8.20 | 6.42–7.37 | 7.47–8.01 | 7.87–8.70 | |
| Merged modelling fitsa | 9 | X: 4.97–5.03 | X: 2.69–2.74 | X: 1.36–1.43 | NA | 6.84–7.14 | 7.81–8.01 | 8.00–8.39 | |
| N: 4.76–4.81 | N: 2.44–2.48 | N: 1.28–1.32 |
a Models that satisfy the R-factor and disulfide filters and the Rg, Rxs-1, and Rxs-2 parameters for both X-rays (X) and neutrons (N) are displayed. Because the R-factor depended on which scattering curve comparison was used, this was therefore denoted as NA for not available.
Figure 11.

Density plots of the best-fit IgG2 models in PBS-137 buffer. The graphics were rendered using Tachyon in VMD. A, density plot for all 123,371 models is shown as a mesh with the Fc region shown as a gray solid surface. This is the reference for B–F. B, models that satisfied an X-ray R-factor cutoff below 5% for the curve at 1 mg/ml in PBS-137 in 100% light water. The two Fab regions are shown in gold and orange (30,088 models). C, models that satisfied a neutron R-factor cutoff of 3.75% for the curve at 1 mg/ml in PBS-137 in 100% heavy water. The two Fab regions are shown in red and purple (10,731 models). D, models that satisfied both the X-ray and neutron R-factors. The two Fab regions are shown in brown and tan (4,866 models). E, models that satisfied using an α-carbon separation of 0.75 nm between each of the four pairs of cysteine residues in the IgG2 hinge. The two Fab regions are shown in cyan and blue (5,242 models). F, 13 final best-fit models for IgG2 in PBS-137 that meet the X-ray and neutron R-factor cutoff and disulfide filters. The two Fab regions are shown in purple and black (13 models).
The α-carbon disulfide distance constraints of 0.75 nm greatly limited the possible Rg values of the structures. When the 123,371 models were filtered for distances of ≤0.75 nm between the four cysteine pairs (blue, Fig. 10, A and B), only 5,242 models remained (cyan and blue, Fig. 11E). After the R-factor filters were applied to the 5,242 models for each of the PBS-50, PBS-137, and PBS-250 curves, this left 1,474, 1,247, and 1,100 models, respectively (Table 2). The fit of the best-fit IgG2 model with the lowest R-factor for each X-ray experimental curve showed good visual agreements out to a Q value of 1.1 nm−1 (Fig. 12, A–C); note that the same best-fit model was identified for the X-ray fits in PBS-50 and PBS-250, in agreement with the observed lack of conformational change in these buffers. The M1 and M2 values of the X-ray best-fit structures in Fig. 12, A–C, were 4.0 and 7.7 nm, in good agreement with the observed values of 4.8 ± 0.3 and 7.6 ± 0.3 nm (Fig. 9C). Also, the P(r) curves for the best-fit models showed a smaller L value of 15–16 nm compared with the experimental L value of 18 nm.
Figure 12.

X-ray and neutron-scattering curve fits and Kratky analyses for the best-fit IgG2 models. The experimental data are indicated by white open circles, and the modeled best-fit curve is indicated in red. The models show 0.75-nm separations between each of the four pairs of cysteines in the IgG2 hinge. The X-ray best fits correspond to 0.5 mg/ml IgG2 in PBS-50 (A), 1 mg/ml IgG2 in PBS-137 (B), and 1.5 mg/ml IgG2 in PBS-250 (C). The neutron best fits correspond to 0.45 mg/ml IgG2 in PBS-50 (D), 1 mg/ml IgG2 in PBS-137 (E), and 1.99 mg/ml IgG2 in PBS-250 (F) in 100% 2H2O. The insets correspond to the experimental (black) and best-fit modeled (red) P(r) curves, in which M1, M2, and L are arrowed. G–L, corresponding Kratky plots for the same six comparisons between experiment and models are shown.
The d1 value represented the separation between the centers of the two Fab regions (Fig. 1). The min(d2,d3) and max(d2,d3) values represented the minimum and maximum separation between the centers of each Fab–Fc pair (d2 and d3). The 123,371 models covered a large range of d1, min(d2,d3), and max(d2,d3) values, in reflection of the asymmetric and symmetric nature of the IgG2 models. After the R-factor and α-carbon disulfide distance filters were applied, the ranges of d1, min(d2,d3), and max(d2,d3) distances were much reduced to similar values of 5.6–7.5, 6.8–8.4, and 7.6–8.9 nm in the three fits (Table 2). The reductions were explained by a convergence to a single best-fit conformational ensemble in the PBS-50, PBS-137, and PBS-250 buffers. The d1 distances were also smaller than the min(d2,d3) distances, indicating that the distance and angle between the two Fab regions are smaller than the Fab–Fc angles (Table 2). The min(d2,d3) and max(d2,d3) ranges overlapped. These considerations indicated that the best-fit IgG2 models adopted a largely symmetric Y-shape structure according to the X-ray modeling fits.
Neutron-scattering modeling fits for IgG2
The 123,371 models were converted to their unhydrated small-sphere representations for comparison with the SANS curves. Their Rg values ranged between 3.77 and 5.70 nm (Fig. 10, C and D; Table 2). The Rxs-1 and Rxs-2 ranges were 1.55–2.96 and 0.05–2.14 nm, respectively. The modeled Rg, Rxs-1, and Rxs-2 values were smaller than those for the corresponding X-ray Rg, Rxs-1, and Rxs-2 values because of the invisibility of the hydration shell using neutrons in heavy water. Following an R-factor examination of the available experimental scattering curves with three to five different concentrations in PBS-50, PBS-137, and PBS-250 in heavy water, the best experimental neutron curves were taken to be 0.45, 1.0, and 1.99 mg/ml IgG2 in PBS-50, PBS-137, and PBS-250, respectively. The R-factor cutoffs were 3.75% for 1 mg/ml IgG2 in PBS-137 and 6% for 0.45 mg/ml IgG2 in PBS-50 when these two curves were compared (Table 2). When the curve for 1.99 mg/ml IgG2 in PBS-250 was compared with 0.45 mg/ml IgG2 in PBS-50 (100% 2H2O), the R-factor cutoff was 8.6%. This R-factor cutoff was too lenient, giving 57,566 models, and thus this was reduced to 8.2%, giving 35,215 models (Table 2). The lowest R-factors for the three scattering curves in PBS-50, PBS-137, and PBS-250 buffers were 4.15, 2.85, and 6.13%, respectively, which were again improved compared with the starting IgG2 structure values of 6.1, 4.0, and 8.6%. The R-factor cutoff filters resulted in 44,835, 10,731, and 35,213 models, respectively (red, Fig. 10, C and D, and Table 2). The 10,731 models for 1 mg/ml IgG2 in PBS-137 showed a broad conformational distribution (Fig. 11C). The modeled Rg minimum was centered on the experimental Rg value, thus showing good agreement (red, Fig. 10, C and D).
The α-carbon disulfide distance constraints of 0.75 nm resulted in only 5,242 models remaining out of 123,371 (see above) (Fig. 11E). After filtering for R-factors, 13, 13, and 19 models remained for the three curves in PBS-50, PBS-137, and PBS-250, respectively (yellow, Fig. 10, C and D; Table 2). The best-fit IgG2 models with the lowest R-factor for the three buffers agreed with the experimental neutron curves up to a Q-value of 1.0 nm−1 (Fig. 12, D–F). The P(r) curves were in good agreement when overlaid, although a smaller L value of 15 nm was seen compared with the experimental L value of 17 nm. The M1 and M2 values of the neutron best-fit structures in Fig. 12, D–F, were 4.0 and 7.5 nm, in good agreement with the observed values of 4.8 ± 0.5 and 7.6 ± 0.3 nm (Fig. 9D). Surprisingly, this turned out to be the same IgG2 model in all three fits. The R-factor of 3.5% for PBS-137 was the lowest of the three.
The application of the joint R-factor cutoff filter and the 0.75-nm α-carbon disulfide distance constraints to the SANS modeling restricted the range of d1, min(d2,d3), and max(d2,d3) distances in a similar fashion to the SAXS modeling (Table 2). After the R-factor and disulfide distance filters were applied, far fewer neutron models were acceptable (13–19 models) compared with the X-ray models (1,100–1,474 models). The ranges of d1, min(d2,d3), and max(d2,d3) distances were altered to similar values of 6.4–7.4, 7.5–8.1, and 7.9–8.7 nm, respectively, in the three fits compared with X-rays (Table 2). The neutron d1 values of 6.4–7.4 nm were higher than those for X-rays of 5.6–7.5 nm, although these ranges overlapped, suggesting that the IgG2 structures with wider Fab regions were favored in the neutron fits. The range of neutron d1 values was narrower than for the X-ray d1 values. Interestingly, the min(d2,d3) and max(d2,d3) distances of the three sets of filtered neutron models were consistent with each other, and the ranges overlapped. It was concluded from the neutron modeling that IgG2 adopted a symmetric Y-shape structure, in agreement with the X-ray modeling.
Joint X-ray and neutron best-fit IgG2 models
The final best-fit models were identified by using both the SAXS and SANS R-factor cutoffs and the disulfide α-carbon distance constraints of ≤0.75 nm as filters. Compared with the distributions of the 30,088 and 10,731 best-fit models for the X-ray and neutron R-factor cutoff filters, respectively (Fig. 11, B and C), the application of both R-factor cutoff filters reduced the best-fit models to 4,866 (brown, Fig. 11D). These 4,866 IgG2 models showed Fab regions that encompassed the majority of conformational space around the Fc region. The α-carbon disulfide distance constraint of ≤0.75 nm had severely restricted the allowed positions of the Fab regions around the Fc region (Fig. 11E). When the SAXS and SANS R-factors and disulfide distance constraints were jointly applied, the number of IgG2 best-fit models were reduced to 13, 13, and 19 models for PBS-50, PBS-137, and PBS-250, respectively (Table 2). The 13 best-fit models for PBS-137 adopted a symmetrical Y-shape (Fig. 11F). Overall, nine best-fit IgG2 models fitted all six SAXS and SANS experimental I(Q) and P(r) curves in PBS-50, PBS-137, and PBS-250. This outcome indicated little or no differences in the IgG2 solution structures in three salt concentrations or between light and heavy water. This agreed with the AUC analyses (Fig. 6A).
Dimensionless Kratky plots of (Q.Rg)2.I(Q)/I(0) vs Q.Rg provided information on the folded state and flexibility of IgG2 (Fig. 12, G–L). They showed a characteristic two-peak curve similar to that shown previously (24). The X-ray data offered better signal–to–noise ratios than the neutron data. Comparison between the best-fit modeled and experimental Kratky plots showed good agreement up to a Q.Rg of 6 for X-rays and 4 for neutrons. The increased X-ray intensities beyond Q.Rg of 6 was attributed to potential flexibility in the IgG2 structure that had not been considered in the modeling. A similar intensity increase beyond Q.Rg of 4 for neutrons may also indicate flexibility, but it may also include a flat background due to an incoherent scattering contribution that had not been discounted.
The s20, w0 values of the nine scattering best-fit models were calculated using HYDROPRO (“Experimental procedures”) to compare these with the experimental values (Fig. 6A). Using the density (1.00529 g/ml) and viscosity (0.01002 poise) parameters for PBS-137 buffer at 20 °C and a partial specific volume of 0.7 ml/g, the mean s20, w0 value was 7.04 ± 0.05 S, which was less than that of 7.32 ± 0.02 S seen experimentally. The energy-minimized IgG2 starting structure prior to the Monte Carlo simulations gave an s20, w0 value of 7.10 S. This difference in s20, w0 values suggested that the best-fit IgG2 scattering model was slightly more elongated in its solution structure than the starting IgG2 structure.
Discussion
The X-ray and neutron-scattering data for human myeloma IgG2, coupled with atomistic Monte Carlo simulations of the dihedral angles in the main-chain backbone, have revealed novel molecular details of its solution structure. Importantly, this provided the first molecular explanation of the different functional IgG2 interactions with its protein ligands. Comprehensive data sets were obtained on human myeloma IgG2 for reason of its availability. Mass spectrometry and AUC showed only a 2% range in mass and a single c(s) peak, respectively, and thus the polydispersity in these samples was low and did not preclude molecular structure analyses. The IgG2 structure was unaffected in scattering experiments in concentration series in three salt concentrations and in light and heavy water. The IgG2 modeling was based on separate crystal structures for the Fab and Fc regions to generate a starting model that was refined by energy minimization and subjected to dihedral angle Monte Carlo modeling. Three filters based on the X-ray data, neutron data, and disulfide distances in the hinge region identified nine best-fit structures. The resulting classical human IgG2 revealed a symmetric Y-shaped conformation in solution that was able to account for its different functional interactions with complement C1q and the FcγR receptors.
Together, solution scattering and Monte Carlo modeling have offered molecular structural information on the IgG1, IgG2, and IgG4 subclasses. The IgG2 starting structure included the full IgG2 hinge 220ERKCCVECPPCPAPPVAGP238. Of the 123,371 physically-realistic IgG2 models derived from this hinge, the joint X-ray and neutron best-fit strategy brought down the number of filtered structures from 30,088 and 10,731 models, respectively, to a joint total of 4,866 models (brown, Fig. 13A). Thus the comparison of hydrated and unhydrated scattering structures proved to be effective. The further filter of ≤0.75 nm for the disulfide bridges between the four α-carbon cysteine pairs in the hinge gave 5,242 permitted structures (blue, Fig. 13A). The 13–19 best-fit models with the lowest R-factors that passed the double-scattering and disulfide filters revealed that human IgG2 adopts a Y-shaped symmetric conformation in solution. The 13 best-fit models for PBS-137 in light and heavy water were shown (black, Fig. 13A), of which nine models at d1 of ∼7 nm fitted all the X-ray and neutron-scattering curves from six different buffers. These nine structures are available in supporting Material, alongside their computed scattering curves and the experimental data.
Figure 13.
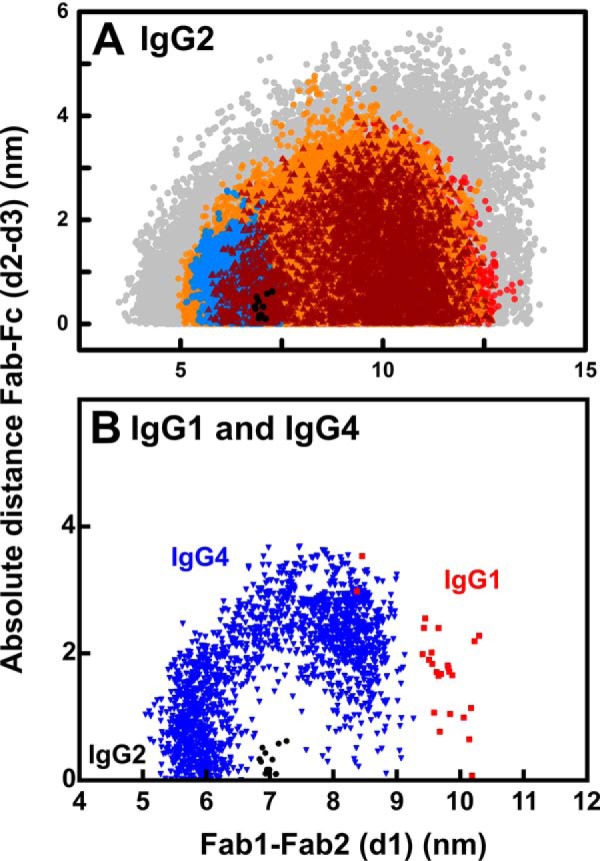
Distribution of the Fab–Fab and Fab–Fc distances in human IgG2. The analyses are shown for 1 mg/ml IgG2 in PBS-137. The inter-Fab distance, d1, between the center–of–mass of the two Fab arms and the absolute difference in Fab to Fc distances, d2–d3, are shown (Fig. 1). A, all 123,731 models from the Monte Carlo simulations are shown in gray. The 30,088 models with an X-ray R-factor below 5% are shown in orange. The 10,731 models with a neutron R-factor below 3.75% are shown in red. The 4,866 models filtered by both the X-ray and neutron R-factor filters are shown in brown. The 5,242 models that have less than 0.75 nm α-carbon separations for each of the four pairs of cysteine residues in the hinge are shown in blue. The 13 best-fit models that satisfy the X-ray and neutron and disulfide filters are shown in black. B, IgG2 models (black) denote those that meet the X-ray and neutron and disulfide filters from A and are compared with those for IgG1 (red) and IgG4 (blue) that were calculated in the same way.
Similar methods were used to determine the solution structures of human monoclonal IgG1 and IgG4 by joint X-ray and neutron Monte Carlo scattering fits.4 That work identified two α and β clusters of symmetric and asymmetric structures, respectively. The clusters were defined by d1 distances of ≤7 or ≥7 nm for the α and β clusters, respectively (Fig. 13B). The best-fit models of IgG1 corresponded to the β cluster of asymmetric structures, and this agreed with the crystal structure of intact human IgG1 b12 (red, Fig. 13B). The best-fit models of IgG4 B72.3 corresponded to both the α cluster of symmetric structures and β cluster of asymmetric structures (blue, Fig. 13B). Interestingly, the IgG2 best-fit models did not correspond to either of the best-fit clusters for IgG1 or IgG4, and instead they were located between these at d1 = 7 nm (black, Fig. 13B). The three analyses indicated that these three IgG subclasses show different conformations. This outcome explains the evolution of the human IgG subclasses such that human IgG1, IgG2, and IgG4 exhibit distinct structural and functional properties. IgG1 and IgG4 have two Cys–Cys bridges in their hinges, whereas IgG2 has four Cys–Cys bridges; in addition, IgG2 lacks a second Gly residue in its hinge that is present in IgG1 and IgG4 (35, 36). Furthermore, IgG2 has a shorter hinge than IgG1 and IgG4 (Fig. 2H). These three features are expected to make the IgG2 hinge more rigid, compared with the IgG1 and IgG4 hinges, and alter its function. For example, it can now be seen why IgG2 is able to perform a unique structural role as the only IgG subclass that binds predominantly to bacterial capsular polysaccharide antigens (2, 3).
The outcome of scattering modeling and the number of best-fit models depends on the inputs, i.e. the quality of the experimental scattering curves, the starting model, and the number of Monte Carlo models and their filtering. Each are discussed in turn. (i) For example, the lowest X-ray R-factors for IgG2 of 4.2–4.7% were higher than those for IgG1 of 2.6–2.9% and IgG4 of 2.5–2.6%.4 This difference is attributable to the larger Q-range of 0.13–2 nm−1 used for IgG2 here (with higher noise at larger Q values), whereas these Q-ranges were lower at 0.09–1.1 and 0.15–1.1 nm−1 for IgG1 and IgG4, respectively. Nonetheless, all three studies resulted in R-factor versus Rg graphs with clear minima that identified an ensemble of best-fit structures. The final Rg values of the IgG2 models of 4.8 nm (Table 2) were similar to those of 4.9 nm for IgG4, but less than that of 5.2 nm for IgG1.4 (ii) The assumptions used for generating the initial models can be important. For example, the earlier neutron-scattering fits for human monoclonal anti-streptavidin IgG2 employed an IgG2 starting model based on the crystal structure of full-length mouse IgG2A with three Cys residues in the hinge and not four (PDB code 1IGT) and two Gly residues in its hinge, and only varied three amino acids in the IgG2A upper hinge to generate 56,511 acceptable models (23). Unsurprisingly, these authors determined an asymmetric IgG2 structure. In this study, Fab and Fc crystal structures for human IgG2 were used alongside variation of the full-length human IgG2 hinge with all 19 residues, including four Cys residues in the hinge and only a single hinge Gly residue (Fig. 2E). This study resulted in symmetric IgG2 structures that well-explained the biological function of IgG2 (see below). Further structural analyses with monoclonal IgG2 will clarify these differences further. (iii) A large number of starting models facilitated the identification of best-fit structures. Starting from 704,000, 700,000, and 400,000 trial models for IgG1, IgG4, and IgG2, respectively, the numbers of evaluated physically realistic models with no steric overlap were 231,492 (IgG1), 190,437 (IgG4), and 123,371 (IgG2). These resulted in final totals of 28, 2,748, and 13 best-fit structures, respectively (Fig. 13B). The joint X-ray and neutron fits were the key filter in reaching the final 28 models for IgG1, whereas the joint X-ray and neutron fits together with the disulfide separation filters were key in reaching the final nine best-fit models for IgG2. The rather larger number of final best-fit IgG4 models resulted from the relatively unrestricted shorter IgG4 hinge conformation that gave many more compatible models.
There are three different isoforms of IgG2, namely IgG2A (classical), IgG2A/B, and IgG2B, which are found in both human monoclonal IgG2 and myeloma-derived IgG2 (8–10). The isoforms vary in the disulfide bond connectivity in the IgG2 hinge, where these studies suggested that the two Cys-223–Cys-223 and Cys-224–Cys-224 disulfide bonds between the two heavy chains (Fig. 1) can be broken with the formation of new disulfide bonds with the Fab regions. The light and heavy chains in the Fab region are connected by a Cys-135–Cys-214 bridge (Fig. 1). In the IgG2B isoform, Cys-223 from one heavy chain can form an inter-chain disulfide bond with the C-terminal Cys-214 in the light chain. Cys-224 from one heavy chain can form an inter-chain disulfide bond with Cys-135 in the other heavy chain (10, 37–40) or an intra-chain disulfide bond with Cys-135 in the same heavy chain (8, 41). Also, Cys-223 can form an intra-chain disulfide bond with Cys-135 in the same heavy chain where Cys-224 forms an inter-chain disulfide bond with Cys-214 in the light chain (9). The disulfide bond variations in IgG2A/B and IgG2B are still not fully understood, and the impact of these different disulfide bond variants upon antigen binding as well as effector functions is currently unknown.
Although no SAXS and SANS data were collected on the individual IgG2A/B and IgG2B isoforms, our IgG2 best-fit models for human IgG2 provided new insight into these two other isoforms formed by potential Cys-223–Cys-214 and Cys-224–Cys-135 bridges. These alternative disulfide arrangements may result in more compact global structures than the classic IgG2A isoform (8, 40). Interestingly, both these disulfide bond variants were indeed found in our library of 123,371 IgG2 models. Thus, 53 models showed α-carbon separations below 0.75 nm for Cys-223–Cys-214, and another 126 showed separations below 0.75 nm for Cys-224–Cys-135. However, none of these models satisfied the joint X-ray/neutron R-factor cutoff filter and the inter-chain disulfide separation of below 0.75 nm for the best-fit IgG2 models, showing that their structures were distinct from that of IgG2 in its IgG2A isoform as studied. The above 53 models gave R-factors of 5.1–16.2% for X-rays and 4.6–15.9% for neutrons, both in PBS-137. The above 126 models gave R-factors of 4.9–8.9% for X-rays and 4.3–11.0% for neutrons. In comparison, the best overall R-factor was lower at 3.5% for IgG2 (Table 2). The s20, w0 values of the 53 and 126 models were 7.2 ± 0.2 and 7.2 ± 0.1 S, respectively, which were not much different from the experimental value of 7.32 ± 0.02 S in PBS-137 and the best-fit modeled value of 7.04 S above. Although a small difference of about 0.3 S was seen between IgG2A and IgG2B, this difference was considered to be low. Overall, even though the IgG2A/B and IgG2B isoforms showed different solution structures, they were not more compact than the classic IgG2A isoform.
A Y-shaped symmetric structure of IgG2 (or IgG2A) had been determined by our atomistic modeling. This outcome differs from the postulated moderately asymmetric structures for the IgG2A and IgG2B isoforms based on comparison with the asymmetric IgG1 crystal structure (8, 11) and the asymmetric structures reported elsewhere from EM and neutron-scattering (18, 23). By EM, the conclusion of asymmetric IgG2 structures was attributed to the study of an assumed IgG2A/B structure with an asymmetric disulfide arrangement at its hinge (18). The differences from the previous neutron-scattering modeling that gave an asymmetric solution structure could arise from the use of the mouse IgG2a crystal structure to fit the neutron data instead of a human IgG2 Fab and Fc structure (23). In that study, the neutron data on human anti-streptavidin IgG2 were measured at a high concentration in nonphysiological buffers containing 10 mm sodium acetate (pH 5.2), which may have caused conformational changes, whereas here we have used more dilute concentrations for our AUC runs as well as our X-ray and neutron data collection, all at pH 7.4.
The atomistic best-fit models for human IgG2 provided new molecular insight into its binding to the FcγRII and FcγRIII receptors, but not to C1q of complement nor to the FcγRI receptor. This key assessment was performed using recently available crystal structures of the Fc region of human IgG1 complexed with these ligands (Fig. 14). The C1q globular head in complex with the IgG1–Fc region (PDB code 6FCZ) (42) was aligned with the nine best-fit models of IgG2 through their Fc regions, giving a satisfactory r.m.s.d. of 0.149 nm in α-carbon positions. Clear steric clashes between the C1q domains and the Fab2 region of IgG2 were visible, explaining why C1q cannot bind to IgG2 (Fig. 14A). The FcγRI (CD64) receptor in complex with the IgG1–Fc region (PDB code 4X4M) (43) was also aligned with the nine IgG2 best-fit models through their Fc regions, resulting in an r.m.s. of 0.168 nm (Fig. 14B). Here, clear steric clashes between the D1 and D3 domains of the three-domain “sea-horse” FcγRI structure were visible with the Fab1 and Fab2 regions of IgG2, explaining why IgG2 cannot bind to FcγRI. The views of Fig. 14, A and B, show that the Fab regions were too close to the Fc region to permit C1q and FcγRI binding. In addition, the IgG2 sequence does not possess the key amino acid contact residues required for the complexes between the IgG2-Fc region and each of C1q and FcγRI as revealed by their recent co-crystal structures (Fig. 2, E and F). In distinction with these first two cases, the dissociation constant KD of IgG2 with the FcγRIIIA Val-158 receptor is 14 μm (44), showing that the IgG2–FcγRIIIA complex is formed, albeit weakly. To examine this, the Fc region of the nine best-fit IgG2 models were aligned with three crystal structures for the Fc–FcγRIII complex (PDB codes 3SGJ, 5VU0, and 5YC5) to give satisfactory low r.m.s. values of 0.100, 0.102, and 0.102 nm, respectively (Fig. 14C) (45–47). The two orthogonal views of the superimposed IgG2 complexes with FcγRIIIA Val-158 show that these IgG2 complexes with the two-domain receptor were sterically compatible with the position of the Fab regions and were therefore allowed. The ability of scattering modeling to generate atomistic structures for the full-length IgG2 molecules has provided important clarifications of the ability of IgG2 to bind to C1q and the Fc receptors.
Figure 14.
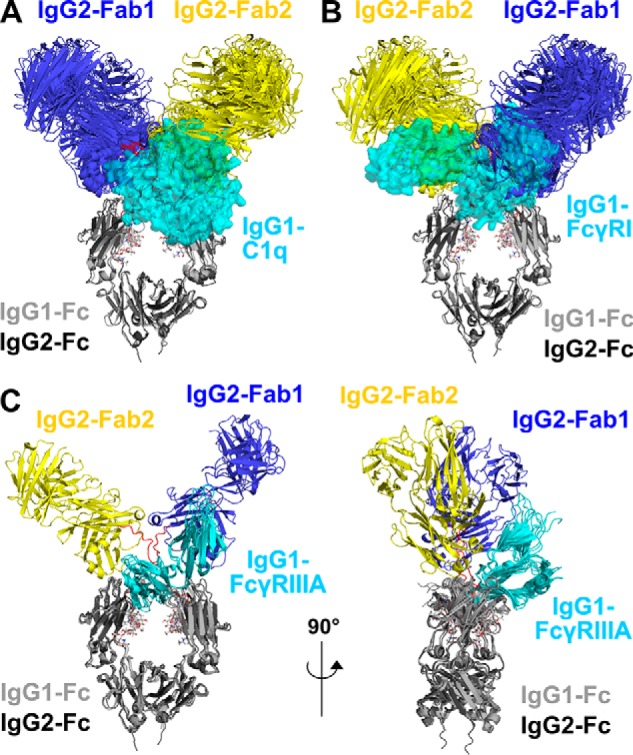
Superimposition of the nine IgG2 best-fit models with complement C1q and two Fc receptors. The nine IgG2 best-fit models are compared with crystal structures for the IgG1–C1q, IgG1–FcγRI, and IgG1–FcγRIII complexes. Superimpositions of the Fc regions of the IgG2 models with the crystal structures of the Fc complexes were achieved using the “align” function of PyMOL. The IgG2 Fab regions are shown in blue (Fab1) and yellow (Fab2) and the Fc region in dark gray as in Fig. 1. The Fc region is seen in the same view in A, B, and C (left), and it is rotated by 90° in C (right). The glycans in the IgG2 Fc region are represented as sticks, and the IgG2 hinge is represented as red loops. A, superimposition of the nine IgG2 best-fit models with the IgG1–Fc complex with the globular head of C1q (PDB code 6FCZ). The IgG1–Fc region is represented as a light gray cartoon, and C1q is represented as a cyan semi-transparent surface. B, superimposition of the nine IgG2 best-fit models with the IgG1–Fc complex with FcγRI (PDB code 4X4M). The IgG1–Fc region is represented as a light gray cartoon, and FcγRI is represented as a cyan semi-transparent surface. C, two orthogonal views at 90° of the superimposition of the IgG2 best-fit model with three crystal structures for the IgG1–Fc complexes with FcγRIIIA Val-158 (PDB codes 3SGJ, 5VU0, and 5YC5). The IgG1–Fc regions are represented as light gray cartoon schematics and FcγRIIIA are represented as cyan cartoons.
Experimental procedures
Purification and composition of IgG2
Purified myeloma IgG2 from human plasma (Athens Research, Athens, GA) was further purified by gel filtration using a Superose 6 10/300 column (GE Healthcare) to remove aggregates, then concentrated using Amicon Ultra spin concentrators (50-kDa molecular mass cutoff), and dialyzed at 4 °C against its ultracentrifugation and scattering buffers (see below). The N-linked oligosaccharides at Asn-297 on the CH2 domains (Fig. 1) were represented as a complex-type biantennary oligosaccharide with a Man3-GlcNAc2 core and two NeuNAc·Gal·GlcNAc antennae (48). Using SLUV2 in the SCT software package, the IgG2 molecular mass was calculated to be 147.4 kDa from the IgG2 sequence (Fig. 2) based on the Fab and Fc crystal structures (PDB codes 3KYM and 4HAF, respectively); its unhydrated volume was 189.9 nm3; its hydrated volume was 231.0 nm3 (based on a hydration of 0.3 g of water per g of glycoprotein and an electrostricted volume of 0.0245 nm3 per bound water molecule); its partial specific volume (v̄) was 0.7294 ml/g, and its absorption coefficient at 280 nm was 15.33 (1%, 1 cm pathlength) (49).
All data were recorded in PBS with different NaCl concentrations. The buffer with 137 mm NaCl, 8.1 mm Na2HPO4, 2.7 mm KCl, and 1.5 mm KH2PO4 (pH 7.4) was termed PBS-137. When 50 mm NaCl or 250 mm NaCl were used, these were termed PBS-50 and PBS-250, respectively. Buffer densities were measured using an Anton Paar DMA 5000 density meter, for comparison with the theoretical values calculated by SEDNTERP (50). This resulted in densities of 1.00529 g/ml for PBS-137 at 20 °C (theoretical, 1.00534 g/ml), 1.00145 g/ml for PBS-50 at 20 °C (theoretical, 1.00175 g/ml), and 1.0098 g/ml for PBS-250 at 20 °C (theoretical, 1.00998 g/ml), all in 100% light water. A buffer viscosity of 0.01002 poise was used for the light water buffers. The densities were increased to 1.11183 g/ml for PBS-137 at 20 °C (theoretical, 1.11247 g/ml), 1.10839 g/ml for PBS-50 at 20 °C (theoretical, 1.10889 g/ml), and 1.116752 g/ml for PBS-250 at 20 °C (theoretical, 1.11711 g/ml), all in 100% 2H2O. A viscosity of 0.01200 poise was used for the heavy water buffers.
Native MS of IgG2
IgG2 was deglycosylated with PNGase F (New England Biolabs, Herts., UK) according to the manufacturer's protocol. The native and deglycosylated IgG2 samples were placed into spin concentrators (Amicon Ultra 500, MWCO 50 kDa) pre-soaked with 200 mm ammonium acetate (LC/MS grade water). The samples were buffer-exchanged into 200 mm ammonium acetate six times at 4 °C in the spin concentrators. The samples were ran on a Synapt G1 instrument (Waters Corp., UK); the spectra were analyzed in MassLynx Version 4.1 MS software (Waters Corp., UK), and mass calculations were performed in Amphitrite Atropos (51). The experimental acquisition parameters were as follows: capillary voltage of 1.3 kV, sampling cone voltage of 175 V, extraction cone voltage of 4.5 V, backing pressure of 7 mbar, trap collision energy of 40 eV, and transfer collision energy of 10 eV. The mass range was 1,000–18,000 m/z. Proteins were sprayed using nano-electrospray ionization from gold-coated capillaries prepared in-house using a Flaming Brown P97 needle puller and a Quorum Q150R S sputter coater.
Sedimentation velocity data for IgG2
Sedimentation velocity data were obtained on two Beckman XL-I analytical ultracentrifuges equipped with AnTi50 rotors for IgG2 samples in PBS-50, PBS-137, and PBS-250 at 20 °C in each of 100% H2O and 100% 2H2O. Data were collected at rotor speeds of 40,000 rpm. in two-sector cells with column heights of 12 mm. Sedimentation analysis was performed using direct boundary Lamm fits of up to 300 scans using SEDFIT (version 14.6) (52, 53). SEDFIT resulted in size-distribution analyses c(s) that assumed all species to have the same frictional ratio f/f0. The final SEDFIT analyses used a fixed resolution of 200 and optimized the c(s) fit by floating f/f0, the meniscus, and bottom of the sedimentation boundaries until the overall r.m.s. deviations (< 0.02) and visual appearance of the fits were satisfactory. The percentage of oligomers in the total loading concentration was derived using the c(s) integration function. Partial specific volumes of 0.73 and 0.70 ml/g were used for samples in 100% H2O and 100% 2H2O, respectively. HYDROPRO version 10 was used to calculate the sedimentation coefficients based on the molecular structure of human IgG2 (54), using an atomic level shell calculation and a hydrodynamic radius of 0.29 nm of each element in the model.
X-ray and neutron-scattering data for IgG2
X-ray scattering data were obtained on Instrument BM29 at the European Synchrotron Radiation Facility, Grenoble, France (55). Data were acquired using a Dectris Pilatus 1M detector with a resolution of 981 × 1043 pixels (pixel size of 172 × 172 μm). Sample volumes of 50 μl were loaded into PCR strip tubes for the BioSAXS automatic sample changer (56). Each sample in the quartz capillary was moved continuously during beam exposure to reduce radiation damage. Sets of 10 time frames, with a frame exposure time of 1 s each, were acquired, alongside real-time checks that confirmed the absence of radiation damage during data acquisition. After this, any frames containing radiation damage were removed, and the remaining frames were averaged. EDNA software provided automatic data processing in which the intensities I(Q) were automatically scaled by concentration (57). The Biosaxs Customized Beamline Environment (BsxCUBE) software was used for control of the automatic sample changer, and the sample settings were loaded from the Information System for Protein Crystallography Beamlines database (ISPyB) (55, 58). IgG2 samples were studied in each of PBS-50, PBS-137, and PBS-250 at 20 °C at eight concentrations between 0.5 and 4.0 mg/ml in a dilution series. Data for samples at above 1.5 mg/ml were not used due to radiation-induced damage.
Neutron-scattering data were obtained on Instrument D22 at the Institut Laue-Langevin, Grenoble, France. The data were acquired using a two-dimensional 3He detector with 128 × 128 pixels of 7.5 × 7.5 mm2 in size. The sample-to-detector and collimation distances were both 5.6 m. The wavelength λ was 0.60 nm. Sample volumes of 400 μl were used. Samples were measured in rectangular Hellma cells of 2 mm thickness in a thermostatted sample rack set at 20 °C. IgG2 was studied in PBS-50, PBS-137, and PBS-250 in 100% 2H2O buffers at 20 °C. The dialyses were performed on site immediately prior to D22 experiments to reduce the risk of aggregate formation. IgG2 concentrations were 0.30, 0.45, 0.59, 1.19 and 2.38 mg/ml for PBS-50, 0.5, 1.0, 2.0, 3.0 and 4.0 mg/ml for PBS-137, and 0.33, 1.99 and 2.66 mg/ml for PBS-250.
In a given solute–solvent contrast, the Rg is a measure of structural elongation if the internal inhomogeneity of scattering densities within the glycoprotein has no effect. Guinier analyses at low Q (where Q = 4π sin θ/λ; 2θ is the scattering angle, and λ is the wavelength) give the Rg and the forward scattering at zero-angle I(0) (59) as shown in Equation 1,
| (Eq. 1) |
this expression is valid in a Q.Rg range up to 1.5. If the structure is elongated, the mean radius of gyration of cross-sectional structure Rxs and the mean cross-sectional intensity at zero-angle (I(Q)Q)Q → 0 are obtained from Equation 2,
| (Eq. 2) |
the cross-sectional plot for Igs exhibits two distinct regions, a steeper innermost one and a flatter outermost one (60), and the two analyses correspond to Rxs-1 and Rxs-2, respectively. The Rg and Rxs analyses were performed using the SCT software package (45). The Q ranges for the Rg, Rxs-1, and Rxs-2 values were 0.15–0.28, 0.31–0.47, and 0.65–1.04 nm−1, respectively. Indirect Fourier transformation of the scattering data I(Q) in reciprocal space into real space to give the distance distribution function P(r) was carried out using the program GNOM version 4.6 (61) as shown in Equation 3,
| (Eq. 3) |
P(r) corresponds to the distribution of distances r between volume elements. This provides the maximum dimension of the antibody L and its most commonly occurring distance M in real space. For this, the X-ray I(Q) curve utilized up to 1,043 data points in the Q range between 0.03 and 4.92 nm−1. The neutron I(Q) curve utilized up to 108 data points in the Q range between 0.1 and 1.7 nm−1. Dimensionless Kratky plots of (Q.Rg)2·I(Q)/I(0) vs Q.Rg were calculated using the Guinier Rg values to provide information on the folded state and flexibility of IgG2 (62–65).
Generation of starting structure of IgG2
A full-sequence starting model was created for human IgG2 using two crystal structures of the separate Fab and Fc regions and that for human mAb IgG2 anti-LINGO1 Li33 represented the IgG2 Fab region (PDB code 3KYM) (15). The human IgG2 Fc region was used directly (PDB code 4HAF) (16). The EU numbering was used here where Asn-297 (Fig. 1) is equivalent to Asn-297 in IgG1 (29, 66, 67).4 In the Fab region, the hinge residues 223CCVECPPCPAPPVAGP238 and the last Cys residue on the light chain (C terminus) were unresolved. In the Fc region, most of the unresolved residues (235VAGP238, 265DVSHEDPE272, 294EQF296, and 325NKGLP329) were on one of the two heavy chains, but they were resolved in the other heavy chain. 445PGK447 on both heavy chains at the C terminus was also unresolved. The missing Fc residues were reconstructed by replacing the entire heavy chain with a duplicate of the complete heavy chain using superimposition using PyMOL version 1.3 (Schrödinger, LCC). The root mean square difference of the superimposition of the newly built and original heavy chains of 209 and 190 residues was low at 0.0852 nm, showing excellent agreement between the two structures. The missing hinge 223CCVECPPCPAPPVAG237 and the C-terminal residues for both the light and heavy chains were modeled with backbone ϕ and ψ angles of 10° using the PyMOL script build_seq (PyMOL Script Repository, Queen's University, Ontario, Canada). All disulfide bonds were retained. Force field parameterizations were generated, and the hydrogen atoms were added to the starting IgG2 structure using the glycan reader component of CHARMM-GUI (68, 69) and the CHARMM36 force field (70–74). This includes the disulfide bond between the light chain and heavy chain. The starting structure was then energy-minimized for 2,000 steps in NAMD (version 2.9) as the simulation engine (https://sassie-web.chem.utk.edu/sassie2/).5
Dihedral Monte Carlo simulations
Dihedral Monte Carlo simulations in SASSIE used the Complex Monte Carlo module whereby 400,000 models were sampled rapidly (75). This module varied backbone dihedral angles for the IgG2 hinge residues 220ERKCCVECPPCPAPPVAGP238. A Metropolis sampling methodology was used to sample the energetically-allowed dihedral angles, using only the dihedral component of the CHARMM potential to determine the energy of each configuration (70). Sterically-overlapping IgG structures were removed during sampling. Overall, from the total of 400,000 generated IgG2 models, 123,371 models were accepted for the scattering fits.
Different conformational searches of IgG2 were as follows.
(i) First, 200,000 simulations were performed without any disulfide bond constraints in which the entire hinge 220ERKCCVECPPCPAPPVAGP238 was varied in the sampling. Maximum rotation angle steps of 30° were used in this simulation. From this search, 106,799 models (53%) were sterically acceptable with no overlaps.
(ii) Using the models from search i, four were selected as new starting structures. These were selected by measuring the α-carbon distances between the hinge residue pairs Cys-223–Cys-223, Cys-224–Cys-224, Cys-227–Cys-227, and Cys-230–Cys-230 to be under 1 nm, and these were the only four models that met this criterion. A simulation of 20,000 structures for each of these four starting structures was performed with the constraint that the four cysteine pairs involved in inter-heavy chain disulfide bonding remained within 1 nm of one another (i.e. “disulfide distance” constraints). The entire hinge 220ERKCCVECPPCPAPPVAGP238 was varied in the simulation with maximum rotation angle steps of 15°. This produced 9,560 accepted structures that retained their 1 nm separation (12%) out of the total of 80,000 simulations.
(iii) Subsequently, a further five starting structures were defined by setting the α-carbon distances between the four pairs of hinge cysteines Cys-223–Cys-223, Cys-224–Cys-224, Cys-227–Cys-227, and Cys-230–Cys-230 to be under 0.75 nm in the best-fit models from the 9,560 structures (as above). Four of the structures showed no crossover at their hinges, meaning that each Fab region remained on the same side of the IgG structure as their CH2 and CH3 domains (Fig. 1). One of the five structures showed a slight crossover of its hinges. The first four starting structures were subjected to disulfide distance constraints of 0.75 nm between each of the four hinge cysteine pairs in a simulation of 80,000 structures, from which 3,108 (4%) were accepted. The fifth structure was subjected to constraints of 1 nm of α-carbon separations between each of the four hinge cysteine pairs in a simulation of 20,000 structures, of which 3,037 models were accepted (15%). The entire hinge 220ERKCCVECPPCPAPPVAGP238 was varied in the sampling with maximum rotation angle steps of 15° used in all five simulations.
(iv) As a control, the same starting structures used for ii and iv were subjected to two different filters of either 1 or 0.75 nm α-carbon separations in the Cys pairs to confirm that the filters in SASSIE were not biased in producing accepted models during the simulation and that the SASSIE simulation produced randomized trial models. One of the starting structures from ii was subjected to a further simulation of 20,000 structures using a constraint of ≤0.75 nm in the α-carbon separation for each of the four cysteine pairs in the hinge. The entire hinge 220ERKCCVECPPCPAPPVAGP238 was varied in the sampling with maximum rotation angle steps of 15°, and 867 models from 20,000 were accepted (4%).
Scattering curve calculations and analyses
The scattering curves for the 123,371 accepted models were calculated using the SCT software package (49). This is a coarse-grained method that converts the atomistic models into small sphere models for use with the Debye equation adapted to spheres to calculate the theoretical scattering curves I(Q) (76). For comparison with neutron data, the sphere models were left unhydrated; however, smearing corrections were applied (wavelength 0.60 nm; wavelength spread 10%; beam divergence of 0.016 radians). For comparison with X-ray data, hydration spheres were added to create a hydration shell corresponding to 0.3 g of water/g of protein (33, 34). The atomic coordinates were converted into small sphere models using a grid with a cube-side length of 0.54298 nm and a cutoff of four atoms, and these parameters were optimized using SCT to reproduce the unhydrated protein volume. The target dry volume was 189.9 nm3 (the modeled dry volume was 189.9 nm3) and the target wet volume was 250.1 nm3 (the modeled hydrated volume was 253.6 nm3). For comparison with the X-ray and neutron-scattering data, each experimental I(Q) value was matched to the theoretical I(Q) value with the closest Q value, after which the R-factor was computed by analogy with crystallography where lower R-factors represent better fits as shown in Equation 4,
| (Eq. 4) |
where η is a scaling factor used to match the theoretical curve to the experimental I(0) value. An iterative search to minimize the R-factor was used to determine η. The theoretical scattering curves that matched the experimental scattering curves were accepted as valid models of the antibody solution structure. The experimental curves were fitted using a Q-range of 0.13–2 nm−1 for the X-ray and neutron curves.
A cutoff R-factor, below which models were assigned as best-fit, depended on the experimental scattering curve, its signal-to-noise ratio, and its Q range. To determine this cutoff, two experimental curves were used to calculate two R-factors for each of the 123,371 curves. The correlation between the two R-factors was assessed using both the Pearson r and Spearman rs coefficients (31). By gradually excluding the models with higher R-factors, this identified the point at which the ranking of the fits was no longer consistently determined for the two curves. The cutoff was chosen as the point where both the r and rs coefficients dropped below 0.5.4 If there is a correlation between the two compared curves where r and rs are not equal to 0.5, then the cutoff R-factor filter selected is the minimum R-factor for that experimental curve plus 1–2%. Here, the number of accepted models was reduced by approximately two-thirds using the R-factor cutoff filter to select for the better models with a lower R-factor. To analyze the models, the distances between the centers of mass of the two Fab regions (d1) and the distances between the centers of mass of each Fab region to the Fc region (d2 and d3) were measured (Fig. 1). Note that because IgG2 is symmetric, the differentiation between Fab1 and Fab2 and the corresponding d2 and d3 values is for clarity. The Fab1 and Fab2 structures in the models were distinguished according to their chain names. The asymmetry of an antibody structure was measured by the absolute difference between the two Fab–Fc distances, abs(d2-d3).
Author contributions
G. K. H., A. D. G., H. B., and C. E. formal analysis; G. K. H. validation; G. K. H., A. D. G., and H. B. investigation; G. K. H., A. D. G., H. B., C. E., K. T., and J. G. methodology; S. J. P. supervision; S. J. P. funding acquisition; G. K. H. and S. J. P. writing-original draft; S. J. P. project administration; G. K. H. and S. J. P. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Petra Pernot and Dr. Adam Round (ESRF, Grenoble, France) and Dr. Anne Martel (ILL, Grenoble, France) for excellent instrumental support; Dr. David W. Wright for computational support; and Dr. Kersti Karu for generous MS support.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting Material, with their computed scattering curves, and supporting experimental data.
D. W. Wright, E. L. K. Elliston, G. K. Hui, and S. J. Perkins, submitted for publication.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- H
- heavy chain
- L
- light chain
- V
- variable
- C
- constant
- AUC
- analytical ultracentrifugation
- SAXS
- small-angle X-ray scattering
- SANS
- small-angle neutron scattering
- PDB
- Protein Data Bank
- r.m.s.
- root mean square
- PNGase
- peptide:N-glycosidase.
References
- 1. Strohl W. R., and Strohl L. M. (eds) (2012) in Therapeutic Antibody Engineering, pp. 37–595, Woodhead Publishing Series in Biomedicine, Woodhead Publishing, Sawston, Cambridge, UK [Google Scholar]
- 2. Siber G. R., Schur P. H., Aisenberg A. C., Weitzman S. A., and Schiffman G. (1980) Correlation between serum IgG-2 concentrations and the antibody response to bacterial polysaccharide antigens. N. Engl. J. Med. 303, 178–182 10.1056/NEJM198007243030402 [DOI] [PubMed] [Google Scholar]
- 3. Barrett D. J., and Ayoub E. M. (1986) IgG2 subclass restriction of antibody to pneumococcal polysaccharides. Clin. Exp. Immunol. 63, 127–134 [PMC free article] [PubMed] [Google Scholar]
- 4. Brezski R. J., and Jordan R. E. (2010) Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? MAbs. 2, 212–220 10.4161/mabs.2.3.11780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brezski R. J., Oberholtzer A., Strake B., and Jordan R. E. (2011) The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. MAbs. 3, 558–567 10.4161/mabs.3.6.18119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seino J., Eveleigh P., Warnaar S., van Haarlem L. J., van Es L. A., and Daha M. R. (1993) Activation of human complement by mouse and mouse/human chimeric monoclonal antibodies. Clin. Exp. Immunol. 94, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoo E. M., Wims L. A., Chan L. A., and Morrison S. L. (2003) Human IgG2 can form covalent dimers. J. Immunol. 170, 3134–3138 10.4049/jimmunol.170.6.3134 [DOI] [PubMed] [Google Scholar]
- 8. Dillon T. M., Ricci M. S., Vezina C., Flynn G. C., Liu Y. D., Rehder D. S., Plant M., Henkle B., Li Y., Deechongkit S., Varnum B., Wypych J., Balland A., and Bondarenko P. V. (2008) Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J. Biol. Chem. 283, 16206–16215 10.1074/jbc.M709988200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinez T., Guo A., Allen M. J., Han M., Pace D., Jones J., Gillespie R., Ketchem R. R., Zhang Y., and Balland A. (2008) Disulfide connectivity of human immunoglobulin G2 structural isoforms. Biochemistry 47, 7496–7508 10.1021/bi800576c [DOI] [PubMed] [Google Scholar]
- 10. Wypych J., Li M., Guo A., Zhang Z., Martinez T., Allen M. J., Fodor S., Kelner D. N., Flynn G. C., Liu Y. D., Bondarenko P. V., Ricci M. S., Dillon T. M., and Balland A. (2008) Human IgG2 antibodies display disulfide-mediated structural isoforms. J. Biol. Chem. 283, 16194–16205 10.1074/jbc.M709987200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saphire E. O., Parren P. W., Pantophlet R., Zwick M. B., Morris G. M., Rudd P. M., Dwek R. A., Stanfield R. L., Burton D. R., and Wilson I. A. (2001) Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science 293, 1155–1159 10.1126/science.1061692 [DOI] [PubMed] [Google Scholar]
- 12. Scapin G., Yang X., Prosise W. W., McCoy M., Reichert P., Johnston J. M., Kashi R. S., and Strickland C. (2015) Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol. 22, 953–958 10.1038/nsmb.3129 [DOI] [PubMed] [Google Scholar]
- 13. Dangl J. L., Wensel T. G., Morrison S. L., Stryer L., Herzenberg L. A., and Oi V. T. (1988) Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. EMBO J. 7, 1989–1994 10.1002/j.1460-2075.1988.tb03037.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Phillips M. L., Tao M. H., Morrison S. L., and Schumaker V. N. (1994) Human/mouse chimeric monoclonal antibodies with human IgG1, IgG2, IgG3 and IgG4 constant domains: electron microscopic and hydrodynamic characterization. Mol. Immunol. 31, 1201–1210 10.1016/0161-5890(94)90034-5 [DOI] [PubMed] [Google Scholar]
- 15. Pepinsky R. B., Silvian L., Berkowitz S. A., Farrington G., Lugovskoy A., Walus L., Eldredge J., Capili A., Mi S., Graff C., and Garber E. (2010) Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci. 19, 954–966 10.1002/pro.372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Teplyakov A., Zhao Y., Malia T. J., Obmolova G., and Gilliland G. L. (2013) IgG2 Fc structure and the dynamic features of the IgG CH2-CH3 interface. Mol. Immunol. 56, 131–139 10.1016/j.molimm.2013.03.018 [DOI] [PubMed] [Google Scholar]
- 17. Vafa O., Gilliland G. L., Brezski R. J., Strake B., Wilkinson T., Lacy E. R., Scallon B., Teplyakov A., Malia T. J., and Strohl W. R. (2014) An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 65, 114–126 10.1016/j.ymeth.2013.06.035 [DOI] [PubMed] [Google Scholar]
- 18. Ryazantsev S., Tischenko V., Nguyen C., Abramov V., and Zav'yalov V. (2013) Three-dimensional structure of the human myeloma IgG2. PLoS ONE 8, e64076 10.1371/journal.pone.0064076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kilár F., Simon I., Lakatos S., Vonderviszt F., Medgyesi G. A., and Závodszky P. (1985) Conformation of human IgG subclasses in solution. Eur. J. Biochem. 147, 17–25 10.1111/j.1432-1033.1985.tb08712.x [DOI] [PubMed] [Google Scholar]
- 20. Gregory L., Davis K. G., Sheth B., Boyd J., Jefferis R., Nave C., and Burton D. R. (1987) The solution conformations of the subclasses of human IgG deduced from sedimentation and small angle X-ray scattering studies. Mol. Immunol. 24, 821–829 10.1016/0161-5890(87)90184-2 [DOI] [PubMed] [Google Scholar]
- 21. Franey H., Brych S. R., Kolvenbach C. G., and Rajan R. S. (2010) Increased aggregation propensity of IgG2 subclass over IgG1: role of conformational changes and covalent character in isolated aggregates. Protein Sci. 19, 1601–1615 10.1002/pro.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mosbæk C. R., Konarev P. V., Svergun D. I., Rischel C., and Vestergaard B. (2012) High concentration formulation studies of an IgG2 antibody using small angle X-ray scattering. Pharm. Res. 29, 2225–2235 10.1007/s11095-012-0751-3 [DOI] [PubMed] [Google Scholar]
- 23. Clark N. J., Zhang H., Krueger S., Lee H. J., Ketchem R. R., Kerwin B., Kanapuram S. R., Treuheit M. J., McAuley A., and Curtis J. E. (2013) Small-angle neutron-scattering study of a monoclonal antibody using free-energy constraints. J. Phys. Chem. B 117, 14029–14038 10.1021/jp408710r [DOI] [PubMed] [Google Scholar]
- 24. Tian X., Langkilde A. E., Thorolfsson M., Rasmussen H. B., and Vestergaard B. (2014) Small-angle X-ray scattering screening complements conventional biophysical analysis: comparative structural and biophysical analysis of monoclonal antibodies IgG1, IgG2, and IgG4. J. Pharm. Sci. 103, 1701–1710 10.1002/jps.23964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian X., Vestergaard B., Thorolfsson M., Yang Z., Rasmussen H. B., and Langkilde A. E. (2015) In-depth analysis of subclass-specific conformational preferences of IgG antibodies. IUCrJ. 2, 9–18 10.1107/S205225251402209X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukuda M., Watanabe A., Hayasaka A., Muraoka M., Hori Y., Yamazaki T., Imaeda Y., and Koga A. (2017) Small-scale screening method for low-viscosity antibody solutions using small-angle X-ray scattering. Eur. J. Pharm. Biopharm. 112, 132–137 10.1016/j.ejpb.2016.11.027 [DOI] [PubMed] [Google Scholar]
- 27. König N., Paulus M., Julius K., Schulze J., Voetz M., and Tolan M. (2017) Antibodies under pressure: a small-angle X-ray scattering study of immunoglobulin G under high hydrostatic pressure. Biophys. Chem. 231, 45–49 10.1016/j.bpc.2017.05.016 [DOI] [PubMed] [Google Scholar]
- 28. Rayner L. E., Hui G. K., Gor J., Heenan R. K., Dalby P. A., and Perkins S. J. (2014) The Fab conformations in the solution structure of human immunoglobulin G4 (IgG4) restrict access to its Fc region implications for functional activity. J. Biol. Chem. 289, 20740–20756 10.1074/jbc.M114.572404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rayner L. E., Hui G. K., Gor J., Heenan R. K., Dalby P. A., and Perkins S. J. (2015) The solution structures of two human IgG1 antibodies show conformational stability and accommodate their C1q and FcγR ligands. J. Biol. Chem. 290, 8420–8438 10.1074/jbc.M114.631002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perkins S. J., Wright D. W., Zhang H., Brookes E. H., Chen J., Irving T. C., Krueger S., Barlow D. J., Edler K. J., Scott D. J., Terrill N. J., King S. M., Butler P. D., and Curtis J. E. (2016) Atomistic modelling of scattering data in the Collaborative Computational Project for small angle scattering (CCP-SAS). J. Appl. Crystallogr. 49, 1861–1875 10.1107/S160057671601517X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Swinscow T. D. V., and Campbell M. J. (2001) Statistics at Square One, 10th Ed., BMJ Books, London [Google Scholar]
- 32. Zhao H., Ghirlando R., Alfonso C., Arisaka F., Attali I., Bain D. L., Bakhtina M. M., Becker D. F., Bedwell G. J., Bekdemir A., Besong T. M., Birck C., Brautigam C. A., Brennerman W., Byron O., et al. (2015) A multilaboratory comparison of calibration accuracy and the performance of external references in analytical ultracentrifugation. PLoS ONE 10, e0126420 10.1371/journal.pone.0126420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perkins S. J. (1986) Protein volumes and hydration effects. Eur. J. Biochem. 157, 169–180 10.1111/j.1432-1033.1986.tb09653.x [DOI] [PubMed] [Google Scholar]
- 34. Perkins S. J. (2001) X-ray and neutron-scattering analyses of hydration shells: a molecular interpretation based on sequence predictions and modelling fits. Biophys. Chem. 93, 129–139 10.1016/S0301-4622(01)00216-2 [DOI] [PubMed] [Google Scholar]
- 35. Hamilton R. G. (2001) The Human IgG Subclasses (Mohan C., ed) pp. 7–11, Calbiochem-Novabiochem Corp., San Diego [Google Scholar]
- 36. Vidarsson G., van Der Pol W.-L., van Den Elsen J. M., Vilé H., Jansen M., Duijs J., Morton H. C., Boel E., Daha M. R., Corthésy B., and van De Winkel J. G. (2001) Activity of human IgG and IgA subclasses in immune defense against Neisseria meningitidis serogroup B. J. Immunol. 166, 6250–6256 10.4049/jimmunol.166.10.6250 [DOI] [PubMed] [Google Scholar]
- 37. Liu Y. D., Chen X., Enk J. Z., Plant M., Dillon T. M., and Flynn G. C. (2008) Human IgG2 antibody disulfide rearrangement in vivo. J. Biol. Chem. 283, 29266–29272 10.1074/jbc.M804787200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang B., Harder A. G., Connelly H. M., Maheu L. L., and Cockrill S. L. (2010) Determination of Fab-hinge disulfide connectivity in structural isoforms of a recombinant human immunoglobulin G2 antibody. Anal. Chem. 82, 1090–1099 10.1021/ac902466z [DOI] [PubMed] [Google Scholar]
- 39. Liu H., and May K. (2012) Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 4, 17–23 10.4161/mabs.4.1.18347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang A., Fang J., Chou R. Y., Bondarenko P. V., and Zhang Z. (2015) Conformational difference in human IgG2 disulfide isoforms revealed by hydrogen/deuterium exchange mass spectrometry. Biochemistry 54, 1956–1962 10.1021/bi5015216 [DOI] [PubMed] [Google Scholar]
- 41. Lightle S., Aykent S., Lacher N., Mitaksov V., Wells K., Zobel J., and Oliphant T. (2010) Mutations within a human IgG2 antibody form distinct and homogeneous disulfide isomers but do not affect Fcγ receptor or C1q binding. Protein Sci. 19, 753–762 10.1002/pro.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ugurlar D., Howes S. C., de Kreuk B.-J., Koning R. I., de Jong R. N., Beurskens F. J., Schuurman J., Koster A. J., Sharp T. H., Parren P. W. H. I., and Gros P. (2018) Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 359, 794–797 10.1126/science.aao4988 [DOI] [PubMed] [Google Scholar]
- 43. Lu J., Chu J., Zou Z., Hamacher N. B., Rixon M. W., and Sun P. D. (2015) Structure of FcγRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc. Natl. Acad. Sci. U.S.A. 112, 833–838 10.1073/pnas.1418812112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bruhns P., Iannascoli B., England P., Mancardi D. A., Fernandez N., Jorieux S., and Daëron M. (2009) Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725 10.1182/blood-2008-09-179754 [DOI] [PubMed] [Google Scholar]
- 45. Ferrara C., Grau S., Jäger C., Sondermann P., Brünker P., Waldhauer I., Hennig M., Ruf A., Rufer A. C., Stihle M., Umaña P., and Benz J. (2011) Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. U.S.A. 108, 12669–12674 10.1073/pnas.1108455108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Falconer D. J., Subedi G. P., Marcella A. M., and Barb A. W. (2018) Antibody fucosylation lowers the FcγRIIIa/CD16a affinity by limiting the conformations sampled by the N162-glycan. ACS Chem. Biol. 13, 2179–2189 10.1021/acschembio.8b00342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kiyoshi M., Caaveiro J. M. M., Tada M., Tamura H., Tanaka T., Terao Y., Morante K., Harazono A., Hashii N., Shibata H., Kuroda D., Nagatoishi S., Oe S., Ide T., Tsumoto K., et al. (2018) Assessing the heterogeneity of the Fc-glycan of a therapeutic antibody using an engineered FcγReceptor IIIa-immobilized column. Sci. Rep. 8, 3955 10.1038/s41598-018-22199-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deisenhofer J. (1981) Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-Å resolution. Biochemistry 20, 2361–2370 10.1021/bi00512a001 [DOI] [PubMed] [Google Scholar]
- 49. Wright D. W., and Perkins S. J. (2015) SCT: a suite of programs for comparing atomistic models with small-angle scattering data. J. Appl. Crystallogr. 48, 953–961 10.1107/S1600576715007062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laue T. M., Shah B. D., Ridgeway T. M., and Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., and Horton J. C., eds) pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 51. Sivalingam G. N., Yan J., Sahota H., and Thalassinos K. (2013) Amphitrite: a program for processing travelling wave ion mobility mass spectrometry data. Int. J. Mass Spectrom. 345, 54–62 10.1016/j.ijms.2012.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schuck P. (1998) Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75, 1503–1512 10.1016/S0006-3495(98)74069-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ortega A., Amorós D., and García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 10.1016/j.bpj.2011.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pernot P., Round A., Barrett R., De Maria Antolinos A., Gobbo A., Gordon E., Huet J., Kieffer J., Lentini M., Mattenet M., Morawe C., Mueller-Dieckmann C., Ohlsson S., Schmid W., Surr J., et al. (2013) Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 10.1107/S0909049513010431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Round A., Felisaz F., Fodinger L., Gobbo A., Huet J., Villard C., Blanchet C. E., Pernot P., McSweeney S., Roessle M., Svergun D. I., and Cipriani F. (2015) BioSAXS sample changer: a robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallogr. D Biol. Crystallogr. 71, 67–75 10.1107/S1399004714026959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Incardona M.-F., Bourenkov G. P., Levik K., Pieritz R. A., Popov A. N., and Svensson O. (2009) EDNA: a framework for plugin-based applications applied to X-ray experiment online data analysis. J. Synchrotron Radiat. 16, 872–879 10.1107/S0909049509036681 [DOI] [PubMed] [Google Scholar]
- 58. De Maria Antolinos A., Pernot P., Brennich M. E., Kieffer J., Bowler M. W., Delageniere S., Ohlsson S., Malbet Monaco S., Ashton A., Franke D., Svergun D., McSweeney S., Gordon E., and Round A. (2015) ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta Crystallogr. D Biol. Crystallogr. 71, 76–85 10.1107/S1399004714019609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Glatter O., and Kratky O. (1982) Small Angle X-ray Scattering, Academic Press, New York [Google Scholar]
- 60. Pilz I., Kratky O., Licht A., and Sela M. (1973) Shape and volume of anti-poly(d-alanyl) antibodies in the presence and absence of tetra-d-alanine as followed by small-angle X-ray scattering. Biochemistry 12, 4998–5005 10.1021/bi00748a028 [DOI] [PubMed] [Google Scholar]
- 61. Semenyuk A. V., and Svergun D. I. (1991) GNOM. A program package for small-angle scattering data processing. J. Appl. Crystallogr. 24, 537–540 10.1107/S002188989100081X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kratky O., and Porod G. (1949) Röntgenuntersuchung Gelöster Fadenmoleküle. Recl. des Trav. Chim. des Pays-Bas. 68, 1106–1122 [Google Scholar]
- 63. Doniach S. (2001) Changes in biomolecular conformation seen by small angle X-ray scattering. Chem. Rev. 101, 1763–1778 10.1021/cr990071k [DOI] [PubMed] [Google Scholar]
- 64. Brennich M., Pernot P., and Round A. (2017) How to analyze and present SAS data for publication. Adv. Exp. Med. Biol. 1009, 47–64 10.1007/978-981-10-6038-0_4 [DOI] [PubMed] [Google Scholar]
- 65. Cordeiro T. N., Herranz-Trillo F., Urbanek A., Estaña A., Cortés J., Sibille N., and Bernadó P. (2017) Structural characterization of highly flexible proteins by small-angle scattering. Adv. Exp. Med. Biol. 1009, 107–129 10.1007/978-981-10-6038-0_7 [DOI] [PubMed] [Google Scholar]
- 66. Edelman G. M., Cunningham B. A., Gall W. E., Gottlieb P. D., Rutishauser U., and Waxdal M. J. (1969) The covalent structure of an entire γG immunoglobulin molecule. Proc. Natl. Acad. Sci. U.S.A. 63, 78–85 10.1073/pnas.63.1.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kabat E. A., Te Wu T., Perry H. M., Gottesman K. S., and Foeller C. (1992) Sequences of Proteins of Immunological Interest. DIANE Publishing, Collingdale, PA [Google Scholar]
- 68. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 69. Jo S., Song K. C., Desaire H., MacKerell A. D. Jr., and Im W. (2011) Glycan Reader: automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J. Comput. Chem. 32, 3135–3141 10.1002/jcc.21886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- 71. MacKerell A. D. Jr., Feig M., and Brooks C. L. 3rd. (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 10.1002/jcc.20065 [DOI] [PubMed] [Google Scholar]
- 72. Guvench O., Hatcher E. R., Venable R. M., Pastor R. W., and Mackerell A. D. (2009) CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 5, 2353–2370 10.1021/ct900242e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Raman E. P., Guvench O., and MacKerell A. D. (2010) CHARMM additive all-atom force field for glycosidic linkages in carbohydrates involving furanoses. J. Phys. Chem. B 114, 12981–12994 10.1021/jp105758h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and MacKerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Curtis J. E., Raghunandan S., Nanda H., and Krueger S. (2012) SASSIE: a program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints. Comput. Phys. Commun. 183, 382–389 10.1016/j.cpc.2011.09.010 [DOI] [Google Scholar]
- 76. Perkins S. J., and Weiss H. (1983) Low-resolution structural studies of mitochondrial ubiquinol:cytochrome c reductase in detergent solutions by neutron-scattering. J. Mol. Biol. 168, 847–866 10.1016/S0022-2836(83)80078-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.