

Abstract
Neurite outgrowth requires coordinated cytoskeletal rearrangements in the growth cone and directional membrane delivery from the neuronal soma. As an essential Rho guanine nucleotide exchange factor (GEF), TRIO is necessary for cytoskeletal dynamics during neurite outgrowth, but its participation in the membrane delivery is unclear. Using co-localization studies, live-cell imaging, and fluorescence recovery after photobleaching analysis, along with neurite outgrowth assay and various biochemical approaches, we here report that in mouse cerebellar granule neurons, TRIO protein pools at the Golgi and regulates membrane trafficking by controlling the directional maintenance of both RAB8 (member RAS oncogene family 8)– and RAB10-positive membrane vesicles. We found that the spectrin repeats in Golgi-resident TRIO confer RAB8 and RAB10 activation by interacting with and activating the RAB GEF RABIN8. Constitutively active RAB8 or RAB10 could partially restore the neurite outgrowth of TRIO-deficient cerebellar granule neurons, suggesting that TRIO-regulated membrane trafficking has an important functional role in neurite outgrowth. Our results also suggest cross-talk between Rho GEF and Rab GEF in controlling both cytoskeletal dynamics and membrane trafficking during neuronal development. They further highlight how protein pools localized to specific organelles regulate crucial cellular activities and functions. In conclusion, our findings indicate that TRIO regulates membrane trafficking during neurite outgrowth in coordination with its GEF-dependent function in controlling cytoskeletal dynamics via Rho GTPases.
Keywords: neurite outgrowth, membrane trafficking, neurodevelopment, guanine nucleotide exchange factor (GEF), Rho (Rho GTPase), Rab, signal transduction
Introduction
After receiving developmental signals, post-mitotic neurons differentiate into mature neurons and establish specific functional structures, including neurites. The area of the plasma membrane of a developing neuron is estimated to increase by 10,000-fold because of the formation of axons and dendrites (1, 2). During this period, a large number of intracellular processes are initiated, including protein and lipid synthesis, cytoskeletal dynamics, membrane production, and trafficking. Cytoskeletal rearrangements and membrane trafficking are both required for neurite outgrowth, with the former providing the driving force for growth cone turning and elongation (3, 4) and the latter providing membrane lipids and proteins at sites located far from the Golgi network (2, 5). Within these highly regulated processes, Rho family GTPases, such as RAC1, CDC42, and RHOA, have been found to regulate cytoskeletal rearrangements (6–8). RAC1 and CDC42 regulate neurite elongation and branch formation, and RHOA induces neurite retraction (9–12). On the other hand, Rab family GTPases regulate membrane trafficking processes (2, 13–15), in which RAB6, 8, 10, 13, and 33 function in trans-Golgi network (TGN)-related3 vesicles; RAB5, 7, 21, and 22 regulate early and late endosomes; and RAB4, 11, and 35 regulate recycling endosomes (13, 16). The GTPase activities of both Rho and Rab are controlled by GTP-/GDP-bound cycles, which are regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins, respectively (8, 13). The orchestrated interactions of these two families of small GTPases are thus essential for neurite development.
As a member of the Dbl family of GEFs, the triple functional domain (TRIO) is a large GEF with multiple functional domains that regulates Rho family GTPases, including RAC1, CDC42, and RHOA (17), and plays a key role in axon growth and guidance in Caenorhabditis elegans and Drosophila (18–23). Deletion of TRIO in mice abolishes neurite outgrowth and axon guidance and produces multiple abnormalities, such as deformities in the skeletal muscle, disorganization of neuronal tissues, and defects in learning and memory (24–28). TRIO also serves a role in excitatory synaptic transmission and long-term potentiation (29). In particular, several TRIO mutants have been identified in individuals with intellectual disability, schizophrenia, and autism (30–33). Biochemically, the N-terminal GEF domain is responsible for the activation of the Rho GTPases RAC1 and RHOG (further activates CDC42), whereas the C-terminal GEF domain contributes to RHOA activation (34). The third functional domain is the C-terminal protein serine/threonine kinase domain, whose function remains unknown. The full-length protein containing this kinase domain is expressed at relatively low levels in the nervous system (31, 35, 36). The TRIO N terminus also contains spectrin repeats and a SEC-14 motif, whose functions are as yet undetermined. Because spectrin is associated to the Golgi apparatus, here we have determined the possible involvement of TRIO in the Golgi apparatus and the Golgi-derived membrane vesicles. The result shows that TRIO is located in the Golgi apparatus through the spectrin repeats in a complex with RABIN8, a common GEF for both RAB8 and RAB10. TRIO facilitates RABIN8 phosphorylation via a direct interaction with RABIN8. TRIO deletion leads to an increase in the frequency of the directional switch of RAB8- and RAB10-positive vesicles and hence a decrease in the traffic distance of the vesicles. However, the velocities of the vesicles are not affected. This observation revealed a novel role of TRIO in membrane trafficking, which is instructive to understand the process coupling of membrane trafficking and cytoskeleton rearrangement during neurite outgrowth.
Results
TRIO is able to localize in the Golgi apparatus
Because TRIO contains spectrin repeats, which are tightly associated with the Golgi apparatus (37), we speculate that TRIO may have the ability to localize at the Golgi apparatus, thereby potentially regulating Golgi-derived membrane trafficking. First, we prepared subcellular fractions of mouse cerebellum using OptiPrep density gradient. The result showed that TRIO protein was detected in the fractions that were positive for Golgi marker GM130 and early endosome marker EEA1 and RAB5, whereas the TRIO signal was much weaker in the fractions positive for ER marker BIP (Fig. 1, A and B). We next studied the subcellular localization of TRIO in the mouse neuroblastoma Neuro-2a cells. Ectopic expression of EGFP-fused TRIO9S and TRIO8, the two main isoforms expressed in cerebellum (35, 36), displayed a strong EGFP fluorescence in the perinuclear region, whereas EGFP alone did not (Fig. S1). Staining the transfected Neuro-2a cells with Golgi marker GM130 or TGN38 suggested that the enriched EGFP fluorescence in the perinuclear region was highly associated with Golgi apparatus (Fig. S1).
Figure 1.

TRIO localizes to the Golgi apparatus in CGNs. A, P10 mouse cerebellum was homogenized, and the postnuclear supernatants were subjected to 2.5–30% OptiPrep density gradient for subcellular fractionation. Fractions were subjected to Western blotting with TRIO, GM130, BIP, EEA1, and RAB5 antibodies. B, relative distribution of blotted proteins in A, with the strongest intensity of each protein considered as 1. C, CGNs isolated from WT mice were cultured for 24–48 h and then stained with the antibody against Golgi markers GM130, TGN46, VAMP4, and RCAS1, as well as the TRIO-N antiserum and DAPI. The scale bar in the left panel represents 20 μm, and the scale bars in the right magnified panels represent 5 μm. Scatter plots and the PCCs of the fluorescence intensities of red and green channels are also shown. D, scatter plots and bar graph showing the PCC of TRIO and Golgi markers fluorescence intensities quantified from 15 to 20 neurons in each group, from three independent experiments. MW, molecular weight.
To determine the endogenous TRIO localization in neurons, we prepared a TRIO-specific antiserum targeting the N-terminal spectrin repeats (Fig. S2A). The specificity of this antiserum was determined using TrioNKO (Trioflox/flox;Nestin-Cre) (27) lysates by Western blotting (Fig. S2B) and cultured TrioWKO (Trioflox/flox;Wnt1-Cre, see below) CGNs by immunofluorescence (Fig. S2C). In addition, fluorescence intensity derived from this antiserum was highly correlated with EGFP intensity in COS-7 cells overexpressing EGFP-TRIO9S or EGFP-TRIO8 (Fig. S2D). We then immunostained endogenous TRIO in cultured CGNs using this antiserum and found that endogenous TRIO was also enriched in the perinuclear region, with lower but still detectable intensity along the neurite and in the growth cone (Fig. 1C). This enrichment of TRIO fluorescence was correlated with GM130 (Fig. 1C), suggesting that the endogenous TRIO also localizes in Golgi apparatus in CGNs. To confirm this observation, co-localization analysis of TRIO with other Golgi markers, including TGN46, VAMP4, and RCAS1, was performed (Fig. 1C). Quantification of the co-localization indicated that TRIO protein was highly correlated with Golgi apparatus, especially trans-Golgi networks (Fig. 1D). To investigate the amino acids required for TRIO's localization in Golgi, we constructed a series of plasmids encoding truncated TRIO proteins fused with EGFP (Fig. 2A). TRIO(1–230) encodes the SEC-14 domain; TRIO(208–673), TRIO(446–909), and TRIO(672–1295) encode the fragments of different spectrin repeats; and TRIO(1296–1909) encodes the N-terminal GEF1 domain. After transfection with the plasmids, the COS-7 cells expressing EGFP-TRIO(208–673) displayed EGFP fluorescence signals highly restricted in the GM130-positive region (Fig. 2B), whereas the COS-7 cells transfected with other plasmids, including the control EGFP plasmid, showed diffused EGFP signals in the cytosol or in the nuclei (Fig. 2B). EGFP-TRIO(208–673) was also enriched in regions positive for other Golgi markers, including TGN46 (Fig. 2C), VAMP4 (Fig. 2D), and RCAS1 (Fig. 2E). Thus, TRIO is able to localize in the Golgi apparatus through the first spectrin fragment.
Figure 2.

TRIO spectrin repeats localizes to the Golgi apparatus. A, schematic diagram of TRIO (1–230), (208–673), (446–909), (672–1295), and (1296–1909) sequences. B, COS-7 cells transfected with pEGFP-C3, pEGFP-TRIO(1–230), pEGFP-TRIO(208–673), pEGFP-TRIO(446–909), pEGFP-TRIO(672–1295), and pEGFP-TRIO(1296–1909) were stained with the Golgi marker GM130. EGFP-TRIO(208–673) fluorescence was highly correlated with GM130 signal. The scale bar represents 20 μm. C–E, COS-7 cells transfected with pEGFP-TRIO(208–673) were stained with Golgi markers TGN46 (C), VAMP4 (D), and RCAS1 (E). The scale bars represent 10 μm.
TRIO regulates membrane trafficking during neurite outgrowth
To study the function of TRIO during neurite outgrowth, we prepared TRIO-deficient CGNs by crossing Trio floxed mice with Wnt1-Cre (27, 38). Wnt1-Cre was expressed in the cerebellum and in cultured CGNs, as illustrated by the Rosa-mTmG reporter (39) (Fig. S2, A and B). Unlike the TrioNKO mice we reported previously, the resultant TrioWKO (Trioflox/flox;Wnt1-Cre) mice live to adulthood, although the body weights were apparently smaller than control mice. We then performed a fluorescence recovery after photobleaching (FRAP) analysis to determine the potential functions of TRIO in membrane trafficking. EGFP-tagged human transferrin receptor (EGFP–hTfR) was transfected to neurons to label newly synthesized membranes (40, 41). Because the membrane recovery from photobleaching reflects the processes of membrane trafficking and synthesis, there are investigators using this method to assess the membrane dynamics during hippocampal axonal growth (41). After photobleaching, TrioWKO neurites showed a slower recovery rate of the fluorescence intensity than the control neurites (p < 0.01) (Fig. 3, A and B). Quantitation of the area under curve after bleaching showed that the recovery percentage of TRIO-deficient CGNs was significantly decreased compared with the control neurons (p < 0.01) (Fig. 3C). Thus, the TRIO-deficient CGNs exhibited abnormal membrane recovery during the neurite growth.
Figure 3.
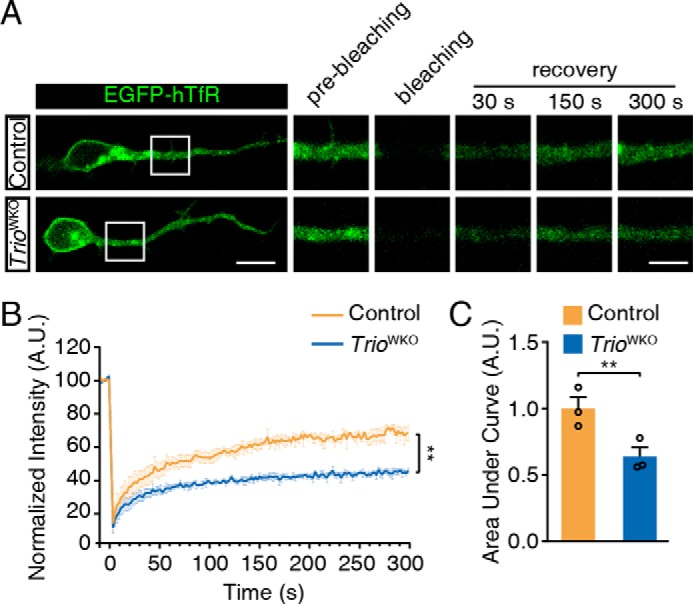
TRIO regulates membrane trafficking in CGNs. A, CGNs were transfected with pEGFP–hTfR and then subjected to the FRAP analysis. The bleached region is labeled with a white box. Representative images captured at the indicated time points are shown in the right panel. The scale bar in the left panel represents 10 μm, and the scale bar in the right panel represents 5 μm. B, statistical analysis of the intensity recovery curve of the normalized fluorescence intensity of control and TrioWKO neurons. Each group included three mice (n = 3) and 31–33 neurons/group; error bars indicate S.E. (two-way ANOVA). **, p < 0.01. C, statistical analysis of the area under curve of the recovery curve in B. The error bars indicate S.E. (Student's t test). **, p < 0.01. A.U., arbitrary units.
Because TRIO is associated with Golgi apparatus, we then determined the role of TRIO in the directional trafficking of TGN-derived membrane vesicles. It has been reported that the directional trafficking of these membrane vesicles to the neuronal growth cone is primarily mediated by RAB8 and RAB10 during axonal growth (42, 43), in which tdTomato fluorescent protein–fused RAB10 is applied to label RAB10-positive membrane vesicles and monitor the trafficking behaviors (42). We here used the tdTomato to label RAB8A and RAB10 to investigate whether TRIO affects RAB8- and RAB10-positive membrane trafficking. We observed the labeled membrane vesicles using time-lapse microscopy, generated the kymographs, and analyzed the trafficking behaviors using KymoAnalyzer (44). TRIO knockout in CGNs decreased the average distance traveled by RAB8A-positive vesicles in both the anterograde and retrograde directions, but the switch frequencies were significantly increased (Fig. 4, A–C, and Movies S1 and S2). However, the average velocities of vesicles traveling in both directions, and the percentages of time spent in motion states were not altered (Fig. 4, D and E, and Movies S1 and S2). In addition, RAB10-positive vesicles in TRIO-deficient CGNs showed a similar behavior to RAB8-positive vesicles in terms of the decreased distance traveled and increased switching frequency and comparable average velocities and percentage of time spent in motion states (Fig. 4, F–J, and Movies S3 and S4). This observation suggested an essential role of TRIO in regulating the switching frequency of the membrane vesicles.
Figure 4.

TRIO regulates RAB8- and RAB10-positive vesicle trafficking. A, representative kymographs of RAB8A-positive vesicles. The scale bars represent 5 μm and 5 s, as indicated in the figure. B–E, quantification of RAB8-positive vesicle movements, including distance traveled along combined and single segments (B), switching frequencies (C), velocities of combined segments (D), and percentage of time in motion (E). Each group included three mice (n = 3) and 20–24 neurons/group. The error bar indicates S.E. *, p < 0.05; **, p < 0.01. F, representative kymographs of RAB10-positive vesicles. The scale bars represent 5 μm and 5 s, as indicated in the graph. G–J, quantification of RAB10-positive vesicle movement parameters, including distance traveled along combined and single segments (G), switching frequencies (H), velocities of combined segments (I), and percentage of time in motion (J). Each group comprised three mice (n = 3) and 35–36 neurons/group. The error bars indicate S.E. (Student's t test). *, p < 0.05; **, p < 0.01. n.s., not significant; Ant, anterograde; Ret, retrograde.
RAB8/RAB10 activation is required for TRIO-mediated neurite outgrowth
The abnormal trafficking of RAB8- and RAB10-positive vesicle prompted us to hypothesize that RAB8 and RAB10 activities were altered in TRIO-deficient neurons. Three siRNAs specifically targeting Rab8a or Rab10 were introduced to knockdown these two GTPases (Fig. S4, A and E). The result showed that knockdown of Rab8a or Rab10 significantly inhibited neurite outgrowth (Fig. S4, B–D and F–H) as in previous reports (43, 45), suggesting a required role for RAB8 and RAB10 in neurite outgrowth in CGNs. We then measured the levels of GTP-bound RAB8 and RAB10 in the developing cerebella. After pulldown with the purified GST–MICAL-L2-C protein (46), the GTP-RAB8 and GTP-RAB10 proteins from the cerebellum were measured by Western blotting. The results showed that the GTP-bound forms of both RAB8 and RAB10 were decreased in TRIO-deficient cerebellar tissues (Fig. 5, A and B), suggesting a requirement of TRIO for RAB8/RAB10 activation. The observation from Neuro-2a cells overexpressing EGFP-TRIO9S or EGFP-TRIO8 displaying increased levels of GTP-RAB8 (Fig. 5, C and D) also supports this conclusion. The activation of RAB10 could not be determined because of the low expression of RAB10 in Neuro-2a cells (data not shown). In parallel, we also performed GST–FIP3–RBD11 pulldown assay (47, 48) and GST–Rabaptin5–R5BD pulldown assay (49) to determine the GTP-bound levels of RAB11 (a recycling endosome-associated Rab) and RAB5 (an early endosome-associated Rab), in TRIO-deficient cerebella or TRIO-overexpressing Neuro-2a cells (Fig. S5). No alteration of either GTP–RAB11 or GTP–RAB5 was observed, implying that TRIO activated RAB8/RAB10 in a selective manner. We then assessed the roles of RAB8 and RAB10 in the neurite outgrowth defect induced by TRIO deletion. After the introduction of RAB8A(Q67L), an active form of RAB8, into TRIO-deficient CGNs, the neurite length was significantly restored (56.9 ± 4.4 μm versus 40.5 ± 2.1 μm, p < 0.05) (Fig. 5, E and F). We also transfected the CGNs with RAB8A(T22N), a dominant-negative RAB8, and analyzed neurite growth. The dominant-negative RAB8 did not decrease the neurite length in TRIO-deficient CGNs further (40.5 ± 2.1 μm versus 44.2 ± 0.7 μm). For control CGN, however, the introduction of the dominant-negative RAB8 led to a significant decrease in neurite length (52.8 ± 6.9 μm versus 67.7 ± 5.8 μm, p < 0.05) (Fig. 5, E and F), and an increased percentage of the neurons with neurites less than 50 μm (Fig. 5G). Introduction of constitutive-active RAB8 to TRIO-deficient neurons led to a decreased percentage of the neuron with neurite less than 50 μm. We also introduced RAB10(T23N) into the control neurons and found that this dominant-negative RAB10 protein dramatically inhibited neurite outgrowth (Fig. 5, H–J), whereas the constitutive-active RAB10 rescued the growth defect of TrioWKO CGN neurites (Fig. 5, H–J). Of noted, these dominant-negative and constitutive active Rab mutants did not modulate the activity of Rho family GTPases, because overexpressing these mutants in Neuro-2a cells did not alter RAC1 activity (Fig. S6). Based on these results, we conclude that both RAB8 and RAB10 are required for neurite outgrowth in CGNs, and these two Rab GTPases may be regulated by TRIO.
Figure 5.

RAB8/RAB10 activation is required for TRIO-mediated neurite outgrowth. A, GST–MICAL-L2-C pulldown assay to determine GTP-RAB8 and GTP-RAB10 level in cerebella isolated from P10 TrioWKO mice and littermate control mice. B, quantification of GTP-RAB8 and GTP-RAB10 levels in A. The error bars indicate S.E. (Student's t test). *, p < 0.05; n = 4. C, GST-JFC1 pulldown assay to determine GTP-RAB8 levels in Neuro-2a cells transfected with either the pEGFP-TRIO9S, pEGFP-TRIO8, or pEGFP-C3 plasmid. D, quantification of GTP-RAB8 levels in C. The error bars indicate S.E. (one-way ANOVA with Bonferroni's test). *, p < 0.05; n = 4. E, CGNs isolated from control or TrioWKO mice were transfected with either pcDNA–EGFP, pcDNA-RAB8A(T22N)-P2A-EGFP, or pcDNA-RAB8A(Q67L)-P2A-EGFP. Transfected neurons were cultured for 2 DIV, followed by staining with DAPI, EGFP, and Tau. The scale bar represents 20 μm. F, quantification of the neurite length in the images shown in E. The error bars indicate S.E. (two-way ANOVA with Tukey's test). *, p < 0.05; n = 4. Numbers of measured neurons are indicated in the columns. G, quantification of neurite length distribution in E. The error bars indicates S.E.; n = 4. H, CGNs isolated from control or TrioWKO mice were transfected with either pEGFP-C3, pEGFP–RAB10(T23N), or pEGFP–RAB10(Q68L) and then cultured for 2 DIV, followed by staining with DAPI, EGFP, and Tau. The scale bar represents 20 μm. I, quantification of the neurite length in the images shown in H. The error bars indicate S.E. (two-way ANOVA with Tukey's test). ****, p < 0.0001; n = 4. Numbers of measured neurons are indicated in the columns. J, quantification of the neurite length distribution in H. The error bars indicate S.E.; n = 4. A.U., arbitrary units; n.s., not significant; MW, molecular weight.
TRIO recruits RABIN8 to traffic vesicles and hence facilitates RABIN8 activation
Because RABIN8 has been identified as the major GEF for RAB8 and RAB10 in neurite outgrowth (46, 50), we hypothesized that TRIO might be necessary for RABIN8 activation. We first measured active RABIN8 (phosphorylated RABIN8) in the protein lysates from the fresh cerebellar tissues from TRIOWKO mice and their control littermates at postnatal day 10. The phosphorylated RABIN8 was immunoprecipitated and blotted with an anti–phospho-mitogen-activated protein kinase/cyclin-dependent kinase substrate antibody (51) (Fig. 6A). The levels of phosphorylated RABIN8 were dramatically decreased in TrioWKO cerebella (Fig. 6, A and B) and CGNs in culture (Fig. 6, C and D). Because TRIO has no kinase domain for RABIN8 phosphorylation, the effect of TRIO on RABIN8 phosphorylation may be mediated through ERK1/2 (51). We thus measured the ERK1/2 protein in the fractions of membrane vesicles and expectedly found that ERK and RABIN8 co-existed in the subcellular fractions that were positive for RAB8 and RAB10 (Fig. 6E).
Figure 6.

TRIO promotes RABIN8 phosphorylation and regulates RABIN8 activity. A, RABIN8 phosphorylation analysis in cerebella tissues. B, quantification of the relative level of phosphorylated RABIN8 normalized to the pelleted RABIN8 in the blots shown in A. The error bars indicate S.E. (Student's t test). *, p < 0.05; n = 3. C, RABIN8 phosphorylation analysis in CGNs. D, quantification of RABIN8 phosphorylation in C. The error bars indicate S.E. (Student's t test). *, p < 0.05; n = 3. E, P10 mouse cerebellum was homogenized, and the postnuclear supernatants were subjected to 2.5–30% OptiPrep density gradient for subcellular fractionation. Fractions were subjected to Western blotting with RABIN8, RAB8, RAB10, and ERK1/2 antibodies. F, CGNs were cultured for 2 DIV and subjected to immunofluorescence using RABIN8 and RAB8 antibodies. The scale bar represents 5 μm. Scatter plots and the PCC of the fluorescence intensities of red and green channels were also shown. G, quantification of RABIN8 and RAB8 co-localization in neurites of CGNs as shown in F. Pearson's correlation coefficient and Manders' coefficient M1 and M2 were analyzed. The error bars indicate S.E. (Student's t test). *, p < 0.05. Each group comprised three mice (n = 3) and 18 neurons/group. A.U., arbitrary units; n.s., not significant; IP, immunoprecipitation; MW, molecular weight.
To understand the interplays of TRIO and RABIN8, we respectively characterized RABIN8 localization in TGN and membrane vesicles. We immunostained COS-7 cells with anti-RABIN8 and anti-TGN38 antibodies. The RABIN8 signal was observed in the cytoplasm and was enriched in the TGN38-positive region (Fig. S3A). In cultured CGNs, the RABIN8 signal was also enriched in the TGN38-positive TGN. This localization was consistent with the observation from cultured hippocampal neurons (52), and the localization pattern of RABIN8 in TGN was not affected apparently by TRIO knockout (Fig. S3B). We then examined the localization of RABIN8 in membrane vesicles by immunostaining. RABIN8 was detected in the RAB8-positive vesicles along the neurite from control CGN (Fig. 6F), but TRIO knockout resulted in a significantly decreased portion of RABIN8 overlapping with RAB8 (0.44 ± 0.03 versus 0.28 ± 0.01 of Pearson's correlation coefficient, and 0.82 ± 0.02 versus 0.65 ± 0.03 of Manders' coefficient 2, p < 0.05) (Fig. 6, F and G), suggesting that TRIO deletion reduced the accessibility of RABIN8 to RAB8-positive vesicles. This result indicated that TRIO was necessary for RABIN8 recruitment to the membrane vesicles where RABIN8 phosphorylation might be possibly catalyzed by ERK1/2.
The spectrin repeats of the TRIO N terminus are required for the interaction with RABIN8
To test whether the recruitment of RABIN8 by TRIO is mediated by physical interaction, we performed a series of binding assays. The P21 cerebellar lysates that expressed TRIO9S and TRIO8 at equivalent levels were subjected to immunoprecipitation using anti-TRIO and anti-RABIN8 antibodies to determine their interaction in vivo. TRIO and RABIN8 were detected in the respective immunoprecipitates by anti-RABIN8 and anti-TRIO antibodies but not by control IgG (Fig. 7A). We also transfected Neuro-2a cells with pEGFP-TRIO8, together with or without a plasmid expressing FLAG-RABIN8 to verify this observation. 48 h after transfection, the cells were lysed, and protein lysates were subjected to immunoprecipitation using a FLAG antibody covalently conjugated to agarose beads. Western blots of these immunoprecipitated samples showed that TRIO8 was detected in the immunoprecipitates of the cells expressing FLAG-RABIN8 (Fig. 7B). This observation was also supported by GST pulldown assay for P21 cerebellar lysates by using purified GST and recombinant GST-RABIN8 fusing protein. Both main isoforms of TRIO were detected in the samples containing GST-RABIN8, but not in the samples containing GST alone (Fig. 7D).
Figure 7.

TRIO directly interacts with RABIN8. A, co-immunoprecipitation of endogenous TRIO and RABIN8 in P21 mouse cerebellum. B, co-immunoprecipitation of EGFP-TRIO8 and FLAG-RABIN8 in Neuro-2a cells. C, co-immunoprecipitation of FLAG-RABIN8 with EGFP-TRIO (1–1295) and TRIO (1296–1909) in COS-7 cells. D, GST-RABIN8 pulldown assay of P21 mouse cerebellum lysates. E, RABIN8 phosphorylation assay to CGNs treated with ITX3. RAC1 activity was decreased. F, quantification of RABIN8 phosphorylation in E. The error bars indicate S.E. (Student's t test); n = 3. G, direct binding assay of His6-RABIN8 to GST–TRIO variants. Coomassie Blue staining of GST–TRIO variants are shown at the bottom. H, COS-7 cells were transfected with FLAG-RABIN8, together with either pEGFP-C3, pEGFP-TRIO(1–230), pEGFP-TRIO(208–673), pEGFP-TRIO(446–909), pEGFP-TRIO(672–1295), or pEGFP-TRIO(1296–1909), and were lysed and subjected to immunoprecipitation. RABIN8 phosphorylation level was determined. I, quantification of the pRABIN8 level in H. The error bars indicate S.E. (one-way ANOVA with Bonferroni's test). *, p < 0.05; **, p < 0.01; n = 6. n.s., not significant; A.U., arbitrary units; IP, immunoprecipitation. MW, molecular weight; WB, Western blot.
To identify the region of TRIO required for RABIN8 interaction, we prepared two truncated TRIO fragments: TRIO(1–1295) containing the spectrin repeats and TRIO(1296–1909) containing the GEF1 domain. Immunoprecipitation showed that EGFP-TRIO(1–1295) was detected in FLAG-RABIN8 precipitates, whereas EGFP-TRIO(1296–1909) did not produce any signal (Fig. 7C). Thus, the N-terminal region rather than the GEF domain of TRIO is required for the interaction with RABIN8. This conclusion was also supported by the result from the application of TRIO GEF1 inhibitor ITX3 (53), which showed no alteration of RABIN8 phosphorylation level in the ITX3-treated CGNs, whereas in which, the GTP-RAC1 levels were decreased (Fig. 7, E and F). To determine whether TRIO directly bind with RABIN8, we next expressed and purified the His6-tagged RABIN8 and then incubated with GST or the GST-fused TRIO variants: TRIO(1–230), TRIO(446–909), and TRIO(672–1295) (Fig. 2A). We also expressed TRIO(208–673) but failed in the purification because of the formation of a tight insoluble inclusion body. The result showed that the spectrin-containing variants were able to strongly bind to His6-RABIN8, and TRIO(1–230) containing SEC-14 domain bound weakly to His6-RABIN8, whereas GST alone did not show any binding (Fig. 7G). These results suggested that TRIO was able to physically bind with RABIN8 through the spectrin repeats. We then transfected COS-7 cells with either plasmid of pEGFP-C3, pEGFP-TRIO(1–230), pEGFP-TRIO(208–673), pEGFP-TRIO(446–909), pEGFP-TRIO(672–1295), or pEGFP-TRIO(1296–1909), together with plasmid encoding FLAG-RABIN8 to investigate whether each TRIO fragment was able to induce RABIN8 phosphorylation. The results suggested that TRIO(672–1295) and TRIO(1296–1909) decreased RABIN8 phosphorylation (Fig. 7, H and I). Because only TRIO(208–673) was observed in Golgi, these data suggested that TRIO(672–1295) or TRIO(1296–1909) may affect RABIN8 phosphorylation in a dominant-negative manner. Thus, both the correct localization of TRIO to Golgi and correct interaction of RABIN8 and TRIO appear to be required for RABIN8 activation.
Discussion
In this report, we reveal that Golgi-resident TRIO essentially regulates the directional membrane trafficking of the developing CGNs. At the trans-Golgi network and membrane vesicles, the pooled TRIO interacts with and activates RABIN8, which is necessary for RAB8 and RAB10 activation. RAB8 and RAB10 activation are key for the membrane vesicles trafficking from neuronal soma to the growth cone, and constitutively active RAB8 and RAB10 restored the impaired outgrowth of TRIO-deficient neurites. Based on these results, we concluded that TRIO mediated neurite growth by coordinating at least two processes: cytoskeletal rearrangement and membrane trafficking. This coordination was established by different pools of TRIO protein. We propose a working model of the role of TRIO in neurite growth. In this model, extracellular signals activate growth cone-resident TRIO and lead to the activation of Rho GTPases, which regulates cytoskeletal dynamics in the growth cone, thereby regulating neurite elongation. Simultaneously, Golgi-resident TRIO recruits RABIN8 to membrane vesicles for activation through a physical interaction of the spectrin repeats within the TRIO N terminus, and the activated RABIN8 drives membrane transport through the activation of Rab GTPases such as RAB8 and RAB10. The cytoskeletal rearrangements mediated by TRIO-activated Rho GTPases provide the driving force for neurite growth, and the membrane vesicles transport lipid cargoes to enable new membrane formation. This working model is further supported by multiple lines of evidence: (1) Deletion of TRIO leads to an impaired response to various signaling molecules, such as Netrin-1 and Semaphorin-6A (27). This implies a common regulatory scenario shared by these signals, similar to the membrane trafficking process required for neurite outgrowth. (2) A point mutation within the spectrin repeat also causes autism, schizophrenia, and intellectual disability (30–33). Because several Dbl family proteins, such as Kalirin, contain spectrin repeats, we predict that these proteins might also follow the similar working model to achieve their corresponding functions. Importantly, because many proteins contain multiple domains and some of them are capable of binding to subcellular structures, the subcellular pool formation might be a general regulatory mechanism for the functional protein machines.
Recently, TRIO mutations were identified in patients with autism, schizophrenia, and intellectual disability (30–33), and the mutations may occur both in TRIO GEFD1 and the spectrin repeat (N1080I) (32). These observations strongly suggest a critical role for TRIO in learning and memory. Based on our result, in addition to the altered cytoskeleton rearrangement mediated by TRIO GEF domains, the altered membrane trafficking mediated by the spectrin repeat may also essentially contribute to the phenotypes of these diseases. This may be instructive in developing therapeutic interventions for this condition. For example, we may use reagents to activate membrane trafficking to restore the impaired neurite growth in subjects carrying a mutation in the spectrin repeats.
In summary, we revealed a function of TRIO in regulating membrane trafficking during neurite outgrowth, in addition to the GEF-dependent function in regulating cytoskeletal dynamics by Rho GTPases. This function is accomplished by Golgi and the Golgi-derived vesicle pool of TRIO, whereby interaction with RABIN8 via the non-GEF domain. TRIO recruits and activates RABIN8 and subsequently activates RAB8 and RAB10 so as to regulate membrane trafficking by controlling the membrane vesicles switch frequency. Our findings enabled us to propose a molecular mechanism depicting the coordination of cytoskeletal rearrangements and membrane-trafficking during neurite growth.
Materials and methods
DNA constructs
The DNA fragments composing the mouse Trio CDS (1–6998) were PCR-amplified from mouse genomic DNA, mouse cerebellar cDNA, and IMAGE clones 6406413 and 6306998 (Thermo Fisher Scientific) and were inserted into the multiple cloning site (MCS)–modified pUC19 vector to generate pUC19–EcoRI–TRIO(1–6998)–XhoI–NotI using a ClonExpress MultiS one-step cloning kit (C113, Vazyme, Nanjing, China) and T4 DNA ligase (TaKaRa), following a series steps of molecular cloning. TRIO9S (6984–7110) was PCR-amplified from IMAGE clone 6406413 (Thermo Fisher Scientific) and inserted into XhoI- and NotI-linearized pUC19–EcoRI–TRIO(1–6998)–XhoI–NotI using a ClonExpress II one-step cloning kit (C112, Vazyme) to construct pUC19–TRIO9S. TRIO8 (4022–5717) was PCR-amplified with a TRIO8-specific sequence included in the reverse primer from Trio CDS (1–6998) and recombined into BglII and NotI linearized pUC19–EcoRI–TRIO(1–6998)–XhoI–NotI to construct pUC19–TRIO8. The resulting pUC19–TRIO9S and pUC19–TRIO8 vectors were digested with EcoRI and NotI, and the corresponding coding sequences were ligated to the EcoRI- and NotI-linearized MCS-modified pEGFP-C3 vector to construct the pEGFP-TRIO9S and pEGFP-TRIO8 plasmids, respectively. Trio8 (1–1295) and Trio8 (1296–1909) were then further PCR-amplified from pEGFP-TRIO8 and back cloned into the same EcoRI and NotI sites to construct the corresponding vectors. Trio (1–230), Trio (208–673), Trio (446–909), and Trio (672–1295) were also PCR-amplified from the pEGFP-TRIO8 plasmid and cloned into pEGFP-C3 to generate EGFP fusion proteins and into pGEX-5X-1 (GE Healthcare) to generate GST fusion proteins, respectively.
The mouse Rab8a CDS was PCR-amplified from mouse cerebellar cDNA and cloned into pMD19T vector (TaKaRa). The RAB8A(T22N) and RAB8A(Q67L) point mutations were generated using a Mut Express II fast mutagenesis kit (C212, Vazyme). PCR-amplified Rab8a(T22N) and Rab8a(Q67L) CDS were recombined with the P2A–EGFP sequence into the pcDNA3.1(+) backbone to construct pcDNA–RAB8A(T22N)–P2A–EGFP and pcDNA–RAB8A(Q67L)–P2A–EGFP, respectively. A control plasmid, pcDNA–EGFP, was also constructed by inserting the EGFP sequence into the same cloning site. The mouse Rab10 CDS was also PCR-amplified from mouse cerebellar cDNA and cloned into the pMD19T vector (TaKaRa). The Rab8a and Rab10 CDS were then cloned into the EcoRI- and NotI-linearized MCS-modified pEGFP-C3 vector to construct pEGFP–RAB8A and pEGFP–RAB10, respectively, using a ClonExpress II one-step cloning kit (C112, Vazyme). The pEGFP–RAB10 was used as template to generate point mutations, pEGFP–RAB10(T23N) and pEGFP–RAB10(Q68L), with a Mut Express II fast mutagenesis kit (C212, Vazyme). The tdTomato CDS was PCR-amplified from a tdTomato-coding plasmid and replaced the EGFP CDS between the AgeI and XhoI sites in pEGFP–RAB8A and pEGFP–RAB10 to construct ptdTomato-RAB8A and ptdTomato-RAB10, respectively. The Rabin8 CDS was PCR-amplified from mouse cerebellar cDNA and cloned into the pGEX-5X-1 vector (GE Healthcare) to construct pGEX-RABIN8, into the pET-32c vector (Novagen) to construct pET32-His6-RABIN8, or into the pCMV-FLAG-N vector to construct pCMV-FLAG-RABIN8.
The mouse MICAL-L2-C (833–1008) sequence was PCR-amplified from mouse cerebellar cDNA, the human Rabaptin5-R5BD (739–862) sequence was PCR-amplified from HEK293T cell cDNA, the human FIP3-RBD11 (649–756) sequence was PCR-amplified from pCMV-FIP3 plasmid (Nanjing Bioworld Biotechnology Co., Ltd., Nanjing, China), the human full-length JFC1 CDS was PCR-amplified from pDONR223-JFC1 plasmid (Nanjing Bioworld Biotechnology Co., Ltd., Nanjing, China), and these four DNA fragments were ligated into the pGEX-5X-1 (GE Healthcare) vector to construct pGEX-MICAL-L2-C, pGEX-RABAPTIN-R5BD, pGEX-FIP3-RBD11, and pGEX-JFC1, respectively. The plasmid expressing GST-PAK-GBD was kindly provided by Hollis T. Cline (54). All of the constructed plasmids mentioned above were verified by Sanger sequencing
siRNAs
Double-stranded oligonucleotides targeting mouse Rab8a and Rab10 were synthesized (GenePharma, Shanghai, China) with the following sequences: siRab8a#1, 5′-GCCUUCAACUCCACAUUCA-3′; siRab8a#2, 5′-CUCGAUGGCAAGAGGAUUA-3′; siRab8a#3, 5′-GGAAUUGGAUUCGGAACAU-3′; siRab10#1, 5′-UGAAGAUGUGGAAAGAAUG-3′; siRab10#2, 5′-UUCGGACGAUGCCUUCAAU-3′; siRab10#3, 5′-GCAUCAUGCUAGUGUAUGA-3′; and control siRNA, 5′-UUCUCCGAACGUGUCACGU-3′.
Animals
The mice used in this study were Trio floxed mice (27), B6.Cg-Tg(Nes-cre)1Kln/J (Nestin-Cre mice, 003771, The Jackson Laboratory), Tg(Wnt1-cre)11Rth Tg(Wnt1-GAL4)11Rth/J (Wnt1-Cre mice, 003829, The Jackson Laboratory), and B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (Rosa-mTmG mice, 007676, The Jackson Laboratory). Trio floxed mice were crossed with Nestin-Cre mice (Trioflox/flox, Nes-Cre, or TrioNKO) and Wnt1-Cre mice (Trioflox/flox, Wnt1-Cre, or TrioWKO) to delete TRIO in the cerebellum. Wnt1-Cre mice were also crossed with Rosa-mTmG mice to examine Cre expression in the cerebellum and cultured CGNs. All animal procedures were performed according to the animal protocol approved by the Institutional Animal Care and Use Committee of the Model Animal Research Center of Nanjing University (Nanjing, China).
Generation of TRIO N terminus-specific antiserum
The CT233 antigen was selected, which was previously used as rat spectrin 5 and 6, to raise a rabbit anti-TRIO polyclonal antibody (35). The codon-optimized DNA sequence encoding mouse TRIO spectrin repeats 5 and 6, from amino acids Val674 to Arg900, was synthesized (Genscript, Nanjing, China) and cloned into the pGEX-5X-1 vector (GE Healthcare). The plasmid was expressed in DH5α cells, production of the GST fusion protein was induced with 0.1 mm IPTG, and the recombinant protein was purified using the batch purification method with GSH–Sepharose 4B (catalog no. 17-0756-01, GE Healthcare), according to the manufacturer's instructions. The eluted protein was then dialyzed against PBS overnight and quantified using a Bio-Rad protein assay kit (catalog no. 500-0006, Bio-Rad). GST was not cleaved, and the fusion protein was diluted to 1 mg/ml in PBS. It was mixed well with an equal volume of Freund's complete adjuvant (F5881, Sigma–Aldrich) by vortexing for 30 min at room temperature. 200 μl of the mixture (100 μg of fusion protein) were used to immunize BALB/c mice via intraperitoneal injections. These mice were immunized a second and third time by repeating the injections of 100 μg of fusion protein mixed with an equal volume of Freund's incomplete adjuvant (F5506, Sigma–Aldrich) after 14 and 28 days, respectively. The mice were sacrificed 40 days after the first injection, and sera were collected, followed by the characterization of each antiserum by Western blotting. Of all the antisera collected, the most specific antiserum was selected and used in the present study.
Cell culture and transfection
Neuro-2a cells were cultured in minimum essential medium (catalog no. 12571-063, Life Technologies), and COS-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, catalog no. 12800-017, Life Technologies), both of which were supplemented with 10% fetal bovine serum (FBS, catalog no. 10099-141, Life Technologies), 100 units/ml penicillin, and 100 μg/ml streptomycin, in an atmosphere of 5% CO2. Before transfection, the medium was replaced with fresh culture medium. Plasmid transfections were performed using LipoMAX (catalog no. 32011, Sudgen Biotech, Nanjing, China), according to the manufacturer's instructions. COS-7 cells overexpressing FLAG-RAB10 were transfected by Rab10-specific siRNAs or control siRNA using Lipofectamine RNAiMAX (catalog no. 13778-030, Life Technologies), according to the manufacturer's instructions.
Neuronal culture
Cerebellar granule neurons were prepared from mice before postnatal day 8, as previously described (27). Briefly, cerebella were isolated and trypsinized in TrypLE (catalog no. 12604-013, Life Technologies) at 37 °C for 10 min and were passed through a fire-polished glass pipette to obtain a single cell suspension. Approximately 4–5 × 104 CGNs were plated onto poly-d-lysine (100 μg/ml, P0899, Sigma–Aldrich)–coated 8-mm × 8-mm glass coverslips in wells of a 24-well plate in DMEM, 10% FBS at 37 °C and incubated for 4 h. The medium was then replaced with Neurobasal medium (catalog no. 21003-049, Life Technologies) supplemented with 2% B-27 (catalog no. 17504-044, Life Technologies), 1× GlutaMAX (catalog no. 35050-061, Life Technologies), and 1× penicillin/streptomycin (catalog no. 15140-122, Life Technologies) and cultured for the indicated times. To inhibit GEF1 activity, 100 μm ITX3 (HY-16663, MedChemExpress) (from 10 mm stock solution in DMSO) was used for 48 h, and its vehicle control was 1% DMSO.
Neuronal nucleofection
CGNs were prepared from P6-8 mice as described above, because a sufficient number of neurons can be obtained from mice at this age for nucleofection. CGNs from each cerebellum were suspended in 200 μl of nucleofection buffer, and each 100-μl cell suspension (∼2 × 106 neurons) was used for a nucleofection reaction using an Amaxa Nucleofector 2b device (Lonza) with program O-005. For the nucleofection of plasmids, 5 μg of each plasmid were used in a single reaction. The cells were then pipetted out of the cuvette in 1 ml of DMEM containing 10% FBS and aliquoted into three wells of a 24-well plate on poly-d-lysine–coated 8-mm × 8-mm glass coverslips (for rescue experiments) or one PDL-coated 35-mm glass-bottomed dish (for live cell image), and the medium was changed after 4 h, as described above.
Immunofluorescence, neurite outgrowth assay, and co-localization assay
Neurons and COS-7 cells grown on coverslips were washed with PBS, fixed with 4% paraformaldehyde (pH 7.4 in PBS) at room temperature for 5 min, and then washed with PBS three times for 5 min each. Neurons were then permeabilized with 0.5% Triton X-100 in PBS for 15 min, blocked with 1% BSA at room temperature for 1 h, and incubated with primary antibodies overnight at 4 °C. The primary antibodies used in the present study were: rabbit anti-TAU (1:200, A390, BS1357, Bioworld), mouse anti-GFP (1:1000, GF28R, MA5–15256, Thermo Fisher Scientific), rabbit anti-GM130 (1:100, ab52649, Abcam), rabbit anti-TGN46 (1:100, ab50595, Abcam), rabbit anti-VAMP4 (1:50, 10738-1-AP, Proteintech), rabbit anti-RCAS1 (1:200, catalog no. 12290S, Cell Signaling Technology), mouse anti-TGN38 (1:100, 2F7.1, NB300-575, Novus Biologicals), mouse anti-TRIO antiserum (1:200), and rabbit anti-RABIN8 (1:50, 12321-1-AP, Proteintech). After a 1-h incubation with the secondary antibodies and DAPI (BioSharp) at room temperature, the coverslips were washed and mounted with ProLong Gold Antifade reagent (P36934, Life Technologies). The secondary antibodies used in the present study were: Alexa Fluor 546–conjugated donkey anti-rabbit IgG (A10040, Life Technologies), Alexa Fluor 546–conjugated donkey anti-mouse IgG (A10036, Life Technologies), Alexa Fluor 488–conjugated goat anti-rabbit IgG (A11008, Life Technologies), and Alexa Fluor 488–conjugated donkey anti-mouse IgG (A21202, Life Technologies). For the neurite outgrowth assay, images were acquired at 512 × 512 pixels using an Olympus FV1000 confocal microscope (Olympus, Japan) by an experimenter who was blinded to the genotypes and/or experimental conditions, and the neurite lengths (i.e. the distance between the center of the soma and the tip of the longest neurite) were measured manually using National Institutes of Health ImageJ software. Only neurites that met the criteria described elsewhere were measured (55): (1) the neurite emerged from an isolated neuron, (2) the neurite was not in contact with other neurons or neurites, and (3) the neurite was longer than the diameter of the soma. For other analyses of immunofluorescence staining, the images were captured using a Zeiss LSM880 confocal microscope (Zeiss, Germany). Co-localization was quantified using the JACoP plugin (56) in ImageJ, including the Pearson's correlation coefficient (Pr) and Manders' coefficients (M1 and M2), using Costes' automatic threshold. Fluorescence intensity scatter plot was generated using the ScatterJn (57) plugin in ImageJ.
Live-cell imaging and kymograph analysis
Nucleofected neurons were cultured in phenol red-free Neurobasal medium (catalog no. 12348-17, Life Technologies) supplemented with 2% B-27 (catalog no. 17504-044, Life Technologies), 1× GlutaMAX (catalog no. 35050061, Life Technologies), and 1× penicillin/streptomycin (catalog no. 15140122, Life Technologies) for 24–30 h and observed under a GE deltaVision Elite microscope (GE healthcare) at 37 °C and 5% CO2. Time-lapse images were acquired every 100 ms for a total of 30 s using a 60× objective lens (PlanApo NA1.42, oil immersion, Olympus). A pixel resolution of 0.108 μm/pixel was generated using this system, and deconvoluted images were generated. Kymographs of RAB8A- and RAB10-positive vesicles were generated using the KymoResliceWide plugin in ImageJ software, with a line width setting of 10 pixels, and the movement parameters, including distance traveled, switch frequencies, velocities, and percentage of time in motion, were semiautomatically analyzed using the KymoAnalyzer macro (44) in ImageJ software.
FRAP analysis
Neurons grown in 35-mm glass-bottomed dishes for 1 DIV were transfected with pEGFP–hTfR with the calcium method using the CalPhos kit (Clontech), according to a previously described procedure (58), and the cells were allowed to express the fusion protein for another 24–30 h. FRAP experiments were performed using a Zeiss LSM880 confocal microscope with a controlled temperature of 37 °C in a humid chamber with a 5% CO2 atmosphere. Five scans of prebleaching images were initially acquired to obtain a baseline intensity measurement. Bleaching was performed on selected neurites expressing EGFP–hTfR using the 488-nm argon laser line at 100% power with 20 iterations. The intensity during fluorescence recovery was measured by acquiring 150 additional scans at 2-s intervals with a 2% laser power at a resolution of 512 × 512 pixels and an optical zoom of 2. Background correction and data normalization were performed using previously described methods (41). Briefly, a group of bleached neurites, a nonbleached region, and background region were first determined, and the intensity of the background region was subtracted from the intensity of the bleached neurites and nonbleached region at each time point for background correction. The corrected intensity of the bleached neurites was divided by the intensity of the nonbleached region and normalized to the average intensity of the five prebleaching scans.
Subcellular fractionation
Subcellular fraction was prepared using OptiPrep Axis-Shield density gradient medium according to the procedure of the application sheet S24 (Alere Technologies AS, Oslo, Norway) with modifications. Briefly, the cerebellum was isolated from P10 mouse and homogenized in 1 ml of homogenization medium (0.25 m sucrose, 1 mm EDTA, and 10 mm HEPES·NaOH, pH 7.4) by Dounce tissue grinder and five passes through a fine syringe needle. Postnuclear supernatant was prepared by centrifuging the homogenate at 1500 × g for 10 min. Discontinuous OptiPrep gradient was prepared using underlayering technique by layering 2 ml of the following solutions: 2.5, 5, 10, 15, 20, 25, and 30% iodixanol in homogenization medium. Approximately 700 μl of postnuclear supernatant was uploaded to the gradient and centrifuged at 88,000 × g at 4 °C for 16 h using SW32 rotor (Beckman, German). Twenty-eight fractions (500 μl each) were collected from the top, supplemented with 4× Laemmli buffer, boiled at 95 °C for 10 min, and subjected to Western blotting.
Purification of the GST fusion protein
GST, GST–RABIN8, GST–JFC1, GST–MICAL-L2-C, GST–RABAPTIN5–R5BD, GST–FIP3–RBD11, and GST–PAK–GBD were expressed in DH5α cells that had been induced with 0.1 mm IPTG at 37 °C for 4 h, and GST–TRIO (1–230), GST–TRIO (446–909), and GST–TRIO (672–1295) were expressed in BL21(DE3) cells that had been induced with 0.1 mm IPTG at 30 °C for 4 h. All GST fusion proteins were purified with GSH-Sepharose 4B (catalog no. 17-0756-01, GE Healthcare) using the gravity flow method, according to the manufacturer's instructions. Eluted GST–TRIO variants were dialyzed against buffer containing 20 mm HEPES·NaOH, pH 7.4, and 150 mm NaCl; other GST proteins were dialyzed against 50 mm Tris·HCl, pH 7.4, overnight; and protein concentrations were determined using a Bio-Rad protein assay kit (catalog no. 500-0006).
GST pulldown assay
The P21 mouse cerebellum was lysed in lysis buffer (50 mm Tris·HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, pH 8.0, 1% Triton X-100, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). The lysate was cleared by centrifugation at 12,000 rpm for 10 min, and equal amounts of cleared lysate were incubated with 40 μl of a 50% slurry of GSH–Sepharose 4B and 20 μg of purified GST or GST-RABIN8 overnight at 4 °C. The beads were then washed three times with 500 μl of lysis buffer prior to the addition of 2× Laemmli buffer. Samples were boiled at 95 °C for 10 min and subjected to Western blotting.
Purification of the His6-RABIN8 and direct binding assay
His6-RABIN8 were expressed in BL21(DE3) cells that had been induced with 1 mm IPTG at 37 °C for 4 h. Bacteria pellets were then sonicated in PBS with 1 mg/ml lysozyme. Proteins were then pelleted by centrifugation at 9000 rpm for 10 min and dissolved in PBS with 8 m urea. Cleared supernatant were then incubated with nickel–nitrilotriacetic acid beads (Life Technologies) at room temperature for 1 h. The beads were first washed by gravity flow with 100 mm Tris·HCl, pH 8.0, 6 m urea, and 20 mm imidazole and then washed another three times by the same wash buffer in which the urea concentration was 4, 2, and 0 m. The purified His6-RABIN8 was eluted with 500 mm imidazole in 100 mm Tris·HCl, pH 8.0, and dialyzed against buffer containing 20 mm HEPES·NaOH, pH 7.4, and 150 mm NaCl. The concentration was determined using a Bio-Rad protein assay kit (catalog no. 500-0006, Bio-Rad). Direct binding of His6-RABIN8 to GST–TRIO variants was assayed by diluting 10 μg of purified His6-RABIN8, 20 μg of purified GST–TRIO variants, and 40 μl of a 50% slurry of GSH–Sepharose 4B (catalog no. 17-0756-01, GE Healthcare) in 600 μl of binding buffer (20 mm HEPES·NaOH, pH 7.4, 150 mm NaCl, 0.1% Triton X-100). The samples were incubated overnight at 4 °C. The supernatants were collected, and the beads were washed three times with binding buffer and boiled at 95 °C for 10 min after the addition of Laemmli buffer. For Western blotting, 1/10 of pellet samples and 1/100 of supernatant samples were loaded to SDS-PAGE.
Immunoprecipitation
Immunoprecipitation was performed to determine the interaction of RABIN8 and TRIO, and RABIN8 phosphorylation. Mouse cerebella, Neuro-2a cells, COS-7 cells, or CGNs cultured in 60-mm dishes were lysed in lysis buffer (50 mm Tris·HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, pH 8.0, 1% Triton X-100, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). For RABIN8 immunoprecipitation to determine the phosphorylation level, 1× PhosSTOP phosphatase inhibitor mixture (catalog no. 04906845001, Roche) was included in the lysis buffer. Lysates were cleared by centrifugation at 12000 rpm for 10 min; some cleared supernatants were retained as the total input, and the remaining lysates were incubated with 20 μl of a 50% slurry of Anti-FLAG M2 affinity gel (A2220, Sigma–Aldrich) or 40 μl of a 50% slurry of protein A–Sephorose (catalog no. 17-0974-01, GE Healthcare) plus the rabbit anti-TRIO antibody (H120, sc-28564, Santa Cruz Biotechnology), the rabbit anti-RABIN8 antibody (12321-1-AP, Proteintech), or equal amount of the normal rabbit IgG (sc-2027, Santa Cruz Biotechnology) overnight at 4 °C. Bound proteins were eluted as described above and subjected to Western blotting.
Rab GTPases activation assay
GST–MICAL-L2-C, GST–Rabaptin5–R5BD, GST–FIP3–RBD11, and GST–JFC1 were purified as described above. Rab GTPases activation assays were performed as previously described (51). For the GST pulldown, cerebella were lysed in 600 μl of lysis buffer (20 mm Tris·HCl, pH 7.4, 100 mm KCl, 5 mm MgCl2, 0.5% Triton X-100, 1 mm DTT, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin), and the Neuro-2a cells cultured in wells of a 6-well plate that had been transfected with pEGFP-C3, pEGFP-TRIO9S, and pEGFP-TRIO8 were serum-starved overnight at 36 h after transfection and were then lysed with 300 μl of lysis buffer. An aliquot of the cleared lysates (60 μl of cerebellar lysates or 30 μl of Neuro-2a lysates) was retained as the total protein sample, and the remaining lysates were incubated with 20 μg of GST-JFC1 and 40 μl of a 50% slurry of GSH–Sepharose 4B (catalog no. 17-0756-01, GE Healthcare) at 4 °C for 4 h, followed by three washes with 400 μl of lysis buffer. The bound GTP-Rabs was eluted by boiling with Laemmli buffer and was detected by Western blotting.
RAC1 activation assay
RAC1 activation assays were performed using a previously described procedure (59), with minor modifications. Neuro-2a cells were lysed in lysis buffer (50 mm Tris·HCl, pH 7.2, 1% Triton X-100, 500 mm NaCl, 10 mm MgCl2, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin), and the cleared lysates were incubated with 20 μg of GST-PAK-GBD and 40 μl of a 50% slurry of GSH–Sepharose 4B (catalog no. 17-0756-01, GE Healthcare) at 4 °C for 45 min. The beads were then washed three times with 400 μl of lysis buffer and were boiled to elute the bound GTP-RAC1. For experiment in Fig. 7E, the supernatant was collected and directly used for RABIN8 immunoprecipitation to detect phosphorylated RABIN8 level.
Protein extraction and Western blotting
The mouse cerebellar tissue was lysed at the indicated time points in modified RIPA buffer (50 mm Tris·HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with a 1× protease inhibitor mixture (catalog no. 04693132001, Roche). Western blotting was performed to detect protein expression, GST pulldowns, direct binding assay, immunoprecipitations, RAB8/RAB10 activation assay, and RAC1 activation assay. The antibodies used were TRIO (1:200, H120, sc-28564, Santa Cruz Biotechnology), the homemade TRIO antiserum (1:2000), GM-130 (1:1000, ab52649, Abcam), BIP (1:1000, clone 40/BiP, 610978, BD Biosciences), EEA1 (1:600, ab2900, Abcam), RAB5, (1:1000, ab18211, Abcam), GFP (1:2000, GF28R, MA5–15256, Thermo Fisher Scientific), RAB8 (1:1000, clone 4/Rab8, 610844, BD Biosciences), RAB10 (1:1000, 11808-1-AP, Proteintech), RAB11 (1:1000, clone 47/Rab11, 610656, BD Biosciences), RABIN8 (1:1000, 12321-1-AP, Proteintech), phospho-mitogen-activated protein kinase/cyclin-dependent kinase substrates (1:1000, 34B2, catalog no. 2325, Cell Signaling), FLAG (1:1000, clone M2, F1804, Sigma–Aldrich), RAC1 (1:1000, clone 23A8, catalog no. 05-389, Millipore), ERK1/2 (1:1000, BS3627, Bioworld), and β-actin (1:5000, clone AC-15, A1978, Sigma–Aldrich). For blotting RABIN8 immunoprecipitations, a mouse secondary antibody specific to rabbit IgG light chain was used (1:5000, ab99697, Abcam).
Statistical analysis
All quantification analysis of Western blotting was performed using ImageJ software. In all experiments using mice, n represents the number of mice used. The data are presented as the means ± S.E. The statistical analysis was performed using Prism 6.05 software. As indicated in the figure legends, Student's t test or one-way ANOVA with Bonferroni's test was used for tests with only one variable, and a two-way ANOVA with Tukey's test was used for tests with two independent variables. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Author contributions
T. T., J. S., and M.-S. Z. conceptualization; T. T., J. S., Y. L., P. W., X. C., W. Z., Y.-Y. Z., L. W., W. W., and Y. Z. investigation; T. T. and J. S. visualization; T. T., Y. P., and M.-S. Z. methodology; T. T., J. S., and M.-S. Z. writing-original draft; T. T., J. S., and M.-S. Z. writing-review and editing; Y. P., J. L., and Y. S. S. resources; M.-S. Z. supervision; M.-S. Z. funding acquisition.
Supplementary Material
Acknowledgments
We thank Xuena Zhang and Juan Liang for technical support.
This work was supported by National Natural Science Foundation of China Grants 31272311 and 31330034 (to M.-S. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Movies S1–S4.
- TGN
- trans-Golgi network
- CDS
- coding sequence
- CGN
- cerebellar granule neuron
- FRAP
- fluorescence recovery after photobleaching
- GEF
- guanine nucleotide exchange factor
- GST
- glutathione S-transferase
- PCC
- Pearson's correlation coefficient
- EGFP
- enhanced green fluorescent protein
- MCS
- multiple cloning site
- IPTG
- isopropyl β-d-thiogalactopyranoside
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- DAPI
- 4′,6′-diamino-2-phenylindole
- DIV
- day(s) in vitro
- PMSF
- phenylmethylsulfonyl fluoride
- ANOVA
- analysis of variance.
References
- 1. Pfenninger K. H. (2009) Plasma membrane expansion: a neuron's Herculean task. Nat. Rev. Neurosci. 10, 251–261 10.1038/nrn2593 [DOI] [PubMed] [Google Scholar]
- 2. Wojnacki J., and Galli T. (2016) Membrane traffic during axon development. Dev. Neurobiol. 76, 1185–1200 10.1002/dneu.22390 [DOI] [PubMed] [Google Scholar]
- 3. Dent E. W., Gupton S. L., and Gertler F. B. (2011) The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol. 3, a001800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalil K., and Dent E. W. (2005) Touch and go: guidance cues signal to the growth cone cytoskeleton. Curr. Opin. Neurobiol. 15, 521–526 10.1016/j.conb.2005.08.005 [DOI] [PubMed] [Google Scholar]
- 5. Tojima T., and Kamiguchi H. (2015) Exocytic and endocytic membrane trafficking in axon development. Dev. Growth Differ. 57, 291–304 10.1111/dgd.12218 [DOI] [PubMed] [Google Scholar]
- 6. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- 7. Hall A., and Lalli G. (2010) Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb. Perspect. Biol. 2, a001818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jaffe A. B., and Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 10.1146/annurev.cellbio.21.020604.150721 [DOI] [PubMed] [Google Scholar]
- 9. Da Silva J. S., Medina M., Zuliani C., Di Nardo A., Witke W., and Dotti C. G. (2003) RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J. Cell Biol. 162, 1267–1279 10.1083/jcb.200304021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garvalov B. K., Flynn K. C., Neukirchen D., Meyn L., Teusch N., Wu X., Brakebusch C., Bamburg J. R., and Bradke F. (2007) Cdc42 regulates cofilin during the establishment of neuronal polarity. J. Neurosci. 27, 13117–13129 10.1523/JNEUROSCI.3322-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Govek E. E., Newey S. E., and Van Aelst L. (2005) The role of the Rho GTPases in neuronal development. Genes Dev. 19, 1–49 10.1101/gad.1256405 [DOI] [PubMed] [Google Scholar]
- 12. Tahirovic S., Hellal F., Neukirchen D., Hindges R., Garvalov B. K., Flynn K. C., Stradal T. E., Chrostek-Grashoff A., Brakebusch C., and Bradke F. (2010) Rac1 regulates neuronal polarization through the WAVE complex. J. Neurosci. 30, 6930–6943 10.1523/JNEUROSCI.5395-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hutagalung A. H., and Novick P. J. (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91, 119–149 10.1152/physrev.00059.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stenmark H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 10.1038/nrm2728 [DOI] [PubMed] [Google Scholar]
- 15. Villarroel-Campos D., Bronfman F. C., and Gonzalez-Billault C. (2016) Rab GTPase signaling in neurite outgrowth and axon specification. Cytoskeleton 73, 498–507 10.1002/cm.21303 [DOI] [PubMed] [Google Scholar]
- 16. Mizuno-Yamasaki E., Rivera-Molina F., and Novick P. (2012) GTPase networks in membrane traffic. Annu. Rev. Biochem. 81, 637–659 10.1146/annurev-biochem-052810-093700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Debant A., Serra-Pagès C., Seipel K., O'Brien S., Tang M., Park S. H., and Streuli M. (1996) The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. U.S.A. 93, 5466–5471 10.1073/pnas.93.11.5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Awasaki T., Saito M., Sone M., Suzuki E., Sakai R., Ito K., and Hama C. (2000) The Drosophila trio plays an essential role in patterning of axons by regulating their directional extension. Neuron 26, 119–131 10.1016/S0896-6273(00)81143-5 [DOI] [PubMed] [Google Scholar]
- 19. Bateman J., Shu H., and Van Vactor D. (2000) The guanine nucleotide exchange factor trio mediates axonal development in the Drosophila embryo. Neuron 26, 93–106 10.1016/S0896-6273(00)81141-1 [DOI] [PubMed] [Google Scholar]
- 20. Liebl E. C., Forsthoefel D. J., Franco L. S., Sample S. H., Hess J. E., Cowger J. A., Chandler M. P., Shupert A. M., and Seeger M. A. (2000) Dosage-sensitive, reciprocal genetic interactions between the Abl tyrosine kinase and the putative GEF trio reveal trio's role in axon pathfinding. Neuron 26, 107–118 10.1016/S0896-6273(00)81142-3 [DOI] [PubMed] [Google Scholar]
- 21. Newsome T. P., Schmidt S., Dietzl G., Keleman K., Asling B., Debant A., and Dickson B. J. (2000) Trio combines with Dock to regulate Pak activity during photoreceptor axon pathfinding in Drosophila. Cell 101, 283–294 10.1016/S0092-8674(00)80838-7 [DOI] [PubMed] [Google Scholar]
- 22. Steven R., Kubiseski T. J., Zheng H., Kulkarni S., Mancillas J., RuizMorales A., Hogue C. W., Pawson T., and Culotti J. (1998) UNC-73 activates the Rac GTPase and is required for cell and growth cone migrations in C. elegans. Cell 92, 785–795 10.1016/S0092-8674(00)81406-3 [DOI] [PubMed] [Google Scholar]
- 23. Zheng C., Diaz-Cuadros M., and Chalfie M. (2016) GEFs and Rac GTPases control directional specificity of neurite extension along the anterior-posterior axis. Proc. Natl. Acad. Sci. U.S.A. 113, 6973–6978 10.1073/pnas.1607179113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Backer S., Hidalgo-Sánchez M., Offner N., Portales-Casamar E., Debant A., Fort P., Gauthier-Rouvière C., and Bloch-Gallego E. (2007) Trio controls the mature organization of neuronal clusters in the hindbrain. J. Neurosci. 27, 10323–10332 10.1523/JNEUROSCI.1102-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Briançon-Marjollet A., Ghogha A., Nawabi H., Triki I., Auziol C., Fromont S., Piché C., Enslen H., Chebli K., Cloutier J. F., Castellani V., Debant A., and Lamarche-Vane N. (2008) Trio mediates netrin-1–induced Rac1 activation in axon outgrowth and guidance. Mol. Cell. Biol. 28, 2314–2323 10.1128/MCB.00998-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Brien S. P., Seipel K., Medley Q. G., Bronson R., Segal R., and Streuli M. (2000) Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 97, 12074–12078 10.1073/pnas.97.22.12074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peng Y.-J., He W.-Q., Tang J., Tao T., Chen C., Gao Y.-Q., Zhang W.-C., He X.-Y., Dai Y.-Y., Zhu N.-C., Lv N., Zhang C.-H., Qiao Y.-N., Zhao L.-P., Gao X., et al. (2010) Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J. Biol. Chem. 285, 24834–24844 10.1074/jbc.M109.096537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zong W., Liu S., Wang X., Zhang J., Zhang T., Liu Z., Wang D., Zhang A., Zhu M., and Gao J. (2015) Trio gene is required for mouse learning ability. Brain Res. 1608, 82–90 10.1016/j.brainres.2015.02.040 [DOI] [PubMed] [Google Scholar]
- 29. Herring B. E., and Nicoll R. A. (2016) Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. U.S.A. 113, 2264–2269 10.1073/pnas.1600179113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ba W., Yan Y., Reijnders M. R., Schuurs-Hoeijmakers J. H., Feenstra I., Bongers E. M., Bosch D. G., De Leeuw N., Pfundt R., Gilissen C., De Vries P. F., Veltman J. A., Hoischen A., Mefford H. C., Eichler E. E., et al. (2016) TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 25, 892–902 10.1093/hmg/ddv618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katrancha S. M., Wu Y., Zhu M., Eipper B. A., Koleske A. J., and Mains R. E. (2017) Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum. Mol. Genet. 26, 4728–4740 10.1093/hmg/ddx355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pengelly R. J., Greville-Heygate S., Schmidt S., Seaby E. G., Jabalameli M. R., Mehta S. G., Parker M. J., Goudie D., Fagotto-Kaufmann C., Mercer C., DDD Study, Debant A., Ennis S., and Baralle D. (2016) Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 53, 735–742 10.1136/jmedgenet-2016-103942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sadybekov A., Tian C., Arnesano C., Katritch V., and Herring B. E. (2017) An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 8, 601 10.1038/s41467-017-00472-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bateman J., and Van Vactor D. (2001) The Trio family of guanine-nucleotide-exchange factors: regulators of axon guidance. J. Cell Sci. 114, 1973–1980 [DOI] [PubMed] [Google Scholar]
- 35. McPherson C. E., Eipper B. A., and Mains R. E. (2005) Multiple novel isoforms of Trio are expressed in the developing rat brain. Gene 347, 125–135 10.1016/j.gene.2004.12.028 [DOI] [PubMed] [Google Scholar]
- 36. Portales-Casamar E., Briançon-Marjollet A., Fromont S., Triboulet R., and Debant A. (2006) Identification of novel neuronal isoforms of the Rho-GEF Trio. Biol. Cell 98, 183–193 10.1042/BC20050009 [DOI] [PubMed] [Google Scholar]
- 37. Beck K. A. (2005) Spectrins and the Golgi. Biochim. Biophys. Acta 1744, 374–382 10.1016/j.bbamcr.2005.04.008 [DOI] [PubMed] [Google Scholar]
- 38. Danielian P. S., Muccino D., Rowitch D. H., Michael S. K., and McMahon A. P. (1998) Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326 10.1016/S0960-9822(07)00562-3 [DOI] [PubMed] [Google Scholar]
- 39. Muzumdar M. D., Tasic B., Miyamichi K., Li L., and Luo L. (2007) A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- 40. Burack M. A., Silverman M. A., and Banker G. (2000) The role of selective transport in neuronal protein sorting. Neuron 26, 465–472 10.1016/S0896-6273(00)81178-2 [DOI] [PubMed] [Google Scholar]
- 41. Gärtner A., Fornasiero E. F., Valtorta F., and Dotti C. G. (2014) Distinct temporal hierarchies in membrane and cytoskeleton dynamics precede the morphological polarization of developing neurons. J. Cell Sci. 127, 4409–4419 10.1242/jcs.149815 [DOI] [PubMed] [Google Scholar]
- 42. Deng C. Y., Lei W. L., Xu X. H., Ju X. C., Liu Y., and Luo Z. G. (2014) JIP1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J. Neurosci. 34, 1710–1723 10.1523/JNEUROSCI.4496-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huber L. A., Dupree P., and Dotti C. G. (1995) A deficiency of the small GTPase rab8 inhibits membrane traffic in developing neurons. Mol. Cell. Biol. 15, 918–924 10.1128/MCB.15.2.918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neumann S., Chassefeyre R., Campbell G. E., and Encalada S. E. (2017) KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic 18, 71–88 10.1111/tra.12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang T., Liu Y., Xu X. H., Deng C. Y., Wu K. Y., Zhu J., Fu X. Q., He M., and Luo Z. G. (2011) Lgl1 activation of rab10 promotes axonal membrane trafficking underlying neuronal polarization. Dev. Cell 21, 431–444 10.1016/j.devcel.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 46. Homma Y., and Fukuda M. (2016) Rabin8 regulates neurite outgrowth in both GEF activity–dependent and –independent manners. Mol. Biol. Cell 27, 2107–2118 10.1091/mbc.E16-02-0091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campa C. C., Margaria J. P., Derle A., Del Giudice M., De Santis M. C., Gozzelino L., Copperi F., Bosia C., and Hirsch E. (2018) Rab11 activity and PtdIns(3)P turnover removes recycling cargo from endosomes. Nat. Chem. Biol. 14, 801–810 10.1038/s41589-018-0086-4 [DOI] [PubMed] [Google Scholar]
- 48. Franco I., Gulluni F., Campa C. C., Costa C., Margaria J. P., Ciraolo E., Martini M., Monteyne D., De Luca E., Germena G., Posor Y., Maffucci T., Marengo S., Haucke V., Falasca M., et al. (2014) PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev. Cell 28, 647–658 10.1016/j.devcel.2014.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qi Y., Liang Z., Wang Z., Lu G., and Li G. (2015) Determination of Rab5 activity in the cell by effector pull-down Assay. In Rab GTPases (Li G., ed) pp. 259–270, Springer, New York: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hattula K., Furuhjelm J., Tikkanen J., Tanhuanpää K., Laakkonen P., and Peränen J. (2006) Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J. Cell Sci. 119, 4866–4877 10.1242/jcs.03275 [DOI] [PubMed] [Google Scholar]
- 51. Wang J., Ren J., Wu B., Feng S., Cai G., Tuluc F., Peränen J., and Guo W. (2015) Activation of Rab8 guanine nucleotide exchange factor Rabin8 by ERK1/2 in response to EGF signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 148–153 10.1073/pnas.1412089112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ultanir S. K., Hertz N. T., Li G., Ge W. P., Burlingame A. L., Pleasure S. J., Shokat K. M., Jan L. Y., and Jan Y. N. (2012) Chemical genetic identification of NDR1/2 kinase substrates AAK1 and Rabin8 Uncovers their roles in dendrite arborization and spine development. Neuron 73, 1127–1142 10.1016/j.neuron.2012.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bouquier N., Vignal E., Charrasse S., Weill M., Schmidt S., Léonetti J.-P., Blangy A., and Fort P. (2009) A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem. Biol. 16, 657–666 10.1016/j.chembiol.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 54. Li Z., Aizenman C. D., and Cline H. T. (2002) Regulation of Rho GTPases by crosstalk and neuronal activity in vivo. Neuron 33, 741–750 10.1016/S0896-6273(02)00621-9 [DOI] [PubMed] [Google Scholar]
- 55. Lagenaur C., and Lemmon V. (1987) An L1-like molecule, the 8D9 antigen, is a potent substrate for neurite extension. Proc. Natl. Acad. Sci. U.S.A. 84, 7753–7757 10.1073/pnas.84.21.7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 57. Zeitvogel F., and Obst M. (2016) ScatterJn: an ImageJ plugin for scatterplot-matrix analysis and classification of spatially resolved analytical microscopy data. J. Open Res. Software 4, e5 10.5334/jors.89 [DOI] [Google Scholar]
- 58. Zheng C. Y., Petralia R. S., Wang Y. X., and Kachar B. (2011) Fluorescence recovery after photobleaching (FRAP) of fluorescence tagged proteins in dendritic spines of cultured hippocampal neurons. J. Vis. Exp. e2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pellegrin S., and Mellor H. (2008) Rho GTPase Activation Assays, John Wiley & Sons, Inc. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.